


Abstract
We describe a new cell-free protein synthesis (CFPS) method for site-specific incorporation of non-natural amino acids (nnAAs) into proteins in which the orthogonal tRNA (o-tRNA) and the modified protein (i.e. the protein containing the nnAA) are produced simultaneously. Using this method, 0.9–1.7 mg/ml of modified soluble super-folder green fluorescent protein (sfGFP) containing either p-azido-l-phenylalanine (pAzF) or p-propargyloxy-l-phenylalanine (pPaF) accumulated in the CFPS solutions; these yields correspond to 50–88% suppression efficiency. The o-tRNA can be transcribed either from a linearized plasmid or from a crude PCR product. Comparison of two different o-tRNAs suggests that the new platform is not limited by Ef-Tu recognition of the acylated o-tRNA at sufficiently high o-tRNA template concentrations. Analysis of nnAA incorporation across 12 different sites in sfGFP suggests that modified protein yields and suppression efficiencies (i.e. the position effect) do not correlate with any of the reported trends. Sites that were ineffectively suppressed with the original o-tRNA were better suppressed with an optimized o-tRNA (o-tRNAopt) that was evolved to be better recognized by Ef-Tu. This new platform can also be used to screen scissile ribozymes for improved catalysis.
INTRODUCTION
Despite the vast functional and structural variety in proteins, the 20 natural amino acids that comprise these proteins are limited in their chemical diversity. Most of the natural amino acids possess inert site chains, and the ones with reactive side chains typically appear multiple times in a given protein. As a result, precise post-translational modification of proteins is usually impractical with the 20 natural amino acids.
This problem is solved by the site-specific incorporation of non-natural amino acids (nnAAs), which contain a diverse array of reactive side chains. Initially, these nnAAs were chemically coupled to the amber suppressor tRNA (i.e. a tRNA that recognizes the UAG amber stop codon), and >70 nnAAs were site-specifically introduced into proteins using this method (1). Partly due to the fact that stoichiometric amounts of acylated tRNA were needed, the modified protein yields were low (∼ 50 μg/ml) (2). Moreover, the nnAAs could only be incorporated in vitro, as acylated tRNAs could not be introduced into cells. Biological acylation, in contrast, mimics the incorporation of natural amino acids, and involves dedicated orthogonal tRNAs (o-tRNAs) and synthetases that are specific to each nnAA and act catalytically. The nnAA is covalently coupled to the 3′ terminus of the o-tRNA by the synthetase specific to the nnAA (nnAARS). The acylated o-tRNA then forms a ternary complex with GTP and the Escherichia coli elongation factor Ef-Tu, enters the aminoacyl (A) site on the ribosome, outcompetes the release factor 1 (RF1), which would otherwise terminate translation, and adds the nnAA to the nascent polypeptide chain at the amber stop codon.
The first orthogonal components for site-specific incorporation of nnAAs in E. coli were derived from the tyrosyl tRNA (MjtRNATyr) and the tyrosyl-tRNA synthetase (MjTyrRS) of the archaeon Methanocaldococcus jannaschii. There was already some orthogonality between the tyrosyl pairs from M. jannaschii and from E. coli due to the difference in the modes of tRNATyr recognition between the M. jannaschii TyrRS and the E. coli TyrRS (3,4). First, the anticodon of the MjtRNATyr was changed from GUA to CUA to suppress the amber (UAG) stop codon. Then, the amber suppressor MjtRNATyr was mutated to be fully orthogonal to the endogenous aminoacyl-tRNA synthetases while retaining the ability to be aminoacylated by the MjTyrRS (5). Finally, the amino acid-binding site of the MjTyrRS was mutated to recognize a nnAA so that the resultant enzyme would acylate the evolved o-tRNA exclusively with the particular nnAA. The absence of cross-reactivity between the orthogonal components and the endogenous ones was crucial for maintaining translational fidelity. The Schultz laboratory used this methodology to site-specifically incorporate >30 nnAAs (6) encompassing a vast range of chemical diversity. Advancements in o-tRNA design (7) and the expression of orthogonal components (8,9) led to accumulation of >100 mg/l modified protein in vivo. These phenylalanine / tyrosine analogues contain, among others, acetyl (10,11), halide (12,13), benzoyl (14), methyl (15), amino (16), azido (17) and alkyne (18) moieties. The latter two, namely p-azido-l-phenylalanine (1) and p-propargyloxy-l-phenylalanine (2, Figure 1), are particularly useful for bioconjugation, as proteins containing these nnAAs can be directly coupled using the copper (I)-catalysed azide–alkyne cycloaddition reaction. More recently, orthogonal pairs derived from the pyrrolysyl tRNA-synthetase pair of the archaea Methanosarcina barkeri and Methanosarcina mazei were used to incorporate a wide variety of pyrrolysine and phenylalanine analogues during in vivo expression in E. coli; some of these analogues also contain azide or alkyne moieties (19–24). In one study, a single pyrrolysyl tRNA-synthetase mutant was used to incorporate as many as 12 different phenylalanine analogues (25).
Figure 1.
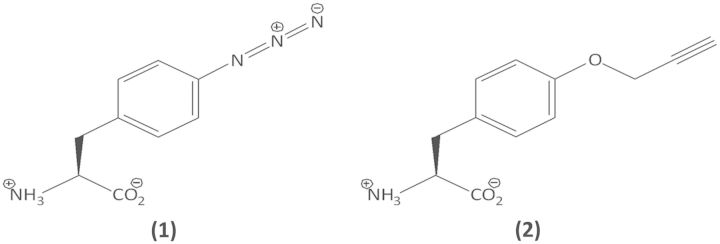
The nnAAs used in this study. (1) p-azido-l-phenylalanine (pAzF), and (2) p-propargyloxy-l-phenylalanine.
Cell-free protein synthesis (CFPS) with bacterial extracts has been successfully used in this laboratory to produce a large variety of active proteins. These proteins include, but are not limited to, metalloproteins such as ferredoxin (26) and [FeFe] hydrogenase (27), virus-like particles (28), membrane proteins (29,30), transcription factors (31) and proteins containing nnAAs (32–35). Recently, the cell-free accumulation of 700 mg/l of human granulocyte-macrophage colony-stimulating factor (hGM-CSF) was demonstrated at 100-litre scale (36). Amino acid stabilization (37,38), a natural chemical environment (39) and the activation of central metabolism (40) were critical in enabling the inexpensive and efficient cell-free synthesis of these proteins.
The open nature and the direct accessibility of the cell-free platform were particularly important for the synthesis of proteins containing nnAAs. Initial experiments were focused on extract strain development and the optimization of orthogonal component supply. The purified orthogonal synthetase and the nnAA, along with standard reagents, were added to an orthogonal extract in which the o-tRNA was constitutively expressed. Using this platform, proteins containing pAzF or pPaF were synthesized at high concentrations (150–930 μg/ml) and suppression efficiencies (25–96%; suppression efficiency is defined as the ratio of the modified protein yield to the natural protein yield) (32,33). Unfortunately, however, the yields and suppression efficiencies varied significantly from protein to protein, extract to extract and position to position (that is, position in the protein at which the nnAA is incorporated). The dependence of modified protein yields on the site of nnAA incorporation is called the position effect; it is also referred to as the codon or mRNA context effect in the literature (41–44). We have recently measured the o-tRNA concentrations, demonstrated their limitation in this platform and developed a new cell-free method in which the o-tRNA is synthesized in vitro, purified and added to the more productive standard extract along with the purified synthetase, nnAA and the standard reagents (45). Despite the improvements in modified protein titers and suppression efficiencies, we needed a more practical and inexpensive method for supplying the o-tRNA, as in vitro transcription and purification were both laborious and expensive.
In this article, we describe a modular, scalable and efficient cell-free platform in which the o-tRNA is synthesized simultaneously with the modified protein (Figure 2). In this new cell-free system, T7 RNA polymerase (RNAP) catalyses the transcription of both the mRNA encoding the modified protein and a ‘transzyme’, which is a fusion of the hammerhead ribozyme (46) and the o-tRNA. The transzyme folds correctly and cleaves itself to liberate the active o-tRNA. The o-tRNA is then acylated with the nnAA by the specific synthetase, and the nnAA is added to the nascent polypeptide chain at the amber stop codon. Using this method, 0.9 to 1.7 mg/ml of super-folder green fluorescent protein (sfGFP) containing either pAzF or pPaF accumulated after 16 h; these yields correspond to 50–88% suppression efficiency. The modified sfGFP yields and suppression efficiencies were measured across 12 different nnAA incorporation sites and using two different o-tRNA sequences. We observed that the position effect could not be explained by any of the reported hypotheses. The variation in modified protein yields and suppression efficiencies across the 12 incorporation sites was reduced when the o-tRNAopt [which was optimized for better recognition by Ef-Tu (9)] was produced in situ, instead of the original o-tRNA. Experiments with Schistosoma ribozymes showed that this platform can also be used for evolving more active scissile ribozymes, as modified sfGFP fluorescence is directly related to ribozyme catalysis. Finally, the o-tRNA can be produced in situ from either a linearized plasmid or a crude PCR product, thereby making the system more practical for large-scale applications. The new cell-free platform can be used to effectively incorporate any of the >30 nnAAs (even poorly soluble ones such as pPaF) site-specifically into any protein.
Figure 2.
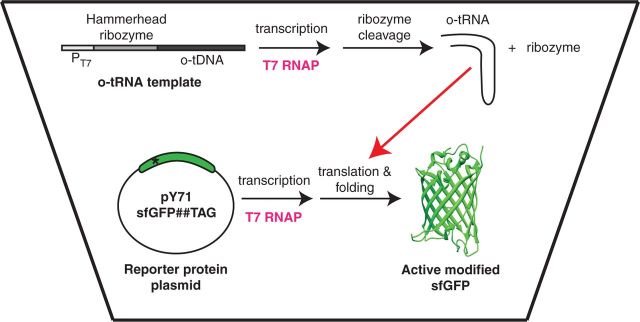
Schematic of the new cell-free platform for site-specific incorporation of nnAAs. The T7 RNA polymerase catalyses the transcription of both the transzyme RNA, which comprises the fusion of the hammerhead ribozyme and the o-tRNA, and the messenger RNA encoding the protein to be modified with the nnAA. Following transcription, the transzyme cleaves itself to release the biologically active o-tRNA needed for nnAA incorporation at the amber stop codon (TAG; depicted by an asterisk). The three-dimensional structure of sfGFP was used to render the modified protein (PDB accession number: 2B3P).
MATERIALS AND METHODS
Plasmid construction
The ‘transzyme’ template (46) consists of the T7 promoter, the hammerhead ribozyme sequence and the o-tRNA sequence. Two different sequences, which contain either the original J17 o-tRNA template (5) or the optimized o-tRNA (o-tRNAopt) template (9), were constructed by PCR using overlapping oligonucleotides. The transzyme sequence encoding the original o-tRNA is: 5′-GCTTTTAGATCTTAATACGACTCACTATAGGGAGACCGGCTGATGAGTCCGTGAGGACGAAACGGTACCCGGTACCGTCCCGGCGGTAGTTCAGCAGGGCAGAACGGCGGACTCTAAATCCGCATGGCGCTGGTTCAAATCCGGCCCGCCGGACCAGGAAGCTTACATCCGTCGACAAAAGC-3′.
The transzyme sequence encoding the o-tRNAopt (‘T-Nap1’ from reference 7) is: 5′-GCTTTTAGATCTTAATACGACTCACTATAGGGAGACCGGCTGATGAGTCCGTGAGGACGAAACGGTACCCGGTACCGTCCCGGCGGTAGTTCAGCAGGGCAGAACGGCGGACTCTAAATCCGCATGGCAGGGGTTCAAATCCCCTCCGCCGGACCAGGAAGCTTACATCCGTCGACAAAAGC-3′. The o-tRNA sequences are given in bold. The transzyme sequence encoding the original o-tRNA was ligated into the pY71 vector (32) at the BglII and SalI restriction sites to create the pY71 HHotDNA. The pY71HHotDNAopt was created by converting the o-tRNA template to the optimized o-tRNA template (o-tDNAopt) using the QuikChange® protocol (Agilent Technologies, Santa Clara, CA).
The Schistosoma transzyme sequence encoding the Schistosoma ribozyme (47) 5′ to the o-tRNA is: 5′-GCTTTTAGATCTTAATACGACTCACTATAGGGAGATGAAGGCACGCCGGCTGATGAGTCCCAAATAGGACGAAATGCCAGAGAGGCATCCCGGCGGTAGTTCAGCAGGGCAGAACGGCGGACTCTAAATCCGCATGGCGCTGGTTCAAATCCGGCCCGCCGGACCAGGGTCGACAAAAGC-3′. The AGAGA loop (underlined) was added to covalently link the Stem III to the gene encoding the cleaved RNA. Three different variants of the Schistosoma transzyme were created by mutating the Stem I ‘bulge’ nucleotides (written in bold). The first variant is given above; the second and third variants contain the AAAA and GTAC sequences, respectively. The secondary structure of the Schistosoma ribozymes was analysed using the Mfold web server (48). All three Schistosoma transzyme variants were assembled by PCR using overlapping oligonucleotides, and ligated into the pY71 at the BglII and SalI restriction sites to create the pY71fullHHotDNA plasmids.
Construction of the plasmid pY71sfGFP216TAG was described previously (32). This plasmid encodes a sfGFP with an amber stop codon at position 216 and a C-terminal Strep-tag. The additional glycine codon at position 215 and the amber stop codon were deleted to obtain pY71sfGFP, which contains the original sfGFP sequence (49,50). Twelve amber stop codons were then individually introduced into the sfGFP sequence using the QuikChange® protocol (Agilent Technologies) to create 12 new coding sequences. A stop codon was introduced at the following positions to replace the codon naturally present at that position (the amino acid normally encoded is indicated): Asn23, Ile39, Lys52, Tyr66, Tyr74, Tyr92, Tyr106, Tyr143, Tyr151, Tyr182, Tyr200 and Tyr223. The DNA polymerase from Pyrococcus furiosus (51) used in the PCRs was produced and purified in our laboratory as described (52).
After the verification of the plasmid sequences by DNA sequencing, E. coli cultures harbouring the plasmids were grown for 16 to 18 h in Terrific Broth (Invitrogen, Carlsbad, CA) and purified using the Qiagen tip-500 columns (Qiagen, Valencia, CA) as per manufacturer instructions. DNA concentrations were calculated from absorbance measurements at 260 nm (1 A260 = 50 μg/ml double-stranded DNA); the absorbance measurements were taken using the HP 8452A diode array spectrophotometer (Agilent Technologies).
Preparation of orthogonal tRNA templates and the synthetases specific to nnAAs
The purified plasmids encoding the o-tRNA and o-tRNAopt were restricted with the BstNI enzyme at 60°C for 2 to 3 h. The plasmid concentration and reaction volume did not exceed 320 μg/ml and 1 ml, respectively; digestion reactions at higher plasmid concentrations or larger volumes were incomplete. After every reaction, 5 μl samples were analysed by agarose gel electrophoresis to ensure complete linearization. The linearized plasmids were subsequently ethanol-precipitated, washed once with 70% v/v ethanol, resuspended in 10 mM Bis-Tris, pH 7.5 buffer overnight at 4°C, and stored at −20°C. Concentrations of the linearized plasmids were calculated from absorbance measurements at 260 nm as described previously.
A linear transzyme template encoding the T7 promoter, the hammerhead ribozyme and the o-tRNAopt was also synthesized by PCR using overlapping oligonucleotides. The sequence for this template is: 5′-GCTTTTAGATCTTAATACGACTCACTATAGGGAGACCGGCTGATGAGTCCGTGAGGACGAAACGGTACCCGGTACCGTCCCGGCGGTAGTTCAGCAGGGCAGAACGGCGGACTCTAAATCCGCATGGCAGGGGTTCAAATCCCCTCCGCCGGACCA-3′. Differently from the previous transzyme template sequences, this sequence terminates with the CCA nucleotides that comprise the 3′ terminus of the o-tRNAopt. This linear DNA was subsequently ethanol-precipitated without purification, washed with 70% ethanol, resuspended in 10 mM Bis-Tris buffer, pH 7.5 overnight, and stored at −20°C. The concentrated PCR product was quantified as explained above.
The synthetases specific to the nnAAs pPaF and pAzF, pPaFRS and pAzFRS, were produced and purified as described (32,33).
Cell extract preparation
The orthogonal S30 cell extract was prepared from KC6 cells (37) harbouring the pDule-tRNA plasmid, as described (33). The lysate was not diluted after homogenization. The specific growth rate of this KC6/o-tRNA culture was 0.57 h−1 during the batch fermentation. The standard KC6 extract was prepared as described (53). After homogenization, 0.2 ml S30 buffer (14 mM magnesium acetate, 60 mM potassium acetate, 10 mM Tris acetate, pH 8.2) was added per ml of lysate, and the diluted lysate was dialysed against 100 volumes of S30 buffer at 4°C four times for 30 min each. Finally, the S30 cell extract was centrifuged at 10 000g and 4°C for 20 min, and the supernatant was aliquoted, flash-frozen and stored at −80°C. The specific growth rate of the KC6 culture was 0.96 h−1 during the fed-batch fermentation.
Cell-free protein synthesis
The PANOx SP (PEP, Amino Acids, NAD, Oxalic Acid, Spermidine and Putrescine) (39) system was used in the cell-free reactions, with several changes in the reagents and reagent concentrations. All of the chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise stated.
Twenty-five-microlitre PANOx SP reaction solutions contained, unless otherwise noted: 10 mM ammonium glutamate, 20 mM magnesium glutamate, 175 mM potassium glutamate, 1.2 mM ATP, 0.86 mM each of CMP, GMP and UMP, 10 mM dibasic potassium phosphate, 34 μg/ml folinic acid, 171 μg/ml E. coli tRNAs (Roche Applied Science, Penzberg, Germany), 33 mM phosphoenolpyruvate (PEP) (Roche Applied Science), 1.5 mM spermidine, 1 mM putrescine, 0.33 mM nicotinamide adenine dinucleotide (NAD), 0.27 mM coenzyme A, 2.7 mM sodium oxalate, 2 mM each of the 20 amino acids, 5 μM l-[14C(U)]-leucine (PerkinElmer, Waltham, MA), 2 mM p-azido-l-phenylalanine (pAzF, Chem-Impex International, Wood Dale, IL) or 4 mM p-propargyloxy-l-phenylalanine (pPaF, synthesized according to reference 54), 0.3 mg/ml pAzFRS or 0.5 mg/ml pPaFRS (prepared as described above), 100 μg/ml T7 RNA polymerase (prepared according to reference 55), 6 nM sfGFP plasmid, 0.2 mg/ml linearized o-tRNA or o-tRNAopt plasmid template and either 0.24 volume of standard KC6 extract or 0.28 volume of orthogonal extract. The glutamate salts were diluted from a 10-fold concentrated (10×) solution. Similarly, a 10× NTMP Master Mix solution contained ATP, the three NMPs, folinic acid and the E. coli tRNAs, and was adjusted to pH 7.3 with dibasic potassium phosphate. The natural amino acids were diluted from a stock solution, which contained a 50 mM concentration of each amino acid. This stock solution was prepared by adding the amino acids in the following order (given in their one-letter code): R, V, W, F, I, L, C, M, A, N, D, E, G, Q, H, K, P, S, T, Y. During the preparation, it was ensured that each amino acid was dissolved before addition of the next, except tyrosine, which is added last and remains suspended in the solution. The reactions were incubated at 30°C for 16 h, unless stated otherwise. CFPS reactions containing pAzF were prepared in a dark room, and the tubes containing the reaction solutions were wrapped in aluminum foil to prevent photodissociation of the aromatic azide (56).
Quantification of natural and modified sfGFPs
Radiolabelled proteins were quantified as follows: The cell-free reactions were stopped by cooling the reaction tubes at −20°C for 4 min. A 4 μl sample from each reaction solution was spotted onto two of three Whatman MM filter papers (Whatman, Springfield Mill, United Kingdom); one of these papers was used to measure the total protein concentration and the other to measure total system radioactivity. The remaining cell-free solution was centrifuged at 20 800g and 4°C for 15 min. Four microlitres of the supernatant was spotted on the third Whatman MM filter paper to measure the soluble protein concentration, and the papers were dried overnight or for 1 h under an incandescent light bulb. Trichloroacetic acid precipitation was used to measure the total and soluble protein concentrations as described (57). The leucine content and the molecular weight of the protein were used to convert radioactivity measurements to protein concentrations. A set of ‘blank’ reactions (which do not contain the protein template) was included in each experiment; the same protocol was applied to these reactions to determine background radioactivity. These measurements were subtracted from the protein radioactivity measurements to obtain accurate protein concentrations. Full-length protein amounts in the soluble fractions were determined by densitometry analysis of the bands on SDS-PAGE gel autoradiograms.
Cell-free samples containing radiolabelled sfGFP were diluted in a sample loading buffer that contains lithium dodecyl sulfate (LDS) (Invitrogen) at pH 8.4 and 50 mM DTT. The solution was subsequently incubated at 95°C for 15 min to ensure complete denaturation of the sfGFP. The samples were loaded onto 10% Bis-Tris SDS-PAGE gels, and run using the MES SDS running buffer (Invitrogen). The gels were subsequently dried and exposed overnight to a storage phosphor screen (Molecular Dynamics, Sunnyvale, CA). Autoradiograms were obtained by imaging the screens on the Typhoon system (GE Healthcare, Uppsala, Sweden), and the band intensities were quantified using the ImageJ software (National Institutes of Health, Bethesda, MD).
The suppression efficiency is defined as the ratio of the yield of full-length modified protein to that of its natural counterpart. In each CFPS experiment, natural sfGFP was synthesized under the same cell-free reaction conditions; the full-length and soluble protein yields were measured via scintillation counting and densitometry.
Characterization of modified sfGFPs
To determine the specific activity of the modified sfGFPs in comparison with that of natural sfGFP, sfGFP23pPaF, sfGFP39pPaF and wild-type sfGFP were produced in CFPS with a C-terminal Strep-tag and purified via Strep-Tactin affinity chromatography (IBA GmbH, Goettingen, Germany) as per manufacturer instructions. The proteins were produced in 0.5 ml CFPS reactions containing all of the mentioned reagents except l-[14C(U)]-leucine. Fractions obtained from Strep-Tactin affinity chromatography were analysed by SDS-PAGE, and those containing sfGFP were pooled and dialysed at 4°C first against 30 volumes of 10 mM potassium phosphate buffer, pH 7.5, then twice against 30 volumes of 20% w/v sucrose dissolved in the same buffer. Proteins were concentrated during the latter dialyses against sucrose-containing phosphate buffer.
Total protein concentrations were determined using the DC protein assay (Bio-Rad, Hercules, CA) with bovine serum albumin (BSA) as the standard for the assay. Full-length and soluble sfGFP concentrations were measured via densitometry analysis on the SDS-PAGE gels of purified proteins. Two-microlitre samples from each protein solution were diluted in 10 mM potassium phosphate buffer at pH 7.4; the fluorescence from these diluted protein solutions was measured between 523 and 548 nm using a Mithras LB940 multimode microplate reader (Berthold Technologies, Bad Wildbad, Germany) with F485 excitation and F535 emission filters. Specific activities were calculated by dividing fluorescence measurements by protein concentrations, measured at different dilutions of the purified proteins.
For mass spectrometry analysis, three additional modified proteins (sfGFP39pPaF, sfGFP143pPaF and sfGFP223pPaF) were produced and purified as explained above. Liquid chromatography/mass spectroscopy (LC/MS) analysis of these proteins was performed at the Stanford University Mass Spectrometry Laboratory. The proteins were precipitated with acetone, reconstituted in a buffer containing 8 M urea and 50 mM ammonium bicarbonate, reduced, alkylated and digested with trypsin overnight at a 1:100 protease-to-protein ratio. The resulting protein fragments were analysed by nano reversed-phase HPLC using an Eksigent 2D NanoLC (Eksigent, Dublin, CA). A fused silica column containing Duragel C18 (Peeke, Redwood City, CA) matrix was used. The initial buffer composition was 98% A (0.1% formic acid in water) / 2% B (0.1% formic acid in acetonitrile), and was increased to 40% B at a flow rate of 600 nl/min. The nanoHPLC was interfaced with a Bruker–Michrom Advance CaptiveSpray (Michrom Bioresources, Auburn, CA), spray source for nanoESI into the mass spectrometer. The LTQ Orbitrap Velos (Thermo Fisher, Waltham, MA) mass spectrometer was set in data-dependent acquisition mode to perform MS/MS on the top 12 most intense multiply charged cations. The .RAW data were searched using Sequest on a Sorcerer platform against the Uniprot database. The data were visualized and validated using Scaffold software (Proteome Software, Portland, OR). Quantification and MS/MS analysis of the identified peptides were conducted using XCalibur (Thermo Fisher) and GPMAW (Lighthouse Data, Odense, Denmark) software.
Measurement of o-tRNA concentrations produced in CFPS
These 25 μl CFPS reactions included 8 μM [3H]-UTP (PerkinElmer) instead of l-[14C(U)]-leucine. A new 10× Master Mix solution was made, in which the UMP was replaced with UTP so that the initial ratio of radioactive to non-radioactive UTP concentrations was known. The sfGFP yields from cell-free reactions containing each of the two Master Mix solutions were comparable. The stock solution of [3H]-UTP was concentrated by evaporation under vacuum at room temperature. The concentrations of tritium-labelled ribonucleic acids were measured using the same protocol as that for radiolabelled proteins. Concentrations of intact o-tRNA and o-tRNAopt were determined by densitometry analysis of the soluble bands on the denaturing polyacrylamide gel autoradiogram.
Six-microlitre samples of cell-free solutions containing tritium-labelled RNA were first diluted in 2× TBE-Urea sample buffer (Invitrogen), then denatured at 95°C for 5 min prior to loading onto a 15% denaturing polyacrylamide gel (TBE-Urea gel, Invitrogen). The gel was run at 180 V for 80 min in 1× TBE buffer, pH 8.3 (Invitrogen), washed with diethyl pyrocarbonate (DEPC)-treated water, dried, and exposed for 4 days to a tritium storage phosphor screen (GE Healthcare). The tritium autoradiogram was generated using the Typhoon system, and the band intensities were quantified using the ImageJ software. For size comparison, the o-tRNA was also transcribed in vitro and purified by denaturing polyacrylamide gel electrophoresis as explained in reference 45. Two-microlitre samples of the purified uncleaved transzyme, o-tRNA and hammerhead ribozyme solutions were run on a 15% denaturing polyacrylamide gel as explained above.
RESULTS
In situ orthogonal tRNA production increases modified protein yields
In the standard cell-free platform for nnAA incorporation, the o-tRNA is produced in the extract source strain, and the nnAA and the purified nnAARS are added as separate reagents. We have previously shown that the o-tRNA is a limiting reagent in this cell-free platform; i.e. that the supplementation with additional o-tRNA increases modified protein yields (45). Since in vitro production and purification of the o-tRNA are both laborious and expensive, we sought a more practical method for supplying the cell-free reaction with adequate o-tRNA.
The hammerhead ribozyme was used to produce the o-tRNA in vitro, because the T7 RNA polymerase does not efficiently transcribe RNAs that start with pyrimidines such as the o-tRNA that requires a cytosine at its 5′ terminus (58). Effective synthesis of transcripts starting with pyrimidines was demonstrated using the ‘transzyme’ approach where the hammerhead ribozyme sequence is inserted between the T7 promoter and the gene encoding the desired transfer RNA (46). The plasmid containing these sequences is restricted using the BstNI enzyme that yields the correct CCA 3′ terminus required for tRNA activity. From this linearized template, the T7 RNA polymerase transcribes the ribozyme–tRNA fusion RNA which subsequently cleaves itself to release the tRNA. Placing the 5′ terminus of the tRNA sequence immediately after the ribozyme cleavage site ensures that the released tRNA has the correct 5′ and 3′ termini required for biological activity (46).
To develop a more practical and inexpensive method of o-tRNA supply, we investigated the in situ production of o-tRNA during the cell-free reaction, where both the o-tRNA and the modified protein are produced simultaneously (Figure 2). The linear template encoding the ribozyme–tRNA (‘transzyme’) fusion and the protein template are added to the cell-free reaction solution along with the nnAA, the nnAARS and the standard reagents for CFPS. During the incubation, T7 RNA polymerase catalyses the transcription of both the mRNA for protein translation and the transzyme (i.e. ribozyme–tRNA fusion). Once the transzyme is transcribed, the o-tRNA is liberated by autocleavage of the hammerhead ribozyme and acylated with the nnAA by the orthogonal synthetase. The acylated o-tRNA then enters the aminoacyl (A) site in the ribosome, suppresses the amber stop codon, and adds the nnAA to the nascent polypeptide chain.
To explore the feasibility of this approach and to identify the non-limiting concentrations of the o-tRNA template, we added different concentrations of the linearized o-tRNA template to the cell-free solution and measured the accumulation of full-length and soluble sfGFP with the nnAA pPaF incorporated at position 39 (sfGFP39pPaF). This new method greatly increased the modified protein yields from 260 μg/ml to ∼1.2 mg/ml of full-length and soluble sfGFP39pPaF. The full-length protein yield was directly proportional to the o-tRNA template concentration up to 150 μg/ml; addition of o-tRNA template in excess of 150 μg/ml did not increase the modified protein yields. The suppression efficiency, which is defined as the ratio of the full-length and soluble modified protein yield to that of the wild-type protein, was increased from 15–70% (Figure 3A, diamonds).
Figure 3.

(A) Titration of linearized o-tRNA and o-tRNAopt plasmid templates in the CFPS reactions containing pY71sfGFP39TAG. Error bars indicate ±1 standard deviation from six reactions. (*P < 0.01, **P = 0.025, ***P = 0.023) (B) Production of sfGFP containing pPaF at position 23 (sfGFP23pPaF) over 24 h. The ‘o-extract’ curve was generated from CFPS reactions in which the cell extract contained the o-tRNA. The ‘transzyme’ values were obtained from reactions, in which the o-tRNA and sfGFP23pPaF were produced simultaneously. A different set of reactions was incubated for each of the time points. Error bars indicate ±1 standard deviation for three reactions.
Next, we wanted to see whether the increase in protein accumulation in the new cell-free platform was due to faster or more prolonged production of the modified protein. Therefore, we measured the accumulation of sfGFP23pPaF (i.e. full-length and modified sfGFP containing pPaF at position 23) over the course of 24 h in two different sets of reactions. In one set, the cell-free reaction solution contained 28% v/v of the orthogonal extract, in which the o-tRNA had been expressed constitutively during the growth of the source strain (Figure 3B, ‘o-extract’). In the other set, the cell-free reactions contained, in addition to the standard reagents, 200 μg/ml linearized o-tRNA template to ensure that the o-tRNA template was not limiting (Figure 3B, ‘transzyme’). A comparison of these two platforms showed that the modified protein was produced faster when it was synthesized simultaneously with the o-tRNA. In the standard CFPS reactions with the orthogonal extract, about half of the protein was produced within the first 2 h, and another 100 μg/ml modified protein accumulated after 14 h. At the end of 16 h, about 250 μg/ml full-length and soluble sfGFP23pPaF accumulated in the CFPS solution, which amounted to a suppression efficiency of ∼15% (Figure 3B, ‘o-extract’). In the ‘transzyme’ platform, there was a burst of protein production within the first hour, fueled by the conversion of phosphoenolpyruvate (PEP) to ATP. This initial period was followed by slower protein production, which was probably fueled by oxidative phosphorylation (40). At the end of 16 h, about 1.2 mg/ml of modified sfGFP had accumulated, corresponding to a suppression efficiency of ∼75% (Figure 3B, ‘transzyme’).
Modified protein production and Ef-Tu recognition of the acylated orthogonal tRNA
The Schultz laboratory recently reported a modified o-tRNA optimized for better recognition by the endogenous elongation factor Ef-Tu (9). This new o-tRNA sequence, dubbed the o-tRNAopt sequence, was among the nine identified from their first tRNA library and enabled more effective site-specific incorporation of several nnAAs including pAzF (7). This step in the nnAA insertion pathway, namely the recognition of the acylated o-tRNA by the E. coli Ef-Tu, was a potential bottleneck that we had not addressed. We thus cloned this o-tRNAopt template 3′ to the hammerhead ribozyme sequence, and compared the performance of the o-tRNAopt with that of the original o-tRNA in our new cell-free platform by titrating each of the two linearized transzyme templates (Figure 3A). At limiting template concentrations (i.e. below 150 μg/ml), Ef-Tu recognition was indeed a limiting factor: cell-free reactions containing the o-tRNAopt template were more productive than the ones containing equal concentrations of the o-tRNA template. However, at template concentrations ≥150 μg/ml, there was no difference in productivity between the two different o-tRNAs; 1.1 to 1.2 mg/ml soluble and full-length sfGFP39pPaF accumulated in both sets of CFPS reactions.
Quantification of in situ orthogonal tRNA synthesis
We then quantified the o-tRNA (or o-tRNAopt) synthesized during cell-free reactions incubated with 20, 40, 60, 80, 100, 150 or 200 μg/ml of the linearized o-tRNA template. Tritiated UTP (3H-UTP) was added to the CFPS reaction solution to label the ribonucleic acids produced during the reaction, and the amount of synthesized o-tRNA was determined by scintillation counting and densitometry, similar to the quantification of modified protein labelled with 14C-leucine (Supplementary Figure S1). UMP was replaced with UTP in these cell-free reactions to increase the accuracy of the synthesized RNA quantification.
As expected, the amount of accumulated o-tRNA and o-tRNAopt was directly proportional to the template concentrations (Table 1). It was surprising to see that the only bands on the tritium autoradiogram were due to the mRNA and the o-tRNA; the uncleaved transzyme and the hammerhead ribozyme are apparently degraded by the ribonucleases in the CFPS solution. We suspect that the hammerhead ribozyme may not cleave itself efficiently in the CFPS reaction due to the difference in optimal temperature and Mg2+ concentrations between those for CFPS (20 mM Mg2+, 30°C) and for ribozyme cleavage (30 mM Mg2+, 60°C) (46). The need for high levels of accumulated o-tRNA and o-tRNAopt is consistent with results from our previous study indicating low turnover for the nnAARS (45).
Table 1.
Accumulation of o-tRNA and o-tRNAopt at different tRNA template concentrations
| Original or optimized o-tRNA | [Template] | [Accumulated o-tRNA] |
|---|---|---|
| Original | 150 µg/ml | 280 ± 40 µg/ml |
| (127 nM) | (12.6 ± 2.0 µM) | |
| Original | 200 µg/ml | 380 ± 20 µg/ml |
| (169 nM) | (17.0 ± 1.0 µM) | |
| Optimized | 150 µg/ml | 170 ± 60 µg/ml |
| (127 nM) | (7.4 ± 2.7 µM) | |
| Optimized | 200 µg/ml | 200 ± 60 µg/ml |
| (169 nM) | (8.9 ± 2.7 µM) |
Error bars indicate ±1 standard deviation for three reactions. The o-tRNA concentrations from the reactions containing less o-tRNA (or o-tRNAopt) template could not be accurately quantified via densitometry because of weak tritium signals from the autoradiograms.
Non-natural amino acid incorporation and the position effect
The position effect (also called the codon context effect in the literature) refers to the dependence of nonsense suppression efficiency (nnAA incorporation, in this case) on the position of the nonsense codon (i.e. amber stop codon, UAG). The nucleotide following the amber stop codon on the mRNA has been suggested to be most influential on the suppression efficiency, with the stop codons followed by a purine being more efficiently suppressed than those followed by a pyrimidine (43,44,59,60). Previous research from our lab also suggested a trend between modified dihydrofolate reductase (DHFR) yields (which is directly related to nnAA incorporation at the UAG codon) and the proximity of the insertion site to the N-terminus of the DHFR protein (33). To further explore these influences, we examined pPaF incorporation in sfGFP at 12 different sites; the locations of these sites in the folded protein are shown in Figure 4A. We replaced, one at a time, all of the tyrosines, which is chemically the most similar natural amino acid to the nnAAs, and several other amino acids on the surface of the protein to minimize chemical or structural perturbations of the natural sfGFP fold. We then quantified the full-length and soluble modified sfGFP that accumulated in five different sets of cell-free reactions (Figure 4B). The first two sets contained all of the reagents except the o-tRNA (‘no o-tRNA’; Figure 4B) or the nnAA (‘no pPaF’; Figure 4B), respectively, and were conducted to assess non-specific suppression of the amber stop codon at different sites; in this case, the full-length protein contained a natural amino acid at the indicated position. The third set (‘o-extract’; Figure 4B) was incubated using the standard platform and contained the orthogonal extract. The fourth (‘transzyme’; Figure 4B) and fifth sets (‘optimized’; Figure 4B) contained 200 μg/ml of the linearized plasmid templates coding for the o-tRNA and the o-tRNAopt, respectively. Natural sfGFP was produced in a different set of reactions, and the modified proteins yields were divided by the natural protein yield to calculate the suppression efficiencies. Finally, the nucleotide following the amber stop codon at each position is indicated below the graph in Figure 4B.
Figure 4.
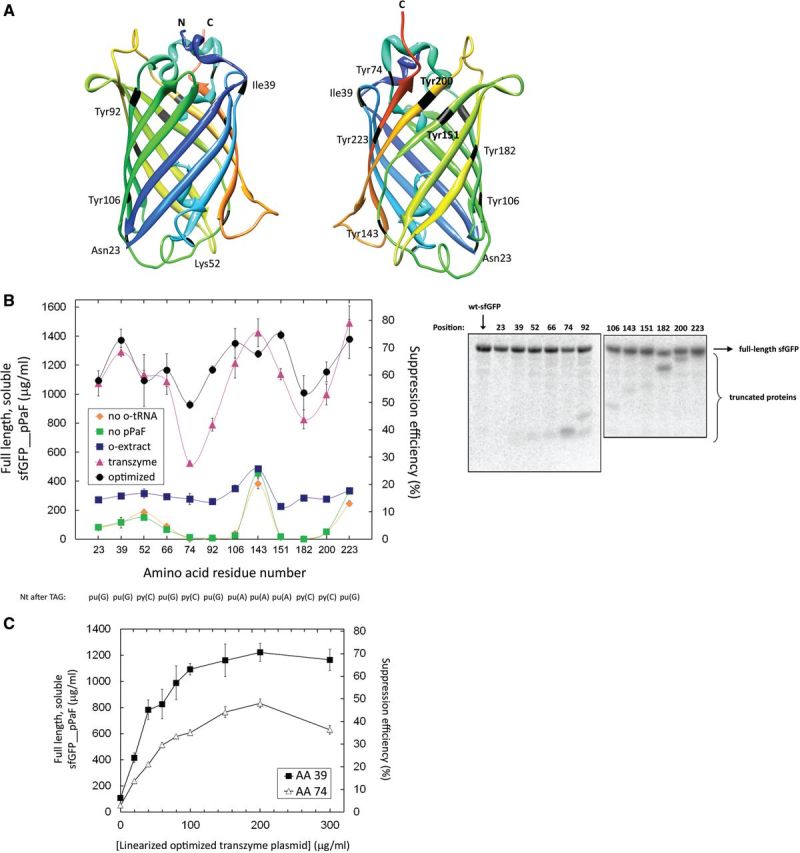
Codon context and nnAA incorporation. (A) sfGFP crystal structure; nnAA incorporation sites are labelled in black. The colour scheme shows the position along the primary sequence, with blue and green indicating the N-terminal regions, and orange and red depicting the C-terminal ones. The chromophore was not given in the crystal structure, hence the absence of Tyr66. (B) Full-length soluble modified protein yields as a function of the incorporation site under different cell-free conditions. ‘No o-tRNA’ and ‘no pPaF’ depict CFPS reactions in which the o-tRNA or the nnAA was omitted from the reaction solutions, respectively. The ‘o-extract’ series includes the reactions in which the orthogonal extract containing the constitutively expressed o-tRNA was used. The ‘transzyme’ and ‘optimized’ reactions contained 200 μg/ml of the linearized transzyme plasmid coding for o-tRNA and o-tRNAopt, respectively. Suppression efficiency is defined as the ratio of accumulated full-length soluble protein containing the nnAA over its natural counterpart (i.e. full-length soluble sfGFP without the nnAA). The bottom line indicates the nucleotides that lie immediately 3′ to the amber stop codon at indicated positions. Error bars depict ±1 standard deviation for three reactions. The right panel shows the SDS-PAGE autoradiogram of radiolabelled sfGFPs. The position of nnAA incorporation is indicated above each lane. (C) Titration of linearized o-tRNAopt plasmid templates in the CFPS reactions containing either pY71sfGFP39TAG or pY71sfGFP74TAG. Error bars indicate ±1 standard deviation for nine sfGFP39pPaF reactions, n = 3 for the sfGFP74pPaF reactions.
In the standard platform, modified sfGFP yields were very similar across all of the 12 positions; the highest modified protein yield was obtained when the UAG codon was at position 143 (‘o-extract’; Figure 4B). The non-specific read-through in the absence of the o-tRNA was also highest at this position. The greatest differences in protein yields across the various UAG codon positions were observed when the cell-free reactions contained the linearized o-tRNA template (‘transzyme’; Figure 4B). Modified, full-length and soluble yields varied between 0.5 mg/ml for sfGFP74pPaF and 1.5 mg/ml for sfGFP223pPaF. Variation in yields and suppression efficiencies was reduced when the o-tRNAopt, instead of the o-tRNA, was simultaneously expressed with the modified protein (‘optimized’, Figure 4B). Full-length and soluble sfGFP74pPaF yields were increased to 0.9 mg/ml, while the yields of modified sfGFP containing pPaF at more readily suppressed sites (such as 39, 151 or 223) remained around 1.4 mg/ml.
Regardless of the manner of o-tRNA supply or the o-tRNA sequence used, we did not observe any correlation between the modified protein yields (i.e. the suppression efficiency) and the residue position along the polypeptide backbone or between the protein yield and the nucleotide following the stop codon. The least efficiently suppressed stop codons (at positions 74 and 182) were followed by a cytosine, which agrees with the published correlation of A = G > C > U for suppression efficiency (44). However, some of the other codons followed by a cytosine (at positions 52 and 200) were just as efficiently suppressed as others followed by purines. Neither did the presence of a uracil at the second position after the UAG codon or the proximity of the codon to the 5′ end lead to better suppression as suggested by earlier studies (33,44). There were no structural factors that contributed to the position effect either. Both residues 74 and 143 are in flexible coils, but position 74 was one of the least efficiently suppressed positions, while position 143 was one of the most efficiently suppressed. Of the most efficiently suppressed positions, residue 39 is in an alpha helix, residue 143 is in a flexible coil, while residues 106 and 151 are in beta strands.
Of the 12 sites, the least efficiently suppressed was at residue 74. We then tested if increasing the o-tRNAopt supply could improve the soluble and full-length sfGFP74pPaF yields. We titrated the linearized o-tRNAopt template and compared sfGFP74pPaF accumulation with the accumulation of sfGFP39pPaF, one of the most efficiently suppressed positions (Figure 4C). For both products, the modified protein yields followed similar trends, peaking with 200 μg/ml of linearized o-tRNAopt template. Thus, using the o-tRNAopt does not avoid the effects of different nnAA incorporation positions.
Substituting the natural amino acids with pPaF at these 12 sites also affected the fluorescence of the modified sfGFP. The incorporation of this nnAA at residue 66 abolished the fluorescence, which was not surprising since replacement of this tyrosine with other nnAAs was shown to alter the spectral properties of GFP (13). The substitution of pPaF for tyrosine at residue 92 reduced the fluorescence of the protein (emission between 523 and 548 nm) by 60–70%. Other substitutions did not alter the specific activity substantially (i.e. reduced by ≤20%). Two modified sfGFP proteins were purified via Strep-Tactin affinity chromatography, and their specific activities were measured and compared with that of natural sfGFP. The specific activities were 95 ± 1 AU/ng for sfGFP, 85 ± 2 AU/ng for sfGFP23pPaF and 86 ± 3 AU/ng for sfGFP39pPaF. Finally, specific activities and soluble yields of pAzF-containing sfGFP proteins were similar to those containing pPaF at the mentioned sites (data not shown).
Mass spectroscopy analysis of the modified protein
In our new platform, the o-tRNA synthesis starts at the same time as the modified protein synthesis; hence there is no o-tRNA in the reaction solution at the beginning of the reaction. Since protein synthesis is fastest in the first hour (Figure 3B), we were concerned that some of the protein initially synthesized may contain a natural amino acid instead of the nnAA due to non-specific read-through of the amber stop codon in the absence of adequate o-tRNA supply. Furthermore, for several modified sfGFPs, full-length and soluble protein accumulated when no o-tRNA was added to the reaction solution (Figure 4B). To examine this potential issue, sfGFP39pPaF, sfGFP143pPaF and sfGFP223pPaF were produced, purified via Strep-Tactin affinity chromatography, digested using trypsin; and the resulting fragments were analysed by LC/MS to ascertain the homogeneity of the product (Supplementary Figure S2). In the cases of sfGFP39pPaF and sfGFP143pPaF, >99% of the protein contained pPaF at the expected position (Supplementary Figure S2A and B). No wild-type peptide was identified in the sfGFP223pPaF sample (Supplementary Figure S2C); hence all of the detectable protein contained pPaF at position 223. Even though non-specific read-through of the amber stop codon was observed in the absence of one of the orthogonal components, only the modified protein (with the nnAA at the expected position) accumulated in the CFPS solutions in the presence of all three orthogonal components.
Comparison of different ribozymes
We also investigated the effect of using different ribozymes for self-cleaving the transzyme, in place of the hammerhead ribozyme, on the modified protein yields. Our hope was to identify a ribozyme that self-cleaves and liberates the o-tRNA more efficiently. The crystal structure of the full-length hammerhead ribozyme from Schistosoma mansoni revealed tertiary interactions that contribute to its 1000-fold higher catalytic efficiency over minimal hammerhead ribozymes such as the one initially used (47). The secondary structures of the three different Schistosoma ribozyme variants are given in Supplementary Figure S3. The changes made to the original sequence are explained below.
We first changed the nucleotides that form hydrogen bonds in Stem I of the Schistosoma ribozyme to allow these nucleotides to base-pair with the 5′ end of the o-tRNA (47). We also added the five-nucleotide AGAGA loop to covalently link Stem III to the sequence encoding the o-tRNA. Finally, we created three variants of this altered Schistosoma ribozyme by mutating the four-nucleotide bulge in Stem I. The first variant contained nucleotides that are able to form additional hydrogen bonds with the o-tRNA. The second variant mimicked the secondary structure of the original Schistosoma ribozyme, and the third variant had the original GUAC sequence at the Stem I bulge. We then replaced the hammerhead ribozyme with these variants, titrated different amounts of the linearized o-tRNA templates (with each template coding for a different ribozyme–tRNA fusion) and measured full-length and soluble sfGFP23pPaF concentrations (Figure 5). The hammerhead ribozyme was substantially more efficient than the Schistosoma ribozymes in liberating the o-tRNA in situ, as evidenced by the modified sfGFP23pPaF accumulation. Among the three Schistosoma ribozyme variants, the second one was the most active; ∼450 μg/ml full-length and soluble sfGFP23pPaF accumulated in the cell-free reactions that contained this variant sequence fused to the o-tRNA template.
Figure 5.

Comparison of modified protein yields obtained with the hammerhead ribozyme and with the three Schistosoma ribozyme variants. The four different linearized o-tRNA plasmid templates (each coding for a different ribozyme) were titrated into cell-free reactions at the indicated concentrations. Error bars indicate ±1 standard deviation for three reactions.
Sequential orthogonal tRNA and protein template additions
Finally, we examined whether we could further increase modified protein yields by accumulating o-tRNA in the cell-free reaction before starting protein synthesis. We included the linearized o-tRNAopt plasmid along with the other reagents in the initial cell-free solution, and incubated the reaction for different periods before adding the modified protein template (i.e. sfGFP gene with an amber stop codon at position 39). All of the reactions were incubated at 30°C for 16 h, and the full-length and soluble sfGFP39pPaF yields were determined via scintillation counting and densitometry (Figure 6).
Figure 6.

Delaying the protein plasmid template addition. 50, 100 or 200 μg/ml of the linearized o-tRNAopt plasmid template was added along with the other standard reagents at the beginning of the reaction; the sfGFP39TAG template was added at the indicated times. Error bars indicate ±1 standard deviation for three reactions.
Delaying the protein template addition by up to 30 min was beneficial at 100 and 200 μg/ml o-tRNAopt template concentrations. At 200 μg/ml o-tRNAopt template, when the protein template was added at t = 15 min, full-length and soluble sfGFP39pPaF yields were increased from 1.37 to 1.67 mg/ml, corresponding to a suppression efficiency of 88%. At 100 μg/ml o-tRNAopt template, modified protein yields were also increased from 1.00 to 1.24 mg/ml, when the protein template was added at t = 30 min. Therefore, we could synthesize almost the same amount of modified protein by adding only half as much o-tRNAopt template if the protein template addition is delayed by 30 min. At 200 μg/ml o-tRNAopt template, the modified protein yields were reduced to 1.11 mg/ml, when the protein template was added 2 h after starting the CFPS incubation. This finding was not surprising, as there is a well-documented burst of protein production early in the PANOx SP cell-free system as was also shown in Figure 3B.
Producing the o-tRNA template by PCR
Several steps are required in the preparation of the linearized o-tRNA template. After the ribozyme–tRNA fusion (i.e. the ‘transzyme’ construct) was cloned into a plasmid, the cells harbouring this plasmid were grown in rich medium. The plasmid was then purified using a commercial kit and linearized using the BstNI restriction enzyme so that the synthesized o-tRNA will have the 3′–CCA terminus required for aminoacylation. Since a very small fraction (<10%) of the plasmid sequence codes for the ‘transzyme’ construct and the concentration of the linearized o-tRNA plasmid in the CFPS solution is high (200 μg/ml), we sought a more convenient method for producing the ‘transzyme’ template. Such a method would be particularly useful for high-throughput screens of different tRNA and ribozyme sequences as well as millilitre-scale cell-free reactions.
We therefore synthesized this o-tRNA template from six overlapping oligonucleotides using PCR. The o-tRNA template could just as easily be synthesized from the plasmid using end primers. The PCR product was subsequently ethanol-precipitated, and, without additional purification, was titrated into the cell-free reaction. In Supplementary Figure S4, the primary abscissa shows the actual o-tRNA template concentrations in the CFPS solution, while the secondary abscissa indicates the calculated equivalent mass concentration of the template if the o-tRNA template were part of the linearized plasmid. At 34 μg/ml of transzyme template (which is the molar equivalent of 400 μg/ml linearized o-tRNAopt plasmid), the full-length and soluble sfGFP39pPaF yields were comparable with those obtained with 200 μg/ml of linearized o-tRNAopt plasmid (Figures 3A and 4B). The addition of the Gam protein (at 6.67 μg/ml), which inhibits the intracellular exonuclease RecBCD (61,62), did not increase the accumulation of the modified sfGFP. (The KC6 extract source strain has the endA gene deleted from its genome (37,38); this gene codes for Endonuclease I, a non-sequence-specific endonuclease.) The suppression efficiencies may have been higher (∼100%) than usual (∼70%) because the wild-type sfGFP yields were slightly lower in this set of cell-free reactions (∼1.2 instead of 1.6–1.9 mg/ml full-length and soluble protein).
Taken together, these results show that the cell-free reactions can be supplied with adequate o-tRNAopt simply by adding a crude PCR product that encodes the hammerhead ribozyme and the o-tRNAopt. Since a few millilitres of this PCR reaction can produce an amount of o-tRNAopt template equivalent to several milligrams of linearized o-tRNA plasmid, this method can be used to quickly produce enough o-tRNAopt template for millilitre-scale cell-free reactions.
DISCUSSION
We describe here a new method of synthesizing modified proteins that contain nnAAs at specific positions. In the previous protocol, the o-tRNA, one of the three reagents required for site-specific incorporation of nnAAs, was constitutively expressed during the growth of the extract source strain (i.e. the orthogonal extract). The nnAA and the purified synthetase enzyme, evolved separately for each nnAA, were then added to the CFPS solutions along with the standard reagents. With this method, the nnAAs pAzF (33) and pPaF (32,63) were site-specifically incorporated into a variety of proteins to allow the modified proteins to be directly conjugated via Click chemistry (32). Despite the substantial improvement over in vivo yields, cell-free yields of modified proteins were almost always lower than their natural counterparts. We previously measured the o-tRNA concentrations and demonstrated that the modified protein yields could be increased by supplementing the cell-free reactions with additional o-tRNA, which was synthesized in vitro and subsequently purified (45). However, in vitro transcription and purification of the o-tRNA are both labour-intensive and costly. We thus sought a more convenient and inexpensive method for providing adequate o-tRNA to the cell-free reactions.
In this new platform, the o-tRNA is synthesized along with the modified protein in the cell-free reaction. T7 RNA polymerase catalyses the transcription of both the o-tRNA and the mRNA encoding the modified protein. The o-tRNA is synthesized as a fusion with a minimal hammerhead ribozyme. This transzyme cleaves itself during the cell-free reaction, and liberates the o-tRNA. The o-tRNA is acylated with the nnAA by the specific aminoacyl-tRNA synthetase; the acylated o-tRNA then enters the ribosome to suppress the amber stop codon, and adds the nnAA to the nascent polypeptide chain. By using the T7 promoter, the o-tRNA is synthesized inexpensively and at adequate concentrations. Because the standard cell extract that does not harbour the o-tRNA is more productive and the o-tRNA is supplied at higher concentrations, the cell-free yields of modified protein and suppression efficiencies were increased several fold. Here, we report the cell-free accumulation of 0.93 ± 0.03 to 1.67 ± 0.16 mg/ml of pPaF-containing sfGFP, which correspond to 50–88% suppression efficiency (Figures 4B and 6). Mass spectrometry analysis of the purified cell-free product demonstrated that only the desired modified protein was synthesized (Supplementary Figure S2). To the best of our knowledge, these yields and suppression efficiencies are the highest reported for proteins containing these nnAAs and are among the highest numbers reported for any of the >30 nnAAs site-specifically incorporated via amber stop codon suppression. Previously, the best reported cell-free yields of modified proteins were 660 μg/ml for E. coli dihydrofolate reductase (eDHFR10pAzF) (33), and 271 μg/ml for sfGFP216pPaF (32). In vivo incorporation of pPaF has been particularly difficult; the best reported modified yields were 2 mg/l for myoglobin (Myo4pPaF) (18). A more recent article describes an optimized in vivo expression system, where researchers were able to site-specifically incorporate pPaF and pAzF into GFP with 10% and 25–85% suppression efficiency, respectively (9). (The large variation in suppression efficiency of pAzF-containing GFP was due to the position effect.) Since absolute yields of modified or wild-type GFP were not reported, we cannot make further comparisons.
In addition, the new platform is modular in two different ways: (i) the experimenter does not need to prepare a special orthogonal extract containing o-tRNA, and this is both more convenient and results in higher protein yields and (ii) the o-tRNA concentration can be adjusted to efficiently incorporate >30 nnAAs site-specifically using E. coli-based protein synthesis platforms (64). The experimenter can now use the E. coli-based commercial cell-free kits and incorporate these nnAAs into virtually any protein by supplementing the cell-free reactions with a crude PCR product that codes for the ribozyme–tRNA fusion, the hexahistidine-tagged synthetase specific to the nnAA (which can be conveniently purified using immobilized metal ion affinity chromatography) and the nnAA itself. In addition, higher concentrations of modified proteins in the crude cell-free solution will increase the yields after downstream purification and covalent coupling processes. This method is now used routinely in our laboratory for site-specific incorporation of the nnAAs pAzF and pPaF into a variety of different proteins.
Adding 200 μg/ml (169 nM) of linearized plasmid template to the CFPS reaction (a plasmid concentration that is much higher than the 6 nM standard concentration for protein templates) produced about 8.9 μM of o-tRNAopt. We believe this high o-tRNA template concentration was required because of limited hammerhead ribozyme activity under the CFPS conditions. The hammerhead ribozyme cleaves itself optimally at 60°C and 30 mM Mg2+ (46); in contrast, the CFPS reactions are incubated at 30°C and contain 20 mM Mg2+. However, this is a very complex mixture, and we do not know what the concentration of free Mg2+ is in CFPS; the free Mg2+ concentration may very well change over the course of the reaction. Under these suboptimal conditions, the uncleaved ribozyme–tRNA transcript would accumulate. We could not verify this hypothesis, as the ribozyme and the uncleaved ribozyme–tRNA fusion were not observed on the tritium radiogram (Supplementary Figure S1). These RNAs were probably digested by the endogenous ribonucleases in the cell-free reaction, and this may also help to explain the high plasmid concentration requirement.
Several other alterations were investigated to further improve this methodology and increase cell-free yields of modified proteins: (i) the protein template addition was delayed to allow for o-tRNA accumulation before starting protein synthesis (Figure 6), (ii) the hammerhead ribozyme was replaced with the catalytically more efficient Schistosoma ribozyme (35, Figure 5) and (iii) an o-tRNA template titration was performed for two different amber stop codons to see whether difficult sites (such as residue 74 on sfGFP) can be better suppressed by providing more o-tRNAopt (Figure 4C). Delaying the protein template addition was beneficial. Full-length and soluble sfGFP39pPaF yields were increased by ∼20% when the protein template was added 15 min after the reaction was initiated with 200 μg/ml o-tRNAopt template. Our results indicated that this strategy can also be used to reduce the concentrations of o-tRNAopt template without reducing modified protein yields.
The hammerhead ribozyme performed better than the three Schistosoma ribozyme variants. We had altered the wild-type Schistosoma sequence substantially so that it would hybridize to the o-tRNA and also added a loop to form a contiguous sequence until the cleavage site (Supplementary Figure S3); these changes most likely reduced the catalytic activity of the resultant ribozymes and led to lower sfGFP39pPaF accumulation (Figure 5). In hindsight, the Schistosoma ribozyme was probably a poor choice, as it hybridizes extensively with its substrate: 10 nucleotides in the Schistosoma ribozyme form hydrogen bonds with the substrate, whereas only 4 nucleotides do so in the minimal hammerhead ribozyme (46). Therefore, when the substrate is changed, these 10 nucleotides in the Schistosoma ribozyme must be altered to preserve the hydrogen bonding, which, in turn, probably affects its catalytic activity.
On the other hand, our platform provides the ability to screen ribozymes for improved performance. Full-length and fluorescent sfGFP is obtained only when the ribozyme–tRNA transcript cleaves itself to release the biologically active o-tRNA; as a result, fluorescence is directly coupled to the catalytic activity of the ribozyme. The dynamic range of the signal can be adjusted by changing the tRNA template concentrations, as demonstrated in Figure 5. Finally, the ethanol-precipitated crude PCR product is nearly as effective as the purified linearized plasmid (Supplementary Figure S4). These attributes are also useful for a protocol in which a library of ribozymes would be fused to the o-tRNAopt, synthesized simultaneously with a modified sfGFP protein in CFPS, and screened for fluorescence. The CFPS reactions producing the highest fluorescence would indicate the most effective ribozyme.
The open nature of the CFPS platform was crucial in both the development of this methodology and the analysis of the position effect as it pertains to nnAA incorporation. We evaluated the site-specific incorporation of pPaF into sfGFP at 12 different sites and investigated the effect of Ef-Tu recognition of the o-tRNA on codon context by comparing two different o-tRNAs (i.e. the o-tRNA and the o-tRNAopt). Our hope was to identify a factor that correlates with the suppression efficiency, to predict the sites most amenable to suppression. Factors reported to be important for the position effect were as follows: the identity of the nucleotide following the stop codon, with purines being better than pyrimidines (G ≥ A > C ≥ U) (43,44,59,60); a beneficial effect of thymidine as the second nucleotide after the stop codon (44); a beneficial effect of lysine as the third amino acid before the stop codon site (65); and the identity of the amino acid two residues before the stop codon site, with acidic amino acids allowing for better suppression than basic ones (66). In addition, previous research in our laboratory showed better nnAA incorporation at amber stop codons closer to the N-terminus of the eDHFR protein (33). Our results for site-specific nnAA incorporation do not directly correlate with any of these factors. They agree with those of Ayer and Yarus, who demonstrated that in vivo suppression of the amber stop codon is independent of base-pairing interactions between the nucleotide after the stop codon and the one preceding the anticodon on the suppressor tRNA (67).
We were puzzled to see in Figure 4B that the position effect was less pronounced when the o-tRNA was limiting in the cell-free reactions containing the orthogonal extract (o-extract). These observations suggest that the position effect is not correlated to the availability of the o-tRNA or rather the ternary complex of aminoacylated o-tRNA, Ef-Tu and GTP. If it were correlated, then we would have expected difficult-to-suppress sites such as position 74 to be much less effectively suppressed at limiting o-tRNA concentrations than observed with the o-extract (Figure 4B). If anything, the o-tRNA limitation would have exacerbated the position effect, as more time would have been available for RF1 to enter the A site of the ribosome and terminate translation at the amber stop codon. Further investigation is thus needed to better understand the position effect and its underlying causes.
When the original o-tRNA was synthesized in situ, we noticed a large difference across the 12 sites, with the suppression efficiencies varying between 28 and 79%. The poor suppression efficiencies were significantly improved to narrow the range (to 49–75%), when the o-tRNAopt [optimized for better recognition by the E. coli elongation factor Ef-Tu (7,9)] and modified sfGFP were simultaneously produced (Figure 4B). These results suggest that the suppression of difficult sites is more effective when the ternary complex between Ef-Tu, GTP and the aminoacylated o-tRNA is formed more effectively.
In addition to the position effect, modified protein yields are also dependent on how well the wild-type protein is synthesized. Therefore, the suppression efficiency, which is defined as the ratio of the modified protein yield to the native protein yield, provides a useful and normalized metric for assessing nnAA incorporation. The suppression efficiency can be expressed as:
| (1) |
where Fraction of RF1 insertion indicates the fraction of the time RF1 suppresses the amber stop codon in place of the ternary complex and prematurely terminates translation. This fraction would be expected to vary directly with the RF1 concentration and inversely with the concentration of the ternary complex.
The ultimate goal in site-specific nnAA incorporation is reaching 100% suppression efficiency, i.e. when the nnAA incorporation pathway would no longer be limiting. Across the 12 incorporation sites, we were able to increase the suppression efficiency from ∼25% to ∼50–75% by removing limitations in orthogonal component supply and in Ef-Tu recognition of the acylated o-tRNA. Even though the RF1 concentration is greatly reduced in the CFPS platform (from about 7.6 μM to 380 nM) due to the dilution of endogenous macromolecules (68), RF1 is still present in the CFPS platform and it can outcompete the aminoacylated o-tRNA as evidenced by the accumulation of prematurely truncated protein products (Figure 4B). We believe this competition with RF1 limits the modified protein yields and suppression efficiencies in the new platform. Elimination of RF1 through various methods has been shown to increase amber stop codon suppression both in vivo and in vitro (69–73). Recently, researchers were able to convert the 314 amber stop codons in the chromosome to TAA codons across different strains using multiplex automated genome engineering (MAGE), which would permit the removal of the prfA gene (which codes for RF1) (74). It would be interesting to repeat these experiments using an extract that is generated from a strain devoid of RF1, and observe its effects on suppression efficiency and the codon context. Apart from competition by RF1, the presence of one or several nnAAs may perturb the three-dimensional fold and induce a limitation in protein folding or may render the modified protein more prone to aggregation, thereby effectively reducing modified protein yields and suppression efficiencies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–4.
ACKNOWLEDGEMENTS
The authors thank Dr Aaron Goerke for the pK7pAzFRS-His6 plasmid, Dr Brad Bundy for the pY71sfGFP216TAG plasmid; Drs William Yang and Kedar Patel for the pPaF, pPaFRS and T7 RNAP reagents; Dr Phil Smith for the Pfu DNA polymerase and the Gam protein; and Dr Chris Adams at the Stanford University Mass Spectrometry Laboratory for LC/MS analysis. The mass spectrometry described was supported by Award Number S10RR027425 from the National Center for Research Resources.
FUNDING
Wallace H. Coulter Foundation; Stanford University School of Engineering. Funding for open access charge: Department of Chemical Engineering or School of Engineering at Stanford University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Cornish VW, Mendel D, Schultz PG. Probing protein structure and function with an expanded genetic code. Angew. Chem. Int. Ed. Engl. 1995;34:621–633. [Google Scholar]
- 2.Ellman J, Mendel D, Anthony-Cahill S, Noren CJ, Schultz PG. Biosynthetic method for introducing unnatural amino acids site-specifically into proteins. Methods Enzymol. 1991;202:301–336. doi: 10.1016/0076-6879(91)02017-4. [DOI] [PubMed] [Google Scholar]
- 3.Bonnefond L, Giegé R, Rudinger-Thirion J. Evolution of the tRNA(Tyr)/TyrRS aminoacylation systems. Biochimie. 2005;87:873–883. doi: 10.1016/j.biochi.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi T, Nureki O, Ishitani R, Yaremchuk A, Tukalo M, Cusack S, Sakamoto K, Yokoyama S. Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat. Struct. Biol. 2003;10:425–432. doi: 10.1038/nsb934. [DOI] [PubMed] [Google Scholar]
- 5.Wang L, Schultz PG. A general approach for the generation of orthogonal tRNAs. Chem. Biol. 2001;8:883–890. doi: 10.1016/s1074-5521(01)00063-1. [DOI] [PubMed] [Google Scholar]
- 6.Liu CC, Schultz PG. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 7.Guo J, Melançon CE, Lee HS, Groff D, Schultz PG. Evolution of amber suppressor tRNAs for efficient bacterial production of proteins containing nonnatural amino acids. Angew. Chem. Int. Ed. Engl. 2009;48:9148–9151. doi: 10.1002/anie.200904035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryu Y, Schultz PG. Efficient incorporation of unnatural amino acids into proteins in Escherichia coli. Nat. Methods. 2006;3:263–265. doi: 10.1038/nmeth864. [DOI] [PubMed] [Google Scholar]
- 9.Young TS, Ahmad I, Yin JA, Schultz PG. An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 2010;395:361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 10.Wang L, Zhang Z, Brock A, Schultz PG. Addition of the keto functional group to the genetic code of Escherichia coli. Proc. Natl Acad. Sci. USA. 2003;100:56–61. doi: 10.1073/pnas.0234824100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Smith BAC, Wang L, Brock A, Cho C, Schultz PG. A new strategy for the site-specific modification of proteins in vivo. Biochemistry. 2003;42:6735–6746. doi: 10.1021/bi0300231. [DOI] [PubMed] [Google Scholar]
- 12.Xie J, Wang L, Wu N, Brock A, Spraggon G, Schultz PG. The site-specific incorporation of p-iodo-L-phenylalanine into proteins for structure determination. Nat. Biotechnol. 2004;22:1297–1301. doi: 10.1038/nbt1013. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Xie J, Deniz AA, Schultz PG. Unnatural amino acid mutagenesis of green fluorescent protein. J. Organic Chem. 2003;68:174–176. doi: 10.1021/jo026570u. [DOI] [PubMed] [Google Scholar]
- 14.Chin JW, Martin AB, King DS, Wang L, Schultz PG. Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc. Natl Acad. Sci. USA. 2002;99:11020–11024. doi: 10.1073/pnas.172226299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang L, Brock A, Herberich B, Schultz PG. Expanding the genetic code of Escherichia coli. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 16.Santoro SW, Wang L, Herberich B, King DS, Schultz PG. An efficient system for the evolution of aminoacyl-tRNA synthetase specificity. Nature Biotechnol. 2002;20:1044–1048. doi: 10.1038/nbt742. [DOI] [PubMed] [Google Scholar]
- 17.Chin JW, Santoro SW, Martin AB, King DS, Wang L, Schultz PG. Addition of p-Azido- L -phenylalanine to the genetic code of Escherichia coli. J. Am. Chem. Soc. 2002;124:9026–9027. doi: 10.1021/ja027007w. [DOI] [PubMed] [Google Scholar]
- 18.Deiters A, Schultz PG. In vivo incorporation of an alkyne into proteins in Escherichia coli. Bioorg. Med. Chem. Lett. 2005;15:1521–1524. doi: 10.1016/j.bmcl.2004.12.065. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen DP, Lusic H, Neumann H, Kapadnis PB, Deiters A, Chin JW. Genetic encoding and labeling of aliphatic azides and alkynes in recombinant proteins via a pyrrolysyl-tRNA Synthetase/tRNA(CUA) pair and click chemistry. J. Am. Chem. Soc. 2009;131:8720–8721. doi: 10.1021/ja900553w. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen DP, Elliott T, Holt M, Muir TW, Chin JW. Genetically encoded 1,2-aminothiols facilitate rapid and site-specific protein labeling via a bio-orthogonal cyanobenzothiazole condensation. J. Am. Chem. Soc. 2011;133:11418–11421. doi: 10.1021/ja203111c. [DOI] [PubMed] [Google Scholar]
- 21.Plass T, Milles S, Koehler C, Schultz C, Lemke EA. Genetically encoded copper-free click chemistry. Angew. Chem. Int. Ed. Engl. 2011;50:3878–3881. doi: 10.1002/anie.201008178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wan W, Huang Y, Wang Z, Russell WK, Pai PJ, Russell DH, Liu WR. A facile system for genetic incorporation of two different noncanonical amino acids into one protein in Escherichia coli. Angew. Chem. Int. Ed. Engl. 2010;49:3211–3214. doi: 10.1002/anie.201000465. [DOI] [PubMed] [Google Scholar]
- 23.Wang YS, Fang X, Wallace AL, Wu B, Liu WR. A rationally designed pyrrolysyl-tRNA synthetase mutant with a broad substrate spectrum. J. Am. Chem. Soc. 2012;134:2950–2953. doi: 10.1021/ja211972x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yanagisawa T, Ishii R, Fukunaga R, Kobayashi T, Sakamoto K, Yokoyama S. Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode N(epsilon)-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem. Biol. 2008;15:1187–1197. doi: 10.1016/j.chembiol.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Wang YS, Fang X, Chen H-Y, Wu B, Wang ZU, Hilty C, Liu WR. Genetic incorporation of twelve meta-substituted phenylalanine derivatives using a single pyrrolysyl-tRNA synthetase mutant. ACS Chem. Biol. 2013;8:405–415. doi: 10.1021/cb300512r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boyer M, Wang C, Swartz J. Simultaneous expression and maturation of the iron-sulfur protein ferredoxin in a cell-free system. Biotechnol. Bioeng. 2006;94:128–138. doi: 10.1002/bit.20830. [DOI] [PubMed] [Google Scholar]
- 27.Boyer ME, Stapleton JA, Kuchenreuther JM, Wang C, Swartz JR. Cell-free synthesis and maturation of [FeFe] hydrogenases. Biotechnol. Bioeng. 2008;99:59–67. doi: 10.1002/bit.21511. [DOI] [PubMed] [Google Scholar]
- 28.Bundy BC, Franciszkowicz MJ, Swartz JR. Escherichia coli-based cell-free synthesis of virus-like particles. Biotechnol. Bioeng. 2008;100:28–37. doi: 10.1002/bit.21716. [DOI] [PubMed] [Google Scholar]
- 29.Hovijitra NT, Wuu JJ, Peaker B, Swartz JR. Cell-free synthesis of functional aquaporin Z in synthetic liposomes. Biotechnol. Bioeng. 2009;104:40–49. doi: 10.1002/bit.22385. [DOI] [PubMed] [Google Scholar]
- 30.Wuu JJ, Swartz JR. High yield cell-free production of integral membrane proteins without refolding or detergents. Biochim. Biophys. Acta. 2008;1778:1237–1250. doi: 10.1016/j.bbamem.2008.01.023. [DOI] [PubMed] [Google Scholar]
- 31.Yang WC, Patel KG, Lee J, Ghebremariam YT, Wong HE, Cooke JP, Swartz JR. Cell-free production of transducible transcription factors for nuclear reprogramming. Biotechnol. Bioeng. 2009;104:1047–1058. doi: 10.1002/bit.22517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bundy BC, Swartz JR. Site-specific incorporation of p-propargyloxyphenylalanine in a cell-free environment for direct protein-protein click conjugation. Bioconjug. Chem. 2010;21:255–263. doi: 10.1021/bc9002844. [DOI] [PubMed] [Google Scholar]
- 33.Goerke AR, Swartz JR. High-level cell-free synthesis yields of proteins containing site-specific non-natural amino acids. Biotechnol. Bioeng. 2009;102:400–416. doi: 10.1002/bit.22070. [DOI] [PubMed] [Google Scholar]
- 34.Patel KG, Ng PP, Kuo C-C, Levy S, Levy R, Swartz JR. Cell-free production of gaussia princeps luciferase–antibody fragment bioconjugates for ex vivo detection of tumor cells. Biochem. Biophys. Res. Commun. 2009;390:971–976. doi: 10.1016/j.bbrc.2009.10.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welsh JP, Patel KG, Manthiram K, Swartz JR. Multiply mutated Gaussia luciferases provide prolonged and intense bioluminescence. Biochem. Biophys. Res. Commun. 2009;389:563–568. doi: 10.1016/j.bbrc.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 36.Zawada JF, Yin G, Steiner AR, Yang J, Naresh A, Roy SM, Gold DS, Heinsohn HG, Murray CJ. Microscale to manufacturing scale-up of cell-free cytokine production-a new approach for shortening protein production development timelines. Biotechnol. Bioeng. 2011;108:1570–1578. doi: 10.1002/bit.23103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calhoun KA, Swartz JR. Total amino acid stabilization during cell-free protein synthesis reactions. J. Biotechnol. 2006;123:193–203. doi: 10.1016/j.jbiotec.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 38.Michel-Reydellet N, Calhoun K, Swartz J. Amino acid stabilization for cell-free protein synthesis by modification of the Escherichia coli genome. Metab. Eng. 2004;6:197–203. doi: 10.1016/j.ymben.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 39.Jewett MC, Swartz JR. Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol. Bioeng. 2004;86:19–26. doi: 10.1002/bit.20026. [DOI] [PubMed] [Google Scholar]
- 40.Jewett MC, Calhoun KA, Voloshin A, Wuu JJ, Swartz JR. An integrated cell-free metabolic platform for protein production and synthetic biology. Mol. Syst. Biol. 2008;4:1–10. doi: 10.1038/msb.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Budisa N. Prolegomena to future experimental efforts on genetic code engineering by expanding its amino acid repertoire. Angew. Chem. Int. Ed. Engl. 2004;43:6426–6463. doi: 10.1002/anie.200300646. [DOI] [PubMed] [Google Scholar]
- 42.Hoesl MG, Budisa N. Recent advances in genetic code engineering in Escherichia coli. Curr. Opin. Biotechnol. 2012;23:751–757. doi: 10.1016/j.copbio.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 43.Phillips-Jones MK, Watson FJ, Martin R. The 3’ codon context effect on UAG suppressor tRNA is different in Escherichia coli and Human Cells. J. Mol. Biol. 1993;233:1–6. doi: 10.1006/jmbi.1993.1479. [DOI] [PubMed] [Google Scholar]
- 44.Stormo GD, Schneider TD, Gold L. Quantitative analysis of the relationship between nucleotide sequence and functional activity. Nucleic Acids Res. 1986;14:6661–6679. doi: 10.1093/nar/14.16.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albayrak C, Swartz JR. Using E. coli-based cell-free protein synthesis to evaluate the kinetic performance of an orthogonal tRNA and aminoacyl-tRNA synthetase pair. Biochem. Biophys. Res. Commun. 2013;431:291–295. doi: 10.1016/j.bbrc.2012.12.108. [DOI] [PubMed] [Google Scholar]
- 46.Fechter P, Rudinger J, Giegé R, Théobald-Dietrich A. Ribozyme processed tRNA transcripts with unfriendly internal promoter for T7 RNA polymerase: production and activity. FEBS Lett. 1998;436:99–103. doi: 10.1016/s0014-5793(98)01096-5. [DOI] [PubMed] [Google Scholar]
- 47.Martick M, Scott WG. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell. 2006;126:309–320. doi: 10.1016/j.cell.2006.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cabantous S, Terwilliger TC, Waldo GS. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005;23:102–107. doi: 10.1038/nbt1044. [DOI] [PubMed] [Google Scholar]
- 50.Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 51.Uemori T, Ishino Y, Toh H, Asada K, Kato I. Organization and nucleotide sequence of the DNA polymerase gene from the archaeon Pyrococcus furiosus. Nucleic Acids Res. 1993;21:259–265. doi: 10.1093/nar/21.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dabrowski S, Kur J. Cloning and expression in Escherichia coli of the recombinant his-tagged DNA polymerases from Pyrococcus furiosus and Pyrococcus woesei. Protein Expr. Purif. 1998;14:131–138. doi: 10.1006/prep.1998.0945. [DOI] [PubMed] [Google Scholar]
- 53.Goerke AR, Swartz JR. Development of cell-free protein synthesis platforms for disulfide bonded proteins. Biotechn. Bioeng. 2008;99:351–367. doi: 10.1002/bit.21567. [DOI] [PubMed] [Google Scholar]
- 54.Deiters A, Cropp TA, Mukherji M, Chin JW, Anderson JC, Schultz PG. Adding amino acids with novel reactivity to the genetic code of Saccharomyces cerevisiae. J. Am. Chem. Soc. 2003;125:11782–11783. doi: 10.1021/ja0370037. [DOI] [PubMed] [Google Scholar]
- 55.Li Y, Wang E, Wang Y. A modified procedure for fast purification of T7 RNA polymerase. Protein Expr. Purif. 1999;16:355–358. doi: 10.1006/prep.1999.1083. [DOI] [PubMed] [Google Scholar]
- 56.Budyka MF. Photochemistry of aromatic azides and nitrenes. High Energy Chem. 2007;41:176–187. [Google Scholar]
- 57.Calhoun KA, Swartz JR. An economical method for cell-free protein synthesis using glucose and nucleoside monophosphates. Biotechnol. Prog. 2005;21:1146–1153. doi: 10.1021/bp050052y. [DOI] [PubMed] [Google Scholar]
- 58.Imburgio D, Rong M, Ma K, McAllister WT. Studies of promoter recognition and start site selection by T7 RNA polymerase using a comprehensive collection of promoter variants. Biochemistry. 2000;39:10419–10430. doi: 10.1021/bi000365w. [DOI] [PubMed] [Google Scholar]
- 59.Miller JH, Albertini AM. Effects of Surrounding Sequence on the Suppression of Nonsense Codons. J. Mol. Biol. 1983;164:59–71. doi: 10.1016/0022-2836(83)90087-6. [DOI] [PubMed] [Google Scholar]
- 60.Pedersen WT, Curran JF. Effects of the Nucleotide 3′ to an Amber Codon on Ribosomal Selection Rates of Suppressor tRNA and Release Factor-1. J. Mol. Biol. 1991;219:231–241. doi: 10.1016/0022-2836(91)90564-m. [DOI] [PubMed] [Google Scholar]
- 61.Sitaraman K, Esposito D, Klarmann G, Le Grice SF, Hartley JL, Chatterjee DK. A novel cell-free protein synthesis system. J. Biotechnol. 2004;110:257–263. doi: 10.1016/j.jbiotec.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 62.Lui BH, Cochran JR, Swartz JR. Discovery of improved EGF agonists using a novel in vitro screening platform. J. Mol. Biol. 2011;413:406–415. doi: 10.1016/j.jmb.2011.08.028. [DOI] [PubMed] [Google Scholar]
- 63.Patel KG, Swartz JR. Surface functionalization of virus-like particles by direct conjugation using azide-alkyne click chemistry. Bioconjug. Chem. 2011;22:376–387. doi: 10.1021/bc100367u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Young TS, Schultz PG. Beyond the canonical 20 amino acids: expanding the genetic lexicon. J. Biol. Chem. 2010;285:11039–11044. doi: 10.1074/jbc.R109.091306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mottagui-Tabar S, Isaksson LA. Only the last amino acids in the nascent peptide influence translation termination in Escherichia coli genes. FEBS Lett. 1997;414:165–170. doi: 10.1016/s0014-5793(97)00978-2. [DOI] [PubMed] [Google Scholar]
- 66.Mottagui-Tabar S, Björnsson A, Isaksson LA. The second to last amino acid in the nascent peptide as a codon context determinant. EMBO J. 1994;13:249–257. doi: 10.1002/j.1460-2075.1994.tb06255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ayer D, Yarus M. The context effect does not require a fourth base pair. Science. 1986;231:393–395. doi: 10.1126/science.3510456. [DOI] [PubMed] [Google Scholar]
- 68.Adamski F, McCaughan K, Jorgensen F, Kurland C, Tate W. The Concentration of Polypeptide Chain Release Factors 1 and 2 at Different Growth Rates of Escherichia coli. J. Mol. Biol. 1994;238:302–308. doi: 10.1006/jmbi.1994.1293. [DOI] [PubMed] [Google Scholar]
- 69.Short GF, Golovine SY, Hecht SM. Effects of release factor 1 on in vitro protein translation and the elaboration of proteins containing unnatural amino acids. Biochemistry. 1999;38:8808–8819. doi: 10.1021/bi990281r. [DOI] [PubMed] [Google Scholar]
- 70.Sando S, Ogawa A, Nishi T, Hayami M, Aoyama Y. In vitro selection of RNA aptamer against Escherichia coli release factor 1. Bioorg. Med. Chem. Lett. 2007;17:1216–1220. doi: 10.1016/j.bmcl.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 71.Mukai T, Hayashi A, Iraha F, Sato A, Ohtake K, Yokoyama S, Sakamoto K. Codon reassignment in the Escherichia coli genetic code. Nucleic Acids Res. 2010;38:8188–8195. doi: 10.1093/nar/gkq707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson DBF, Xu J, Shen Z, Takimoto JK, Schultz MD, Schmitz RJ, Xiang Z, Ecker JR, Briggs SP, Wang L. RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nat. Chem. Biol. 2011;7:779–786. doi: 10.1038/nchembio.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Loscha KV, Herlt AJ, Qi R, Huber T, Ozawa K, Otting G. Multiple-site labeling of proteins with unnatural amino acids. Angew. Chem. Int. Ed. Engl. 2012;51:2243–2246. doi: 10.1002/anie.201108275. [DOI] [PubMed] [Google Scholar]
- 74.Isaacs FJ, Carr PA, Wang HH, Lajoie MJ, Sterling B, Kraal L, Tolonen AC, Gianoulis TA, Goodman DB, Reppas NB, et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science. 2011;333:348–353. doi: 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]