


Abstract
The DNA damage response (DDR) involves both the control of DNA damage repair and signaling to cell cycle checkpoints. Therefore, unraveling the underlying mechanisms of the DDR is important for understanding tumor suppression and cellular resistance to clastogenic cancer therapeutics. Because the DDR is likely to be influenced by chromatin regulation at the sites of DNA damage, we investigated the role of heterochromatin protein 1 (HP1) during the DDR process. We monitored double-strand breaks (DSBs) using the γH2AX foci marker and found that depleting cells of HP1 caused genotoxic stress, a delay in the repair of DSBs and elevated levels of apoptosis after irradiation. Furthermore, we found that these defects in repair were associated with impaired BRCA1 function. Depleting HP1 reduced recruitment of BRCA1 to DSBs and caused defects in two BRCA1-mediated DDR events: (i) the homologous recombination repair pathway and (ii) the arrest of cell cycle at the G2/M checkpoint. In contrast, depleting HP1 from cells did not affect the non-homologous end-joining (NHEJ) pathway: instead it elevated the recruitment of the 53BP1 NHEJ factor to DSBs. Notably, all three subtypes of HP1 seemed to be almost equally important for these DDR functions. We suggest that the dynamic interaction of HP1 with chromatin and other DDR factors could determine DNA repair choice and cell fate after DNA damage. We also suggest that compromising HP1 expression could promote tumorigenesis by impairing the function of the BRCA1 tumor suppressor.
INTRODUCTION
Mammalian genomes are characterized by heterochromatin, regions that are compact and transcriptionally silent, and euchromatin, regions that have a looser structure and are associated with active gene transcription. Chromatin structure is actively regulated by various epigenetic mechanisms, including modifications of histone proteins during gene expression, DNA replication and the DNA damage response (DDR) (1,2). The classical heterochromatin factor, heterochromatin protein 1 (HP1), is a key component of heterochromatin in diverse organisms (3–6). The three human HP1 isoforms, HP1α, HP1β and HP1γ, all share a characteristic N-terminal chromodomain, a central hinge domain and a C-terminal chromoshadow domain. Through their chromodomains, HP1 proteins interact with di-methylated or tri-methylated lysine residues 9 of histone H3 (H3K9me2 and H3K9Me3) (7). The chromoshadow domain of HP1 interacts with various protein factors, via interactions with its PxVxL-containing motifs. The hinge domain of HP1, which is the least conserved region among three subtypes of HP1, is responsible for binding to RNA molecules (8–10).
DNA damage is frequently generated by the collapse of replication forks or by genotoxic agents, including ionizing radiation (IR). Cells respond to DNA damage by activating the DDR network, which includes DNA repair, cell cycle arrest, senescence and apoptosis (11,12). These DDR pathways are activated by numerous protein factors in a dynamic and highly ordered manner. Protein factors involved in DDR include the p53 and BRCA1 tumor suppressors, cell cycle regulators, apoptosis regulators and DNA repair factors, such as the ATM/ATR (Ataxia-telangiectasia/ATM and Rad3-related) kinase and 53BP1. Specifically, DNA double-stranded breaks (DSBs) lead to cell cycle arrest at cell cycle checkpoints to provide the time needed for repair by either the homologous recombination (HR) or non-homologous end-joining (NHEJ) repair pathways (13). Cells with extensive DNA damage often undergo cell death by apoptosis or other mechanisms. If cells’ responses to damaged DNA are incomplete or aberrant, it is harmful to them and often leads to mutations, genomic instability and carcinogenesis.
The biological roles of HP1 in regulating DDR signaling and repair are not fully understood. HP1 is reported to have diverse cellular functions including transcription regulation, chromatin remodeling, DNA replication, non-coding RNA binding and others (14). Recent studies have revealed that HP1 is also involved in various DDR processes (15–17). However, the spatial and temporal regulation of the association and dissociation of HP1 with chromatin in response to DNA damage is still unclear. In some studies, DNA damage induces the transient removal of HP1 proteins from DSB sites to facilitate the binding of DDR factors to chromatin for DNA repair (15,18,19). However, other studies have indicated that DNA damage induces the association of HP1 with DSB sites, suggesting HP1 is dynamically mobilized and recruited to play an active role for in DDR processes (16,17,20). In support of this, nematodes that are deficient for an isoform of HP1 (HLP-2) have a higher sensitivity to irradiation than their wild-type counterparts (16). There are also discrepancies relating to the function of HP1 in DDR processes that arise due to the isoform of HP1 that is studied and the cell systems and experimental conditions used (4,21,22). At this time, the contributions of each HP1 subtype to the DDR signaling and DSB repair pathways are not clearly understood.
We wished to determine the molecular mechanisms underlying the roles of HP1 in regulating DSB repair and promoting genome stability. Here, we show that HP1 played critical roles in DDR pathways by promoting BRCA1 function and recruitment to DSB sites. BRCA1 is a known essential DDR factor involved in HR repair and control of the G2/M cell cycle checkpoint. In this report, we demonstrate that the three subtypes of human HP1 were important not just for HR DNA repair but also for control of the G2/M cell cycle checkpoint, through their effect on BRCA1.
MATERIALS AND METHODS
Mammalian cells
U2OS, MCF7 and HCC1937 cells were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (FBS). U2OS and MCF7 cells transduced with shRNAs were maintained by the addition of puromycin (10 µg/ml). U2OS-HR and U2OS-NHEJ cells were obtained from Dr Jeremy Stark (City of Hope). AsiSI-ER-U2OS cells were a kind gift from Dr Gaëlle Legube (University of Toulouse).
RNA interference
Two sets of HP1-specific siRNAs were purchased from Dharmacon and Santa Cruz Biotechnology. The siRNAs were transfected into mammalian cells using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) according to manufacturer’s protocol. The shRNA (short hairpin RNA) plasmids pLKO.1 for respective HP1 subtypes were obtained from Academia Sinica. Plasmids were co-transfected into HEK293FT cells together with pΔ8.7 and pVSV-G plasmids. Lentiviral supernatants were harvested on the fifth day. For lentiviral infection, U2OS or MCF-7 cells were plated the day before infection and cultured overnight. Lentiviruses carrying the shRNAs against HP1 were added to cells in culture medium containing polybrene (8 μg/ml) and incubated for 24 h. The transduced cells were enriched and pooled by culturing with puromycin (2 μg/ml). The knockdown of HP1 by RNA interference was confirmed by western blot analyses with specific HP1 antibodies (Abcam).
Western blot analysis
Whole-cell lysates were prepared by lysing cells in RIPA buffer [25 mM Tris–HCl, 125 mM NaCl, 1% Nonidet-P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS) and a Complete Protease Inhibitor Cocktail (Roche)]. Equal amounts of cell lysates were separated by SDS–polyacrylamide gel electrophoresis then transferred to membrane and immunoblotted with antibodies against HP1 subtypes (Abcam), γH2AX (Millipore) and actin (Santa Cruz Biotechnology). Blots were stained with an enhanced chemiluminescence detection kit (ECL-Plus, Amersham Biosciences).
Foci formation assay
MCF7 and HP1-depleted cells were cultured on slides for 20 h and then fixed with a 1:1 mixture of ethanol and methanol at −20°C overnight. U2OS cells and the respective HP1-depleted cells were incubated with pre-extraction buffer (20 mM Tris–HEPES, pH 7.5, 50 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 3 mM MgCl2, 300 mM sucrose and 0.5% Triton X100) for 5 min and fixed with 2% paraformaldehyde at room temperature for 15 min. Fixed MCF7 and U2OS cells were blocked with blocking buffer (PBS with 0.05% Tween 20 and 2% bovine serum albumin) for 30 min. The slides were stained with anti-γH2AX antibody (Millipore), anti-BRCA1 (Santa Cruz Biotechnology) and anti-53BP1 (Millipore) for 1 h at 37°C in a humidified chamber. The slides were washed twice with PBST buffer and incubated with secondary antibodies (Alexa 488 or 568 from Invitrogen) in the humidified chamber for 1 h. For double staining, the slides were incubated with two different primary and then two different secondary antibodies. The slides were washed twice with PBST, and the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Foci were observed using an inverted fluorescence microscope (IX81, Olympus) with a ×60 objective magnification. The images were captured and analyzed by image-pro 6.3 software.
HR/NHEJ reporter assay
HR/NHEJ assays were performed as previously described (23). U2OS-HR and U2OS-NHEJ cells (1 × 105) were first transfected with individual siRNAs (10 pmol) specific for each respective HP1 subtype. The next day, JS20-I-SceI expression constructs (1 µg) were transfected into the U2OS reporter cells using Lipofectamine 2000 (Invitrogen). Green fluorescent protein (GFP)-positive cells were quantified by flow cytometry using a Gallios Flow cytometer/cell sorter (Beckman) 2 days after transfection.
Colony formation assay
MCF7 and HP1-depleted MCF7 cells are cultured under the same conditions before cell seeding. Equal numbers (100 cells) of MCF7 and HP1-depleted MCF7 cells were plated in triplicate in six-well plates. Cells were grown for 10 days before staining. Cells were washed with ice-cold phosphate-buffered saline (PBS) and fixed with ice-cold methanol for 10 min. The methanol was removed and replaced by crystal violet solution (0.2%). After 10-min incubation, the crystal violet solution was removed, and the cells were washed with ddH2O and then dried at room temperature before analyses.
G2/M checkpoint assay
The distribution of cells in various stages of the cell cycle was determined by propidium iodide (PI) staining. For the G2/M checkpoint analysis, cells were double stained with PI and an anti-H3 phospho-serine 10 histone antibody (Millipore). U2OS cells and HCC1937 cells were transfected with siRNAs targeting the HP1 subtypes and incubated for 3 days. Cells were irradiated (4 Gy) and then incubated for additional 3 h before harvesting. Cells were fixed with 70% ethanol for at least 24 h. Fixed cells were washed twice with PBS buffer and pre-incubated with blocking buffer (PBS with 0.1% Tween 20 and 2% bovine serum albumin) for 30 min. Cells were collected by centrifugation and incubated with antibody for histone H3 (serine 10 phosphorylation, Millipore) for 2 h. Cells were washed with PBST buffer and incubated with a fluorescein isothiocyanate (FITC)-conjugated anti-rabbit secondary antibody (Millipore) for 1 h. The stained cells were washed with PBST and resuspended in PBS with PI (Sigma, 2 µg/ml) and RNase A solution (Sigma, 10 µg/ml). Cellular fluorescence (FL1 and FL2 values for FITC and PI) was measured using a Gallios Flow cytometer/cell sorter (Beckman).
Apoptosis assay
Apoptotic cells were measured using the FITC annexin V Apoptosis Detection Kit I (BD Pharmingen) according to manufacturer’s protocol. MCF7 cells and HP1-depleted MCF7 cells were cultured and then harvested before or after irradiation. The cells were then washed twice with ice-cold PBS and resuspended in 1× binding buffer [0.1 M HEPES/NaOH (pH 7.4), 1.4 M NaCl and 25 mM CaCl2. at a concentration of 1 × 106 cells/ml. 100 microliters of cells (1 × 105 cells) was transferred to a 5-ml culture tube and incubated with FITC-conjugated annexin V (5 µl) for 15 min in the dark at room temperature, and then 1× binding buffer (400 µl) was added to each tube, and the stained cells were analyzed by flow cytometry using a Gallios Flow cytometer/cell sorter (Beckman). Approximately 0.5–1 × 105 cells (ungated) were analyzed by blue laser (488 nm) to measure the FL1 parameter. Data were processed by using the Gallios software provided by the manufacturer.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed according to previous published procedures with minor modifications (24). AsiSI-ER-U2OS cells were treated with 4-OHT (4-hydroxytamoxifen), and the soluble chromatin was prepared by sonication. Antibodies against BRCA1 (SantaCruz Biotechnology), 53BP1 (Millipore), RAD51 (SantaCruz Biotechnology) and HP1 (Abcam) were incubated with the fractions of soluble chromatin at 4°C overnight. Immunoprecipitated pellets were washed with washing buffer, and the bound chromatin was eluted with elution buffer. After the cross-linking of the immunoprecipitated chromatin was reversed, the DNA was purified by phenol–chloroform extraction and ethanol precipitation. The purified DNA was dissolved in 30 µl of 10 mM Tris–HCl, pH 8.0 and 1 mM ethylenediaminetetraacetic acid, and 1 µl was used for each polymerase chain reaction (PCR) analysis. The primers were proximal to the cleavage sites by AsiSI (chr1_89231183: FW: 5′-GATTGGCTATGGGTGTGGAC-3′, REV: 5′-CATCCTTGCAAACCAGTCCT-3′; and chr6_90404906: FW: 5′-TGCCGGTCTCCTAGAAGTTG-3′, REV: 5′-GCGCTTGATTTCCCTGAGT-3′) or were distal to the AsiSI site on chr21_21292316 (FW: 5′-TGGCTGGAACTGCTTTCTTT-3′, REV: 5′-GGTGAGTGAATGAGCTGCAA-3′) (25,26). PCR products were analyzed by quantitative PCR (BioRad). The results of the quantitative ChIP assays are the mean ± SD of duplicate PCR reactions from a single experiment. One representative of multiple independent experiments is shown.
RESULTS
HP1 deficiency causes genotoxic stress and reduced repair of IR-induced DNA damage
To understand the roles of HP1 in the DDR pathway, we first investigated whether reducing HP1 levels caused genotoxic stress. We depleted HP1 expression using RNA interference; shRNAs (small hairpin RNAs) against the three subtypes of HP1 were designed and packaged into lentivirus. U2OS (osteosarcoma; data not shown) and MCF7 cells (breast cancer; Supplementary Figure S1, left panels) were infected with the recombinant lentiviruses, and the transduced cells were selected and maintained by growth in media containing puromycin.
To examine genotoxic stress, we monitored focal accumulation of γH2AX by immunofluorescence. The γH2AX (histone variant of H2AX) is phosphorylated at serine 139, and its accumulation in foci is a marker of chromosomal breaks (27,28). We found that the γH2AX foci were dramatically increased in number in U2OS nuclei after irradiation (IR, 4 Gy) (Figure 1A, last panel, first row). Using this marker for genotoxic stress (Figure 1A, first panel, first row), we saw that U2OS cells infected with shRNAs against each HP1 subtype showed increased formation of γH2AX foci, even without any treatment by exogenous DNA-damaging agents (Figure 1A, second to fourth panels, first row). Although there were fewer (20–30%) HP1-depleted MCF7 breast cancer cells that had increased γH2AX foci compared with the U2OS cells, enough cells showed the phenotype to indicate that depleting endogenous HP1 induced γH2AX foci formation, and that this phenomenon was not limited to one cell type (Supplementary Figure S2). Western blot assays confirmed the reduced expression of each HP1 subtype by the respective shRNAs and the increased basal γH2AX level in HP1-depleted MCF7 cells (Supplementary Figure S1, right panels). To confirm that the elevated γH2AX signals were due to endogenous stress before treatment with DNA-damaging agents (29), we used 53BP1, another marker for DNA damage, to test HP1-depleted MCF7 cells. Indeed, 53BP1 foci were also elevated in the HP1-depleted cells (Figure 1A, third row). Interestingly, we often observed larger sized 53BP1 foci in HP1-depleted MCF7 cells, presumably reflecting DSBs that resulted from replication stresses (30,31). Moreover, most of γH2AX foci and 53BP1 foci co-localized, with and without irradiation, suggesting γH2AX the foci in Figure 1 are associated with DSBs (Supplementary Figure S3). The data suggest that the DDR processes that respond to endogenous DNA breaks are defective and could contribute to the accumulation of γH2AX and 53BP1 foci in the non-irradiated HP1-depleted cells.
Figure 1.

HP1 depletion induces γH2AX and 53BP1 foci formation. (A) U2OS cells or HP1-depleted U2OS cells were stained with an anti-γ2AX antibody (Millipore). The γ2AX images from irradiated U2OS cells (4 Gy) are included as a positive control. MCF7 cell or HP1-depleted MCF7 cells were also stained with anti-γ2AX antibody and anti-53BP1 antibody (Millipore). The images of γH2AX foci and 53BP1 foci were captured on the same areas of the slide for comparison (200 ms by fluorescence microscopy). Scale Bar = 10 µm. (B) MCF7 cell or HP1-depleted MCF7 cells were irradiated (4 Gy), and foci formation was analyzed at 0, 1 and 8 h post-IR. Foci numbers in each nucleus were calculated by image-pro software. The histogram summarizes the average numbers of foci in the nuclei from at least 10 individual cells (right panel). P < 0.001 for MCF7; P < 0.04 for MCF7-shHP1α, β and γ.
Because HP1 seemed to be important for the DDR to repair endogenous DNA damage, we tested the possibility that HP1 would also be important for the repair of IR-induced chromosomal breaks. We examined the kinetics of DNA damage recovery in MCF7 cells that were irradiated with 4 Gy and then allowed to recover for 1 or 8 h (Figure 1B, first row). Very low levels of γH2AX foci formation were observed in the sham-irradiated control MCF7 cells. However, in the irradiated cells, γH2AX foci were dramatically elevated at 1 h. By 8 h post-IR, they had returned to basal levels, indicating there are efficient DSB repair processes in MCF7 cells. MCF7 cells were depleted of HP1α, HP1β or HP1γ (as described earlier in the text) and again we observed a higher level of basal γH2AX foci (Figure 1B, second to fourth rows). IR-treatment (4 Gy) of the HP1-depleted MCF7 cells induced additional foci formation at 1 h post-IR. At 8 h post-IR, the γH2AX level returned to the basal level in the parental MCF7 cells, but not in the HP1-depleted MCF7 cells. The numbers of γH2AX foci in these cells still remained high after 8 h (Figure 1B, right panels). Altogether, the data indicate that HP1 plays important roles in suppressing basal DNA damage and in promoting efficient DNA repair of DSBs when they occur.
Knockdown of HP1 increases apoptosis after irradiation
The elevated basal γH2AX foci level observed in the HP1-depleted mammalian cells could be the result of apoptosis because apoptotic cells also exhibit elevated γH2AX signals (32,33). To determine whether depleting HP1 promoted cell apoptosis, MCF7 and HP1-depleted MCF7 cells were stained with annexin V, which binds to phospholipid phosphatidylserine from disrupted plasma membranes and is a marker for early stage apoptotic cells. Before irradiation, the annexin V-positive MCF7 and HP1-depleted MCF7 cells were very few (2–5%), indicating that HP1 depletion did not induce apoptosis in the resting cells (Figure 2). This was further strengthened by the observation that there was no marked change in the sub-G1 population and overall cell cycle profile in non-irradiated MCF7 cells (Supplementary Figure S4). As expected, IR (4 Gy) increased the annexin V-stained fraction of MCF7 cells to 8.6%. Notably, depleting HP1, and especially depleting HP1γ, dramatically enhanced the percentage of annexin V-positive cells (10.2–20.1%) after irradiation. Although we may be underestimating the apoptotic proportions because of cell debris, our observations suggest that HP1 proteins were required to suppress IR-induced apoptosis. However, because the number of apoptotic MCF7 cells was very low before irradiation, the elevated numbers of γH2AX foci visualized in basal non-irradiated HP1-depleted cells (Figure 1) was presumably not because of increased apoptosis. Rather, it suggests endogenous DNA damage accumulated in HP1-depleted cells before irradiation. The defect in the DDR pathway in HP1-depleted cells most likely contributed to the enhanced apoptosis and the increased genomic instability observed after irradiation. We concluded that the increased number of basal γH2AX foci observed in HP1-depleted cells (Figure 1) did not result from apoptosis, even though it is also marked by γH2AX staining (27,28).
Figure 2.

HP1 is involved in IR-induced apoptosis. MCF7 cells and HP1-depleted MCF7 cells were untreated (upper panels) or irradiated (lower panels), then incubated for 20 h after IR-exposure. The cells were collected, stained with annexin V and then 1 × 105 cells (or ∼0.5 × 105 cells for the MCF7-shHP1γ post-IR) from each sample were analyzed by flow cytometry.
Although each subtype of HP1 has conserved domains and common functions, HP1 isoforms cannot effectively compensate for each other’s role in regulating DNA repair and apoptosis in the context of DDR. Therefore, these three subtypes of HP1 may play non-redundant roles in DDR pathway despite their conserved domains.
HP1 is involved in the foci formation of BRCA1 and 53BP1
In response to DNA damage, the ATM/ATR kinase is activated and phosphorylates γH2AX. This event is usually followed by the recruitment of a series of DDR factors, including BRCA1, 53BP1, MDC1, RNF8, RNF168 and others, to the DSB sites (11,12). To identify which DDR factors or which steps of the DDR pathways were affected by depleting HP1, we used irradiation-induced foci-forming assays. Many DDR proteins form microscopically visible mega complexes, the so-called foci, around DSB sites in response to genotoxic stress. Because the DDR factor BRCA1 associates and co-localizes with HP1 in cells (34), and is required for HR, the formation of BRCA1 foci was analyzed. U2OS cells were transfected with either control siRNA or a siRNA designed to target one of the three HP1 subtypes (Supplementary Figure S2, left panels). Two sets of commercially available siRNA specific for HP1 were used to confirm target specificity (Supplementary Figure S5) and the resulting phenotype (data not shown) to rule out a secondary target effect. Transfected cells were irradiated (4 Gy) and then stained with an anti-BRCA1 antibody. BRCA1 foci were easily observed in U2OS cells (transfected with a control siRNA) after IR-treatment (Figure 3A), and 69.1% of the irradiated U2OS cells had >10 foci per cell. In contrast, significantly less (19–25%; P < 0.03) HP1-depleted U2OS cells had >10 foci per cell after IR compared with the control, indicating HP1 played a critical role in forming BRCA1 foci that were induced by genotoxic stress.
Figure 3.

Depletion of HP1 reduces BRCA1 foci formation, but it enhances 53BP1 foci formation. U2OS cells were transfected with the indicated siRNAs and then irradiated (4 Gy). The immunofluorescence images of BRCA1 (A and B) and 53BP1 (B) were taken at 4 h after irradiation. The percentages of cells that have >10 BRCA1 foci are plotted on the graph. These were obtained from three independent experiments that analyzed >100 cells similar to those shown in (A). (B) Double immunofluorescence assay: in the lower panel, the green foci are the BRCA1 foci and red foci are the 53BP1 foci.
Next, we looked at foci formation by 53BP1, which is another DDR factor that is involved in NHEJ, and we compared it with BRCA1 foci. Control U2OS and HP1-depleted U2OS cells were irradiated and double stained with anti-53BP1 and anti-BRCA1 antibodies. Both 53BP1 (red foci) and BRCA1 foci (green foci) formed in U2OS cells in response to IR-treatment. Foci of BRCA1 and 53BP1 did not notably overlap, as previously reported (35,36). Although most control U2OS cells contained >10 BRCA1 and 10 53BP1 foci per cell after irradiation, similar to the previous result, the number of BRCA1 foci (green foci) was clearly reduced in HP1-depleted cells (Figure 3B). Unexpectedly, the number of 53BP1 foci seemed to have increased aberrantly in HP1-depleted cells (Figure 3B and Supplementary Figure S6). Furthermore, the 53BP1 foci also looked much larger and the shape was irregular in HP1-depleted cells. Together with our initial results (Figure 1), these findings indicate that HP1-depleted cells, even in the absence of exogenous DNA-damaging agents, were under elevated genotoxic stress. This was most likely because of endogenous DNA damage that could not be properly repaired because of defects in the DDR pathways. It also suggests that HP1 could balance the number of IR-induced foci formed by BRCA1 and 53BP1 in U2OS cells. We surmise that HP1 was required both for the efficient formation of BRCA1 foci and to prevent uncontrolled large numbers of 53BP1 foci from forming in response to IR. When the roles of BRCA1 in HR repair and 53BP1 in NHEJ repair are considered, depleting HP1 would dysregulate both BRCA1 and 53BP1 foci formation, thereby leading to defective DSB repair or an inappropriate choice of DNA repair pathway by the DDR.
HP1 is required for the association of BRCA1 with chromatin
To define the mechanism of HP1 regulation of BRCA1 and 53BP1 recruitment at the molecular level, we performed ChIP assays on U2OS cells containing the 4-OHT-induced AsiSI-dependent DNA damage system (25,26). Because the restriction enzyme AsiSI is fused to the estrogen receptor ligand-binding domain (ER LBD), the restriction enzyme activity of AsiSI-ER can be induced by 4-OHT treatment. Sheared chromatin was prepared from 4-OHT- or vehicle-treated AsiSI-ER-U2OS cells. The chromatin was immunoprecipitated with an anti-BRCA1 or anti-53BP1 antibody. Specific primer pairs for two DNA breaks sites created by AsiSI [two proximal regions of Chr 1 (chr1_89231183) and Chr 6 (chr6_90404906) and a distal region (chr21_21292316) of Chr 21] were used to amplify the immunoprecipitated chromatin. Both BRCA1 and 53BP1 were recruited to AsiSI-restricted DSB sites (Figure 4A and B). Notably, occupancy of BRCA1 at DSB sites located at Chr 1 and Chr 6 was clearly induced (Figure 4A). In addition, the recruitment of 53BP1 to Chr 1 and Chr 6 DSB sites was also detected (Figure 4B, insets), supporting increased occupancy of BRCA1 and 53BP1 at DSB sites. To determine the role played by HP1 in BRCA1 and 53BP1 recruitment, specific siRNAs against the three HP1 isoforms were transfected into AsiSI-ER-U2OS cells 48 h before 4-OHT-treatment. The depletion of HP1 had marked effects on the recruitment of BRCA1 and 53BP1 to the DSB sites. BRCA1 was no longer recruited to damaged chromatin (Chr 1 and Chr 6) in HP1-depleted cells, even after 4-OHT treatment (Figure 4A). Instead, 53BP1 recruitment to Chr 1 and Chr 6 was drastically enhanced (Figure 4B). In contrast, the recruitment of BRCA1 and 53BP1 to the DSB site in the distal region of Chr 21 was only mildly affected by depleting HP1 on DSB induction (Figure 4A and B), serving a negative control. The nature of BRCA1 and 53BP1 binding to distal Chr 21, although at a lower level, is not yet known, but it might suggest other functional roles for BRCA1, such as transcription regulation in response to DSB induction (37). Consistent with our foci formation assay data (Figure 3), HP1 played a critical role in regulating the association of the BRCA1 and 53BP1 DDR proteins with chromatin. HP1 facilitated BRCA1 recruitment to the DSB sites, but impedes the recruitment of 53BP1.
Figure 4.

HP1-dependent recruitment of BRCA1 to DSB sites. AsiSI-ER-U2OS cells and HP1-depleted AsiSI-ER-U2OS cells were cultured and treated with 4-OHT (4-hydroxytamoxifen; 100 µM) for 4 h. Soluble chromatins were prepared from each culture and immunoprecipitated with specific antibodies (A: BRCA1, B: 53BP1, C: Rad51 and D: HP1). Immunoprecipitated chromatins were analyzed by quantitative PCR using specific primer pairs located at Chr 1: chr1_89231183, Chr 6: chr6_90404906, Chr 21: distal region of chr21_21292316. The results shown are relative signals from the individual precipitated DNA compared with input chromatin. siHP1 is siHP1 α, β and γ combined. Figure 4B includes two insets with enlarged scales showing the increased 53BP1 occupancy at DSB sites of Chr 1 and Chr 6 on 4-OHT treatment.
Because BRCA1 is required for HR repair, we used an anti-RAD51 antibody in the ChIP assay to further probe the role played by HP1 in HR repair. RAD51 is a human homolog of the RecA protein that forms nucleoprotein filaments to catalyze the homologous strand invasion step during recombination (38). DSB induction by 4-OHT/AsiSI-ER led to the increased association of RAD51 to Chr 1 and Chr 6 DSB sites, but not to the distal region of Chr 21 in AsiSI-ER-U2OS cells (Figure 4C). Furthermore, depleting HP1 abrogated the RAD51 recruitment to the DNA lesions on Chr 1 and Chr 6. These results indicate that HP1 regulated the recruitment of the RAD51/BRCA1 complex to the DSB sites and might regulate HR repair.
To determine whether HP1 was associated with chromatin before and after DSB induction, individual sheared chromatin was immunoprecipitated using a specific anti-HP1α antibody (Figure 4D). Different amounts of HP1α proteins were associated with three regions of chromatin we examined. More HP1α protein was associated with Chr 1 and Chr 6 than Chr 21. Interestingly, although HP1α dissociated from the chromatin after 4-OHT/AsiSI-ER-mediated induction of DSBs, the occupancy of chromatin-associated HP1α protein remained at 50–60% of the original levels. Similar to HP1α, HP1β and HP1γ associated more with the Chr 1 and Chr 6 regions than the region of Chr 21 (Supplementary Figure S7), and their occupancy at Chr 1 and Chr 6 was also reduced on DSB induction. Based on these results, we propose that only a portion of HP1 dissociates from chromatin on DSB induction, and that the remaining chromatin-bound HP1 is still capable of regulating the DDR pathway.
HP1 is specifically important for HR repair, but not NHEJ repair
The aforementioned findings indicate that HP1α, HP1β and HP1γ were required to promote BRCA1 recruitment to the sites of DNA damage. Given that BRCA1 is important for the HR repair pathway, we tested whether HP1 promoted HR. We used I-SceI-based, GFP U2OS-HR and U2OS-NHEJ reporter systems to determine the respective roles of HP1 in the HR and NHEJ repair pathways (23). I-SceI is a rare-cutting restriction endonuclease that recognizes a specific 18-bp sequence and leaves a 4-bp 5′-overhang on I-SceI-mediated cutting. The individual reporters were engineered to produce GFP-positive cells only if HR or NHEJ could repair the I-SceI-induced DSBs. Transfection of the I-SceI expression constructs into the reporter cell lines produced significant numbers of GFP-positive cells, which provided the baseline frequencies for HR and NHEJ (Figure 5). Transfection of the cells with siRNA targeting each HP1 subtypes (Supplementary Figure S2, right panels) led to a significant (P < 0.002) reduction in the number of GFP positive cells in the HR reporter cells for all HP1 subtypes (Figure 5, left panel). In contrast, depleting HP1 did not significantly affect the number of GFP-positive cells in U2OS–NHEJ reporter cells (Figure 5, right panel). These results indicate that all three HP1 subtypes were important for HR-dependent DNA repair, but they were not required for NHEJ repair. Because BRCA1 is an important factor in HR repair (39), we suggest that depleting HP1 dramatically reduced BRCA1 recruitment and foci formation, thereby impairing HR repair.
Figure 5.
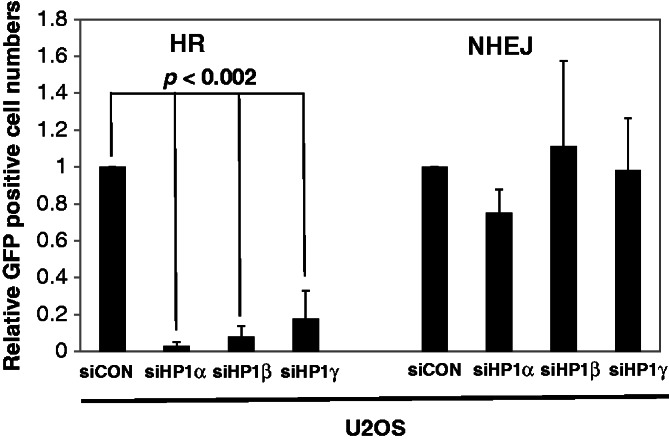
HP1 is required for HR repair. U2OS-NHEJ cells and U2OS-HR cells were transfected by individual siRNAs targeting the three respective HP1 subtypes on day one. The I-SecI expression construct was transfected at 24 h after siRNA transfection, and cells were incubated for additional 48 h. The numbers of GFP-positive cells were determined by fluorescence-activated cell sorting (FACS). GFP- and DSred-expression constructs were co-transfected to normalize for transfection efficiency in the FACS assays. The assays were repeated at least four times (n = 5), and the average relative numbers are presented as the mean ± SD.
Impaired cell cycle checkpoint control in HP1-depleted cells
It seems likely that HP1 promoted HR repair by recruiting BRCA1 to the DSB sites. The tumor suppressor activity of BRCA1 can also be assayed through its regulatory role in the cell cycle checkpoint. Accordingly, we sought to define the role of HP1 in the control of the cell cycle checkpoint. U2OS cells or HP1-depleted U2OS cells were irradiated (4 Gy), then fixed and permeabilized. Fixed cells were then stained with PI and analyzed using flow cytometry. Depleting HP1 affected the overall U2OS cell cycle profile by increasing the sub-G1 population and decreasing the G1 population (Figure 6A). U2OS cells had ∼11% of cells in sub-G1 cell, which serves as a baseline control. Notably, the sub-G1 populations of cells depleted of HP1α, HP1β and HP1γ increased to 20, 23 and 30%, respectively, which is consistent with the elevated apoptosis levels observed in HP1-depleted cells after irradiation (Figure 2). However, the G1 population of U2OS cells (27%) decreased to 12.8, 21 and 6.4% in U2OS cells depleted for HP1α, HP1β and HP1γ, after irradiation. A similar cell cycle profile was obtained using HP1-depleted AsiSI-ER-U2OS cells after 4-OHT treatment (Supplementary Figure S8). Based on the observations of increased sub-G1 populations in HP1-depleted U2OS cells, we propose that HP1 could be involved in regulating the control of apoptosis and the cell cycle.
Figure 6.

HP1 plays an important role in apoptosis and cell cycle checkpoint control. (A) U2OS cells or HP1-depleted U2OS cells were cultured in low serum medium for 18 h and irradiated at 4 Gy. The cells were cultured for another 3 h, stained with PI and 5 × 104 were analyzed by flow cytometry. (B) U2OS, HCC1937 (BRCA1-deficient or supplemented) cells and HP1-depleted MCF7 and HCC1937 cells were cultured in low serum medium for 18 h. Irradiated and sham-irradiated cells were cultured for 3 h before fixing, then stained with an anti-histone H3 phospho-serine 10 antibody (Millipore) and analyzed by FACS.
Because BRCA1 reportedly plays a critical role in control of the G2→M checkpoint during DDR (40), we investigated whether HP1 affects the phosphorylation of histone H3 at serine 10, a hallmark of mitosis. The U2OS mitotic cell population, as measured by H3 serine 10 phosphorylation, was ∼4% before irradiation, but dropped to 0.5% after irradiation, in agreement with a G2/M cell cycle arrest (Figure 6B, first panel, second row). In HP1-depleted U2OS cells, the mitotic cell population was between 2 and 3%, which was somewhat lower than that of the parental U2OS cells. However, the HP1-depleted cells, particularly the HP1α- and HP1β-depleted cells, showed little reduction in mitotic cells after irradiation. Altogether, the data suggest that depleting HP1 caused defects in the arrest of the G2/M cell cycle that resulted in increased apoptosis and impaired G2/M arrest in U2OS cells after irradiation.
To validate this hypothesis, the same experiments were performed using isogenic BRCA1-supplemented and BRCA1-negative HCC1937 breast cancer cells (41). Irradiation of BRCA1-supplemented HCC1937 cells decreased the mitotic cell population by 75% (Figure 6B, second panel, second row). However, BRCA1-supplemented HCC1937 cells depleted of HP1 showed little, if any, change in the size of the mitotic cell population. These data also suggest the arrest of G2/M cell cycle was defective in HP1-depleted cells. These data confirm that similar to BRCA1, HP1 is required for G2/M cell cycle checkpoint control. Next, we analyzed the G2/M cell populations in BRCA1-negative HCC1937 cells. In contrast to the BRCA1-supplemented HCC1937 cells, depleting HP1 from BRCA1-negative HCC1937 cells increased the mitotic cell population (Figure 6B, third panel, second row). Furthermore, the mitotic cell population of BRCA1-negative HCC1937 cells was not reduced after irradiation, supporting a critical role for BRCA1 in control of the G2/M cell cycle checkpoint. Even depleting HP1 did not affect the size of the mitotic cell population in the irradiated BRCA1-negative HCC1937 cells. These data suggest that HP1-mediated control of the G2/M cell checkpoint is dependent on BRCA1 function, indicating that some kind of interaction between HP1 and BRCA1 is required for proper arrest of the G2/M cell cycle checkpoint.
HP1 depletion increases the cell proliferation potential of MCF7 breast cancer cells
Our aforementioned results demonstrate that HP1 played essential roles during DDR. Because defects in the DDR pathways frequently lead to genome instability in mammalian cells, we performed colony formation assays to examine the role of HP1 in cell proliferation in vitro, a hallmark of tumorigenesis. Equal numbers (100 per well) of MCF7 cells and HP1-depleted MCF7 cells were cultured for 10 days, and the colonies were stained with crystal violet. On average, MCF7 cells formed 20 colonies (Figure 7A). In contrast, HP1-depleted MCF7 cells formed more colonies of larger sizes. In particular, the MCF7 cells depleted of HP1α and HP1β formed 2- to 3-fold more colonies than the control MCF7 cells. The data suggest that depleting HP1 enhances the clonogenic potential of MCF7 breast cancer cells. Therefore, deregulation or repression of HP1 might be a key event in tumorigenesis for some types of cancers.
Figure 7.

HP1 depletion enhances colony formation of MCF7 breast cancer cells. Equal numbers (100) of MCF7 cells or HP1-depleted MCF7 cells were cultured in each well of six-well plates. Colonies were stained with crystal violet on Day 10. A representative photo is shown (left panel), and the results were collected from three independent experiments, presented as mean ± SD (histogram, right panel). The plating efficiency was between ∼20 and 65%.
DISCUSSION
Dynamic interaction of HP1 with chromatin and other DDR factors in DDR pathway
The DDR pathway is a multi-step pathway that involves numerous protein factors (11,12). However, the molecular roles of the HP1 subtypes in DDR pathway were unclear. This report demonstrates that HP1 plays essential roles in DDR pathways, including DNA HR repair, control of cell cycle checkpoint and induction of apoptosis. HP1 was originally identified as an important regulator of chromatin formation and gene expression (4,14). However, HP1 may have more diverse functions in DNA repair and tumorigenesis (4,21,22). Recently, several groups showed that HP1 can dynamically associate or dissociate with chromatin in response to DNA damage. Results from several independent studies have also shown that HP1 dynamically associates and dissociates from chromatin in response to DNA damage (15–19). For HP1β, threonine-51 phosphorylation of may be involved in the dynamics (15). Our data showed that HP1α and other HP1 subtypes dissociated from chromatin after DNA damage (Figure 4D and Supplementary Figure S7). However, our ChIP assay also showed that 40–60% of HP1 remained associated with chromatin after DNA damage, suggesting a dynamic association of HP1 during DNA damage repair processes.
The exact mechanisms governing HP1 association with chromatin during DDR is not yet completely understood. Critically, our results suggest that two of the essential roles played by HP1 are to promote recruitment of BRCA1 to DSB sites to form foci and to inhibit the recruitment of 53BP1 to DSBs in response to irradiation (Figures 3 and 4). One possible mechanism for HP1 in the DDR pathway is that it acts as an adapter protein for the efficient assembly of BRCA1, KAP1 and other DDR proteins at the sites of DNA damage. Previously, laser microirradiation assays showed that depleting HP1α impairs the localization of BRCA1, 53BP1, Rad51 and CAF1 to damaged chromatin (20), suggesting active roles for HP1α in the DDR pathway. Baldeyron et al. (20) suggested that HP1α regulates HR repair, especially through its interaction with CAF1. However, they did not elaborate why their knockdown of HP1α specifically affected HR, but not NHEJ, repair (please see later in the text). Another possibility is that HP1 acts through remodeling of the chromatin structure. HP1 associates mostly with heterochromatic regions through its binding to methylated lysine 9 residues of histone H3 (H3K9Me2 or Me3), a heterochromatin histone mark (7). HP1 may also increase the methylation level of H3K9 through the direct recruitment of the Suv39H1 enzyme to chromatin (14). A recent report showed that the tumor suppressor function of BRCA1 is dependent on heterochromatin structure (42). BRCA1-deficient cells frequently show de-repression of genes and the reduced numbers of heterochromatin centers. Together with our results, it suggests that both HP1 and the heterochromatin structure may contribute to the tumor suppressor and HR repair function of BRCA1.
Unexpectedly, the chromatin association and formation of 53BP1 foci were increased in HP1-depleted cells. Recent reports show that BRCA1 foci and 53BP1 foci do not significantly overlap (36). Furthermore, enrichment of BRCA1 at foci antagonizes recruitment of 53BP1 and efficient 53BP1-dependent NHEJ repair (35). The recruitment of 53BP1 to chromatin occurs after γH2AX phosphorylation by ATM, binding of MDC1 and binding of RNF8 to damaged chromatin (43). Furthermore, the tudor domain of 53BP1 associates with methylated histone H3K79 (H3K79Me2) and histone H4K20 (H4K20Me2) (43–46). As many protein–protein interactions and histone modifications are required for the recruitment of 53BP1 to chromatin, HP1 may control the level of histone methylation and protein interaction of 53BP1 with other factors directly or indirectly. For example, HP1 may suppress 53BP1 recruitment and NHEJ repair by controlling the level of histone H3 lysine 36 methylation (H3K36Me2) and KDM4A demethylase activity (47–49). It seems that increased 53BP1 recruitment could represent a compensatory mechanism in BRCA1-deficient or HP1-deficient cells. Thus, the dynamic interaction of HP1 with chromatin and other DDR factors may determine the DNA repair pathway choices and cell fate in response to DNA damage.
HP1 is required for the induction of HR repair
Here, we also showed that HP1 was required for HR DNA repair, but not NHEJ repair. This result implies that HP1 may be a factor that influences the choice between the HR and NHEJ repair pathways for DSBs. Mechanisms for cells to choose between the two DNA repair pathways have not been clear until now. Published literature indicates that the repair choice could depend on cell cycle status, chromatin structure and other factors (50). HR repair takes place during the S and G2 phases, whereas NHEJ repair is more prevalent in cells at the G1 stage of the cell cycle. Recent reports suggest that BRCA1 is involved in the HR DNA repair pathway, and BRCA1 associates more with heterochromatic regions (42,51,52). However, HR repair by BRCA1 is inhibited by the DDR factor 53BP1, and 53BP1 may enhance the activity of the NHEJ DNA repair pathway (51,52). Our results show, unequivocally, that HP1 promoted HR and recruits BRCA1 to form foci. In contrast, depleting HP1 increased 53BP1 association with DSB sites and 53BP1 foci formation. It is plausible that the local concentration of HP1 could determine the occupancy of BRCA1 or 53BP1 on chromatin, supporting the hypothesis that HP1 is a critical determinant involved in the choice between HR and NHEJ repair. Our study suggests that different local concentrations of HP1 on chromatin could influence the outcome of the DDR (Figure 8). Irradiating cells caused the HP1 protein to partially dissociate from chromatin but the remaining HP1 facilitated the recruitment of BRCA1 to the damaged DNA sites (Figures 4D and 8). DNA lesions in HP1 abundant chromatin are repaired during G2/M cell cycle arrest by BRCA1-mediated HR repair. However, more 53BP1 was clearly recruited to DSB sites in HP1-depleted and BRCA1-deficient cells, which used NHEJ for repair or apoptosed. Very recently, Soria and Almouzni (53) reported differential contribution of HP1 subtypes to HR, during the time our manuscript was under review. Notably, results from their sister chromatid exchange (SCE) assay that HP1 depletion reduced SCE activities have independently validated our observations reported herein. However, there is a discrepancy between Soria’s and our data with respect to the role of HP1γ in HR repair as determined by reporter assays. Nonetheless, it does not affect our main conclusion on that HP1 promotes HR but not NHEJ repair. Therefore, we propose that differential local concentrations of HP1 could play a critical role in determining which DSB repair pathway is used (e.g. HR versus NHEJ).
Figure 8.

The roles of HP1 for BRCA1 function and the choice between HR and NHEJ repair. HP1 is a chromatin-associated protein, but the distribution of HP1 on chromatin is not even. Irradiation partially removes HP1 from chromatin, but the remaining HP1 molecules have active roles in recruiting BRCA1 to the damaged DNA sites. HP1 facilitates BRCA1 recruitment to chromatin after DNA damage, thereby activating G2/M cell cycle arrest and error-free HR repair. However, if the chromatin is HP1- or BRCA1-deficient, 53BP1 should be recruited resulting in error-prone NHEJ repair of the DNA damage. The loss of both the G2/M checkpoint control and HR repair pathway may increase the likelihood of apoptosis.
HP1 proteins are not evenly distributed on chromatin. Higher concentrations of HP1 proteins are usually associated with heterochromatin, and lower concentrations of HP1 proteins are associated with euchromatin. However, there are reports showing that HP1 also associates with euchromatic regions (54,55). The association of HP1 with chromatin may also differ depending on the HP1 subtype. However, we found that all three subtypes of HP1 were involved in suppressing DNA damage, probably by regulating HR repair (Figure 5). Although there were subtle differences between the HP1 subtypes in cell cycle checkpoint control, apoptosis and colony formation (Figures 2, 6 and 7), each subtype of HP1 could not compensate for another in foci formation and HR repair (Figures 1, 3 and 5). The unique functions of each subtype of HP1 in the DDR pathway need to be further investigated. Studies of post-translational modifications or specific interactions with other proteins or non-coding RNAs may reveal the unique role of each HP1 subtype (56–58).
HP1 is an important regulator for BRCA1 in cell cycle checkpoint, apoptosis control and tumor suppression
Two consequences of the DDR are cell cycle arrest and apoptosis. Cell cycle arrest provides time for the DNA repair pathways to repair DNA lesions. More severely damaged cells are usually removed from the population by internal cell death mechanisms, such as apoptosis. Interestingly, HP1 may be involved in both these processes. HP1 was a key factor in cell cycle checkpoint control, including G2/M arrest, but HP1 also suppressed apoptosis after DNA damage (Figures 2 and 5). Probably HP1 has a role in facilitating cell cycle arrest and DNA repair and in delaying apoptosis. Previous studies support a role for HP1 in cell cycle control and apoptosis (59). They observed that depleting HP1 from Drosophila cells led to a decrease in the S and G2/M cell phases and a dramatic increase in apoptotic cells. These authors suggested that many cell cycle regulators were also misregulated in HP1-depleted Drosophila cells.
Because HP1 was an important factor for recruiting BRCA1 to chromatin and forming (Figures 3 and 4), it could control other cellular functions, including the cell cycle checkpoint and apoptosis, through its regulation of BRCA1. BRCA1 is required for G1/S, intra S and G2/M cell cycle checkpoint control and apoptosis (24,40,60). Thus, HP1 may be a critical partner protein for the tumor suppressor function of BRCA1. Although both MCF7 cells and U2OS cells have wild-type endogenous BRCA1, the endogenous BRCA1 is not functional in the DDR when HP1 is depleted. This BRCAness phenomenon includes impairment of the HR DNA repair pathway, which is an error-free repair mechanism (61). Accordingly, in HP1-depleted cells, DNA damage may only be repairable by the error-prone NHEJ pathway. Furthermore, cell cycle arrest after DNA damage is essential for maintaining genomic integrity and cell survival. A defect in the HR repair mechanism and cell cycle checkpoint control in HP1-depleted cells may lead to chromatin instability and/or carcinogenesis. This suggests that HP1 could be a critical factor in suppressing tumorigenesis in some cancers, such as breast cancer and ovarian cancer.
Previously, several groups showed that the expression of HP1 subtypes is altered in some cancer cells and tissues (62). HP1 expression levels are usually decreased in cancer cells (62). For example, HP1α expression is decreased in metastatic and aggressive breast cancer cells (63). In contrast, another group demonstrated HP1α expression is upregulated in certain breast cancer patients (64). These studies indicate the potential significance of HP1 protein in breast cancer tumorigenesis. Our study provides a novel molecular mechanism for the role of HP1 in the tumorigenesis process. Further analysis of HP1 in cancer cells may provide additional clues about the roles HP1 might play in carcinogenesis. Germ line mutations or altered expression of HP1 may be involved in dysregulation of DDR pathways and the tumorigenesis process. The significance of HP1 in the DDR pathway suggests that HP1 expression could serve as a biomarker for prognosis and as a promising target for cancer therapy. Because dysregulation of DDR also affects patient responses to anti-cancer therapies, the analysis of HP1 in cancer samples may contribute to the design of more effective therapy regimens.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online. Supplementary Figures 1–8.
ACKNOWLEDGEMENTS
The authors thank Dr Gaëlle Legube (University of Toulouse) and the RNAi consortium at Academia Sinica for valuable reagents and helpful advice, members of the Ann laboratory for helpful discussions and Dr Margaret Morgan for editing.
FUNDING
National Institute of Health Research [R01DE10742 and R01DE14183 to D.K.A., R01CA120954 to J.M.S.]; National Science Council [101-2321-B-001-029, 101-2325-B-001-033 to H.M.S.]. Funding for open access charge: National Institute of Health Research [R01DE10742 and R01DE14183].
Conflict of interest statement. None declared.
REFERENCES
- 1.Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Cheung P, Allis CD, Sassone-Corsi P. Signaling to chromatin through histone modifications. Cell. 2000;103:263–271. doi: 10.1016/s0092-8674(00)00118-5. [DOI] [PubMed] [Google Scholar]
- 3.Hiragami K, Festenstein R. Heterochromatin protein 1: a pervasive controlling influence. Cell. Mol. Life Sci. 2005;62:2711–2726. doi: 10.1007/s00018-005-5287-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeng W, Ball AR, Jr, Yokomori K. HP1: heterochromatin binding proteins working the genome. Epigenetics. 2010;5:287–292. doi: 10.4161/epi.5.4.11683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 6.Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 7.Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 8.Eskeland R, Eberharter A, Imhof A. HP1 binding to chromatin methylated at H3K9 is enhanced by auxiliary factors. Mol. Cell. Biol. 2007;27:453–465. doi: 10.1128/MCB.01576-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31:2305–2312. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature. 2002;416:103–107. doi: 10.1038/nature722. [DOI] [PubMed] [Google Scholar]
- 11.Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007;19:238–245. doi: 10.1016/j.ceb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 12.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 14.Maison C, Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 2004;5:296–304. doi: 10.1038/nrm1355. [DOI] [PubMed] [Google Scholar]
- 15.Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453:682–686. doi: 10.1038/nature06875. [DOI] [PubMed] [Google Scholar]
- 16.Luijsterburg MS, Dinant C, Lans H, Stap J, Wiernasz E, Lagerwerf S, Warmerdam DO, Lindh M, Brink MC, Dobrucki JW, et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J. Cell Biol. 2009;185:577–586. doi: 10.1083/jcb.200810035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zarebski M, Wiernasz E, Dobrucki JW. Recruitment of heterochromatin protein 1 to DNA repair sites. Cytometry A. 2009;75:619–625. doi: 10.1002/cyto.a.20734. [DOI] [PubMed] [Google Scholar]
- 18.Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV, Karpen GH. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell. 2011;144:732–744. doi: 10.1016/j.cell.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, Jeggo PA. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 20.Baldeyron C, Soria G, Roche D, Cook AJ, Almouzni G. HP1alpha recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J. Cell Biol. 2011;193:81–95. doi: 10.1083/jcb.201101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ball AR, Jr, Yokomori K. Revisiting the role of heterochromatin protein 1 in DNA repair. J. Cell Biol. 2009;185:573–575. doi: 10.1083/jcb.200904033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dinant C, Luijsterburg MS. The emerging role of HP1 in the DNA damage response. Mol. Cell. Biol. 2009;29:6335–6340. doi: 10.1128/MCB.01048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gunn A, Bennardo N, Cheng A, Stark JM. Correct end use during end joining of multiple chromosomal double strand breaks is influenced by repair protein RAD50, DNA-dependent protein kinase DNA-PKcs, and transcription context. J. Biol. Chem. 2011;286:42470–42482. doi: 10.1074/jbc.M111.309252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee YH, Bedford MT, Stallcup MR. Regulated recruitment of tumor suppressor BRCA1 to the p21 gene by coactivator methylation. Genes Dev. 2011;25:176–188. doi: 10.1101/gad.1975811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, Legube G. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010;29:1446–1457. doi: 10.1038/emboj.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Massip L, Caron P, Iacovoni JS, Trouche D, Legube G. Deciphering the chromatin landscape induced around DNA double strand breaks. Cell Cycle. 2010;9:2963–2972. doi: 10.4161/cc.9.15.12412. [DOI] [PubMed] [Google Scholar]
- 27.Lowndes NF, Toh GW. DNA repair: the importance of phosphorylating histone H2AX. Curr. Biol. 2005;15:R99–R102. doi: 10.1016/j.cub.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 28.Pinto DM, Flaus A. Structure and function of histone H2AX. Subcell. Biochem. 2010;50:55–78. doi: 10.1007/978-90-481-3471-7_4. [DOI] [PubMed] [Google Scholar]
- 29.Cleaver JE. gammaH2Ax: biomarker of damage or functional participant in DNA repair “all that glitters is not gold!”. Photochem. Photobiol. 2011;87:1230–1239. doi: 10.1111/j.1751-1097.2011.00995.x. [DOI] [PubMed] [Google Scholar]
- 30.Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011;193:97–108. doi: 10.1083/jcb.201011083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011;13:243–253. doi: 10.1038/ncb2201. [DOI] [PubMed] [Google Scholar]
- 32.Lu C, Zhu F, Cho YY, Tang F, Zykova T, Ma WY, Bode AM, Dong Z. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell. 2006;23:121–132. doi: 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 2000;275:9390–9395. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- 34.Maul GG, Jensen DE, Ishov AM, Herlyn M, Rauscher FJ., III Nuclear redistribution of BRCA1 during viral infection. Cell Growth Differ. 1998;9:743–755. [PubMed] [Google Scholar]
- 35.Chapman JR, Sossick AJ, Boulton SJ, Jackson SP. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012;125:3529–3534. doi: 10.1242/jcs.105353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mok MT, Henderson BR. A comparison of BRCA1 nuclear localization with 14 DNA damage response proteins and domains: identification of specific differences between BRCA1 and 53BP1 at DNA damage-induced foci. Cell. Signal. 2010;22:47–56. doi: 10.1016/j.cellsig.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 37.Gorski JJ, Savage KI, Mulligan JM, McDade SS, Blayney JK, Ge Z, Harkin DP. Profiling of the BRCA1 transcriptome through microarray and ChIP-chip analysis. Nucleic Acids Res. 2011;39:9536–9548. doi: 10.1093/nar/gkr679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cousineau I, Abaji C, Belmaaza A. BRCA1 regulates RAD51 function in response to DNA damage and suppresses spontaneous sister chromatid replication slippage: implications for sister chromatid cohesion, genome stability, and carcinogenesis. Cancer Res. 2005;65:11384–11391. doi: 10.1158/0008-5472.CAN-05-2156. [DOI] [PubMed] [Google Scholar]
- 39.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol. Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 40.Xu B, Kim S, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol. Cell. Biol. 2001;21:3445–3450. doi: 10.1128/MCB.21.10.3445-3450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 42.Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, Verma IM. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature. 2011;477:179–184. doi: 10.1038/nature10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.FitzGerald JE, Grenon M, Lowndes NF. 53BP1: function and mechanisms of focal recruitment. Biochem. Soc. Trans. 2009;37:897–904. doi: 10.1042/BST0370897. [DOI] [PubMed] [Google Scholar]
- 44.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huyen Y, Zgheib O, Ditullio RA, Jr, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- 46.Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–614. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 47.Fnu S, Williamson EA, De Haro LP, Brenneman M, Wray J, Shaheen M, Radhakrishnan K, Lee SH, Nickoloff JA, Hromas R. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl Acad. Sci. USA. 2011;108:540–545. doi: 10.1073/pnas.1013571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin CH, Li B, Swanson S, Zhang Y, Florens L, Washburn MP, Abmayr SM, Workman JL. Heterochromatin protein 1a stimulates histone H3 lysine 36 demethylation by the Drosophila KDM4A demethylase. Mol. Cell. 2008;32:696–706. doi: 10.1016/j.molcel.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, Sixma TK, Richard S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012;31:1865–1878. doi: 10.1038/emboj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 51.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soria G, Almouzni G. Differential contribution of HP1 proteins to DNA end resection and homology-directed repair. Cell Cycle. 2013;12:422–429. doi: 10.4161/cc.23215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kwon SH, Workman JL. The changing faces of HP1: from heterochromatin formation and gene silencing to euchromatic gene expression: HP1 acts as a positive regulator of transcription. Bioessays. 2011;33:280–289. doi: 10.1002/bies.201000138. [DOI] [PubMed] [Google Scholar]
- 55.Piacentini L, Fanti L, Berloco M, Perrini B, Pimpinelli S. Heterochromatin protein 1 (HP1) is associated with induced gene expression in Drosophila euchromatin. J. Cell Biol. 2003;161:707–714. doi: 10.1083/jcb.200303012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maison C, Bailly D, Roche D, Montes de Oca R, Probst AV, Vassias I, Dingli F, Lombard B, Loew D, Quivy JP, et al. SUMOylation promotes de novo targeting of HP1alpha to pericentric heterochromatin. Nat. Genet. 2011;43:220–227. doi: 10.1038/ng.765. [DOI] [PubMed] [Google Scholar]
- 57.Keller C, Adaixo R, Stunnenberg R, Woolcock KJ, Hiller S, Buhler M. HP1(Swi6) mediates the recognition and destruction of heterochromatic RNA transcripts. Mol. Cell. 2012;47:215–227. doi: 10.1016/j.molcel.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 58.Brower-Toland B, Findley SD, Jiang L, Liu L, Yin H, Dus M, Zhou P, Elgin SC, Lin H. Drosophila PIWI associates with chromatin and interacts directly with HP1a. Genes Dev. 2007;21:2300–2311. doi: 10.1101/gad.1564307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Lucia F, Ni JQ, Vaillant C, Sun FL. HP1 modulates the transcription of cell-cycle regulators in Drosophila melanogaster. Nucleic Acids Res. 2005;33:2852–2858. doi: 10.1093/nar/gki584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Somasundaram K, Zhang H, Zeng YX, Houvras Y, Peng Y, Wu GS, Licht JD, Weber BL, El-Deiry WS. Arrest of the cell cycle by the tumour-suppressor BRCA1 requires the CDK-inhibitor p21WAF1/CiP1. Nature. 1997;389:187–190. doi: 10.1038/38291. [DOI] [PubMed] [Google Scholar]
- 61.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer. 2004;4:814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 62.Dialynas GK, Vitalini MW, Wallrath LL. Linking heterochromatin protein 1 (HP1) to cancer progression. Mutat. Res. 2008;647:13–20. doi: 10.1016/j.mrfmmm.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kirschmann DA, Lininger RA, Gardner LM, Seftor EA, Odero VA, Ainsztein AM, Earnshaw WC, Wallrath LL, Hendrix MJ. Down-regulation of HP1Hsalpha expression is associated with the metastatic phenotype in breast cancer. Cancer Res. 2000;60:3359–3363. [PubMed] [Google Scholar]
- 64.De Koning L, Savignoni A, Boumendil C, Rehman H, Asselain B, Sastre-Garau X, Almouzni G. Heterochromatin protein 1alpha: a hallmark of cell proliferation relevant to clinical oncology. EMBO Mol. Med. 2009;1:178–191. doi: 10.1002/emmm.200900022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.