


Abstract
RNA is a major drug target, but the design of small molecules that modulate RNA function remains a great challenge. In this context, a series of structurally homologous ‘polyamide amino acids’ (PAA) was studied as HIV-1 trans-activating response (TAR) RNA ligands. An extensive thermodynamic study revealed the occurence of an enthalpy–entropy compensation phenomenon resulting in very close TAR affinities for all PAA. However, their binding modes and their ability to compete with the Tat fragment strongly differ according to their structure. Surprisingly, PAA that form loose complexes with TAR were shown to be stronger Tat competitors than those forming tight ones, and thermal denaturation studies demonstrated that loose complexes are more stable than tight ones. This could be correlated to the fact that loose and tight ligands induce distinct RNA conformational changes as revealed by circular dichroism experiments, although nuclear magnetic resonance (NMR) experiments showed that the TAR binding site is the same in all cases. Finally, some loose PAA also display promising inhibitory activities on HIV-infected cells. Altogether, these results lead to a better understanding of RNA interaction modes that could be very useful for devising new ligands of relevant RNA targets.
INTRODUCTION
In the past decade, numerous studies have emphasized the huge potential of small non-coding hairpin RNA fragments as drug targets. Indeed, such small RNA elements are often involved in crucial biological processes such as, for example, in transcription and translation steps of viral and bacterial replication cycles, through specific interactions with proteins or nucleic acids. Synthetic molecules able to bind specifically to such relevant RNA targets and to disrupt their association with cognate partners can modulate their biological functions. Therefore, such RNA ligands represent very attractive tools in molecular biology and/or pharmacological agents for treating infectious diseases.
Small molecular structures targeting viral/bacterial hairpin RNA fragments have already been identified using standard and virtual screening approaches (1–4). However, thus far, these molecules have met little clinical success as compared with those targeting proteins and, to a less extent, DNA. A deeper understanding of RNA recognition processes by both cognate partners and synthetic ligands is essential for developing new structurally optimized molecules capable of both binding specifically to RNA targets and competing strongly with the cognate partners. While RNA recognition processes have been extensively studied at the structural level, only few works have been devoted to thermodynamic studies of RNA/small ligand associations (5–11), in contrast to protein/ligand or DNA/ligand ones. Nevertheless, identification of the main molecular forces that govern RNA/ligand associations would be of great interest for a complete understanding of RNA binding modes and for drug design (12,13). Among the great number of potential hairpin RNA targets, extensive research has been dedicated to the highly conserved HIV-1 trans-activating response (TAR) RNA fragment in view of preventing its complexation with the trans-activator of transcription (Tat) protein and cellular factors (1,2,14,15). This complex plays a crucial role in the viral transcription step of HIV. We have previously reported a new type of molecules named α- polyamide amino acids (PAA) that represents promising structures as HIV TAR ligands (16). These molecules are constituted by a poly-(2-aminoethylglycyl) backbone onto which are condensed l-α-amino acids. From a small library of α-PAA trimers, we identified several compounds able to bind to TAR RNA and to prevent the Tat/TAR association at a micromolar level (17). Some of them displayed an anti-HIV activity in peripheral blood mononuclear cells (PBMC). However, as these compounds have a short half-life in cell culture media, structural modifications have been considered to improve stability. Thus, starting from seven tri-α-PAA previously identified as ‘lead’ compounds (Figure 1, 1a-7a), two new series of tri-PAA analogs modified either on the polyamide backbone or on the amino acid moiety have been elaborated. In the first series, the α-amino acid residues are changed for their corresponding β-analogs (β-PAA series, 1b–7b), whereas for the second series, the (2-aminoethylglycyl) unit of the backbone is replaced by a 2-aminoethyl β-alanyl one (C-α-PAA series, 1c–7c).
Figure 1.

Structures of the three series of PAA.
Besides the synthesis of the newly designed PAA analogs, we report here a detailed study on the TAR binding of the three series of PAA, in the absence or presence of a Tat fragment competitor. For the three PAA series, beyond affinity and specificity criteria, each PAA/TAR equilibrium has been characterized by an extensive thermodynamic study, providing new insights on the binding modes that led to ‘structure/activity’ relationships. To support our hypotheses concerning the distinctive binding modes of PAA, the structural TAR RNA changes induced on binding of PAA have been investigated by circular dichroism (CD) studies, and the TAR interaction sites of two PAA models have also been identified through NMR experiments. Important enough, we also report here our results about anti-HIV activity and toxicity on infected and non-infected PBMC.
MATERIALS AND METHODS
Experimental procedures
Unless otherwise stated, all reagents and solvents were of analytical grade and from Sigma (St Louis, MO, USA). HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid] and all inorganic salts for buffers were purchased from Calbiochem (molecular biology grade). RNA and DNA oligonucleotides were purchased from IBA GmbH and used without further purification. A mixture of yeast pre- and mature tRNAs (containing >30 different species) was purchased from Sigma (type X-SA). The labeled Tat peptide was purchased from EZBiolab (Carmel, CA, USA) and used without further purification. All buffers were filtered through 0.22-μm Millipore filters (GP ExpressPLUS membrane). A small aliquot (50–100 ml) was first filtered and then discarded to avoid any contaminants that might have been leached from the filter. Solutions used for fluorescence experiments were prepared by diluting the concentrated stocks in Milli-Q water and filtered again as described above.
Fluorescence binding assays
Ligand solutions were prepared as serial dilutions by an epMotion automated pipetting system (eppendorf) in buffer A [20 mM HEPES (pH 7.4 at 25°C), 20 mM NaCl, 140 mM KCl and 3 mM MgCl2] at a concentration twice higher than the desired final concentration that will be reached after addition of the RNA solution. The appropriate ligand solution (30 µl) was then added to a well of a non-treated black 384-well plate (Nunc 237105), in triplicate. Refolding of the RNA was performed using a thermocycler (ThermoStatPlus Eppendorf) as follows: the RNA, diluted in 1 ml of buffer A, was first denatured by heating to 90°C for 2 min and then cooled to 4°C for 10 min, followed by incubation at 20°C for 15 min. After refolding, the RNA was diluted to a working concentration of 10 nM through addition of the appropriate amount of buffer A. The tube was mixed, and 30 µl of the RNA solution was added to each well containing ligand. This subsequent dilution lowered the final RNA concentration to 5 nM. The fluorescence was measured on a GeniosPro (Tecan) with an excitation filter of 485 ± 10 nm and an emission filter of 535 ± 15 nm. Each point was measured five times with a 500-μs integration time and averaged. Binding was allowed to proceed for at least 30 min at room temperature to achieve equilibrium.
To study the temperature dependence, the plates were incubated after 30-min equilibrium at different temperatures ranging from 5°C to 35°C. The salt dependence was studied in 20 mM HEPES (pH 7.4 at 25°C), 20 mM NaCl and 3 mM MgCl2, with the KCl concentration varying from 70 to 250 nM. For competitive experiments in the presence of a dsDNA, a 15-mer sequence (5′-CGTTTTTATTTTTGC-3α) and its complement, annealed beforehand, were added to buffer A to obtain a 100-fold nucleotide excess over TAR RNA (900 nM duplex; 5 nM RNA). For competitive experiments in the presence of tRNA, a mixture of pre- and mature yeast tRNAs (containing >30 different species from baker’s yeast (Saccharomyces cerevisiae, Sigma, type X-SA) was added to buffer A to obtain a 100-fold nucleotide excess over TAR RNA. Stock solutions of tRNA were prepared in water and quantified using an extinction coefficient of 9640 cm−1 M−1 per base (18).
Fluorescence resonance energy transfer (FRET) displacement assays
Ligand solutions and RNA (40 nM working solution) were prepared as described above in buffer B [50 mM tris buffer (pH 7.4 at 25°C), 20 mM KCl and 0.005% Tween 20]. Labeled Tat peptide was prepared at 40 nM in buffer B and mixed to an equal volume of TAR RNA for 20 min at room temperature to form the Tat/TAR complex before adding the ligand. The appropriate ligand solution (30 µl) was then added to a well of a non-treated black 384-well plate, in triplicate, and 30 µl of the Tat/TAR solution was added. This subsequent dilution lowered the final Tat/TAR concentration to 10 nM. Fluorescence was measured as described above after 30 min of incubation at room temperature.
Temperature-dependent UV spectroscopy (UV melting)
Thermal denaturation scans were obtained using a Cary 300 (Varian) spectrophotometer equipped with an electrothermal multicell holder. A quartz cell of 700 µl with 1-cm path length was used for all the absorbance studies. Absorbance versus temperature profiles were recorded at 260 nm. After structuration of TAR RNA and incubation (1 h) with the corresponding ligand, the temperature was raised from 25 to 90°C, with a heating rate of 0.5°C/min. Thermal denaturation studies were carried out at 1 μM TAR RNA and PAA concentrations of 0 (TAR alone), 1 and 5 μM. The experiments were performed in buffer D [10 mM sodium cacodylate, 10 mM NaCl (pH 7.5) and 0.1 mM EDTA]. The melting temperature (Tm) value was taken as the midpoint of the melting transition as determined by the maximum of the first-derivative plot with Prism software.
CD study
CD measurements were performed with a Jasco J-810 spectropolarimeter equipped with a Jasco PTC 423S Peltier temperature controller. Samples were prepared in buffer C [50 mM tris buffer (pH 7.4 at 25°C), 20 mM KCl and 0.005% Tween 20]. Spectra were obtained at 3 μM RNA or PAA (for individual spectra) or a molar ratio of 1:1, 1:5 and 1:10 RNA:PAA for the complexes in a 1-mm path length cell after 1 h incubation time in the case of RNA/PAA complexes. RNA samples were heat denatured and allowed to refold as described above before measurement. Spectra were recorded at 20°C from 360 to 200 nm at 1-nm intervals, with an integration time of 4 s and a 50-nm/min speed. CD scans were repeated five times and then averaged and corrected by the subtraction of buffer background.
Data analysis
Binding data (KD and FRET experiments) were analyzed using Prism 5 (GraphPad Software) by non-linear regression following the equation
KD values were converted to ΔG° values as ΔG° = −RTln KD.
Salt dependence of KD was analyzed by the following equation:
| (1) |
where Knel is the dissociation constant at the standard state in 1 M KCl, Z is the number of ions displaced from the nucleic acid and ψ is the fractional probability of a counterion being thermodynamically associated with each phosphate of the RNA. Knel and Zψ were treated as fitting parameters.
For thermodynamic analysis, ΔG° values were plotted versus temperature (T). Non-linear regression using the three-parameter fit in Prism 5 was used to fit the following equation to the data:
| (2) |
where Tr is a constant reference temperature (in our study Tr = 293.15 K), and the three fit parameters are as follows: ΔH°Tr, the change in enthalpy on binding at Tr; ΔS°Tr, the change in entropy on binding at Tr; and ΔCp, the change in heat capacity. ΔCp was assumed to be independent of temperature; inclusion of a ΔCp/ΔT term in the analysis did not improve the quality of the fits and gave larger standard errors for the returned parameters.
ΔH°T and ΔS°T were calculated from the results obtained from the fitting of the curve ΔG° values versus T by the Equation (2) using the following equation:
| (2.1) |
and
| (2.2) |
where 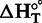 is the change in enthalpy on binding at T (25°C), and
is the change in enthalpy on binding at T (25°C), and 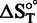 is the change in entropy on binding at T (25°C).
is the change in entropy on binding at T (25°C).
For enthalpy–entropy compensation (EEC) analysis, TΔS° values were plotted versus ΔH°. The linear regression in Prism 5 was used to fit the following equation to the data:
| (3) |
Chemistry, characterization (HPLC, HRMS ESI+) of new compounds, NMR and biological assay experiments as well as melting temperature data are given in supporting information.
RESULTS AND DISCUSSION
Chemistry
Solid-phase synthesis of the seven α-PAA hits (Figure 1, 1a–7a) was previously described (17). β-PAA (1b–7b) and C-α-PAA (1c–7c) analogs were prepared following the same procedure, i.e. on a β-alanine-functionalized 4-methylbenzhydrylamine (MBHA) resin, starting from fully and orthogonally N-protected β-PAA or C-α-PAA monomers, respectively. This procedure, illustrated in Figure 2A, includes (i) three successive elongation/deprotection steps involving selected monomers, (ii) acetylation of the last residue and (iii) cleavage of the protecting groups and simultaneous release of the trimeric compounds from the resin. PAA derivatives 1b–c/7b–c were obtained in high HPLC purity (from 83 to 98%) and were purified by semi-preparative HPLC. Structures were confirmed by HRMS experiments (see Supplementary Table S1 in supporting information).
Figure 2.

Synthesis of (A) β-PAA and (B) C-α-PAA protected monomers. (i) HBTU, DIEA, DMF; (ii) for 10b–c and 11b–c: LiOH 1 N, H2O, THF; for 12b–c: Pd(PPh3)4/DEA/DMF; and (iii) CH3CN, Δ, 9 h.
The protected β-PAA monomer synthons 13b–15b were prepared using a two-step procedure (Figure 2B), involving the condensation of the protected β-amino acid residues onto the N-Boc methyl or allyl ester backbone 8 or 8’, and then carboxyl deprotection, i.e. saponification of methyl esters 10b–11b or reaction with catalytic Pd0(PPh3)4 in the case of allyl ester 12b. Similarly, C-α-PAA monomers 13c–15c were synthesized starting from protected α-amino acid residues and from N-Boc methyl or allyl ester backbone 9 or 9’ (Figure 2B). Backbones 9–9’ were obtained in one step, following a Michael addition of the Boc-ethylenediamine on methyl or allyl acrylate, respectively.
Interaction studies: affinity and specificity
Dissociation constants (KD; Table 1) associated with TAR/PAA 1a–c/7a–c equilibria were determined by monitoring the fluorescence change of a TAR18–44 fragment labeled with a fluorescent group (Alexa 488), as previously described (16). Analysis of the TAR/α-PAA previously revealed a one-to-one stoichiometry of the binding (17), and representative curves of the new compounds could perfectly be fitted with a one-to-one model using the non-linear least-squares numerical solver–based binding data global analysis program BIOEQS (19) (data not shown). Whatever the series and the sequence, all PAA derivatives strongly bind to TAR with similar submicromolar affinities (0.2 µM < KD < 0.9 µM). In most cases, C-α-PAA (1c–7c) and β-PAA (1b–7b) compounds display slightly higher affinities than their corresponding α-PAA (1a–7a) (2.5-fold maximum), and within each series, the FKR sequence shows the weakest TAR affinity.
Table 1.
TAR affinity (KD), specificity (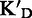 and
and 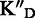 ) and inhibitory constant (IC50) associated with tri-PAA at 25°C (in µM)a
) and inhibitory constant (IC50) associated with tri-PAA at 25°C (in µM)a
| PAA sequence | α-PAA (a) |
β-PAA (b) |
C-α-PAA (c) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KD | IC50 | KD | IC50 |
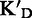 (+ tRNA mixb) (+ tRNA mixb) |
Kd’/ Kd |
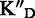 (+ dsDNA mixc) (+ dsDNA mixc) |
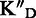 / KD / KD
|
KD | IC50 |
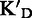 (+ tRNA mixb) (+ tRNA mixb) |
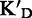 / KD / KD
|
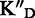 (+ dsDNA mixc) (+ dsDNA mixc) |
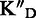 / KD / KD
|
|
| FFK 1 | 0.49 ± 0.02 | 340.5 ± 0.4 | 0.24 ± 0.02 | 42.4 ± 0.7 | 0.4 ± 0.1 | 1.7 | 0.5 ± 0.1 | 2.1 | 0.37 ± 0.02 | 122.8 ± 0.5 | 0.9 ± 0.2 | 2.4 | 0.9 ± 0.3 | 2.4 |
| FFR 2 | 0.47 ± 0.03 | 103.8 ± 0.4 | 0.25 ± 0.01 | 28.7 ± 0.5 | 0.26 ± 0.12 | 1 | 0.39 ± 0.05 | 1.6 | 0.2 ± 0.01 | 108.1 ± 0.8 | 0.83 ± 0.19 | 4.3 | 0.39 ± 0.04 | 2 |
| RFF 3 | 0.46 ± 0.04 | 132.8 ± 0.7 | 0.23 ± 0.01 | 22.8 ± 0.8 | 0.6 ± 0.1 | 2.6 | 0.30 ± 0.05 | 1.3 | 0.36 ± 0.03 | 66.6 ± 0.7 | 0.70 ± 0.15 | 1.9 | 0.70 ± 0.05 | 1.9 |
| FRF 4 | 0.73 ± 0.10 | 599 ± 5.0 | 0.28 ± 0.02 | 8.7 ± 0.4 | 1.1 ± 0.4 | 6.7 | 0.82 ± 0.16 | 2.9 | 0.26 ± 0.02 | 74 ± 0.5 | 1.70 ± 0.44 | 6.5 | 0.60 ± 0.06 | 2.3 |
| FKR 5 | 0.86 ± 0.09 | 12.1 ± 0.8 | 0.70 ± 0.05 | 1.5 ± 0.6 | 0.7 ± 0.15 | 1 | 0.6 ± 0.1 | 0.9 | 0.91 ± 0.11 | 18 ± 0.8 | 1.52 ± 0.54 | 1.7 | n.d. | n.d. |
| FRR 6 | 0.47 ± 0.05 | 3.8 ± 0.8 | 0.31 ± 0.02 | 0.8 ± 0.3 | 0.34 ± 0.13 | 1.1 | 0.27 ± 0.04 | 0.9 | 0.34 ± 0.02 | 2.7 ± 0.8 | 0.80 ± 0.15 | 2.4 | 0.60 ± 0.15 | 1.8 |
| RRF 7 | 0.72 ± 0.07 | 7.6 ± 0.7 | 0.32 ± 0.02 | 0.7 ± 0.1 | 0.5 ± 0.1 | 1.6 | 0.32 ± 0.05 | 1 | 0.34 ± 0.02 | 5.4 ± 0.8 | 0.7 ± 0.2 | 2.1 | 0.6 ± 0.1 | 1.8 |
aAll standard fluorescence measurements were performed in buffer A (20 mM HEPES (pH 7.4 at 25°C), 20 mM NaCl, 140 mM KCl and 3 mM MgCl2).
bMeasured in the presence of a 100-fold nucleotide excess of a mixture of natural tRNA (tRNAmix).
cMeasured in the presence of a 100-fold nucleotide excess of a 15-mer duplex DNA.
To assess the ligand specificity, KD values of C-α-PAA and β-PAA were measured in the presence of a large excess of tRNA (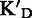 ) and dsDNA (
) and dsDNA (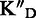 ). We have previously shown that tetra- and tri-α-PAA display a high TAR specificity in the presence of tRNA competitors, whereas only tetramers retain this high specificity in the presence of dsDNA (16,17). As shown in Table 1, most C-α-PAA and β-PAA retain a good specificity for TAR in the presence of both tRNA and dsDNA. Nevertheless, PAA containing F-rich sequences are less specific than PAA containing more cationic K/R-rich sequences (see PAA 1–4 versus PAA 5–7 in Table 1). This suggests that cationic residues are preferentially involved in specific interactions, presumably through hydrogen bonding rather than ionic interactions.
). We have previously shown that tetra- and tri-α-PAA display a high TAR specificity in the presence of tRNA competitors, whereas only tetramers retain this high specificity in the presence of dsDNA (16,17). As shown in Table 1, most C-α-PAA and β-PAA retain a good specificity for TAR in the presence of both tRNA and dsDNA. Nevertheless, PAA containing F-rich sequences are less specific than PAA containing more cationic K/R-rich sequences (see PAA 1–4 versus PAA 5–7 in Table 1). This suggests that cationic residues are preferentially involved in specific interactions, presumably through hydrogen bonding rather than ionic interactions.
FRET displacement assays
The ability of PAA to displace a Tat fragment from a preformed TAR/Tat complex was assessed via a FRET assay, developed from a previously described procedure (20), using a fluoresceinated Tat peptide fragment (amino acids 48–57) and a Dabcyl-labeled TAR fragment (18–44). In the absence of PAA, association of these two partners results in an efficient quenching of the dyes. On addition of increasing amounts of PAA, the fluorescence rises in all cases, demonstrating that PAA can displace the Tat fragment from a preformed Tat/TAR complex. IC50 values associated with each PAA are given in Table 1. Based on these disparate values, ranging from 0.7 to 600 µM, one can infer that the ability to displace Tat strongly depends on both the nature and sequence of PAA, in contrast to KD values, which are similar for all PAA. All β-PAA emerge as stronger Tat/TAR inhibitors than their corresponding C-α- and α-PAA (from 3- to 12-fold and 3.5- to 69-fold, respectively), although KD values differ, at best, by a factor 3. For each series, derivatives containing two phenylalanine residues [F-rich PAA: (1–4)a–c] display a weaker activity than those containing two basic ones [R/K-rich PAA: (5–7) a–c]. The best F-rich PAA within each series (α-FFR 2a, β-FRF 4b and C-α-RFF 3c) is, respectively, 25-, 27- and 13-fold less active than the best R/K-rich PAA within the same series (α-FRR 6a, β-RRF 7b and C-α-FRR 6c), although they all display a similar TAR affinity. These results point out that the determination of a KD value is not enough to predict the ability of a TAR ligand to compete with the Tat protein. One hypothesis would be that even if two PAA display the same affinity for TAR, they could interact at different sites of the hairpin RNA, in such a way that the competitive binding of the Tat protein could be more or less hampered. However, because all PAA (except FFK sequences) contain at least one arginine residue, it is likely that they interact at the bulge region of TAR, as previously reported for several guanidinium-containing compounds (21,22). NMR studies were therefore conducted to identify the TAR binding site and to compare the mode of interaction for two PAA taken as models of weak F-rich (β-FRF 4b) and strong R/K-rich (β-FRR 6b) inhibitors.
NMR studies
Two distinct NMR experiments were implemented. A 2D-TOCSY experiment was monitored to identify the TAR pyrimidine residues that experience noticeable chemical shift changes on addition of PAA 4b and 6b (C and U residues of TAR yield distinctive H5–H6 cross-peaks in 2D-TOCSY spectrum). In parallel, the monitoring of the imino resonance region of TAR on titration of β-PAA led to the identification of the G and U residues involved in the interaction. Figure 3 shows the overlaid TOCSY spectra of the pyrimidine region of TAR in the absence (in black) and presence (in red) of five equivalents of β-FRR (Figure 3A1) or β-FRF (Figure 3A2), respectively. One-dimensional imino spectra in Figure 3B show the selective changes in chemical shifts as β-FRR (Figure 3B1) or β-FRF (Figure 3B2) is added to free TAR (from 0.5 to 10 equivalents). Figure 3C underscores the residues exhibiting either a significant pyrimidine H5–H6 proton chemical shift change (in blue; see Supplementary Table S2 in supporting information) or a significant imino resonance change (in green) on addition of β-PAA. Altogether, these two NMR experiments indicate that the binding site of both compounds is centered around the bulge. In addition, for both β-PAA, the interaction seems to be very specific because other residues located either in the lower stem or the loop remained unaffected even in the presence of a 10-fold excess of β-PAA (data not shown for 2D-TOCSY). The main chemical shift changes observed for the residues of the bulge region are likely due to a folding transition in TAR as previously reported for other ligands carrying a guanidinium group (23–27). In free TAR RNA, a partial stacking of the three bulge nucleotides induces a bending in the overall helix axis. On complexation, the two helical stems coaxially stack to form one continuous helix; the two bulge nucleotides C24 and U25 become unstacked and adopt a highly flexible looped-out conformation. Even if our global NMR data display very similar results for both compounds, a more detailed comparison between 1D imino spectra of the two complexes reveals some differences. In the case of the β-FRR binding, the cluster corresponding to U42, G36 and/or G44 residues is shifted, whereas it is not clearly disrupted in the case of the β-FRF binding (Figure 3B). Moreover, one can notice the appearance of an extra signal at 13.9 ppm (asterisked in Figure 3B1) on addition of high concentrations of β-FRR (>2 eq.), which disappears with increasing temperature and reappears when the temperature decreases (data not shown). Although it is impossible to identify clearly this peak through 1D imino spectra, it could be hypothesized that it corresponds to the imino proton of the U23 residue involved in a base triple with A27/U38, as previously proposed for a few TAR complexes (23–25). In contrast, no extra signal appears around 14 ppm in the case of the β-FRF binding. Therefore, even if the mode of interaction is not strictly the same because distinct local environments can be observed, these results indicate that the TAR binding site of F-rich and K/R-rich PAA is centered around the bulge.
Figure 3.

A/, top: 2D-TOCSY spectra showing pyrimidine H5–H6 cross-peaks for TAR. Black: free TAR (100 µM); red: PAA/TAR complex with β-FRR 6b (A1/) or β-FRF 4b (A2/) at a ratio 5:1. Arrows indicate chemical shift changes on PAA binding. The spectra were acquired at 35°C in a D2O buffer (50 mM NaCl, 20 mM phosphate, pH 7.4). B/, bottom: stacked plot of 1D NMR spectra of the imino region of 50 µM TAR RNA with increasing concentration of β-FRR (B1/) or β-FRF (B2/). The spectra were collected at 286K in a H2O/D2O (90/10) buffer (20 mM phosphate and 50 mM NaCl, pH 7.4). C/, bottom: secondary structure of the 27-mer TAR RNA fragment. Residues shown in blue are those exhibiting a pyrimidine H5–H6 proton chemical shift change (>0.1 ppm, see Table S2 in supplementary data) on addition of β-PAA 4b or 6b. Residues in green are those experiencing an imino resonance change (>0.1 ppm).
Thermodynamic studies
To gain further insights on the TAR binding modes of the three PAA series, thermodynamic binding profiles associated with each PAA/TAR equilibrium were determined. Free energies of Gibbs (ΔG°) were first calculated from the dissociation constants (ΔG° = −RTlnKD) and were found very similar whatever the PAA (from −35 to −38 kJ/mole). The Gibbs energy can be split into two components: the pure electrostatic (polyelectrolyte) contribution ΔG°el and the non-electrostatic (non-polyelectrolyte) contribution ΔG°nel. While ΔG°el is correlated to ionic interactions occurring between two groups of opposite charge and is mainly an entropic effect, ΔG°nel reflects non-ionic events such as the hydrophobic effect that is principally an entropic event, and non-electrostatic interactions (H bonds, Van der Waals, π-stacking, etc.).
Electrostatic and non-electrostatic contributions to the Gibbs energy
To determine electrostatic (ΔG°el) and non-electrostatic (ΔG°nel) contributions associated with each PAA/TAR equilibrium, the KD dependency on the ionic strength of the solution was studied, over a range of KCl concentration from 70 to 250 mM. In all cases, a linear relationship following Equation (1) was obtained between log(KD) and log[KCl], the affinities decreasing with increasing salt concentration, as generally observed for charged molecules. This ionic strength dependence may be explained by the displacement of inorganic salt ions from both the PAA and the RNA, to form an intermolecular ‘ion pair’. According to the polyelectrolyte theory developed to describe DNA–polylysine interactions (28), it can be assumed that the energetic effect of ion displacement from polylysine (or in the present case a PAA) is negligible, and that ion displacement from the nucleic acid energetically predominates. From Equation (1), the slope of log(KD) versus log[KCl] is equal to ZΨ, where Z is the number of ions displaced from the nucleic acid (essentially the number of intermolecular ion pairs) and Ψ is the fractional probability of a counterion being thermodynamically associated with each phosphate of the RNA. For all TAR/PAA interactions, the slopes vary from 0.5 to 1.5. Using estimates for Ψ ranging from 0.68 to 0.89 for single- or double-stranded nucleic acids, respectively (29), Z ranges approximately from 1 to 2. This indicates that regardless of the sequence and nature of PAA, no more than two positive counterions are released on binding. These results seem to demonstrate that the majority of ammonium and guanidinium groups of PAA interact with TAR via hydrogen bonding and/or π-cation interactions rather than via ionic interactions with the phosphate backbone.
Non-electrostatic parts of Gibbs energies (ΔG°nel) were then obtained by extrapolation of Equation (1) to an ionic strength of [KCl] = 1 M, where electrostatic interactions are effectively masked (Table 2). This study highlights that, whatever the nature and sequence of the PAA, ionic interactions with the phosphodiester backbone are not the major driving force for the formation of PAA/TAR complexes, as non-electrostatic interactions dominate the binding by contributing to 75 to 95% of the total binding free energy (cf. ΔG°nel/ΔG° ratio, Table 2).
Table 2.
Thermodynamic parameters for PAA/TAR complexesa
| PAA sequence | FFK 1 | FFR 2 | RFF 3 | FRF 4 | FKR 5 | FRR 6 | RRF 7 | |
|---|---|---|---|---|---|---|---|---|
| α-PAA (a) | ΔG° | −36.0 ± 1.9 | −36.1 ± 1.4 | −36.1 ± 1.4 | −35.0 ± 2.5 | −34.7 ± 3.0 | −36.0 ± 1.9 | −34.9 ± 1.2 |
| ΔH° | −54.2 ± 1.4 | −51.7 ± 0.5 | −53.5 ± 1 | −55.2 ± 1.7 | −66.4 ± 2.1 | −60.3 ± 1.8 | −54.5 ± 1.1 | |
| TΔS° | −18.2 ± 1.4 | −15.6 ± 0.6 | −17,4 ± 1 | −20.2 ± 1.7 | −31.8 ± 2.1 | −24.3 ± 0.5 | −19.6 ± 0.5 | |
| ΔCp | −2.82 ± 0.15 | −2.09 ± 0.22 | −2,01 ± 0,19 | −2.11 ± 0.21 | −2.9 ± 0.38 | −2.61 ± 0.28 | −1.98 ± 0.23 | |
| ΔG°el | −5.7 ± 2 | −2.4 ± 0.8 | −4.5 ± 1.5 | −4.6 ± 2.5 | −8.82 ± 3.1 | −4.9 ± 1.9 | −7.4 ± 1.3 | |
| ΔG°nel | −30.4 ± 0.1 | −33.7 ± 0.3 | −31.6 ± 0.2 | −30.4 ± 0.1 | −25.84 ± 0.7 | −31.1 ± 0.4 | −27.5 ± 0.2 | |
| TΔS°nel | −23.8 ± 1.4 | −17.1 ± 0.6 | −21.9 ± 1 | −24.8 ± 1.7 | −40.6 ± 2.2 | −29.2 ± 1.9 | −27 ± 1.2 | |
| ΔG°nel/ΔG° (%) | 0.84 | 0.93 | 0.88 | 0.87 | 0.75 | 0.86 | 0.79 | |
| β-PAA (b) | ΔG° | −37.8 ± 1.4 | −37.7 ± 1.2 | −37.9 ± 1.1 | −37.3 ± 1.2 | −35.1 ± 1.3 | −37.1 ± 2.8 | −37.0 ± 2.5 |
| ΔH° | −31.6 ± 1.3 | −29.3 ± 1.1 | −26.9 ± 0.8 | −30.6 ± 1.1 | −45.9 ± 0.9 | −45.0 ± 2.5 | −37.6 ± 2.5 | |
| TΔS° | 6.1 ± 0.7 | 8.4 ± 0.5 | 10.9 ± 0.8 | 6.8 ± 0.5 | −10.8 ± 0.9 | −7.9 ± 1.3 | −0.5 ± 1.1 | |
| ΔCp | −1.04 ± 0.19 | −1.35 ± 0.16 | −1.6 ± 0.1 | −1.66 ± 0.16 | −1.85 ± 0.14 | −1.99 ± 0.28 | −1.58 ± 0.14 | |
| ΔG°el | −5.5 ± 1.9 | −4.2 ± 1.5 | −3.78 ± 1.1 | −3.18 ± 1.5 | −4.4 ± 1.3 | −6.4 ± 2.6 | −7.04 ± 3.3 | |
| ΔG°nel | −32.3 ± 0.4 | −33.4 ± 0.2 | −34.1 ± 0.2 | −34.2 ± 0.3 | −30.7 ± 0.4 | −30.7 ± 0.3 | −30 ± 0.5 | |
| TΔS°nel | 0.7 ± 1.3 | 4.2 ± 1.1 | 7.2 ± 0.8 | 3.6 ± 1.1 | − 15.2 ± 0.9 | −14.3 ± 2.5 | −7.6 ± 2.3 | |
| ΔG°nel/ΔG° (%) | 0.86 | 0.89 | 0.90 | 0.92 | 0.88 | 0.83 | 0.81 | |
| C-α-PAA (c) | ΔG° | −36.6 ± 1.2 | −38.2 ± 1.7 | −36.7 ± 1.2 | −37.5 ± 2.9 | −34.4 ± 1.5 | −36.8 ± 1.4 | −36.8 ± 1.4 |
| ΔH° | −50.8 ± 1.2 | −33.3 ± 1.7 | −35.7 ± 1.1 | −36.7 ± 2.9 | −65.9 ± 1.1 | −53.6 ± 1.3 | −57.8 ± 1.3 | |
| TΔS° | −14.2 ± 0.3 | 4.9 ± 0.5 | 1.0 ± 0.6 | 0.8 ± 0.3 | −31.5 ± 1.1 | −16.8 ± 0.5 | −21.0 ± 0.5 | |
| ΔCp | −2.91 ± 0.38 | −1.54 ± 0.25 | −2.0 ± 0.4 | −2.34 ± 0.43 | −3.07 ± 0.16 | −2.79 ± 0.34 | −2.81 ± 0.28 | |
| ΔG°el | −2.7 ± 1.3 | −5.42 ± 2.4 | −2 ± 1.3 | −6.46 ± 4.1 | −6.67 ± 1.6 | −2.3 ± 1.4 | −2.2 ± 1.4 | |
| ΔG°nel | −33.9 ± 0.4 | −32.8 ± 0.6 | −34.7 ± 0.4 | −31 ± 0.3 | −27.8 ± 0.6 | −34.5 ± 0.2 | −34.6 ± 0.4 | |
| TΔS°nel | −16.9 ± 1.2 | −0.5 ± 1.8 | −1.02 ± 1.1 | −5.7 ± 2.9 | −38.2 ± 1.2 | −19.1 ± 1.3 | −23.3 ± 1.3 | |
| ΔG°nel/ΔG° (%) | 0.93 | 0.86 | 0.95 | 0.83 | 0.81 | 0.94 | 0.94 |
aΔG°, ΔH°, TΔS°, ΔG°el, ΔG°nel and TΔS°nel are expressed in kJ/mol. ΔCp is expressed in kJ/mol/K.
Enthalpy and entropy parameters
Enthalpy (ΔH°) and entropy changes (ΔS°) associated with each PAA/TAR equilibrium were determined by Equation (2) after determination of ΔG°T at several temperatures (278–308K). The results of these thermodynamic analyses are summarized in Table 2. In all cases, the formation of PAA/TAR complexes is enthalpy driven, demonstrating that non-covalent interactions (H-bonds, π-stacking, π-cation, etc.) dominate the binding. While binding processes for α-PAA are highly disfavored entropically, TΔS° parameters for β- and C-α-PAA are less unfavorable or even favorable in the case of F-rich compounds (1b–4b and 2c–4c). For each of the three series, ΔH° and TΔS° are found strongly correlated, according to Equation (3), with correlation coefficients higher than 0.99 (Figure 4A) and slopes ‘a’ near unity. This is indicative of an almost perfect EEC phenomenon for which any decrease in binding enthalpy is compensated by a similar decrease in binding entropy, and vice versa (see supporting information for comment on statistical relevance). The consequence is that even if binding enthalpies and entropies are distributed over wide intervals in a given series, ΔG° values along this series are very close. For the three series, the slopes are very similar, underscoring that from a thermodynamic point of view all PAA are homologous molecules. Considering this, it seems unlikely that any tri-PAA containing this set of amino acids (α or β Phe, Arg, Lys) can bind to TAR with higher affinities than those observed in this study.
Figure 4.

A/Enthalpy/entropy compensation plot: ΔH° versus TΔS° for the three series of PAA/TAR equilibria. B/Variation in ΔH° and TΔS° with the PAA sequence for α-PAA (circle), β-PAA (triangle) and C-α-PAA (square).
EEC is a general feature of many biological processes based on molecular associations implying non-covalent interactions. Often observed in the context of protein–ligand interactions, it is the first time, to our knowledge, that EEC is reported for a series of RNA–small ligand binding events. The physical origin of this EEC phenomenon, theoretically (30,31) and experimentally (32,33) supported, is based on the relationship opposing ‘bonding’ and ‘motion’. The stronger is an interaction at a dimeric interface, the lower is the mobility at this interface.
β-PAA and C-α-PAA versus α-PAA
As shown in Figure 4B, β-PAA are associated with higher ΔH° and ΔS° values than their corresponding α-PAA, but the trends in ΔH° (and TΔS°) along PAA sequences are roughly parallel for β-PAA and α-PAA. This likely demonstrates that the structural modification that consists in adding a methylene group into the side chains of α-amino acids of α-PAA leads to the same effect regardless of the PAA sequence. On average, this structural modification has an enthalpy cost of approximately 24 kJ/mole in the case of the four F-rich compounds and of 18 kJ/mole for K/R-rich ones, overweighted by an entropic gain of 26 and 19 kJ/mole, respectively. Because this increase in both ΔH° and ΔS° does not depend on the PAA sequence, it can be suggested that each isosequential β-PAA/α-PAA pair binds to TAR following the same interaction network. In α-PAA, the high magnitude of both enthalpy and entropy values would reveal the establishment of tight complexes, while in contrast, the less favorable ΔH° and less unfavorable TΔS° values observed for β-PAA would be associated with the formation of looser complexes (31). These differences could be linked to the higher flexibility of β-PAA vs α-PAA in the unbound state. Indeed, to keep the same interaction network than α-PAA while avoiding a conformational entropic penalty, one can suppose that the β-PAA/TAR complexes relax to a more stable state by decreasing the interaction tightness at the interface (34). Such a behavior would result both in weakening non-covalent interactions (i.e. less favorable ΔH°) and in increasing mobility of the β-PAA side chains (i.e. less unfavorable TΔS°).
Concerning C-α-PAA structures, thermodynamic profiles demonstrate that the introduction of three methylene groups in the backbone of a given α-PAA impacts differently according to the PAA sequence (Figure 4B). In the case of K/R-rich C-α-PAA 5c-7c and of C-α-FFK 1c, ΔH° and ΔS° values are very close to those associated with the respective α-PAA 5a–7a and 1a. In contrast, the TAR binding of F-rich C-α-PAA 2c–4c occurs with an average enthalpy cost of 18 kJ/mole comparatively with their corresponding α-PAA 2a–4a. Therefore, for C-α-PAA 2c–4c, it could be suggested that increasing the length of the α-PAA backbone does not modify, on complexation, the interaction network with TAR but rather induces a structural loosening of the complex.
F-rich PAA versus K/R-rich PAA
Within each series, F-rich sequences are associated with higher entropy and enthalpy values than R/K-rich ones (except for C-α-FFK) (Table 2 and Figure 4B). This specific behavior of F-rich PAA is probably because they establish fewer non-electrostatic interactions with TAR. Comparatively with R/K-rich PAA, F-rich PAA contain only one cationic residue able to give strong H-bonds via its side chain and two aromatic motives prone to establish weaker π-stacking interactions. It is reasonable thereby to observe for F-rich PAA less enthalpy changes but more favorable entropy values.
Finally, for all PAA/TAR complexes, heat capacities (ΔCp) were found negative, as observed for a variety of small molecules binding to nucleic acids (35,36) (Table 1). However, ΔCp values are significantly higher for loose β-PAA binders than for the corresponding tight α-PAA. Many parameters could account for this difference (changes in solvent-accessible surface area, changes in protonation state, ion release, etc.), but, owing to the structural similarity of the ligands, it may result preferentially from differences in RNA conformational mobility changes arising on binding of the two kinds of PAA (37,38). Because α-PAA are associated with more negative ΔCp values than β-PAA, it would be expected that they induce a more significant RNA conformational change. However, CD data do not seem to support this hypothesis (see vide infra).
Overall, molecular flexibility is one of the key parameters of ligand efficiency, but thermodynamic consequences on binding are not yet well understood. Our results are in line with a recently reported computer modeling study that predicts that varying molecular flexibility while keeping the same interaction network leads to near-linear EEC (39). Moreover, when the receptor is flexible, increasing ligand flexibility leads to stronger binding affinity. Such a trend is observed in our study because more flexible β-PAA and some C-α-PAA display on average a better affinity for TAR than their corresponding α-PAA.
CD and UV melting studies
To compare the TAR structural changes occurring on binding of α-, β- and C-α-PAA, CD studies were implemented. F-rich PAA 2a–c and K/R-rich 7a–c, representatives of the three series, were chosen as models. CD spectra of PAA alone were preliminary measured. Negligible CD intensity in the 200- to 350-nm region was observed, suggesting the lack of any structuration or secondary shape. In contrast, the CD spectrum of TAR alone is characteristic of an A-form double helix, with strong positive and negative bands at 260 nm and 210 nm, respectively, and a weak negative band at 240 nm (40,41). While the 260-nm CD band, very sensitive to base stacking, may vary on binding of a ligand without affecting the general nucleic acid structure, the 210-nm band is more related to the A-form RNA helical structure. CD profiles (see Supplementary Figure S1 in supporting information) associated with tight binders (α-PAA 2a and 7a and C-α-PAA 7c) differ from those associated with loose ones (β-PAA 2b, 7b and C-α-PAA 2c), indicating that TAR conformational changes induced on binding of the two classes of PAA are distinct. In the case of tight binders (2a, 7a, 7c), only a slight decrease in ellipticity at 260 nm was observed, whereas the intensities of the negative bands at 210 nm and 240 nm did not vary. This CD signature is similar to the one reported for binding of Tat derivatives and arginine-containing compounds, the decrease in ellipticity at 260 nm reflecting the externalization of the C24 and U25 residues of the bulge and their concomitant destacking (40–45). Loose binders (2b-c, 7b) change the CD signal of TAR to a more substantial extent. A stronger reduction in ellipticity at 260 nm was observed together with a marked decrease in the intensity of the negative band at 210 nm and to a less extent at 240 nm. Because such modifications may be indicative of a destabilization of the nucleic acid structure (46,47), melting temperature and ultraviolet absorbance spectroscopy studies were conducted. A destabilization of the TAR structure on binding should result in both a decrease of the TAR melting temperature and an increase in its absorbance at 260 nm (48). Our results clearly refute this hypothesis. Indeed, the TAR melting temperature was increased on loose β-PAA 7b binding by 4.1°C and 6.6°C at 1:1 and 5:1 PAA/TAR ratios, respectively, demonstrating a stabilizing effect on the TAR structure (Figure 5; see Supplementary Figure S2 and Supplementary Table S3 in supporting information). In addition, a decrease in absorbance at 260 nm, particularly notable at 5:1 molar ratio, was observed. By comparison, the binding of C-α-PAA 7c increased the TAR melting temperature by 2.5°C and 4.1°C at 1:1 and 5/1 PAA/TAR ratios, respectively, and had no effect on the TAR UV spectrum. Thus, even if loose 7b binder displays the same TAR affinity as tight 7c one (Table 1), its binding induces distinct RNA conformational changes, leading to a higher stabilization of the TAR structure.
Figure 5.
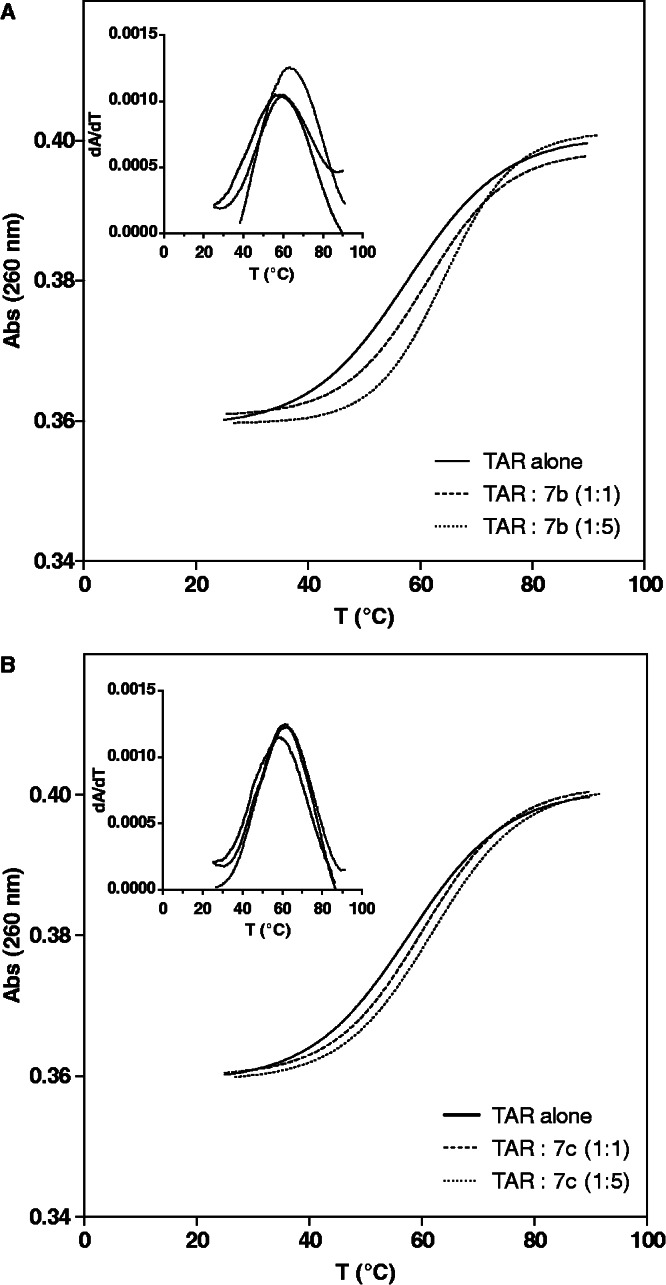
UV melting curves at 260 nm of TAR RNA and its complex with β-PAA 7b (A) or C-α-PAA 7c (B). Inset: first-derivative UV melting curves of TAR and its complex with β-PAA 7b (A) or C-α-PAA 7c (B).
Given these results, the variations observed between the CD spectra are likely due to TAR conformational differences. While the overall A-form of the TAR helix is globally conserved in the two cases, the substantial ellipticity decrease at 210 nm and 260 nm only observed in the case of loose PAA 7b likely indicates a decrease in the A character of the TAR helix on binding (49). One hypothesis could be to assign this change to a distorsion of the A-form. In response to the entropic constraint imposed by the binding of the highly flexible β-PAA structure, the β-PAA/TAR complex would relax to a more stable state not only by decreasing the interaction tightness at the PAA/TAR interface but also by modifying the TAR helicity. However, we have, at this stage, no evidence for such a scenario. Complementary studies will be envisaged to resolve the dynamics of PAA/TAR interactions.
Antiviral activity
Finally, the antiviral activity and the cellular toxicity of C-α and β-PAA were evaluated on PBMC, infected (or not) by the HIV-1 LAI-strain, first at a concentration of 1 mM. AZT was taken as a reference. When the ratio (%inhibition/%toxicity) was >2, ED50, CC50 and selectivity indexes (SI = CC50/ED50) were determined. Results are given in Table 3.
Table 3.
Anti-HIV activity and toxicity of C-α and β-PAA
| PAA | Inhibition (%) | Toxicity (%) | %Inhibition/ %Toxicity | ED50* (μM) | CC50** (μM) | SI*** |
|---|---|---|---|---|---|---|
| FRF 4c | 67 ± 5 | 29 ± 2 | >2.3 | 869 | >1000 | >1.2 |
| FFK 1b | 80 ± 2 | 37 ± 8 | >2.2 | 254 | id | >4 |
| FFR 2b | 83 ± 4 | 38 ± 2 | >2.2 | 130 | id | >8 |
| RFF 3b | 96 ± 1 | 36 ± 3 | >2.7 | 85 | id | >12 |
| FRF 4b | 92 ± 4 | 18 ± 7 | >5.1 | 56 | id | >18 |
| FKR 5b | 85 ± 2 | 29 ± 6 | >2.9 | 219 | id | >4.5 |
| FRR 6b | 95 ± 1 | 53 ± 5 | >1.8 | 60 | id | >17 |
| RRF 7b | 95 ± 1 | 36 ± 4 | >2.6 | 95 | id | >10.5 |
*ED50: Effective Dose. Dose of PAA required to inhibit 50% of the viral replication.
**CC50: Cytotoxic Concentration. Concentration of PAA required to reduce the cell number by 50%.
***SI: Selectivity Index (CC50/ED50).
C-α-PAA derivatives are less efficient to inhibit HIV replication and more toxic than β-PAA. The best C-α-PAA inhibitor, i.e. C-α-FRF 4c, is able to inhibit at 1 mM 67% of the viral replication but with 29% of cellular toxicity, giving a ratio (%inhibition/%toxicity] of only 2.1 and an ED50 value of 869 µM. The three C-α-PAA 5c–7c containing K/R-rich sequences give the worse results (data not shown), although they are among the best inhibitors of the Tat/TAR interaction in free-cell assays.
The β-PAA series is more promising. Indeed, all β-PAA inhibit at 1 mM from 80 to 96% of the viral replication, with a ratio [%inhibition/%toxicity] from 2.2 to 5.1. The best inhibitors, β-FRF 4b and β-FRR 6b display ED50 values of 56 and 60 µM, respectively, and no toxicity up to 1 mM, thus leading to a promising SI values higher than 17. The two β-PAA containing a lysine residue (β-FFK 1b and β-FKR 5b) are the weakest inhibitors. The correlation observed between IC50 and ED50 values for F-rich β-PAA and for R/K-rich β-PAA (R2 = 0.89, see Supplementary Figure S3 in supporting information) tends to further demonstrate that the antiviral activity is due, at least in part, to the inhibition of the Tat/TAR complex.
In conclusion, the HIV-1 TAR RNA binding of three series of homologous molecules (α-PAA, β-PAA, C-α-PAA) was studied, and their ability to displace the TAR/Tat complex was evaluated. Whereas KD values were found roughly similar, great differences were observed in IC50 values, indicating a dependency both on the nature and sequence of PAA for the TAR/Tat inhibition. Our thermodynamic study revealed the occurrence of an EEC phenomenon that explains the KD values’ similarity. Using the interfacial mobility model, we highlighted that the TAR binding of α-PAA and of K/R-rich C-α-PAA results in the establishment of tight complexes, while looser ones are formed with β-PAA and F-rich C-α-PAA. In most cases, these latter are more efficient Tat/TAR inhibitors than their corresponding α-PAA. This could be due to a higher stabilization of the TAR structure on binding.
Altogether, our results give new insights into the comprehension of RNA binding modes that open a wide field of investigation in the design of new RNA ligands. Finally, β-PAA and C-α-PAA constitute new lead compounds from which structure/activity studies can be envisaged to increase their promising antiviral activity.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–3, Supplementary Figures 1–3, Supplementary Methods and Supplementary References [50–52].
ACKNOWLEDGEMENTS
We thank Jean-Marie Guigonis for mass analyses.
FUNDING
SIDACTION; the Agence Nationale de Recherche sur le SIDA and the Caisse Primaire d’Assurance Maladie des Professions Libérales. L. Pascale is recipient of a MENRT Ph.D. fellowship. Funding for open access charge: University fund.
Conflict of interest statement. None declared.
REFERENCES
- 1.Aboul-Ela F. Strategies for the design of RNA-binding small molecules. Future. Med. Chem. 2010;2:93–119. doi: 10.4155/fmc.09.149. [DOI] [PubMed] [Google Scholar]
- 2.Guan L, Disney MD. Recent advances in developing small molecules targeting RNA. ACS Chem. Biol. 2012;7:73–86. doi: 10.1021/cb200447r. [DOI] [PubMed] [Google Scholar]
- 3.Thomas JR, Hergenrother PJ. Targeting RNA with small molecules. Chem. Rev. 2008;108:1171–1224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 4.Daldrop P, Reyes FE, Robinson DA, Hammond CM, Lilley DM, Batey RT, Brenk R. Novel ligands for a purine riboswitch discovered by RNA-ligand docking. Chem. Biol. 2011;18:324–335. doi: 10.1016/j.chembiol.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar S, Bose D, Suryawanshi H, Sabharwal H, Mapa K, Maiti S. Specificity of RSG-1.2 peptide binding to RRE-IIB RNA element of HIV-1 over Rev peptide is mainly enthalpic in origin. PLoS One. 2011;6:e23300. doi: 10.1371/journal.pone.0023300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suryawanshi H, Sabharwal H, Maiti S. Thermodynamics of peptide-RNA recognition: the binding of a tat peptide to TAR RNA. J. Phys. Chem. B. 2010;114:11155–11163. doi: 10.1021/jp1000545. [DOI] [PubMed] [Google Scholar]
- 7.Thomas JR, Liu X, Hergenrother PJ. Biochemical and thermodynamic characterization of compounds that bind to RNA hairpin loops: toward an understanding of selectivity. Biochemistry. 2006;45:10928–10938. doi: 10.1021/bi0607296. [DOI] [PubMed] [Google Scholar]
- 8.Pilch DS, Kaul M, Barbieri CM, Kerrigan JE. Thermodynamics of aminoglycoside-rRNA recognition. Biopolymers. 2003;70:58–79. doi: 10.1002/bip.10411. [DOI] [PubMed] [Google Scholar]
- 9.Bernacchi S, Freisz S, Maechling C, Spiess B, Marquet R, Dumas P, Ennifar E. Aminoglycoside binding to the HIV-1 RNA dimerization initiation site: thermodynamics and effect on the kissing-loop to duplex conversion. Nucleic Acids Res. 2007;35:7128–7139. doi: 10.1093/nar/gkm856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burnouf D, Ennifar E, Guedich S, Puffer B, Hoffmann G, Bec G, Disdier F, Baltzinger M, Dumas P. kinITC: a new method for obtaining joint thermodynamic and kinetic data by isothermal titration calorimetry. J. Am. Chem. Soc. 2012;134:559–565. doi: 10.1021/ja209057d. [DOI] [PubMed] [Google Scholar]
- 11.McLaughlin KJ, Jenkins JL, Kielkopf CL. Large favorable enthalpy changes drive specific RNA recognition by RNA recognition motif proteins. Biochemistry. 2011;50:1429–1431. doi: 10.1021/bi102057m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bissantz C, Kuhn B, Stahl M. A medicinal chemist's guide to molecular interactions. J. Med. Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garbett NC, Chaires JB. Thermodynamic studies for drug design and screening. Expert. Opin. Drug Discov. 2012;7:299–314. doi: 10.1517/17460441.2012.666235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richter SN, Palu G. Inhibitors of HIV-1 Tat-mediated transactivation. Curr. Med. Chem. 2006;13:1305–1315. doi: 10.2174/092986706776872989. [DOI] [PubMed] [Google Scholar]
- 15.Thomas JR, Hergenrother PJ. Targeting RNA with small molecules. Chem. Rev. 2008;108:1171–1224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 16.Bonnard V, Azoulay S, Di Giorgio A, Patino N. Polyamide amino acids: a new class of RNA ligands. Chem. Commun. (Camb.) 2009;17:2302–2304. doi: 10.1039/b815324h. [DOI] [PubMed] [Google Scholar]
- 17.Bonnard V, Pascale L, Azoulay S, Di Giorgio A, Rogez-Kreuz C, Storck K, Clayette P, Patino N. Polyamide amino acids trimers as TAR RNA ligands and anti-HIV agents. Bioorg. Med. Chem. 2010;18:7432–7438. doi: 10.1016/j.bmc.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 18.Luedtke NW, Liu Q, Tor Y. RNA-ligand interactions: affinity and specificity of aminoglycoside dimers and acridine conjugates to the HIV-1 Rev response element. Biochemistry. 2003;42:11391–11403. doi: 10.1021/bi034766y. [DOI] [PubMed] [Google Scholar]
- 19.Rosales T, Royer CA. A graphical user interface for BIOEQS: a program for simulating and analyzing complex biomolecular interactions. Anal. Biochem. 2008;381:270–272. doi: 10.1016/j.ab.2008.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murchie AI, Davis B, Isel C, Afshar M, Drysdale MJ, Bower J, Potter AJ, Starkey ID, Swarbrick TM, Mirza S, et al. Structure-based drug design targeting an inactive RNA conformation: exploiting the flexibility of HIV-1 TAR RNA. J. Mol. Biol. 2004;336:625–638. doi: 10.1016/j.jmb.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 21.Davis B, Afshar M, Varani G, Murchie AI, Karn J, Lentzen G, Drysdale M, Bower J, Potter AJ, Starkey ID, et al. Rational design of inhibitors of HIV-1 TAR RNA through the stabilisation of electrostatic "hot spots". J. Mol. Biol. 2004;336:343–356. doi: 10.1016/j.jmb.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 22.Yang M. Discoveries of Tat-TAR interaction inhibitors for HIV-1. Curr. Drug Targets. Infect. Disord. 2005;5:433–444. doi: 10.2174/156800505774912901. [DOI] [PubMed] [Google Scholar]
- 23.Davidson A, Leeper TC, Athanassiou Z, Patora-Komisarska K, Karn J, Robinson JA, Varani G. Simultaneous recognition of HIV-1 TAR RNA bulge and loop sequences by cyclic peptide mimics of Tat protein. Proc. Natl Acad. Sci. USA. 2009;106:11931–11936. doi: 10.1073/pnas.0900629106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davidson A, Patora-Komisarska K, Robinson JA, Varani G. Essential structural requirements for specific recognition of HIV TAR RNA by peptide mimetics of Tat protein. Nucleic Acids Res. 2011;39:248–256. doi: 10.1093/nar/gkq713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Puglisi JD, Tan R, Calnan BJ, Frankel AD, Williamson JR. Conformation of the TAR RNA-arginine complex by NMR spectroscopy. Science. 1992;257:76–80. doi: 10.1126/science.1621097. [DOI] [PubMed] [Google Scholar]
- 26.Bardaro MF, Jr, Shajani Z, Patora-Komisarska K, Robinson JA, Varani G. How binding of small molecule and peptide ligands to HIV-1 TAR alters the RNA motional landscape. Nucleic Acids Res. 2009;37:1529–1540. doi: 10.1093/nar/gkn1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aboul-Ela F, Karn J, Varani G. The structure of the human immunodeficiency virus type-1 TAR RNA reveals principles of RNA recognition by Tat protein. J. Mol. Biol. 1995;253:313–332. doi: 10.1006/jmbi.1995.0555. [DOI] [PubMed] [Google Scholar]
- 28.Record MT, Jr, Zhang W, Anderson CF. Analysis of effects of salts and uncharged solutes on protein and nucleic acid equilibria and processes: a practical guide to recognizing and interpreting polyelectrolyte effects, Hofmeister effects, and osmotic effects of salts. Adv. Protein Chem. 1998;51:281–353. doi: 10.1016/s0065-3233(08)60655-5. [DOI] [PubMed] [Google Scholar]
- 29.Record MT, Jr, Anderson CF, Lohman TM. Thermodynamic analysis of ion effects on the binding and conformational equilibria of proteins and nucleic acids: the roles of ion association or release, screening, and ion effects on water activity. Q. Rev. Biophys. 1978;11:103–178. doi: 10.1017/s003358350000202x. [DOI] [PubMed] [Google Scholar]
- 30.Dunitz JD. Win some, lose some: enthalpy-entropy compensation in weak intermolecular interactions. Chem. Biol. 1995;2:709–712. doi: 10.1016/1074-5521(95)90097-7. [DOI] [PubMed] [Google Scholar]
- 31.Ford DM. Enthalpy-entropy compensation is not a general feature of weak association. J. Am. Chem. Soc. 2005;127:16167–16170. doi: 10.1021/ja054519c. [DOI] [PubMed] [Google Scholar]
- 32.Li L, Dantzer JJ, Nowacki J, O'Callaghan BJ, Meroueh SO. PDBcal: a comprehensive dataset for receptor-ligand interactions with three-dimensional structures and binding thermodynamics from isothermal titration calorimetry. Chem. Biol. Drug Des. 2008;71:529–532. doi: 10.1111/j.1747-0285.2008.00661.x. [DOI] [PubMed] [Google Scholar]
- 33.Williams DH, Stephens E, O'Brien DP, Zhou M. Understanding noncovalent interactions: ligand binding energy and catalytic efficiency from ligand-induced reductions in motion within receptors and enzymes. Angew. Chem. Int. Ed. Engl. 2004;43:6596–6616. doi: 10.1002/anie.200300644. [DOI] [PubMed] [Google Scholar]
- 34.Krishnamurthy VM, Bohall BR, Semetey V, Whitesides GM. The paradoxical thermodynamic basis for the interaction of ethylene glycol, glycine, and sarcosine chains with bovine carbonic anhydrase II: an unexpected manifestation of enthalpy/entropy compensation. J. Am. Chem. Soc. 2006;128:5802–5812. doi: 10.1021/ja060070r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Islam MM, Pandya P, Kumar S, Kumar GS. RNA targeting through binding of small molecules: Studies on t-RNA binding by the cytotoxic protoberberine alkaloid coralyne. Mol. Biosyst. 2009;5:244–254. doi: 10.1039/b816480k. [DOI] [PubMed] [Google Scholar]
- 36.Stolarski R. Thermodynamics of specific protein-RNA interactions. Acta Biochim. Pol. 2003;50:297–318. [PubMed] [Google Scholar]
- 37.McElroy CA, Manfredo A, Gollnick P, Foster MP. Thermodynamics of tryptophan-mediated activation of the trp RNA-binding attenuation protein. Biochemistry. 2006;45:7844–7853. doi: 10.1021/bi0526074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spolar RS, Record MT., Jr Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- 39.Forrey C, Douglas JF, Gilson MK. The Fundamental Role of Flexibility on the Strength of Molecular Binding. Soft. Matter. 2012;8:6385–6392. doi: 10.1039/C2SM25160D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loret EP, Georgel P, Johnson WC, Jr, Ho PS. Circular dichroism and molecular modeling yield a structure for the complex of human immunodeficiency virus type 1 trans-activation response RNA and the binding region of Tat, the trans-acting transcriptional activator. Proc. Natl Acad. Sci. USA. 1992;89:9734–9738. doi: 10.1073/pnas.89.20.9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Metzger AU, Schindler T, Willbold D, Kraft M, Steegborn C, Volkmann A, Frank RW, Rosch P. Structural rearrangements on HIV-1 Tat (32-72) TAR complex formation. FEBS Lett. 1996;384:255–259. doi: 10.1016/0014-5793(96)00314-6. [DOI] [PubMed] [Google Scholar]
- 42.Frankel AD. Peptide models of the Tat-TAR protein-RNA interaction. Protein Sci. 1992;1:1539–1542. doi: 10.1002/pro.5560011202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang S, Huber PW, Cui M, Czarnik AW, Mei HY. Binding of neomycin to the TAR element of HIV-1 RNA induces dissociation of Tat protein by an allosteric mechanism. Biochemistry. 1998;37:5549–5557. doi: 10.1021/bi972808a. [DOI] [PubMed] [Google Scholar]
- 44.Goel T, Kumar S, Maiti S. Thermodynamics and solvation dynamics of BIV TAR RNA-Tat peptide interaction. Mol. Biosyst. 2013;9:88–98. doi: 10.1039/c2mb25357g. [DOI] [PubMed] [Google Scholar]
- 45.Kumar S, Kellish P, Robinson WE, Jr, Wang D, Appella DH, Arya DP. Click dimers to target HIV TAR RNA conformation. Biochemistry. 2012;51:2331–2347. doi: 10.1021/bi201657k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gregoire CJ, Gautheret D, Loret EP. No tRNA3Lys unwinding in a complex with HIV NCp7. J. Biol. Chem. 1997;272:25143–25148. doi: 10.1074/jbc.272.40.25143. [DOI] [PubMed] [Google Scholar]
- 47.Ranjbar B, Gill P. Circular dichroism techniques: biomolecular and nanostructural analyses- a review. Chem. Biol. Drug Des. 2009;74:101–120. doi: 10.1111/j.1747-0285.2009.00847.x. [DOI] [PubMed] [Google Scholar]
- 48.Beltz H, Azoulay J, Bernacchi S, Clamme JP, Ficheux D, Roques B, Darlix JL, Mely Y. Impact of the terminal bulges of HIV-1 cTAR DNA on its stability and the destabilizing activity of the nucleocapsid protein NCp7. J. Mol. Biol. 2003;328:95–108. doi: 10.1016/s0022-2836(03)00244-4. [DOI] [PubMed] [Google Scholar]
- 49.Kypr J, Kejnovska I, Renciuk D, Vorlickova M. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res. 2009;37:1713–1725. doi: 10.1093/nar/gkp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharp K. Entropy-enthalpy compensation: fact or artifact? Protein Sci. 2001;10:661–667. doi: 10.1110/ps.37801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baleux F, Loureiro-Morais L, Hersant Y, Clayette P, renzana-Seisdedos F, Bonnaffe D, Lortat-Jacob H. A synthetic CD4-heparan sulfate glycoconjugate inhibits CCR5 and CXCR4 HIV-1 attachment and entry. Nat. Chem. Biol. 2009;5:743–748. doi: 10.1038/nchembio.207. [DOI] [PubMed] [Google Scholar]
- 52.Barre-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, xler-Blin C, Vezinet-Brun F, Rouzioux C, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220:868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.