


Abstract
COHCAP (City of Hope CpG Island Analysis Pipeline) is an algorithm to analyze single-nucleotide resolution DNA methylation data produced by either an Illumina methylation array or targeted bisulfite sequencing. The goal of the COHCAP algorithm is to identify CpG islands that show a consistent pattern of methylation among CpG sites. COHCAP is currently the only DNA methylation package that provides integration with gene expression data to identify a subset of CpG islands that are most likely to regulate downstream gene expression, and it can generate lists of differentially methylated CpG islands with ∼50% concordance with gene expression from both cell line data and heterogeneous patient data. For example, this article describes known breast cancer biomarkers (such as estrogen receptor) with a negative correlation between DNA methylation and gene expression. COHCAP also provides visualization for quality control metrics, regions of differential methylation and correlation between methylation and gene expression. This software is freely available at https://sourceforge.net/projects/cohcap/.
INTRODUCTION
Methylation of CpG sites in upstream CpG islands is a well-established method of epigenetic regulation of gene expression, and there are a number of methods for quantifying DNA methylation in promoter regions (1–4). One popular, high-quality technique for measuring methylation of CpG sites is the Illumina methylation array (5,6), which has been used for large patient cohorts (7–16) in addition to smaller-scale experiments (17–24). Although there are a number of algorithms to analyze Illumina methylation array data (25–30), most of these algorithms [with the exception of Illumina Methylation Analyzer (IMA) (30)] focus on defining differentially methylated CpG sites without providing statistics to define differentially methylated regions (e.g. CpG islands). Similarly, integration with gene expression data is an important tool for biological interpretation of results (31), and COHCAP (City of Hope CpG Island Analysis Pipeline) is currently the only methylation package that provides tools for such data integration with differentially methylated regions (not just CpG sites). To meet the common need for this type of analysis of differentially methylated regions using single-nucleotide resolution methylation data, we developed COHCAP.
COHCAP is a pipeline that covers most user needs for differential methylation and integration with gene expression data (Figure 1, Supplementary Figure S1 and S2; Supplementary Table S1). This includes quality control metrics, defining differentially methylated CpG sites, defining differentially methylated CpG islands and visualization of methylation data. Although IMA has one method for providing statistics for differentially methylated regions, COHCAP contains two different methods of CpG island analysis. With the exception of MethLAB (25), COHCAP is the only algorithm to provide a graphical user interface for users without programming experience. Additionally, COHCAP is the only package with flexible analysis of one-group (or more-than-two-group) comparisons. Finally, bisulfite sequencing (BS-Seq) is another method of measuring methylation of CpG sites (32,33), and there are some methods to assist with analysis of BS-Seq data (34,35). However, COHCAP is the only package designed to analyze either Illumina methylation array or BS-Seq data.
Figure 1.

COHCAP workflows for integrative genomic analysis. (A) Average by Site workflow: CpG sites showing differential methylation are selected, and the average beta values for the two groups shown (red versus blue) are calculated per CpG site. Next, the consistency of signal between CpG sites within a CpG island is quantified to determine regions showing significant differential methylation. Finally, if the user has a corresponding gene expression dataset, COHCAP looks for differentially expressed genes that show inverse overlap with differentially methylated regions (e.g. increased methylation with decreased expression, and decreased methylation with increased expression). (B) Average within CpG Island workflow: this is the default workflow for COHCAP. CpG sites showing differential methylation are selected, and the average beta values are calculated for significant sites within a CpG island for each sample. Next, these averaged beta values for each CpG island are compared for the samples between the two groups (red versus blue). If the user has paired gene expression data, integration is performed by looking for a significant negative correlation between beta values and gene expression levels.
To test the utility of COHCAP, we have applied the algorithm to publicly available Illumina array and BS-Seq data (10,17,36) as well as novel cell line datasets (Supplementary Figure S3 and S4). COHCAP is applied to cell line datasets as well as the large The Cancer Genome Atlas (TCGA) breast cancer dataset (10) to study how heterogeneity affects the quality of COHCAP results (Supplementary Figure S3E). The results of COHCAP and IMA (30) for two-group comparisons of both cell line and patient data are compared to test the ability for COHCAP to improve on existing algorithms (Supplementary Figure S3D). The accuracy of the one-group workflow is accessed by comparing the signal for a sample analyzed using the Illumina 450k methylation array as well as the Methylated-CpG Island Recovery Assay (MIRA) protocol on a tiling array (Supplementary Figure S3A). Finally, the ability to apply COHCAP to BS-Seq data is tested by comparing HCT116 cell line data across different samples and DNA methylation technologies (Supplementary Figure S3B and S3C, Figure S4A), as well as comparing simulated two-group analysis for COHCAP and methylKit (35) (Supplementary Figure S4B). In short, this study shows that COHCAP is an accurate unique tool for single-nucleotide resolution DNA methylation analysis.
MATERIALS AND METHODS
COHCAP algorithm
Although COHCAP does not provide methods for data normalization, the minimal input format for COHCAP is very simple, and users can easily apply additional normalization using tools other than Genome Studio (37–42). Additionally, COHCAP does not provide alignment of raw BS-Seq data, but instead uses the output of the Bismark alignment pipeline (43). A Perl script template for creating BS-Seq data that can be analyzed in COHCAP can be found at https://sites.google.com/site/cwarden45/scripts. This site also contains a template for creating a custom annotation file for targeted BS-Seq data, if needed. In general, answers to frequently asked questions are available on the COHCAP wiki (http://sourceforge.net/p/cohcap/wiki/Home/).
The CpG site analysis is based on the method described in Sproul et al. (44), where sites are defined as methylated if they show a percentage of methylation (beta) greater than a certain value (0.7 for cell line data, 0.3 for patient data) and sites are unmethylated if they have beta values <0.3 (by default). We extended this algorithm to include a P-value and false-discovery rate [FDR, using the method of Benjamini and Hochberg (45)] value as cutoffs for differential expression. The method of P-value calculation varies based on the number of groups considered for the analysis (one group, two groups, three or more groups; Supplementary Table S2, Supplemental Methods). Although we do not explicitly use delta-beta values in the COHCAP algorithm, the values are provided in the output files for CpG site and CpG island if users wish to use that metric for prioritization.
The basic assumption for COHCAP’s CpG island analysis is that genomic annotations can be used to define regions of interest, which is a principle that has been applied in other algorithms (30,46). However, the results in this study show that COHCAP optimizes the method of summarization in a way that allows maximal concordance with gene expression changes, especially for the Illumina 450k methylation array. COHCAP contains two workflows to handle CpG island analysis and gene expression integration (Figure 1, Supplemental Methods). Both workflows start after filtering for differentially expressed CpG sites.
The ‘Average by Site’ workflow calculates the average methylation values for each group for each CpG site and then tests the consistency of the signal among the CpG sites within a CpG island. Integration is then performed by comparing overlapping gene lists from the COHCAP analysis and gene expression analysis performed separately (to determine fold change, P-value and FDR values for expression change). Users can specify a minimum number of sites to filter the CpG island results for integration analysis. COHCAP presents a list of genes with an inverse relationship between methylation and gene expression. Average beta values per CpG site for each group can also be exported as .wig files, for visualization in tools like Integrative Genomic Viewer (47) or the University of California Santa Cruz (UCSC) Genome Browser (48).
The ‘Average by Island’ workflow (the default workflow) averages the signal from all the differentially methylated sites within a CpG island and then compares methylation between islands in an identical manner to the comparison of CpG sites. Users can also specify a minimum number of sites to define a CpG island. Integration is performed by testing for a significant negative correlation between CpG island methylation and gene expression. Regions of differential methylation can be visualized via box plot, and genes showing negative correlations between methylation and gene expression can be visualized via scatter plot.
IMA comparison
IMA was tested on the HCT116 cell line dataset from this study as well as the TCGA breast cancer dataset (10). To use criteria most similar to COHCAP, an adjusted P-value of 0.05 was used to compare mutant versus parental HCT116 methylation as well as breast tumor versus normal methylation. Although IMA does not allow users to specify methylated and unmethylated thresholds, we assumed that the closest approximation to the cell line comparison (methylated >0.7, unmethylated <0.3) was a delta-beta value of 0.3, and no delta-beta cutoff was used for the TCGA dataset comparison (because no delta-beta cutoff is equivalent to methylated > 0.3 and unmethylated <0.3).
methylKit comparison
The HCT116 cell line dataset from this study was used to test the ability to apply methylKit to Illumina array data. We randomly assigned coverage to the 450k beta values (uniform 100×, uniform 10×, uniform 5× and 10× with a random variation of ±5 reads) to convert beta values on the Illumina array to corresponding counts of methylated and unmethylated nucleotides per CpG site. To use criteria most similar to COHCAP, the q-value threshold was set to 0.05. Similar to IMA, methylKit does not allow users to specify methylated and unmethylated thresholds, so we assumed that the closest approximation (for methylated >0.7, unmethylated < 0.3) was a delta-beta value of 0.3. For the closest approximation to the CpG island analysis, we used the tiling window analysis function where the window size and step size was set to 1000 base pairs.
RESULTS
COHCAP CpG islands show strong concordance with gene expression data
To study the utility of COHCAP for integrative analysis of cell line data, we compared methylation and expression differences between an HCT116 cell line and a derived mutant strain. The COHCAP quality control metrics (Figure 2) all show clear differences in methylation between the biological replicates for these two groups. Both COHCAP workflows were tested on this dataset, and we found that the ‘Average by Island’ workflow produced the list of regions with the best concordance with gene expression (38.4% versus 11.6%; Supplementary Table S3; Supplementary Figure S5) with a comparable run-time (Supplementary Table S4). We also found that calculating correlation between methylation beta values and gene expression levels (significant with FDR <0.05) indicated that more than half of the differentially methylated regions may influence the differences in gene expression to some extent (52.1%; Supplementary Table S3; Supplementary Figure S5).
Figure 2.
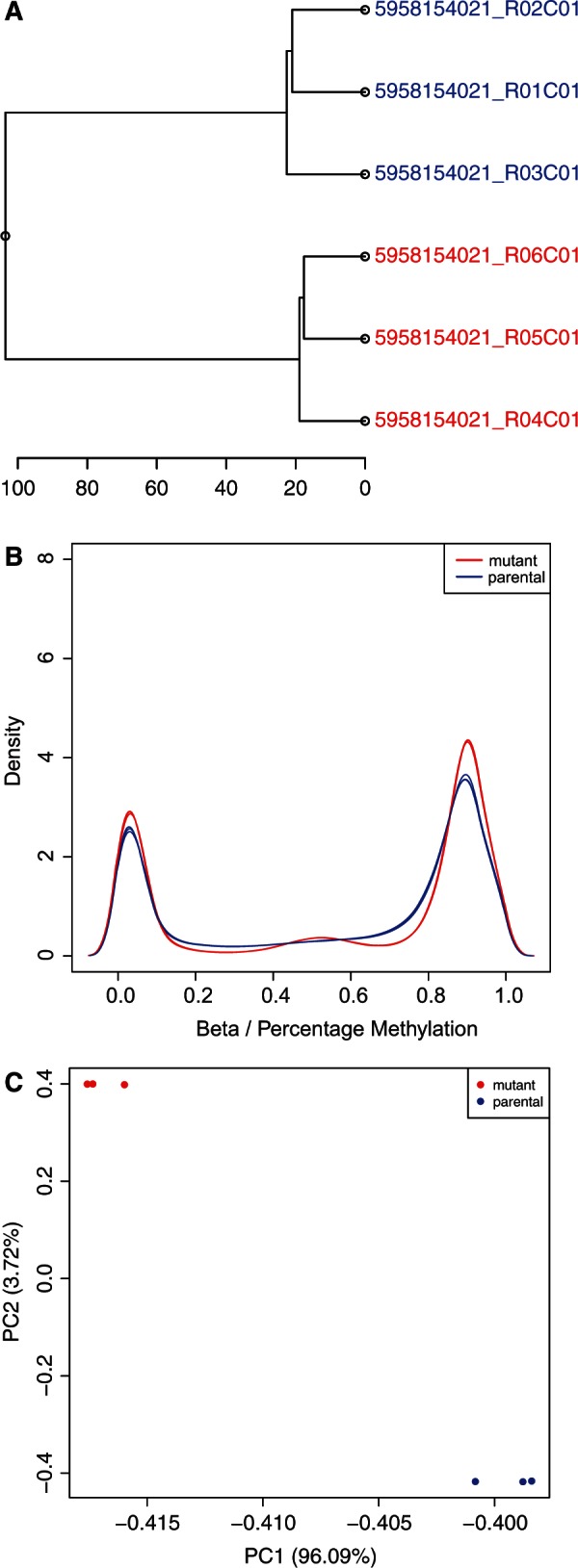
COHCAP quality control metrics. (A) Dendrogram: the sample ID for each sample is shown in the dendrogram representing the hierarchical clustering of the genome-wide beta values for each sample. Sample IDs are colored based on the sample grouping (in this case, the parental HCT116 strain is shown in blue and the mutant strain is shown in red). Notice that the samples in each group cluster together. (B) Sample histogram: density distribution for all the samples in a COHCAP project is shown in the histogram. Again, the color for each sample is determined by the sample grouping. Notice the strong bimodal distribution, corresponding to methylated and unmethylated CpG sites. Sample statistics (median, top quartile, bottom quartile, minimum and maximum) are provided in a text file. (C) Principal component analysis (PCA) plot: samples are plotted based on their coordinates defined by the first two principal components. All the principal component values can be found in a text file. Samples are colored based on sample grouping. Notice that the groups show clear clustering from one another in the PCA plot.
Additionally, we selected the top four regions with the strongest inverse correlation in gene expression and methylation for low-throughput validation (RAB34, NEDD4L, TCF7L2 and VSNL1; Supplementary Figure S6). We found that both expression and methylation differences could be confirmed for three out of the four genes. All four of these genes also showed differential methylation based on the ‘Average by CpG Site’ workflow (Supplementary Table S5), and NEDD4L (the gene with discordant validation results) also qualitatively passed visual inspection of the region (Supplementary Figure S7). The only evidence that we could find to potentially disqualify this region is that it is not upstream of the RefSeq transcription start site (which does not relate to the statistical analysis in COHCAP). Visualization of the COHCAP wiggle (.wig) file is necessary for optimizing the genomic coordinates for a differentially methylated region, so we would also recommend users to check whether the mapping from the Illumina array provides a plausible genomic location for epigenetic regulation of the target gene. Nevertheless, it is likely that COHCAP did an accurate job of summarizing the data provided by the 450k array, and the false positive for NEDD4L may be due to a more general limitation in the interpretation of these results. For example, it is possible that nearby CpG sites not represented on the 450k array showed opposite or random changes in methylation, weakening the significance of the eight probes showing differential methylation on the array. In short, these results indicate that COHCAP is likely to produce candidate regions/genes that are likely to show successful validation in follow-up experiments.
There is one other Illumina methylation array algorithm that provides statistics for differentially methylated regions (30), so we compared the regions predicted from COHCAP with those predicted by IMA. IMA summarizes methylation differences within 11 different genomic regions. Differential methylation in transcription start site regions (TSS200 and TSS1500) and CpG islands (ISLAND, NSHELF, SSHELF, NSHORE, SHORE) were compared with the COHCAP results, and the overlap with differences in gene expression were compared for the TSS200 and TSS1500 regions (Supplementary Table S3, Supplementary Figure S8). As might be expected, the IMA ISLAND regions showing differential methylation had the best concordance with the COHCAP results. The IMA TSS1500 regions of differential methylation showed better overlap with differentially expressed genes compared with the TSS200 regions (9.6% versus 1.8%). The gene expression overlap for the TSS1500 regions was roughly similar to the COHCAP results for the ‘Average by Site’ results (9.6% versus 11.6%), but it was lower than the overlap for the ‘Average by Island’ COHCAP results (38.4% versus 9.6%). Although we tried to make the criteria as similar as possible (FDR <0.05, delta-beta >0.3 for IMA, methylated beta >0.7 for COHCAP and unmethylated beta <0.3 for COHCAP), it is possible that this may be due to differences in the thresholds used for comparison. Therefore, we decided to compare overlap with gene expression while varying FDR cutoffs (with and without beta thresholds, using the ‘Average by Island’ results). The overlap with gene expression was very robust to FDR cutoff values (Supplementary Figure S9). Without the use of beta thresholds, the COHCAP and IMA expression overlaps were roughly similar. However, COHCAP always showed a greater overlap with gene expression than IMA when beta thresholds were applied (regardless of FDR cutoff). The overall concordance with gene expression was highest when using correlation to integrate the methylation and gene expression data (with beta thresholds), and correlation consistently showed better concordance with gene expression than overlap whenever all other parameters were kept constant (Supplementary Figure S10). This comparison with IMA shows that COHCAP contains the tools (e.g. methylated/unmethylated thresholds, integration via correlation) that allow for identifying regions of differential methylation with maximal concordance with gene expression.
COHCAP can robustly analyze heterogeneous patient data
We have showed that COHCAP provides accurate results that outperform IMA on a small homogenous cell line dataset. To show scalability for large heterogeneous datasets, we used COHCAP to integrate the Illumina 450k methylation data and RNA-Seq gene expression data available for TCGA breast cancer patients (10). This dataset provides genomic data for tumors and paired normal tissue, so we used COHCAP to identify regions of differential methylation between cancerous and normal breast tissue (Supplementary Table S6). It was necessary to specify the normal/tumor pairing to detect regions of differential methylation (with FDR <0.05). In fact, increasing the sample size ∼4 × (N=562 versus 134) had a minimal effect on detecting regions of differential methylation (12.6–23.4% increase) when considering all samples compared with only the samples with paired normal data. Because of the considerably shorter run-time (Supplementary Table S7) and nearly identical gene lists, only the paired samples were considered for subsequent analysis.
Given the heterogeneity of the patient samples, it was no longer reasonable to define groups where the mean beta values lie in separate methylated/unmethylated peaks (as done for the mutant HCT116 dataset), so we used 0.3 as both the methylated and unmethylated threshold [similar to Sproul et al. (44)] and compared the COHCAP results with the IMA results (without the use of any beta threshold) for detecting regions of differential methylation. The integration via overlap results were similar (Supplementary Table S6B), but the integration via correlation showed a slightly higher rate (36.2%). The concordance rate (for both the TCGA and HCT116 datasets) is even higher (55.8%) when the number of genes is considered instead of the number or regions because many CpG islands do not map to genes (Supplementary Figure S11). This concordance rate is comparable with the cell line results, where the number of genes was more similar to the number of regions. In fact, this high concordance rate is robust against a wide variety of thresholds for the TCGA analysis, but the HCT116 analysis was much more sensitive to the size of the gene/island lists (Supplementary Figure S11). The reason for this robust concordance in the TCGA data is the considerably larger number of samples per group, which allows for greater statistical power to detect covariation between DNA methylation and gene expression (Supplementary Figure S12). Finally, it is worth noting that the negative correlation rate is much higher than the positive correlation rate, regardless of which thresholds are used for analysis (Supplementary Figure S13). This emphasizes COHCAP is capable of capturing true biological regulation: if the signal was random noise, we would expect approximately equal rates of positive and negative correlations. All these results emphasize the value of integrating via correlation, which is not possible unless an analysis package directly handles both DNA methylation and gene expression data.
The expression overlap was still similar for COHCAP and IMA, but this metric only represents the sensitivity of the algorithm to detect regions that may affect gene expression. When the gene expression overlap is visualized in a Venn diagram (Supplementary Figure S14), it becomes clear that COHCAP shows similar sensitivity to IMA, with greater specificity (as determined by comparing the inverse overlap with the matched overlap). More specifically, IMA listed 2052 (25.1%) regions with an inverse overlap (expression up and methylation down, and vice versa) between methylation and expression lists, but it also listed an even larger number of (2147, 26.3%) regions with a matched overlap (e.g. expression and methylation up, expression and methylation down). In contrast, COHCAP identified 186 (21.1%) regions with inverse overlap and 102 (7.9%) regions with matched overlap (which we assume correlates with a lower false-positive rate). We hypothesize that this is due to the inclusion of CpG shores and CpG shelves as a single functional unit for the CpG island in COHCAP (compared with five different genomic regions in IMA), greater flexibility in mapping CpG islands to target genes based on the Illumina annotation file, implementation of a minimum number of varying CpG sites in COHCAP (but not IMA) and/or COHCAP using methylated and unmethylated thresholds (both set to 0.3, in this case) for CpG site and CpG island filtering.
One of the genes showing differential methylation with a corresponding change in expression was estrogen receptor (ESR1; Figure 3), which is a therapeutically relevant target known to be over-expressed in breast cancer patients (49), which has also been shown to be demethylated in a smaller breast cancer patient cohort (44). One of the advantages to using the ‘Average by Island’ COHCAP workflow is the correlation in gene expression will not just represent average differences in populations but also covariation within populations (for large heterogeneous datasets like the TCGA dataset). Therefore, the estrogen receptor scatter plot (Supplementary Figure 3B) also does a good job of showing that the correlation between DNA methylation and gene expression not only is limited to population-level differences between two groups (such as shown in Supplementary Figure 3A) but also can detect covariance within groups (especially for large heterogeneous datasets like the TCGA dataset). As expected, ESR1 methylation levels are significantly negatively correlated with gene expression levels in tumors alone (r = −0.63, P = 4.1 × 10−38, Supplementary Figure S11). In fact, the correlation is stronger among tumor samples than when tumor and normal samples combined (r = −0.63 versus −0.36). Additionally, ESR1 shows decreased methylation in estrogen receptor positive (ER+) patients compared with estrogen receptor negative (ER−) patients (delta-beta = −0.11, P = 2.3 × 10−5, Supplementary Figure S15). This is important because normal and tumor cells may have different type of cell populations—for example, the tumor cells will often have a greater proportion of epithelial cells (50). Therefore, integration via correlation in the COHCAP ‘Average by Island’ workflow can be a useful tool when working with heterogeneous patient datasets.
Figure 3.

Estrogen receptor over-expression is correlated with decreased methylation. All the images in this figure represent ways that users can visualize regions of differential methylation (using estrogen receptor, ESR1, in the TCGA breast cancer dataset). (A) Box plot: the average beta value for a normal sample is higher than the primary sample, indicating that this CpG island (mapped to ESR1) shows decreased methylation in breast tumors. The box plot shows the median, minimum, maximum and quartiles for beta values for each group. This figure was produced using the ‘Average by Island’ workflow. (B) Scatter plot: methylation levels of ESR1 are negatively correlated with RNA-Seq expression levels. Individual samples are colored based on their sample grouping. This figure was produced using the ‘Average by Island’ workflow. (C) Visualization of.wig files in Integrative Genomics Viewer: normal, primary and delta-beta values (primary beta − normal beta) are exported from COHCAP as.wig files. A.bed file of UCSC CpG islands is also included in the visualization. Visualization of the ESR1 gene shows that a large number of CpG sites show a consistent decrease in methylation in tumor samples, and the peak is centered around the ESR1 translation start site. This figure was produced using the ‘Average by Site’ workflow.
It is important to note that COHCAP and IMA use different mapping systems, so the resulting gene lists will be different no matter what parameters are used (Supplementary Figure S6). This can be seen clearly when comparing overlapping genes from COHCAP versus IMA analysis (Supplementary Figure S16). More specifically, IMA maps CpG sites that lie in either the TSS200 or TSS1500 regions (200 or 1500 bp upstream of the transcription start site, respectively). Therefore, you will never see conflicting trends for methylation patterns for a given gene list, even for the very large IMA TSS1500 gene lists. However, COHCAP defines regions based on the UCSC CpG island annotations (including the shelves and shores), and gene mappings are based on proximity to any part of the gene. For example, COHCAP identified regions of both increased and decreased methylation mapped to the PLEC1 gene (Figure S17). However, it should be noted that COHCAP is relatively conservative in its predictions, so it is rare to encounter genes with opposite trends (and they will probably be cases like PLEC1, where the CpG islands are located in the gene body rather than the promoter).
Moreover, Ingenuity Pathway Analysis (Ingenuity® Systems, www.ingenuity.com) was used to identify other breast cancer biomarkers showing differential methylation with negatively correlated gene expression; However, the IMA and COHCAP biomarkers show little overlap (Supplementary Table S8 and S9). Some COHCAP biomarkers also overlap genes that were previously shown to be regulated by DNA methylation in breast cancer (Supplementary Table S10). So, regardless of the estimated accuracy, it is clear that COHCAP and IMA can be combined to provide maximal biomarker candidates for a particular patient cohort. In short, the biomarker analysis shows that COHCAP can provide unique clinically relevant results.
Another interesting technical observation is that the COHCAP regions show a larger absolute delta-beta value than the IMA regions (0.22 versus 0.087 for the Ingenuity Pathway Analysis biomarkers, and 0.24 versus 0.073 for the entire list of differentially methylated regions before to integration with gene expression data). This is important because it emphasizes the validity to using methylated and unmethylated thresholds. If categorizing regions into those with an average beta value <0.3 and >0.3 had no biological meaning, then you would expect a larger number of regions with very small delta-beta values (just slightly above or below an arbitrary cutoff). However, the strong bimodal distribution of beta values (Figure 2B) indicates that populations that are more consistently methylated or unmethylated should have densities that are biased toward one peak over the other. It should be noted that this can be a conservative requirement. For example, the ER+ versus ER− comparison does not correctly identify ESR1 as showing differential methylation with a methylated and unmethylated threshold of 0.3 (however, ESR1 could be identified if 0.5 was used as the methylated and unmethylated threshold). Nevertheless, we think this is an important feature provided by COHCAP but not IMA.
COHCAP one-group workflow provides accurate reproducible results
The HCT116 mutant cell line comparison and TCGA breast cancer comparison showed the utility of COHCAP for two-group comparisons. However, one of the advantages of COHCAP is that it contains workflows for any number of groups. For example, this can be useful when there is no appropriate control for comparison (or analyzing a single sample). To access the accuracy of the one-group workflow in COHCAP, we analyzed a sample using COHCAP (for the Illumina 450k array) and MIRA (2). The ‘Average by Site’ workflow was used because this is the only way to calculate P-values for a one-group comparison. Good concordance between these technologies indicates that COHCAP provides CpG island metrics that appropriately represent coordinated hyper-methylation of nearby CpG sites. If standard thresholds are used (COHCAP FDR <0.05, NimbleScan P < 0.05), there is clearly greater overlap with the MIRA peaks for the hyper-methylated COHCAP islands (40.2% overlap, Supplementary Figure S18) compared with the hypo-methylated COHCAP islands (2.4%). This is to be expected because the MIRA assay only detects hyper-methylated regions. In fact, practically all overlap is with the 450k methylated regions (622 peaks resulting in 65.8% overlap, compared with 1 peak resulting in 0.1% overlap), if sufficiently conservative thresholds are used to define the MIRA peaks. These results also match the good concordance previously reported for MeDIP-Seq versus the Illumina 450k array (as well as BS-Seq) (51). Moreover, the top 10 COHCAP CpG islands all overlapped a MIRA peak (Supplementary Table S11A, Supplementary Figure S19), which provides an indication that COHCAP can prioritize the regions with consistent patterns of differential methylation.
In contrast, if CpG sites are used to approximate CpG island behavior, the consistency with MIRA peaks is weakened (Supplementary Figure S18). Whereas COHCAP CpG islands have between 94 and 99% concordance with overlapping MIRA peaks, differentially methylated regions defined using individual CpG sites show 44–81% concordance with overlapping MIRA peak. Additionally, use of a single CpG site to define a differentially methylated region causes a substantial number of CpG islands (20–43%) to have conflicting results (i.e. they have at least one methylated site and one unmethylated site). Similarly, the top 10 CpG sites with the highest average beta value within a UCSC CpG island do not show good overlap with the MIRA peaks, whereas the CpG island analysis yielded perfect concordance with these top 10 COHAP regions (Supplementary Table S11B). In short, this comparison provides evidence that COHCAP can accurately identify regions of hyper-methylation with the one-group workflow, and that analysis of CpG islands is more likely to yield accurate results than relying on individual CpG sites.
The high accuracy of the one-group COHCAP workflow estimated using the MIRA data is further validated by comparing HCT116 BS-Seq and 450k array data. Targeted BS-Seq and 450k array data for HCT116 is publicly available (12,36), so we compared the results of the HCT116 data from this study with these publicly available datasets. More specifically, the one-group workflow was first used to determine the overlap of methylated and unmethylated regions in the HCT116 using the two 450k datasets (Figure 4A), and it was clear that regions with corresponding methylation designations (e.g. methylated versus methylated, unmethylated versus unmethylated) show much greater overlap than unrelated regions (e.g. methylated versus unmethylated). As a technical note, the dataset from this study was used as a benchmark to calculate run-time (Supplementary Table S12); users should be aware that one-group analysis typically takes longer than two-group analysis (for comparable-size datasets) because the CpG site filter usually removes a much smaller proportion of the CpG sites for CpG island analysis. This 450k comparison indicates that non-random overlap can be seen with the same technology even though the samples were produced at different times by different laboratories. This is not trivial because methylation patterns can diverge over time (52,53), so an accurate method of analysis would still show some inconsistency between samples. Similarly, the data from this study also shows non-random overlap with COHCAP regions detected using BS-Seq (Figure 4B). Concordance between BS-Seq and Illumina methylation array has been previously reported (21,51,54), but it was not clear whether the specific regions identified by COHCAP would show good concordance for these samples produced from different laboratories. Furthermore, the BS-Seq data lacked replicates, and there are clear differences in signal distribution for BS-Seq data compared with Illumina array data (Figure S20). Therefore, we believe the COHCAP results for the 450k array and BS-Seq results show very strong overlap. Most importantly, the concordance of the independent HCT116 datasets for the 450k/BS-Seq comparison further emphasizes the accuracy of the one-group COHCAP workflow (for both hyper- and hypo-methylated regions).
Figure 4.

Overlapping signal from HCT116 Illumina array and BS-Seq data. (A) Illumina array overlap: two independently produced samples (17, this study) measuring hyper- and hypo-methylated regions in the HCT116 cell are used to compare similarity in signal for CpG islands between these two studies. Each study had triplicate samples, which were used for COHCAP analysis. The matched overlap is clearly much greater than the inverse overlap, indicating that there is non-random overlap due to concordance of the COHCAP results. (B) Illumina array versus BS-Seq: two independently produced samples for HCT116 methylation for the Illumina array (this study) and targeted BS-Seq (36) were used to compare the similarity of COHCAP hyper- and hypo-methylated regions between these two different technologies. The matched overlap is clearly much greater than the inverse overlap, indicating that there is non-random overlap due to the good concordance and reproducibility of the COHCAP results.
COHCAP analysis shows high concordance with methylKit on simulated BS-Seq data
To further justify that COHCAP is appropriate for analysis of BS-Seq data, we used methylKit to analyze the HCT116 mutant versus parental 450k data from this study (35) that has been simulated as BS-Seq data (by assigning various coverage values to the 450k probes). The goal of this comparison is to test the interchangeability of 450k and BS-Seq algorithms (e.g. application of COHCAP to BS-Seq data and application of methylKit to 450k data). The methylKit algorithm enables users to search for windows (in this case, 1000-bp windows) showing enrichment of hyper- and hypo-methylated CpG sites. If all 450k probes are assumed to have 100× coverage, there is very strong overlap between the COHCAP and methylKit results: 139 of 140 COHCAP (99.3%) CpG Islands showing differential methylation were found to show at least 50% overlap with at least one of the 1-kb methylKit windows showing differential methylation (out of 14 449 total methylKit windows). These results are similar using lower coverage values that are more typically observed in BS-Seq data (such as 5× and 10× coverage), and the majority of the COHCAP regions rank among the top 5% of the methylKit regions (Supplementary Table S13). In short, this two-group methylKit comparison and the one-group HCT116 450k/BS-Seq comparison emphasize that COHCAP can provide accurate analysis of BS-Seq data.
DISCUSSION
This study emphasizes that methylation-expression correspondence depends highly on the method of integration. Namely, this study shows that lists of differentially methylated CpG islands can show an approximately 50% concordance rate with the gene expression changes, if the appropriate methodology is used (Supplementary Figure S10, S11 and S13). To be clear, it should be emphasized that the goal is not to broadly characterize or predict the role of DNA methylation on gene expression; for example, COHCAP does not support or refute the results of studies like Bell et al. (55). Instead, the goal of COHCAP is to provide lists of differentially methylated CpG islands with optimal concordance with gene expression data, and we have shown that the ‘Average by Island’ workflow in COHCAP achieves this goal by outperforming the two-group ‘Average by Site’ workflow in COHCAP as well as the TSS1500 analysis in IMA (30).
COHCAP analysis shows that estrogen receptor, a gene known to be over-expressed in breast cancer patients, also shows a decrease in methylation in breast tumors compared with normal tissue (Figure 3). Interestingly, the region with the clearest differential methylation is located near the translation start site for ESR1 but not the RefSeq transcription start site (although that also shows a corresponding change in expression, albeit with weaker resolution on the Illumina array; Figure S21). Hence, this may be why the region could be detected with COHCAP and not IMA. It should also be noted that none of these biomarkers showing differential methylation with correlated gene expression was described in the corresponding article for the TCGA data (10), which may emphasize the ability for COHCAP to facilitate epigenetic discoveries.
This study shows that COHCAP is able to accurately detect regions of differential methylation with maximal concordance with corresponding gene expression levels. COHCAP provides a number of unique workflows, such as integration with gene expression, application to both Illumina methylation array and targeted BS-Seq data and the ability to analyze a study design with any number of groups. The accuracy of COHCAP was verified by validation of differentially methylated genes in the HCT116 cell line, good reproducibility of COHCAP CpG islands using different technologies (such as MIRA and BS-Seq) and recovery of known breast cancer biomarkers (some of which have been previously shown to be under epigenetic regulation). Overall, COHCAP has been shown scalable for high-quality integrative analysis of cell line data as well as large heterogeneous patient samples.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–13, Supplementary Figures 1–21, Supplemental Methods and Supplementary References [10,17,43,45,56–64].
ACKNOWLEDGEMENTS
The authors would like to thank Altuna Akalin for suggesting how to apply methylKit to Illumina 450k methylation array data; Xutao Deng, Xiwei Wu, Zheng Liu and Marc Jung for discussions regarding the COHCAP algorithm; Susan Neuhausen, Yuan Chun Ding, Linda Malkas, Long Gu and Thanh Dellinger for discussions regarding application of COHCAP to other datasets; and Shiuan Chen and Edward Newman for discussions regarding the breast cancer estrogen receptor analysis. The authors are also grateful for the insightful comments from the anonymous reviewers of this manuscript.
FUNDING
The National Institutes of Health [Comprehensive Cancer Center Grant P30 CA33572, R01 CA115674 to R.J., R01 CA122976 to H.Y.]; City of Hope National Medical Center [institutional funding for Y.C.Y. and A.D.R.]. Funding for open access charge: City of Hope National Medical Center institutional funds.
Conflict of interest statement. None declared.
REFERENCES
- 1.Irizarry RA, Ladd-Acosta C, Carvalho B, Wu H, Brandenburg SA, Jeddeloh JA, Wen B, Feinberg AP. Comprehensive high-throughput arrays for relative methylation (CHARM) Genome Res. 2008;18:780–790. doi: 10.1101/gr.7301508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rauch T, Li H, Wu X, Pfeifer GP. MIRA-assisted microarray analysis, a new technology for the determination of dna methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006;66:7939–7947. doi: 10.1158/0008-5472.CAN-06-1888. [DOI] [PubMed] [Google Scholar]
- 3.Bock C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012;13:705–719. doi: 10.1038/nrg3273. [DOI] [PubMed] [Google Scholar]
- 4.Barrera V, Peinado MA. Evaluation of single CpG sites as proxies of CpG island methylation states at the genome scale. Nucleic Acids Res. 2012;40:11490–11498. doi: 10.1093/nar/gks928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roessler J, Ammerpohl O, Gutwein J, Hasemeier B, Anwar S, Kreipe H, Lehmann U. Quantitative cross-validation and content analysis of the 450k DNA methylation array from Illumina, Inc. BMC Res. Notes. 2012;5:210. doi: 10.1186/1756-0500-5-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The Cancer Genome Atlas Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The Cancer Genome Atlas Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dedeurwaerder S, Desmedt C, Calonne E, Singhal SK, Haibe-Kains B, Defrance M, Michiels S, Volkmar M, Deplus R, Luciani J, et al. DNA methylation profiling reveals a predominant immune component in breast cancers. EMBO Mol. Med. 2011;3:726–741. doi: 10.1002/emmm.201100801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Eijk K, de Jong S, Boks M, Langeveld T, Colas F, Veldink J, de Kovel C, Janson E, Strengman E, Langfelder P, et al. Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMC Genomics. 2012;13:636. doi: 10.1186/1471-2164-13-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell. 2012;49:359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, Belvisi MG, Brown R, Vineis P, Flanagan JM. Epigenome-wide association study in the European Prospective Investigation into cancer and nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum. Mol. Genet. 2013;22:843–851. doi: 10.1093/hmg/dds488. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, Reinius L, Acevedo N, Taub M, Ronninger M, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotech. 2013;31:142–147. doi: 10.1038/nbt.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the infinium methylation 450K technology. Epigenomics. 2011;3:771–784. doi: 10.2217/epi.11.105. [DOI] [PubMed] [Google Scholar]
- 18.Kim JW, Kim ST, Turner AR, Young T, Smith S, Liu W, Lindberg J, Egevad L, Gronberg H, Isaacs WB, et al. Identification of new differentially methylated genes that have potential functional consequences in prostate cancer. PLoS One. 2012;7:e48455. doi: 10.1371/journal.pone.0048455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duncan CG, Barwick BG, Jin G, Rago C, Kapoor-Vazirani P, Powell DR, Chi JT, Bigner DD, Vertino PM, Yan H. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 2012;22:2339–2355. doi: 10.1101/gr.132738.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén SE, Greco D, Söderhäll C, Scheynius A, Kere J. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7:e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pan H, Chen L, Dogra S, Ling Teh A, Tan JH, Lim YI, Lim YC, Jin S, Lee YK, Ng PY, et al. Measuring the methylome in clinical samples: improved processing of the infinium human methylation450 beadchip array. Epigenetics. 2012;7:1173–1187. doi: 10.4161/epi.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011;6:692–702. doi: 10.4161/epi.6.6.16196. [DOI] [PubMed] [Google Scholar]
- 23.Lee ST, Xiao Y, Muench MO, Xiao J, Fomin ME, Wiencke JK, Zheng S, Dou X, de Smith A, Chokkalingam A, et al. A global DNA methylation and gene expression analysis of early human B-cell development reveals a demethylation signature and transcription factor network. Nucleic Acids Res. 2012;40:11339–11351. doi: 10.1093/nar/gks957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grafodatskaya D, Chung B, Butcher D, Turinsky A, Goodman S, Choufani S, Chen YA, Lou Y, Zhao C, Rajendram R, et al. Multilocus loss of DNA methylation in individuals with mutations in the histone H3 Lysine 4 demethylase KDM5C. BMC Med. Genomics. 2013;6:1. doi: 10.1186/1755-8794-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kilaru V, Barfield RT, Schroeder JW, Smith AK, Conneely KN. MethLAB: a graphical user interface package for the analysis of array-based DNA methylation data. Epigenetics. 2012;7:225–229. doi: 10.4161/epi.7.3.19284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emes RD, Wessely F. Identification of DNA methylation biomarkers from Infinium arrays. Front. Genet. 2012;3:161. doi: 10.3389/fgene.2012.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barfield RT, Kilaru V, Smith AK, Conneely KN. CpGassoc: an R function for analysis of DNA methylation microarray data. Bioinformatics. 2012;28:1280–1281. doi: 10.1093/bioinformatics/bts124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun H, Wang S. Penalized logistic regression for high-dimensional DNA methylation data with case-control studies. Bioinformatics. 2012;28:1368–1375. doi: 10.1093/bioinformatics/bts145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baek SJ, Yang S, Kang TW, Park SM, Kim YS, Kim SY. MENT: methylation and expression database of normal and tumor tissues. Gene. 2013;518:194–200. doi: 10.1016/j.gene.2012.11.032. [DOI] [PubMed] [Google Scholar]
- 30.Wang D, Yan L, Hu Q, Sucheston LE, Higgins MJ, Ambrosone CB, Johnson CS, Smiraglia DJ, Liu S. IMA: An R package for high-throughput analysis of Illumina’s 450K Infinium methylation data. Bioinformatics. 2012;28:729–730. doi: 10.1093/bioinformatics/bts013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kulis M, Heath S, Bibikova M, Queiros AC, Navarro A, Clot G, Martinez-Trillos A, Castellano G, Brun-Heath I, Pinyol M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet. 2012;44:1236–1242. doi: 10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- 32.Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, Milne TA, Huang Y, Biswas D, Hess JL, et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 2012;8:e1002781. doi: 10.1371/journal.pgen.1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484:339–344. doi: 10.1038/nature10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansen K, Langmead B, Irizarry R. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13:R83. doi: 10.1186/gb-2012-13-10-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akalin A, Kormaksson M, Li S, Garrett-Bakelman F, Figueroa M, Melnick A, Mason C. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee EJ, Pei L, Srivastava G, Joshi T, Kushwaha G, Choi JH, Robertson KD, Wang X, Colbourne JK, Zhang L, et al. Targeted bisulfite sequencing by solution hybrid selection and massively parallel sequencing. Nucleic Acids Res. 2011;39:e127. doi: 10.1093/nar/gkr598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium humanmethylation450 beadchips. Genome Biol. 2012;13:R44. doi: 10.1186/gb-2012-13-6-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Touleimat N, Tost J. Complete pipeline for Infinium® Human methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics. 2012;4:325–341. doi: 10.2217/epi.12.21. [DOI] [PubMed] [Google Scholar]
- 39.Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S. A Beta-Mixture Quantile Normalisation method for correcting probe design bias in illumina infinium 450k DNA methylation data. Bioinformatics. 2012;29:189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 41.Teschendorff AE, Zhuang J, Widschwendter M. Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics. 2011;27:1496–1505. doi: 10.1093/bioinformatics/btr171. [DOI] [PubMed] [Google Scholar]
- 42.Leek JT, Storey JD. A general framework for multiple testing dependence. Proc. Natl Acad. Sci. USA. 2008;105:18718–18723. doi: 10.1073/pnas.0808709105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sproul D, Nestor C, Culley J, Dickson JH, Dixon JM, Harrison DJ, Meehan RR, Sims AH, Ramsahoye BH. Transcriptionally repressed genes become aberrantly methylated and distinguish tumors of different lineages in breast cancer. Proc. Natl Acad. Sci. USA. 2011;108:4364–4369. doi: 10.1073/pnas.1013224108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 46.Gu H, Bock C, Mikkelsen TS, Jager N, Smith ZD, Tomazou E, Gnirke A, Lander ES, Meissner A. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat. Methods. 2010;7:133–136. doi: 10.1038/nmeth.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat. Biotech. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hayashi SI, Eguchi H, Tanimoto K, Yoshida T, Omoto Y, Inoue A, Yoshida N, Yamaguchi Y. The expression and function of estrogen receptor alpha and beta in human breast cancer and its clinical application. Endocr. Relat. Cancer. 2003;10:193–202. doi: 10.1677/erc.0.0100193. [DOI] [PubMed] [Google Scholar]
- 50.Keller P, Lin A, Arendt L, Klebba I, Jones A, Rudnick J, DiMeo T, Gilmore H, Jefferson D, Graham R, et al. Mapping the cellular and molecular heterogeneity of normal and malignant breast tissues and cultured cell lines. Breast Cancer Res. 2010;12:R87. doi: 10.1186/bcr2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clark C, Palta P, Joyce CJ, Scott C, Grundberg E, Deloukas P, Palotie A, Coffey AJ. A Comparison of the whole genome approach of MeDIP-Seq to the targeted approach of the infinium humanmethylation450 BeadChip(®) for methylome profiling. PLoS One. 2012;7:e50233. doi: 10.1371/journal.pone.0050233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li A, Walling J, Kotliarov Y, Center A, Steed ME, Ahn SJ, Rosenblum M, Mikkelsen T, Zenklusen JC, Fine HA. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol. Cancer Res. 2008;6:21–30. doi: 10.1158/1541-7786.MCR-07-0280. [DOI] [PubMed] [Google Scholar]
- 53.Landan G, Cohen NM, Mukamel Z, Bar A, Molchadsky A, Brosh R, Horn-Saban S, Zalcenstein DA, Goldfinger N, Zundelevich A, et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat. Genet. 2012;44:1207–1214. doi: 10.1038/ng.2442. [DOI] [PubMed] [Google Scholar]
- 54.Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotech. 2010;28:1097–1105. doi: 10.1038/nbt.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bell J, Pai A, Pickrell J, Gaffney D, Pique-Regi R, Degner J, Gilad Y, Pritchard J. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011;12:R10. doi: 10.1186/gb-2011-12-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 57.Kanaya N, Kubo M, Liu Z, Chu P, Wang C, Yuan YC, Chen Y. Protective effects of white button mushroom (Agaricus bisporus) against hepatic steatosis in ovariectomized mice as a model of postmenopausal women. PLoS One. 2011;6:e26654. doi: 10.1371/journal.pone.0026654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tompkins JD, Hall C, Chen VC, Li AX, Wu X, Hsu D, Couture LA, Riggs AD. Epigenetic stability, adaptability, and reversibility in human embryonic stem cells. Proc. Natl Acad. Sci. USA. 2012;109:12544–12549. doi: 10.1073/pnas.1209620109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Current Protocols in Molecular Biology. John Wiley & Sons, Inc; 2010. Galaxy: a web-based genome analysis tool for experimentalists. Chapter 19: Unit 19.10.1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goecks J, Nekrutenko A, Taylor J, Team TG. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sabatier R, Finetti P, Adelaide J, Guille A, Borg JP, Chaffanet M, Lane L, Birnbaum D, Bertucci F. Down-regulation of ECRG4, a candidate tumor suppressor gene, in human breast cancer. PLoS One. 2011;6:e27656. doi: 10.1371/journal.pone.0027656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.How Kit A, Nielsen HM, Tost J. DNA methylation based biomarkers: Practical considerations and applications. Biochimie. 2012;94:2314–2337. doi: 10.1016/j.biochi.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 64.Faryna M, Konermann C, Aulmann S, Bermejo JL, Brugger M, Diederichs S, Rom J, Weichenhan D, Claus R, Rehli M, et al. Genome-wide methylation screen in low-grade breast cancer identifies novel epigenetically altered genes as potential biomarkers for tumor diagnosis. FASEB J. 2012;26:4937–4950. doi: 10.1096/fj.12-209502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.