


Abstract
Restriction endonucleases are highly specific in recognizing the particular DNA sequence they act on. However, their activity is affected by sequence context, enzyme concentration and buffer composition. Changes in these factors may lead to either ineffective cleavage at the cognate restriction site or relaxed specificity allowing cleavage of degenerate ‘star’ sites. Additionally, uncharacterized restriction endonucleases and engineered variants present novel activities. Traditionally, restriction endonuclease activity is assayed on simple substrates such as plasmids and synthesized oligonucleotides. We present and use high-throughput Illumina sequencing-based strategies to assay the sequence specificity and flanking sequence preference of restriction endonucleases. The techniques use fragmented DNA from sequenced genomes to quantify restriction endonuclease cleavage on a complex genomic DNA substrate in a single reaction. By mapping millions of restriction site–flanking reads back to the Escherichia coli and Drosophila melanogaster genomes we were able to quantitatively characterize the cognate and star site activity of EcoRI and MfeI and demonstrate genome-wide decreases in star activity with engineered high-fidelity variants EcoRI-HF and MfeI-HF, as well as quantify the influence on MfeI cleavage conferred by flanking nucleotides. The methods presented are readily applicable to all type II restriction endonucleases that cleave both strands of double-stranded DNA.
INTRODUCTION
Type II restriction endonucleases cleave double-stranded DNA at a constant position with respect to a short (3–8 bp) recognition sequence (1). Their exquisite specificity has rendered them among the most useful tools in molecular biology (1,2). However, the impact of additional variables such as organic solvent, ion, small molecule and enzyme concentrations has large effects on the specificity of restriction endonucleases, often leading to cleavage at non-cognate sites (termed star activity) (3–7). Many commonly used restriction endonucleases show some star activity even under standard reaction conditions (3). The DNA substrate itself can also modulate cleavage. It has been noted that nucleotides flanking the recognition site confer large contributions to the energetics of cleavage (8–12). Quantitative analysis of star activity and flanking effects will help to elucidate the structure–function rules for restriction enzymes, define the window of optimal restriction endonuclease specificity as well as tailor reaction conditions toward novel target sequences.
Despite the conserved functionality among the restriction endonuclease family, these enzymes show great divergence in both sequence and mechanism (1,9,13,14). Apart from isoschizomers, most members show little sequence homology to each other or other known proteins (1). Additionally, the variable distribution of base-contacting residues among the restriction endonucleases has confounded recognition sequence prediction (9,15,16). Consequently, restriction endonuclease characterization must be carried out empirically for each enzyme. Star activity (4,17–21) and flanking preference (8–12) have been investigated for several enzymes. These experiments have been performed on homogeneous substrates. A series of oligonucleotides containing different star or flanking sequences are synthesized, annealed, cleaved and analyzed one by one, making exhaustive studies difficult. Recognition site determination is typically carried out by digestion of a homogeneous plasmid or virus DNA substrate followed by agarose gel visualization of cleavage products (6,22–25). This technique is lacking both in its substrate complexity and sensitivity. A given cognate or star site could occur few times in these substrates, and at times, not at all. This limits the ability to accurately quantify activity at different cleavage sites owing to a lack of diversity of flanking nucleotides. Star activity is often several orders of magnitude lower than cleavage at the cognate site (3,17). Consequently a large component of star activity will remain cryptic when cleavage products must be of sufficient abundance to be visualized on an agarose gel.
The growing amount of prokaryotic genomic sequence putatively coding for uncharacterized restriction endonucleases (26,27) in conjunction with ongoing efforts to engineer altered specificities (22–25,28–30) will be aided by high-throughput methods to quantify restriction endonuclease activity instead of the methods currently available. For example, to characterize the genome-wide digestion patterns of the methylation-specific restriction endonuclease AbaSDFI (31), genomic rat brain DNA was digested with AbaSDFI to map 5-hydroxymethylcytosines, the digestion products were cloned into plasmids and Sanger sequenced one by one to map 122 cleavage sites to the rat genome. A similar strategy was used to demonstrate the relaxed specificity of the restriction enzyme TspGWI in the presence of sinefungin by Sanger sequencing 218 clones (5).
High-throughput sequencing has become a valuable tool for analyzing DNA–protein interactions. The ability to experimentally pair a DNA–protein interaction to a sequencing event has enabled techniques such as ChIP-seq (32) to provide sensitive statistics on transcription factor–DNA binding. We use derivations of the RAD-seq (33) method to quantitatively measure restriction endonuclease activity across the sequenced Drosophila melanogaster and Escherichia coli genomes. This method specifically prepares DNA adjacent to restriction sites for Illumina sequencing, allowing the relative sequence counts of sites with different flanking nucleotides to be determined. The RAD-seq protocol was carried out with serial enzyme dilutions to identify flanking motif enrichment in enzyme-limiting reactions. Modifications were made to the protocol to sequence all cleavage events regardless of overhang to generate a complex profile of relative activities at cognate and star sites in a single experiment. We apply these methods to quantify the cleavage patterns of EcoRI and MfeI, to compare star activity with their engineered high-fidelity counterparts and to quantify the effect of flanking nucleotides on MfeI activity.
MATERIALS AND METHODS
All enzymes and buffers used in this study were contributed by New England BioLabs.
Star activity assay
To assay restriction enzyme activity on a genome-wide scale, we designed an unbiased strategy to sequence all digested fragments regardless of overhang (see Figure 1). In all, 1000–1500 bp fragments of E. coli strain REL606 DNA were digested under star conditions, and smaller 300–500 bp fragments whose decreased molecular mass indicated digestion were separated and Illumina sequenced. Both 1 000 000 reads from EcoRI digests and 650 000 reads from MfeI digests were mapped back to the REL606 genome, and adjacent cleavage sites were computationally analyzed.
Figure 1.

The path of a single restriction site–containing genomic locus is shown for both the RAD-seq protocol (left) and the modified overhang-independent RAD-seq protocol used in the star activity assay (right).
(1) Generation of random 1000–1500 bp digestion templates: 3 μg of REL606 genomic DNA were randomly sheared by sonication (Bioruptor). DNA fragments between 1000 and 1500 bp were then separated and purified by agarose gel electrophoresis. The DNA was then blunt-end repaired using Quick Blunting Kit and 3′ adenylated using Klenow exo−. To distinguish the sheared DNA ends, non-divergent Illumina end 2 adapters composed of annealed oligonucleotides 5′-Phos-GATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCGATCTCGTATGCCGTCTTCTGCTTG-3′ and 5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT-3′ were ligated to the 1000–1500 bp pool using concentrated T4 DNA ligase. Ten nanograms of this sample was used in a 20-cycle Phusion polymerase chain reaction (PCR) with the Illumina PE primer 2.0 (5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT-3′) following Phusion product guidelines to select 1000–1500 bp fragments with the Illumina end 2 sequence on each end.
(2) Star condition digest: To generate a complex cleavage activity profile, DNA from the previous step was digested with an excess of restriction enzyme. Fifty-two nanogram of DNA was digested with 50 U of MfeI (GenBank accession number SRR652142) or high-fidelity MfeI (MfeI-HF; accession SRR652141) in a 50 μl reaction containing 1 × NEB4 and 5% glycerol for 24 h at 37°C. Thirty-two nanograms of DNA was digested with 200 U of EcoRI (accession SRR652140) in a 50 μl reaction containing 1× NEB1 and 10% glycerol for 24 h at 37°C. Thirty-two nanograms of DNA was digested with 200 U of high-fidelity EcoRI (EcoRI-HF; accession SRR652139) in a 50 μl reaction containing 1× NEB4 and 10% glycerol for 24 h at 37°C.
(3) Tagging of cleaved end with Illumina end 1 adapter: The digested DNA was blunt-end repaired using Quick Blunting Kit to neutralize all potential overhangs. The DNA was then 3′ adenylated using Klenow exo− and ligated using concentrated T4 DNA Ligase to barcoded divergent first-end Illumina adapters composed of annealed oligos 5′-GGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-barcode-T-3′ and 5′-Phos-barcode-AGATCGGAAGAGCGTCGTGTACTACGTT-3′. Ten nanograms of DNA from the ligation reaction was then used as template for an 18-cycle Phusion PCR with Illumina primers PE PCR Primer 2.0 (5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT-3′) and a shortened PE PCR Primer 1.0 (5′-AATGATACGGCGACCACCGA-3′) following Phusion product guidelines. Use of a divergent first-end adapter requires the paired-end primer to anneal first for amplification to occur. This eliminates the first end sequence on the sheared side. We used change in molecular mass to select for digested molecules. As the predigestion sample ranged from 1000 to 1500 bp, we agarose gel–purified 300–500-bp PCR fragments for sequencing to assure cleavage.
This final library exclusively contained molecules with a first end Illumina sequence on the cleaved side and a second end sequence on the sheared side. The experimental samples were sequenced on an Illumina HiSeq 2000 to generate 100 bp single-end reads beginning at the cleavage site. The samples were separated by barcode and the reads were mapped back to the E. coli genome to infer the cleavage site.
Fidelity index determination
The fidelity index (FI) was determined for EcoRI-HF and MfeI-HF by the standard method (3). The substrate used for all FI determinations was lambda DNA.
Flanking sequence preference assay
To determine the flanking sequence preferences of MfeI, D. melanogaster genomic DNA was digested in saturating and enzyme-limiting conditions. The DNA adjacent to the restriction site was then PCR amplified and Illumina sequenced as per the RAD-seq protocol (33) depicted in Figure 1. From each digest, 550 000 reads were mapped to the Drosophila genome. Flanking sequence preference was inferred from motif enrichment in enzyme-limiting conditions.
(1) MfeI digests: Digests were carried out in 50 μl reactions containing 786 ng of D. melanogaster strain Oregon-R genomic DNA, 1× NEB4, 1% glycerol and varying amounts of MfeI for 15 min at 37°C. A range of partial digest conditions was achieved by varying the amount of enzyme through 12 serial dilutions each, decreasing enzyme concentration by a factor of two as follows: Reaction 1 contained 10 U (GenBank accession number SRR652186), Rxn 2: 5 U (accession SRR652187), Rxn 3: 2.5 U (accession SRR652188), Rxn 4 : 1.25 U (accession SRR652189), Rxn 5: 0.63 U (accession SRR652190), Rxn 6: 0.31 U (accession SRR652191), Rxn 7: 0.16 U (accession SRR652192), Rxn 8: 0.08 U (accession SRR652193), Rxn 9: 0.04 U (accession SRR652194), Rxn 10: 0.02 U (accession SRR652195), Rxn 11: 0.01 U (accession SRR652196), Rxn 12: 0.005 U (accession SRR652197).
(2) RAD-seq library preparation: RAD-seq libraries were prepared according to Baird et al. (33) with the following parameters. MfeI restriction site–associated DNA (RAD) adapters were composed of annealed oligonucleotides of the form 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-barcode-3′ and 5′-Phos-AATT-barcode-AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT-3′. Each of the 12 experimental digests was ligated to an MfeI RAD adapter with a unique barcode to allow sequencing on the same Illumina HiSeq 2000 lane. Before amplification, reactions 1–4 (high enzyme), 5–8 (mid enzyme) and 9–12 (low enzyme) were pooled to increase the sequence contribution of the lower enzyme samples as the concentration of digested fragments was expected to be much greater in the higher enzyme samples. The final step in the RAD-seq library preparation protocol is a PCR enrichment of DNA restriction fragments flanked by both sequences necessary for Illumina sequencing. Ten nanograms of the high-enzyme ligation were PCR amplified using Phusion polymerase with PE PCR Primer 1.0 and a shortened PE PCR Primer 2.0 (5′-CAAGCAGAAGACGGCATACGA-3′) for 15 cycles. This was increased to 17 cycles with 15-ng ligation template for mid-enzyme libraries and 20 cycles with 20-ng template for low-enzyme libraries.
Fragments averaging 550 bp were agarose gel purified from each reaction and sequenced on an Illumina HiSeq 2000 to generate single-end 100-bp reads.
Data processing
Sequence reads were aligned to D melanogaster genome build 5.4.52, or E. coli genome REL606 using Novoalign v2.07 (Novocraft.com). Custom Perl scripts (available from EAJ on request) counted the sequence reads at each genomic location. For the flanking nucleotide assay, the flanking nucleotides were inferred from the genome reference sequence for each aligned read, and the total counts of reads for each flanking sequence tracked. For the star activity assay, the reads found for each recognition sequence were normalized by their count in the genome.
RESULTS
Star activity assay
Restriction enzymes are known to digest DNA at non-cognate sequences called star sites. We developed a star activity assay for quantifying the relative activity of restriction enzymes at cognate and non-cognate sites using genomic DNA as a substrate. The star activity assay comprises shearing genomic DNA to a defined length, digestion with a restriction enzyme and selecting amplified fragments much smaller than the original sheared fragments for sequencing. Because the DNA fragments are blunted after digestion, the sequencing adapters ligate equally well to cognate and non-cognate sites. The full sequence of the digested site can be recovered after alignment of the sequence read back to the reference genome. Thus, the relative sequencing coverage of each genomic locus can be quantified, and the normalized sequencing coverage of each particular site sequence motif, represented many times across a genome, can be determined.
MfeI star activity
After digestion of E. coli genomic DNA with MfeI in star activity conditions for 24 h, non-cognate sequences with single base pair changes from the cognate CAATTG were seen at digested sites. The bulk of non-cognate reads came from CAACTG and its reverse complement CAGTTG, and a small number of additional reads were created by digestion of CAATTA, CAATTC, CACTTG and their reverse complements TAATTG, GAATTG and CAAGTG (see Table 1). These star sites were also seen after digestion with an engineered high-specificity version of MfeI (MfeI-HF, NEB), although at much lower coverage compared with wild-type MfeI (see Table 1). For example, the percent of total reads for the most abundant star site, CAACTG, was more than 6-fold higher for MfeI compared with MfeI-HF. MfeI-HF also showed a substantial reduction in star activity compared with the wild-type enzyme when FI was used as the metric. The FI determined in this study of MfeI-HF in NEB4 is ≥500, while the previously determined FI of wild-type MfeI in NEB4 is only 32 (3), demonstrating a ≥16-fold reduction in star activity on a simple substrate. While both of these assays demonstrate the increased fidelity of the engineered MfeI-HF, their results cannot be quantitatively compared owing to differences in substrate and reaction conditions.
Table 1.
Percent of reads at star sites after digestion with MfeI
| Site | MfeI | MfeI-HF |
|---|---|---|
| CAACTG | 3.03 | 0.46 |
| CAATTA | 0.14 | 0.07 |
| CAATTC | 0.08 | 0.07 |
| CAAGTG | 0.04 | 0.11 |
EcoRI star activity
After digestion of E. coli genomic DNA with EcoRI in star activity conditions for 24 h, six non-cognate sequences with single base pair changes from the cognate GAATTC were seen at digested sites, although three of these made up a small fraction of the reads (see Table 2). These star sites comprised a significant portion of all sequences from the EcoRI digestion, with >31% of all reads coming from GAATTT sites, and GAAGTC and GAATTA sites having 4 and 2% of all reads, respectively. The sites GAACTC, GAATTG and GAATGC together made up only ∼0.4% of all reads. The high fidelity version of EcoRI had much improved specificity in star activity conditions. The percent of total reads coming from star sites was 3000-fold lower in EcoRI-HF compared with EcoRI (see Table 2). As in the comparison of MfeI with MfeI-HF, the coverage difference between EcoRI and EcoRI-HF was less pronounced with the minor-frequency star sites. FI testing also showed a drastic improvement in specificity for the engineered EcoRI-HF. The FI determined in this study of EcoRI-HF in NEB4 is ≥16 000, while the previously determined FI of wild-type EcoRI in NEB4 is only 4 (3), demonstrating a ≥4000-fold reduction in star activity on lambda DNA.
Table 2.
Percent of reads at star sites after digestion with EcoRI
| Site | EcoRI | EcoRI-HF |
|---|---|---|
| GAATTT | 31.58 | 0.01 |
| GAAGTC | 4.17 | 0.01 |
| GAATTA | 2.64 | 0.01 |
| GAACTC | 0.31 | 0.01 |
| GAATTG | 0.05 | 0.01 |
| GAATGC | 0.04 | 0.01 |
Flanking sequence preference of MfeI at cognate site CAATTG
We also examined how the digestion of cognate sites is affected by the flanking nucleotide sequence. The simpler RAD method was used to generate short DNA tags at each cognate cleavage site. As in the previous assay, the number of tags found at each locus was used as a measure of digestion efficiency. By calculating a normalized coverage for each particular flanking sequence the influence of these sequences on restriction enzyme activity could be determined.
We digested genomic DNA from D. melanogaster with the restriction enzyme MfeI using enzyme concentrations that ranged from fully saturating to limiting (10–0.005 U). We reasoned that the highest enzyme concentrations would digest every available cognate site to near completion, whereas enzyme preferences for particular sequences in the flanking nucleotides would be apparent at the lowest concentrations. RAD libraries were made for each enzyme concentration and sequenced to an average of ∼3× coverage for all sites. The sequence reads were mapped to the genomic sequence and the flanking nucleotides extracted for each site. The read counts for sites or half sites sharing a flanking sequence were binned, and the average coverage calculated.
The single nucleotide adjacent to MfeI had a strong effect on site preference (see Figure 2A). As the amount of MfeI was diluted, the sequencing reads became concentrated on preferred sites, creating higher coverage depth for preferred sites and lower coverage depth for sites that were digested less efficiently. If the site sequences are ranked by the change in sequencing coverage from the most enzyme to the least, the greatest increase in coverage is the palindromic GCAATTGC, and the greatest decrease is the palindromic ACAATTGT. In general, there is strong concordance in the coverage change for sites that are reverse complements of each other, as would be expected (see Table 3). All the sites with a 5′ G base or 3′ C base have an increase in coverage under dilute conditions, demonstrating that MfeI has a strong preference for these nucleotides adjacent to the cognate cut site. A 5′ T base or 3′ A base has a near neutral effect on coverage, and 5′ A or C bases or 3′ T or G bases have a negative effect on coverage, demonstrating that their presence in the flanking sequence makes an MfeI restriction site less likely to be cleaved in dilute enzyme conditions.
Figure 2.
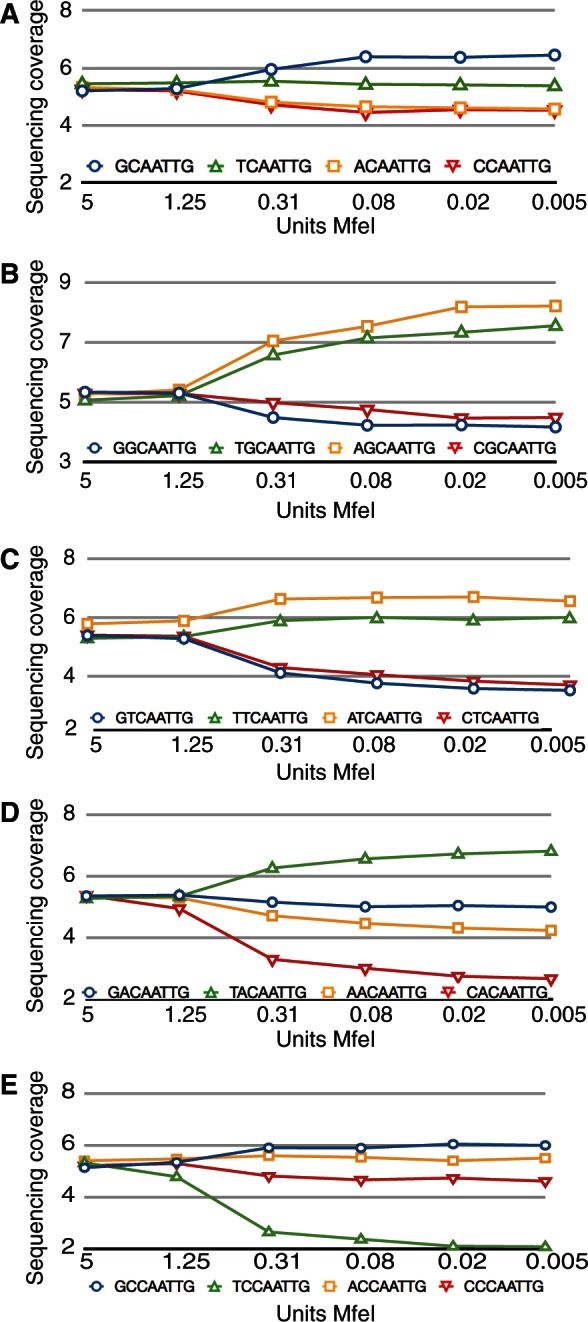
MfeI activity is affected by flanking base preference. All graphs plot normalized sequencing coverage (y-axis) versus units of enzyme (x-axis). Blue circles, G base; green triangles, T base; yellow squares, A base; red triangles, C base. (A) Changes in sequencing coverage for the different bases adjacent to the MfeI half site, i.e. N-CAA. (B–E) Changes in sequencing coverage for the different distal bases of the dinucleotide adjacent to the MfeI half site, for the dinucleotide NG-CAA (graph B), NT-CAA (graph C), NA-CAA (graph D), NC-CAA (graph E).
Table 3.
The change in sequencing coverage from enzyme saturating to limiting conditions for each of the 16 single-nucleotide flanking pairs surrounding the cognate MfeI site
| Site | Change in coverage |
|---|---|
| GCAATTGC | 2.6 |
| TCAATTGC | 1.3 |
| GCAATTGA | 1.1 |
| GCAATTGG | 0.5 |
| CCAATTGC | 0.4 |
| ACAATTGC | 0.2 |
| GCAATTGT | 0.1 |
| TCAATTGA | −0.1 |
| CCAATTGA | −0.8 |
| ACAATTGA | −0.8 |
| TCAATTGT | −0.9 |
| TCAATTGG | −0.9 |
| CCAATTGT | −1.0 |
| ACAATTGG | −1.2 |
| CCAATTGG | −1.4 |
| ACAATTGT | −1.4 |
This preference for certain sequences by the MfeI restriction enzyme extends beyond the single adjacent base. The 5′ G base preference becomes even more pronounced when the dinucleotide is 5′ (A/T)G, but the 5′ (G/C)G dinucleotide has a reduced sequencing coverage (see Figure 2B). The preference for A or T bases in the second 5′ position away from the cut site is also true for the (A/T)T versus (G/C)T dinucleotides (see Figure 2C), but dinucleotides with a 5′ A or C base adjacent to the restriction cut site have more complicated interactions. The TA dinucleotide has a strong positive effect on sequencing coverage, while (A/C/G)A are all weakly to strongly negative (see Figure 2D). The TC dinucleotide has the lowest sequencing coverage of all dinucleotides, while (A/C/G)C have only a weak effect on coverage (see Figure 2E). Our data show the flanking effects of each dinucleotide to operate independently of the sum of its parts.
Flanking sequence preference of MfeI at star site CAACTG
The abundant non-cognate site CAACTG identified in wild-type MfeI star activity conditions was analyzed for flanking nucleotide preferences to compare flanking effects in star versus cognate activity. There was a wide range of site sequencing coverage depending on the flanking nucleotide sequence of the CAACTG site. There was a 2.3-fold difference in coverage between sites with a 5′ T base and the poorly cut star sites with a 5′ A base (see Figure 3), which is of larger magnitude than the single-base flanking effects seen at the cognate site. Interestingly, the effect of particular flanking sequences differed between the cognate and star sites. The 5′ G base was most preferred by the cognate site, whereas a 5′ T base was most preferred by the star site. The effect of flanking sequences also differed for the two distinct half sites of the CAACTG star site. Whereas palindromic 5′ and 3′ flanking sequences about the cognate MfeI sites confer the same effect, the distinct star half sites CAA and CAG respond differently. While both preferred a 5′ T base and a 5′ A base reduced sequencing coverage the most, the next most preferred 5′ base was a C base for the CAA half site and a G base for the CAG half site (see Figure 3). Our data show that MfeI star site flanking preferences are distinct from those of cognate sites and that each asymmetric star half site may have distinct flanking preferences as well.
Figure 3.
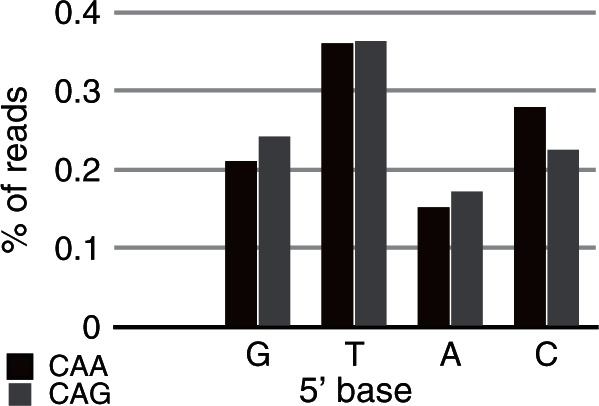
MfeI activity is affected by flanking base preference at CAACTG star sites. Bars represent the percentage of wild-type MfeI star activity assay reads mapping to CAACTG sites having a particular 5′ adjacent base, with a higher percentage indicating that adjacent base creates a favourable context for digestion. Because the star site is asymmetric, adjacent base preferences are shown for the two half sites, CAA (blue) and CAG (green).
DISCUSSION
The power of next-generation sequencing has typically been applied to the characterization of the sequence or function of a genome. Here we use the massively parallel nature of next-generation sequencing to assay the enzymatic activity of restriction endonucleases that cleave both strands of double-stranded DNA. We developed a novel assay to allow the characterization of restriction enzyme recognition sites without any prior knowledge, and also used the related RAD-Seq method to assay the effect of flanking sequence on restriction enzyme cleavage.
We first quantitatively assayed the activity of both EcoRI and MfeI and their high-fidelity counterparts (EcoRI-HF and MfeI-HF) by mapping cleavage events to the E. coli reference genome. For each enzyme, the majority of reads mapped to the cognate sites, demonstrating the correlation between cleavage efficiency and read count as well as highlighting the method’s utility in de novo recognition site discovery. This unbiased detection method also simultaneously quantified star activity over all DNA configurations present in the E. coli genome. The star activity occurred at sites with 1-bp substitutions with respect to the cognate sites as has been previously observed (19). For both enzymes, only a subset of the possible single substitution sites produced sequence reads, which effectively identified those degenerate sites capable of generating appreciable star activity. Different star sites showed a wide range of activity indicating the degree to which specific base changes are tolerated by the restriction enzyme. In the case of EcoRI, the three most abundant star sites in our data (GAATTT, GAAGTC and GAATTA) have been previously shown to be the three most efficiently cleaved (17,18). The high-fidelity restriction enzymes developed by New England Biolabs showed drastically reduced star activity compared with wild type. The assay was able to quantify this reduction across all potential cleavage sites and validate that no major cryptic DNA sequences are cleaved by the engineered high-fidelity variants.
Massively parallel sequencing was also used to quantify the flanking preferences of MfeI. The relative presence of flanking nucleotides in sequence reads generated from the same complex substrate was compared across 12 enzyme concentrations using RAD-Seq. Drosophila genomic DNA was used as substrate to provide sufficient diversity of MfeI sites. Under enzyme saturation, an equal contribution of reads from sites was observed regardless of flanking sequences. As enzyme concentration was decreased, flanking nucleotide preferences of progressively larger magnitude were observed. When reads were binned by a single flanking nucleotide, G-CAATTG sites were shown to be favourable, A-CAATTG and C-CAATTG were shown to be unfavourable and T-CAATTG was relatively neutral. Binning reads by flanking dinucleotides showed even larger effects. While the general trends seen when examining single flanking nucleotides were still apparent, the dinucleotide analysis underscored the unique energetic contributions to cleavage of each unique sequence context. This was shown in our data by the ability of a given nucleotide in the second position away from the cut site to confer either a positive or negative effect on cleavage depending on the identity of the adjacent nucleotide. For example, the thymine nucleotide in the second position away from the cut site led to increased cleavage for TG-CAATTG, TA-CAATTG and TT-CAATTG but decreased cleavage for TC-CAATTG.
We also analyzed MfeI star activity from the first set of experiments with respect to flanking sequence. While the E. coli genome is not of sufficient complexity for exhaustive flanking sequence analysis, it does provide enough diversity to confidently investigate effect of the adjacent base. Because digestion at the CAACTG star site was incomplete, we expected flanking preferences to be apparent much as they were in enzyme-limiting conditions at cognate MfeI sites. Indeed, we found that the flanking sequence affected CAACTG star site cleavage as well. In contrast to the palindromic MfeI cognate site, flanking preferences differed on each side of the asymmetric star sites. Notably, the MfeI star site flanking preferences are distinct from the cognate site flanking preferences, which is consistent with biophysical work suggesting star site–enzyme complexes are profoundly different from their cognate counterparts (2,34).
In this article, we present new high-throughput methods to characterize restriction endonuclease activity. The two techniques link Illumina sequence reads to cleavage events of highly complex substrate provided by sequenced genomes to assay enzyme activity in a highly parallel fashion. The data acquired from their application to MfeI and EcoRI is consistent with previously described principles regarding restriction enzyme activity. These techniques are easily applied to both previously characterized and newly discovered type II restriction endonucleases. Genome sequencing has yielded many thousands of putative restriction endonucleases (26), so the ability to quickly characterize their activity over all possible recognition sites will yield novel target specificities at a much higher rate than is currently possible. Additionally, the structure–function relationship of restriction enzymes has been long-studied; these methods provide a rapid way to generate data about target specificity and activity for enzymes in altered conditions or altered protein structure. Thus, the methodology presented and validated in this study will serve as a basis for applying the power of massively parallel analysis to the active and essential field of restriction enzymology.
ACCESSION NUMBERS
SRR652142, SRR652141, SRR652140, SRR652139, SRR652186, SRR652187, SRR652187, SRR652188, SRR652189, SRR652190, SRR652191, SRR652192, SRR652193, SRR652194, SRR652195, SRR652196, SRR652197
ACKNOWLEDGEMENTS
The authors thank Paul Etter for insightful discussions regarding the protocols developed herein and Doug Turnbull for advice on Illumina sequencing. The authors also thank Richard Roberts and William Jack for their review of the manuscript.
FUNDING
This work was supported by the National Institutes of Health (NIH) [HG006036 to E.A.J.]; and New England BioLabs, Inc. Funding for open access charge: NIH and New England BioLabs, Inc.
Conflict of interest statement. None declared.
REFERENCES
- 1.Orlowski J, Bujnicki JM. Structural and evolutionary classification of Type II restriction enzymes based on theoretical and experimental analyses. Nucleic Acids Res. 2008;36:3552–3569. doi: 10.1093/nar/gkn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jen-Jacobson L. Protein-DNA recognition complexes: conservation of structure and binding energy in the transition state. Biopolymers. 1997;44:153–180. doi: 10.1002/(SICI)1097-0282(1997)44:2<153::AID-BIP4>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 3.Wei H, Therrien C, Blanchard A, Guan S, Zhu Z. The Fidelity Index provides a systematic quantitation of star activity of DNA restriction endonucleases. Nucleic Acids Res. 2008;36:e50. doi: 10.1093/nar/gkn182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kolesnikov VA, Zinoviev VV, Yashina LN, Karginov VA, Baclanov MM, Malygin EG. Relaxed specificity of endonuclease BamH1 as determined by identification or recognition sites in SV40 and pBR322 DNAs. FEBS Lett. 1981;132:101–104. doi: 10.1016/0014-5793(81)80437-1. [DOI] [PubMed] [Google Scholar]
- 5.Zylicz-Stachula A, Zołnierkiewicz O, Jeżewska-Frąckowiak J, Skowron PM. Chemically-induced affinity star restriction specificity: a novel TspGWI/sinefungin endonuclease with theoretical 3-bp cleavage frequency. BioTechniques. 2011;50:397–406. doi: 10.2144/000113685. [DOI] [PubMed] [Google Scholar]
- 6.Saravanan M, Vasu K, Nagaraja V. Evolution of sequence specificity in a restriction endonuclease by a point mutation. Proc. Natl Acad. Sci. USA. 2008;105:10344–10347. doi: 10.1073/pnas.0804974105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malyguine E, Vannier P, Yot P. Alteration of the specificity of restriction endonucleases in the presence of organic solvents. Gene. 1980;8:163–177. doi: 10.1016/0378-1119(80)90035-9. [DOI] [PubMed] [Google Scholar]
- 8.Alves J, Pingoud A, Haupt W, Langowski J, Peters F, Mss G, Wolff C. The influence of sequences adjacent to the recognition site on the cleavage of oligodeoxynucleotides by the EcoRI endonuclease. Eur. J. Biochem. 1984;140:83–92. doi: 10.1111/j.1432-1033.1984.tb08069.x. [DOI] [PubMed] [Google Scholar]
- 9.Wolfes H, Fliess A, Pingoud A. A comparison of the structural requirements for DNA cleavage by the isoschizomers HaeIII, BspRI and BsuRI. Eur. J. Biochem. 1985;150:105–110. doi: 10.1111/j.1432-1033.1985.tb08994.x. [DOI] [PubMed] [Google Scholar]
- 10.Horton NC, Perona JJ. Recognition of flanking DNA sequences by EcoRV endonuclease involves alternative patterns of water-mediated contacts. J. Biol. Chem. 1998;273:21721–21729. doi: 10.1074/jbc.273.34.21721. [DOI] [PubMed] [Google Scholar]
- 11.Engler LE, Sapienza P, Dorner LF, Kucera R, Schildkraut I, Jen-Jacobson L. The energetics of the interaction of BamHI endonuclease with its recognition site GGATCC. J. Mol. Biol. 2001;307:619–636. doi: 10.1006/jmbi.2000.4428. [DOI] [PubMed] [Google Scholar]
- 12.Jen-Jacobson L, Engler LA, Ames JT, Kurpiewski MR, Grigorescu A. Thermodynamic Parameters of Specific and Nonspecific Protein-DNA Binding. Supramol. Chem. 2000;12:143–160. [Google Scholar]
- 13.Bujnicki JM. Phylogeny of the restriction endonuclease-like superfamily inferred from comparison of protein structures. J. Mol. Evol. 2000;50:39–44. doi: 10.1007/s002399910005. [DOI] [PubMed] [Google Scholar]
- 14.Engler LE, Welch KK, Jen-Jacobson L. Specific Binding by EcoRV Endonuclease to its DNA Recognition Site GATATC. J. Mol. Biol. 1997;269:82–101. doi: 10.1006/jmbi.1997.1027. [DOI] [PubMed] [Google Scholar]
- 15.Deibert M, Grazulis S, Janulaitis A, Siksnys V, Huber R. Crystal structure of MunI restriction endonuclease in complex with cognate DNA at 1.7 A resolution. EMBO J. 1999;18:5805–5816. doi: 10.1093/emboj/18.21.5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lesser DR, Kurpiewski MR, Waters T, Connolly BA, Jen-Jacobson L. Facilitated distortion of the DNA site enhances EcoRI endonuclease-DNA recognition. Proc. Natl Acad. Sci. USA. 1993;90:7548–7552. doi: 10.1073/pnas.90.16.7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lesser DR, Kurpiewski MR, Jen-Jacobson L. The energetic basis of specificity in the Eco RI endonuclease-DNA interaction. Science. 1990;250:776–786. doi: 10.1126/science.2237428. [DOI] [PubMed] [Google Scholar]
- 18.Thielking V, Alves J, Fliess A, Mss G, Pingoud A. Accuracy of the EcoRI restriction endonuclease: binding and cleavage studies with oligodeoxynucleotide substrates containing degenerate recognition sequences. Biochemistry. 1990;29:4682–4691. doi: 10.1021/bi00471a024. [DOI] [PubMed] [Google Scholar]
- 19.Sidorova NY, Rau DC. Differences between EcoRI nonspecific and “star” sequence complexes revealed by osmotic stress. Biophys. J. 2004;87:2564–2576. doi: 10.1529/biophysj.104.042390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horton NC, Perona JJ. Role of protein-induced bending in the specificity of DNA recognition: crystal structure of EcoRV endonuclease complexed with d(AGAT) + d(ATCTT) J. Mol. Biol. 1998;277:779–787. doi: 10.1006/jmbi.1998.1655. [DOI] [PubMed] [Google Scholar]
- 21.Nasri M, Thomas D. Relaxation of recognition sequence of specific endonuclease HindIII. Nucleic Acids Res. 1986;14:811–821. doi: 10.1093/nar/14.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samuelson JC, Xu SY. Directed evolution of restriction endonuclease BstYI to achieve increased substrate specificity. J. Mol. Biol. 2002;319:673–683. doi: 10.1016/S0022-2836(02)00343-1. [DOI] [PubMed] [Google Scholar]
- 23.Jurenaite-Urbanaviciene S, Serksnaite J, Kriukiene E, Giedriene J, Venclovas C, Lubys A. Generation of DNA cleavage specificities of type II restriction endonucleases by reassortment of target recognition domains. Proc. Natl Acad. Sci. USA. 2007;104:10358–10363. doi: 10.1073/pnas.0610365104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joshi HK, Etzkorn C, Chatwell L, Bitinaite J, Horton NC. Alteration of sequence specificity of the type II restriction endonuclease HincII through an indirect readout mechanism. J. Biol. Chem. 2006;281:23852–23869. doi: 10.1074/jbc.M512339200. [DOI] [PubMed] [Google Scholar]
- 25.Guan S, Blanchard A, Zhang P, Zhu Z. Alteration of sequence specificity of the type IIS restriction endonuclease BtsI. PLoS One. 2010;5:e11787. doi: 10.1371/journal.pone.0011787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE-a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010;38:D234–D236. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu SY, Zhu Z, Zhang P, Chan SH, Samuelson JC, Xiao J, Ingalls D, Wilson GG. Discovery of natural nicking endonucleases Nb.BsrDI and Nb.BtsI and engineering of top-strand nicking variants from BsrDI and BtsI. Nucleic Acids Res. 2007;35:4608–4618. doi: 10.1093/nar/gkm481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kostiuk G, Sasnauskas G, Tamulaitiene G, Siksnys V. Degenerate sequence recognition by the monomeric restriction enzyme: single mutation converts BcnI into a strand-specific nicking endonuclease. Nucleic Acids Res. 2011;39:3744–3753. doi: 10.1093/nar/gkq1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Y, Lunnen KD, Kong H. Engineering a nicking endonuclease N.AlwI by domain swapping. Proc. Natl Acad. Sci. USA. 2001;98:12990–12995. doi: 10.1073/pnas.241215698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samuelson JC, Morgan RD, Benner JS, Claus TE, Packard SL, Xu SY. Engineering a rare-cutting restriction enzyme: genetic screening and selection of NotI variants. Nucleic Acids Res. 2006;34:796–805. doi: 10.1093/nar/gkj483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Guan S, Quimby A, Cohen-Karni D, Pradhan S, Wilson G, Roberts RJ, Zhu Z, Zheng Y. Comparative characterization of the PvuRts1I family of restriction enzymes and their application in mapping genomic 5-hydroxymethylcytosine. Nucleic Acids Res. 2011;39:9294–9305. doi: 10.1093/nar/gkr607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 33.Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3:e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sapienza PJ, Rosenberg JM, Jen-Jacobson L. Structural and thermodynamic basis for enhanced DNA binding by a promiscuous mutant EcoRI endonuclease. Structure. 2007;15:1368–1382. doi: 10.1016/j.str.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]