

Background: Conserved Asp-137 destabilizes the hydrophobic core of the coiled-coil tropomyosin.
Results: Leu substitution of Asp-137 decreases flexibility of tropomyosin and causes long range structural rearrangements; mouse hearts expressing this variant show altered function.
Conclusion: Residue Asp-137 is important for regulatory function of tropomyosin in the heart.
Significance: Our data support the hypothesis that tropomyosin flexibility regulates cardiac function in vivo.
Keywords: Actin, Cardiac Muscle, Cardiovascular Disease, Muscle, Tropomyosin, Troponin, Flexibility, Mouse Model, Thin Filament, Transgenic
Abstract
α-Tropomyosin (α-TM) has a conserved, charged Asp-137 residue located in the hydrophobic core of its coiled-coil structure, which is unusual in that the residue is found at a position typically occupied by a hydrophobic residue. Asp-137 is thought to destabilize the coiled-coil and so impart structural flexibility to the molecule, which is believed to be crucial for its function in the heart. A previous in vitro study indicated that the conversion of Asp-137 to a more typical canonical Leu alters flexibility of TM and affects its in vitro regulatory functions. However, the physiological importance of the residue Asp-137 and altered TM flexibility is unknown. In this study, we further analyzed structural properties of the α-TM-D137L variant and addressed the physiological importance of TM flexibility in cardiac function in studies with a novel transgenic mouse model expressing α-TM-D137L in the heart. Our NMR spectroscopy data indicated that the presence of D137L introduced long range rearrangements in TM structure. Differential scanning calorimetry measurements demonstrated that α-TM-D137L has higher thermal stability compared with α-TM, which correlated with decreased flexibility. Hearts of transgenic mice expressing α-TM-D137L showed systolic and diastolic dysfunction with decreased myofilament Ca2+ sensitivity and cardiomyocyte contractility without changes in intracellular Ca2+ transients or post-translational modifications of major myofilament proteins. We conclude that conversion of the highly conserved Asp-137 to Leu results in loss of flexibility of TM that is important for its regulatory functions in mouse hearts. Thus, our results provide insight into the link between flexibility of TM and its function in ejecting hearts.
Introduction
α-Tropomyosin (α-TM)3 is an α-helical parallel coiled-coil protein that cooperatively binds actin filaments and serves as a nodal point in control of Ca2+-regulated cardiac muscle dynamics. The position of TM is shifted on the actin filament in response to Ca2+ binding to troponin (Tn) (1, 2). According to current models of thin filament regulation of the actin-myosin interaction, Ca2+ triggers contraction from a blocked thin filament state to a weak cross-bridge binding or closed state, and full activation requires an open state induced by strong cross-bridge binding (3–5). Rapid regulatory relocations of TM are key to these transitions; thus, it is plausible that structural flexibility of TM is important in its regulatory movements.
As with typical coiled-coils, the amino acid sequence of α-TM contains a heptad repeat of the form a-b-c-d-e-f-g with non-polar residues found at the hydrophobic core of the two helices at positions a and d. Hydrophobic residues at positions a and d provide stability to the protein. Interestingly, in α-TM there is a highly conserved, Asp-137 residue that is unexpectedly found at the position d. The presence of two negatively charged residues in the hydrophobic core of a parallel coiled-coil structure is expected to increase flexibility of the molecule by causing destabilization at the region. A previous database search study reported that although exceptional polar and charged residues are found at a and d positions of coiled-coil proteins, Asp is alone in being excluded from d positions of all parallel coiled-coil proteins identified in the Protein Data Bank (6).
TM has breaks in the periodicity of the heptad repeat that are predicted to introduce functionally important flexibility to protein structure. Regulatory functions of TM are believed to be enabled by this flexibility (7–10). In a previous study by Sumida et al. (11), Asp-137 was converted to Leu, which is a highly preferred residue for position d, and as a result a reduced proteolytic susceptibility of the variant associated with its reduced flexibility was reported. Therefore, this study showed involvement of Asp-137 in increased flexibility of TM at the central part. Additionally, a higher S1 ATPase activity of reconstituted filaments regulated by α-TM-D137L was shown.
In the present study, we extended these studies by analyzing structural features of the α-TM-D137L variant, employing solution NMR and Differential scanning calorimetry (DSC). Our 1H-13C NMR spectra of reductively methylated α-TM suggest long range structural perturbations by the D137L conversion. As determined with DSC, the α-TM-D137L variant has higher thermostability than WT, suggesting decreased flexibility. In the description of the α-TM and α-TM-D137L variant, we have used the term “flexibility” as has been common in previous papers (8, 9, 11, 12). However, it should be recognized that in our case describing a protein as flexible is an interpretation of determinations of stability that may have different interpretations.
Accordingly, the highly conserved, unusual Asp-137 of TM would be expected to contribute to key aspects of control mechanisms involving TM. However, the importance of flexibility of TM provided by Asp-137 in ejecting hearts remains unknown. We addressed this question with functional studies on a novel α-TM-D137L transgenic (TG) mouse model. Our results revealed the first evidence demonstrating that a loss of TM flexibility due to Leu substitution of Asp-137 induces altered in situ cardiac function with impaired systolic and diastolic function. There was also reduced cardiomyocyte contractility and myofilament Ca2+ sensitivity along with increased Hill n values with no change in maximum tension, rigor cross-bridge dependent activation, intracellular Ca2+ transients, or post-translational modification (PTM) of major myofilament proteins.
EXPERIMENTAL PROCEDURES
Expression of Recombinant TMs
All recombinantly expressed TMs used in this study carry an Ala-Ser N-terminal extension to mimic N-terminal acetylation of the native TM (13). T7-based pET-3d rat-striated muscle α-TM vector was a gift from Dr. Larry Tobacman (University of Illinois, Chicago, IL). The amino acid sequences of rat and mouse α-TM are identical. This vector was used as a template for preparation of α-TM-D137L plasmid. The QuikChange Lightning site-directed mutagenesis kit (Stratagene) was used with the following primers to produce the α-TM-D137L clone: forward (5′-CATTGAAAGCCGAGCCCAAAAACTTGAAGAAAAGATGGAGATTCAG-3′) and reverse (5′-CTGAATCTCCATCTTTTCTTCAAGTTTTTGGGCTCGGCTTTCAATG-3′). The coding sequences of the expression plasmids were confirmed by DNA sequencing. For expression, BL21(DE3) Escherichia coli cells (Stratagene) were transformed with clones and purified as described previously but with minor modifications (13). The extinction coefficient, which was used for protein concentration determinations, was calculated according to a model developed by Gill and von Hippel (14). The coefficient used for α-TM and α-TM-D137L was E280 nm = 1.56 × 104 m−1 cm−1.
NMR Spectroscopy
Lysine residues and N-terminal primary amine groups of recombinant α-TM and α-TM-D137L proteins were chemically modified with reductive methylation as described (15). Reductive methylation converts Lys residues to 13C-labeled dimethyl-Lys. Thus, the side chain retains its positively charged nature at neutral pH. For NMR experiments, methylated protein samples were dialyzed into 25 mm sodium phosphate buffer, pH 7.0, 0.1 m NaCl, 1 mm EDTA, and 2 mm DTT. The concentration of TMs used in the NMR experiments was 5 μm. 13C methyl signals of α-TM and α-TM-D137L were collected with NMR 1H-13C-edited heteronuclear single quantum coherence (HSQC) experiments at 25 °C with a 900-MHz Bruker spectrometer equipped with a cryogenic probe. The data were processed and analyzed using NMRPipe software (16).
DSC Measurements
DSC experiments were performed on a VP-DSC (MicroCal) at the Center for Structural Biology, University of Illinois (Chicago, IL). All measurements were carried out using reduced recombinant TM species (1.8 mg/ml) in 20 mm MOPS, pH 7.0, 1 mm EDTA, 0.1 m NaCl, and 1 mm β-mercaptoethanol, at a scanning rate of 1 °C/min. The state of TM species (reduced or not) was checked by SDS-PAGE; all TMs used in DSC measurements were in the fully reduced state (data not shown). The reversibility of thermal transitions was tested by a second heating of the sample immediately after cooling from the first scan. The calorimetric traces were corrected for instrumental background by subtracting a scan with buffer in both cells. The DSC data were analyzed using Origin version 7.0 (MicroCal) as described (17). The heat capacity (Cp) versus temperature curve was analyzed to determine the transition temperature (Tm) and the calorimetric enthalpy of transition (ΔHcal). Tm was used to compare thermal stability of TM species. ΔHcal, which is calculated by integrating the area under the peak, was used to compare the size of the transition for each calorimetric domain.
Animals
The experiments were carried out according to guidelines of the Animal Care and Use Committee at the University of Illinois (Chicago, IL).
Generation of α-TM-D137L TG Mouse
The mouse α-TM striated muscle-specific cDNA (1.1 kb) (accession number X64831) was cloned into the pBluescript vector. Three nucleotide changes (GAT > CTG) corresponding to an amino acid substitution at codon 137 (D137L) were carried out using the QuikChange site-directed mutagenesis kit (Stratagene). The sequence was verified by automated DNA sequencing and compared with the published sequence. The α-TM-D137L cDNA was cloned into a vector (18), which contained the cardiac specific α-myosin heavy chain promoter and the human growth hormone poly(A) signal sequence. The transgene construct was purified to generate TG mice (FVB/N strain) as described (19). Founder mice were identified by PCR, and the mutation in the TG lines was verified by nucleotide sequencing of TG mouse genomic DNA.
Genotyping
DNA samples were extracted from tail clips of 17-day-old mice, and PCR was employed for genotyping the α-TM-D137L transgene. The following primers were used: α-myosin heavy chain forward (5′-GCCCACACCAGAAATGACAGA-3′); α-TM reverse (5′-TCCAGTTCATCTTCAGTGCCC-3′); control forward (5′-AGCGAGCTCAGGACATTCTGG-3′) and control reverse (5′-CTCCTAACCACGCTCCTAGCA-3′). An untargeted site of genomic DNA amplification was used as an internal control for the PCRs.
Echocardiography
Echocardiography of TG and non-transgenic (NTG) mice was performed using a Vevo 770 high resolution in Vivo imaging system and RMVTM 707B scan head with a center frequency of 30 MHz (Visual Sonics), as described previously (20). M-mode images of the left atrium were taken from the left parasternal long axis view. Pulsed Doppler was performed with the apical four-chamber view. The mitral inflow was recorded with the Doppler sample volume at the tip of the mitral valve leaflets in the center of the mitral valve orifice. In order to measure time intervals, the Doppler sample volume was moved toward the left ventricular outflow tract, and both the mitral inflow and left ventricular outflow were obtained in the same recording. All measurements and calculations were averaged from three consecutive cycles and performed according to the American Society of Echocardiography guidelines (21, 22). Data analysis was performed with Vevo 770 analytic software.
Measurements of Ventricular Myocyte Shortening and Calcium Transients
Mouse ventricular myocytes were isolated from TG and NTG mice as described in detail previously elsewhere (23, 24).
Fura-2 fluorescence and shortening of cells were monitored simultaneously as described previously (23). Following cardiomyocyte isolation, an aliquot of cells was transferred to a perfusion chamber mounted on the stage of an inverted Nikon microscope and allowed to settle onto the glass. Background fluorescence recordings were taken daily for noise reduction. Following this recording, another aliquot of myocytes were loaded for 15 min with the Ca2+ indicator fura-2 (3 μm from Invitrogen) prepared from a DMSO stock solution. Following loading, cells were perfused using a dye-free 1.2 mm Ca2+ solution to allow for de-esterification of the indicator. Single intact ventricular myocytes were field-stimulated by applying a 4-ms square suprathreshold voltage pulse at 0.5 Hz to the bath through parallel platinum electrodes. For [Ca2+]i transient measurements, myocytes were alternatively excited at wavelengths of 340 and 380 nm, and the emitted fluorescence was collected at a wavelength of 505 nm by a photomultiplier tube and stored in the acquisition software (Felix 32, Photon Technology International) for later offline analysis. Cell shortening was assessed by using edge detection. The cell image was collected by the ×40 Nikon objective and transmitted to a multi-image module. A video edge detector (Crescent Electronics) was used to monitor cell length, and recordings were stored using an acquisition software program (Felix 32, Photon Technology International) for later analysis. [Ca2+]i measurements were reported as background-subtracted 340/380 ratio (fura-2 ratio).
Measurements of Cross-bridge and Ca2+-dependent Activation of Skinned Fiber Bundles
Detergent-extracted fiber bundles were dissected from left ventricular papillary muscles of TG and NTG mice, and measurements of pCa-tension relations at a sarcomere length of 2.2 μm were conducted as described previously with minor changes (25). Skinned fiber bundles were initially placed in high relaxing solution (HR; 10 mm EGTA, 41.89 mm potassium propionate, 6.57 mm MgCl2, 100 mm BES, 6.22 mm ATP, 10 mm creatine phosphate sodium salt, 5 mm sodium azide at pH 7.0) and then maximally activated in activating solution at pCa 4.5 (10 mm EGTA, 9.9 mm CaCl2, 22.16 mm potassium propionate, 6.2 mm MgCl2, 100 mm BES, 6.29 mm ATP, 10 mm creatine phosphate sodium salt, 5 mm sodium azide at pH 7.0) followed by relaxation in HR. Fiber bundles were then subjected to sequential increase in [Ca2+]. Solutions with varying [Ca2+] were prepared by mixing HR with activating solution at pCa 4.5.
Measurements of pMgATP-tension relation were conducted similarly to pCa-tension measurements described above, but the skinned fiber bundles were subjected to a sequential decrease in [MgATP] using a procedure modified from that of Brandt et al. (26). The fiber bundles were initially placed in HR and then subjected to decreases in [MgATP] (10−4 to 10−6 m). Solutions with varying [MgATP] were prepared by mixing pMgATP 3 solution (10 mm EGTA, 63.45 mm potassium propionate, 2.8 mm MgCl2, 1.07 mm ATP, 12 mm creatine phosphate sodium salt, 20 mm BES, 5 mm sodium azide at pH 7.0) with pMgATP 8 solution (10 mm EGTA, 68.98 mm potassium propionate, 1.8 mm MgCl2, 1 × 10−5 mm ATP, 12 mm creatine phosphate sodium salt, 20 mm BES, 5 mm sodium azide at pH 7.0).
In both pCa-tension and pMgATP-tension measurements, all solutions contained the following protease inhibitors: pepstatin A (2.5 μg/ml), leupeptin (1 μg/ml), and phenylmethylsulfonyl fluoride (PMSF) (50 μmol/liter). 10 IU/ml creatine phosphokinase was freshly added to all solutions before use. Ionic strength of all solutions (150 mm), and [MgATP] and free [Ca2+] were calculated with a computer program by A. Fabiato (27) using binding constants listed by Godt and Lindley (28). Data were fit to the Hill equation using Prism software (GraphPad version 5).
Myofibrillar Protein Preparation
The hearts of NTG and TG mice were excised as described (29). Myofibrillar proteins were prepared from the left ventricular myocardium of NTG and TG mice, in the presence of protease (Sigma-Aldrich) and phosphatase (Calbiochem) inhibitor mixtures, according to methods described previously (30). In brief, myofibrils were purified and solubilized from ventricular homogenates in UTC buffer (8 m urea, 2 m thiourea, and 4% (w/v) CHAPS) at a 1:20 (w/v) ratio using Dounce homogenizers. Samples were clarified with centrifugation (18,000 × g for 10 min, 4 °C), and the supernatant fraction was kept at −80 °C until use. Myofibrillar protein sample concentrations were determined using the RC-DC assay kit (Bio-Rad). Samples used for PTM analyses were treated with the 2D clean-up kit from GE Healthcare and then resuspended in UTC buffer.
Two-dimensional Difference Gel Electrophoresis (2D-DIGE)
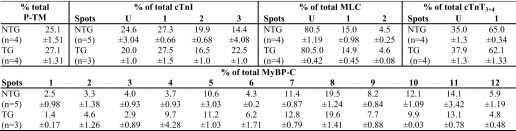
We performed 2D-DIGE as described previously (29–32). Myofibrillar proteins were prepared as described above. Samples from NTG and TG mice and an internal standard (mixture of all NTG and TG samples used in the run) were randomly labeled with fluorescent cyanine dyes from GE Healthcare as described (30). Isoelectric focusing was performed using the following immobilized pH gradient strips in the first dimension: for TM, 24 cm, pH 3.9–5.1 (Bio-Rad); for cardiac troponin-I (cTnI), 18 cm, pH 7–11; for cardiac troponin-T (cTnT) and myosin light chain (MLC), 18 cm, pH 4–7; for myosin-binding protein-C (MyBP-C), 24 cm, pH 3–11 (all from GE Healthcare). The second dimension was run on a 12% SDS-polyacrylamide gel (30) in the Criterion gel system (Bio-Rad). Gels were then imaged on a Typhoon 9410 imager (GE Healthcare). Images were analyzed using PDQuest software (version 8.0 advanced, Bio-Rad). The spot density as a percentage of totals was calculated by dividing the density of a particular spot by the total of all spot densities for that protein.
Analysis of Myosin Heavy Chain Isoform Expression
We separated cardiac MyHC isoforms (α and β) of myofibrillar protein samples from left ventricles of 1-day-old neonatal mice and 4-month-old NTG and TG mice by use of a special SDS-PAGE system described previously (33). Quantification of band densities was done using ImageLab software (version 3.0, Bio-Rad).
In Vitro ATPase Assay
Thin filaments were prepared and reconstituted as described (34) with cTnC (recombinant, mouse), cTnT (recombinant, mouse), cTnI (recombinant, mouse), actin (isolated from tissue, rabbit skeletal muscle), and myosin subfragment-1 (isolated from tissue, rabbit psoas muscle) and with the following variations in TM: control α-TM-WT, α-TM-D137L, reductively methylated α-TM-WT, and reductively methylated α-TM-D137L. ATPase measurements were conducted as described elsewhere (34). The reaction conditions were 0.2 μm subfragment-1, 5 μm actin, 1.5 μm TM, 1.5 μm Tn in 35 mm NaCl, 5 mm MgCl2, 20 mm MOPS, pH 7.0, 1 mm ATP, and either 0.1 mm CaCl2 or 2 mm EGTA.
Statistical Analysis
Measurements of isolated cardiomyocytes, tension-pCa, tension-pMgATP, heart weight/tibia length ratio, 2D-DIGE, and in vitro ATPase were analyzed with an unpaired Student's t test. In addition, echocardiography data from 4- and 8-month-old mice were examined together with analysis of variance followed by Bonferroni post hoc analysis. p ≤ 0.05 was considered statistically significant. Data are expressed as ± S.E.
RESULTS
Long Range Rearrangements Were Observed in α-TM-D137L Structure
NMR spectra of reductively methylated recombinant α-TM and α-TM-D137L were compared with study effects of the D137L conversion on TM structure. Reductive methylation of Lys residues is a highly valuable method that allows characterization of high molecular mass proteins (>30 kDa), such as TM, which is not achievable with traditional solution NMR approaches due to the well known molecular weight limitation problem (35).
Thirty-nine lysine residues of TM are spread along the protein, which allowed us to compare global structural features of α-TM-D137L with α-TM. An overlay of 1H-13C-edited HSQC spectra of reductively methylated α-TM (blue) and α-TM-D137L (red) is shown in Fig. 1. Significant signal overlap was observed in the spectra of WT and D137L TM. Overall, 26 of 39 signals were observed in either spectrum. Comparison of spectra of α-TM and α-TM-D137L demonstrated methyl chemical shift perturbations in all resonances. This result indicated long range structural rearrangements in the structure of α-TM-D137L compared with α-TM.
FIGURE 1.

An overlay of 1H-13C-edited HSQC spectra of reductively methylated WT α-TM (blue) and α-TM-D137L (red). The spectra of 5 μm proteins were obtained at 25 °C on a 900-MHz Bruker NMR spectrometer. Conditions were 25 mm sodium phosphate buffer, pH 7.0, 0.1 m NaCl, 1 mm EDTA, and 2 mm DTT.
Previously, it was shown that reductive methylation does not alter protein structures and native protein-protein interactions (36–38). As a control, we tested whether reductive methylation of TM alters the effects of Leu for Asp-137 substitution in TM function. We compared ATPase rates of thin filament preparations reconstituted with α-TM-WT, α-TM-D137L, reductively methylated α-TM-WT, or reductively methylated α-TM-D137L. The ATPase rate in relaxing conditions (EGTA) was not affected by the presence of modified TM or TM-D137L. Ca2+ switched on the ATPase rate in WT TM (0.26 ± 0.02 s−1) and α-TM-D137L (0.36 ± 0.02 s−1) as well as in modified WT TM (0.72 ± 0.03 s−1) and modified α-TM-D137L (0.81 ± 0.04 s−1). Although the maximum ATPase rate was higher in the modified TM variants, the difference in ATPase rates at maximum Ca2+ between α-TM-WT and α-TM-D137L (0.1 ± 0.01 s−1) was the same as that for modified WT TM and α-TM-D137L (0.09 ± 0.03 s−1).
α-TM-D137L Exhibited Increased Thermal Stability Compared with WT α-TM
In order to further assess the effects of D137L conversion on TM structure, we compared the thermal stability of α-TM (Fig. 2A) with that of α-TM-D137L (Fig. 2B) using DSC. The thermal unfolding character of the variant was noticeably different from that of WT. This was reflected in pronounced increases in Tm values for the two calorimetric domains of the variant (Table 1). Calorimetric domains of the α-TM were seen at maxima 43.35 °C (domain-1) and 49.99 °C (domain-2). Calorimetric enthalpy values for these domains were 91.13 kcal/mol (70.5% of total) and 38.07 kcal/mol (38.0% of total), respectively. In the case of the α-TM-D137L, the same two calorimetric domains were observed, but thermal transitions were at higher maxima, 45.68 °C (domain-1) and 52.19 °C (domain-2), with ΔHcal values of 94.58 kcal/mol (36.5% of total) and 168 kcal/mol (63.0% of total), respectively. The reversibility of thermal transitions was confirmed by a second heating of the sample immediately after cooling from the first scan (data not shown).
FIGURE 2.

Deconvolution analysis of the excess heat capacity (Cp) function of reduced WT α-TM (A) and α-TM-D137L (B). Solid lines represent experimental curves after subtraction of chemical and instrumental base lines. The dotted lines show individual thermal transitions (domains; 1 and 2) obtained from fitting the data to the non-two-state model. The heating rate was 1 °C/min. Concentration of reduced protein samples was 1.8 mg/ml. Buffer conditions were 20 mm MOPS, pH 7.0, 1 mm EDTA, 0.1 m NaCl, and 1 mm β-mercaptoethanol. Samples were heated up to 90 °C, but only a temperature region below 65 °C, where thermal transitions occurred, is depicted (see Table 1 for parameters).
TABLE 1.
Calorimetric parameters obtained from DSC data (Fig. 2) of recombinantly expressed WT α-TM andα-TM-D137L
| Domain-1 (C-terminal region) |
Domain-2 (N-terminal region) |
|||
|---|---|---|---|---|
| Tma | ΔHcalb | Tma | ΔHcalb | |
| °C | kcal/mol (% of total) | °C | kcal/mol (% of total) | |
| α-TM | 43.35 | 91.13 (70.5%) | 49.99 | 38.07 (29.5%) |
| α-TM-D137L | 45.68 | 94.58 (36.5%) | 52.19 | 168 (63%) |
a The error of the given Tm (transition temperature) values did not exceed ±0.2 °C.
b The relative error of the given values of ΔHcal (calorimetric enthalpy) did not exceed ±10%.
Previous studies revealed that the least thermostable domain-1 corresponds to the thermal unfolding of the C-terminal part, whereas the domain-2 corresponds to the N-terminal unfolding of striated α-TM (17, 39–41). Thus, our results suggested that the D137L conversion increases thermal stability of both the N and C terminus of TM with almost equal increases in Tm values of domain-1 and -2. A prominent increase in calorimetric enthalpy of N-terminal calorimetric domain compared with C-terminal domain indicated a higher stabilization effect of Leu substitution at position 137 on the N-terminal domain.
Thermal instability is thought to correlate with protein flexibility. Accordingly, the detected increased coiled-coil stability of α-TM-D137L suggested a decrease in the structural flexibility of the protein.
Generation of α-TM-D137L TG Mice
To examine the physiological importance of the residue Asp-137 and decreased TM flexibility in ejecting hearts, we generated α-TM-D137L TG mice as described above. The transgene construct used to generate the α-TM-D137L TG mice is shown in Fig. 3A. The α-myosin heavy chain promoter drives cardiac specific expression of the α-TM-D137L cDNA.
FIGURE 3.

A, the α-TM-D137L construct used to generate the TG mice. HGH, human growth hormone; B, TM region from a representative two-dimensional difference gel of myofibrillar proteins from 4-month-old NTG and TG mouse hearts. Myofibrillar fractions were labeled separately and equally mixed. Channel 1, Cy5-labeled α-TM (green); channel 2, Cy3-labeled α-TM-D137L (red); channel 3, merged image of channels 1 and 2. 81.3 ± 1.5% replacement (total α-TM-D137L/total α-TM) in TG mouse hearts was detected (n = 11). P-, phosphorylated protein.
Expression Profile and Phosphorylation of α-TM-D137L in TG Mouse Hearts
Multiple TG lines were generated, but only the highest α-TM-D137L-expressing line was studied. We were not able to separate endogenous α-TM and variant α-TM-D137L protein bands using traditional SDS-PAGE (data not shown). Therefore, to quantify expression levels of α-TM-D137L in TG mice, myofibrillar preparations from hearts of age-matched NTG and TG mice were subjected to 2D-DIGE. Results from NTG samples displayed one unphosphorylated and one phosphorylated (P-) TM spot (Fig. 3B, channel 1) that were well characterized previously in our laboratory (30). As anticipated, we detected four TM spots from TG samples (Fig. 3B, channel 2). An overlay of differentially labeled NTG and TG channels allowed us to clearly identify these four spots (Fig. 3B, channel 3). Conversion of a negatively charged Asp residue to a neutral Leu resulted in a rightward shift (toward the minus end of the pH strip) in TM spots due to loss of charge in the protein. Quantification of two-dimensional difference gels showed a 81.3 ± 1.5% replacement (total α-TM-D137L/total α-TM) of WT TM with the variant TM in TG mouse hearts. There was a concomitant decrease in NTG TM levels, maintaining 100% total TM levels. Myofibrillar protein preparations from NTG and TG mouse hearts were run on an SDS-polyacrylamide gel as a control and showed that all other major cardiac myofilament proteins were expressed at a stoichiometric ratio in TG mouse hearts compared with NTG ones (data not shown).
It has been recently reported in both in vitro and in vivo studies that TM phosphorylation plays a role in modulation of muscle force generation (42, 43). Thus, we determined the percentage of total TM phosphorylation (total phosphorylated TM/total TM) in myofibrillar protein samples from 4-month-old NTG and TG mouse hearts using the same 2D-DIGE approach. No statistically significant difference between total phosphorylated TM levels in TG and NTG mouse hearts were detected (see Table 5). We repeated the same phosphorylation analysis of TM with samples from 8-month-old NTG and TG mice and again observed no statistically significant differences (supplemental Table S3).
TABLE 5.
Quantification of two-dimensional difference gels of myofilament proteins from 4-month-old NTG and TG mouse hearts
Representative 2D-DIGE images are shown in Fig. 6.
TG Mice Exhibited Systolic and Diastolic Dysfunction
Echocardiographic analyses of TG and NTG mouse hearts were employed to compare their cardiac function and detect any changes due to α-TM-D137L expression. Results from 4-month-old TG mice predominantly demonstrated systolic dysfunction with a significant depression in velocity of circumferential shortening, fractional shortening, ejection fraction, and mitral annulus systolic velocity (Table 2). Diastolic dysfunction coexisted with systolic dysfunction in TG mouse hearts, as demonstrated by prolongation of isovolumetric relaxation time and an increase in ratio of early pulsed Doppler left ventricular filling and early tissue-Doppler mitral annular velocity. Additionally, a mild left ventricular dilation was present in TG mouse hearts with increases in end-systolic and end-diastolic left ventricular dimensions. Stroke volume and cardiac output were maintained. There was no change in heart rate of TG mice compared with NTG ones. Functional defects observed in 4-month-old TG mouse hearts were maintained at 8-month-old animals without any statistically significant, age-dependent worsening (supplemental Table S1).
TABLE 2.
Cardiac function of NTG and α-TM- D137L TG mice at 4 months of age as assessed by echocardiography
| Parameters | NTG 4 months (n = 11) | TG 4 months (n = 12) |
|---|---|---|
| Systolic | ||
| Fractional shortening (%) | 34.9 ± 0.91 | 30.3 ± 1.16a |
| Ejection fraction (%) | 64.7 ± 1.26 | 57.9 ± 1.76a |
| Velocity of circumferential shortening (circ/s) | 6.4 ± 0.23 | 5.4 ± 0.29a |
| Mitral annulus systolic velocity (mm/s) | 22.3 ± 0.58 | 16.2 ± 0.80a |
| Cardiac output (ml/min) | 19.5 ± 1.48 | 20.8 ± 0.96 |
| Heart rate (beats/min) | 439.0 ± 9.33 | 462.5 ± 10.88 |
| Stroke volume (μl) | 44.7 ± 2.30 | 42.6 ± 2.23 |
| Diastolic | ||
| E/A ratiob | 1.8 ± 0.11 | 1.8 ± 0.19 |
| E/Em ratioc | 31.0 ± 1.47 | 43.3 ± 3.95a |
| Isovolumetric relaxation time (ms) | 13.0 ± 0.42 | 18.51 ± 1.01a |
| E wave DTd (ms) | 22.7 ± 1.19 | 22.2 ± 1.07 |
| Morphology | ||
| LAe (mm) | 2.0 ± 0.04 | 2.0 ± 0.08 |
| LVIDsf (mm) | 2.6 ± 0.09 | 3.1 ± 0.10a |
| LVIDdg (mm) | 3.9 ± 0.10 | 4.2 ± 0.08 |
| Left ventricular mass (mg) | 74.9 ± 3.43 | 88.2 ± 5.00 |
| RWTh (mm) | 0.3 ± 0.02 | 0.3 ± 0.01 |
a p < 0.05 versus NTG.
b Ratio of maximal velocity of E (early LV filling) and A (atrial contraction) waves.
c Ratio of early pulsed Doppler LV filling and early tissue-Doppler mitral annular velocity.
d E wave deceleration time.
e Left atrium.
f End-systolic left ventricular dimension.
g End-diastolic left ventricular dimension.
h Relative wall thickness.
In agreement with morphology data obtained from echocardiography, the heart weight/tibia length ratio, which is an independent measure of hypertrophy, was not significantly different in 4- and 8-month-old NTG and TG mouse hearts (supplemental Fig. S1A). Additionally, survival analyses suggested no significant difference between viability of TG and NTG mice over the course of 8 months (supplemental Fig. S1B).
Contractile Mechanics of Cardiomyocytes from TG Mouse Hearts Were Diminished without Any Changes in Intracellular Ca2+ Transients
The regulation of cardiac contractility is dependent upon the changes in the functional state of the myofilaments in addition to the characteristics of the intracellular calcium transient. Therefore, we examined cell shortening and the Ca2+ transient relations in ventricular cardiomyocytes from 4-month-old NTG and TG mice using video edge detection and fura-2 fluorescence. The representative recordings of cell shortening (Fig. 4A), the kinetics of contraction and relaxation (Fig. 4B), and intracellular Ca2+ transients expressed as fura-2 fluorescence ratio (Fig. 4C) are demonstrated. Cardiomyocytes isolated from TG mice exhibited a statistically significant 30% reduction in the extent of unloaded cell shortening (NTG = 8.5% ± 0.34 versus TG = 6.0 ± 0.30%). The peak rate of contraction (NTG = 131.4 ± 5.3 versus TG = 87.22 ± 3.0 μm/s) and the peak rate of relaxation (NTG = 92.9 ± 5.1 versus TG = 61.75 ± 2.7 μm/s) were also diminished in cells expressing α-TM-D137L (Fig. 4D). The observed changes in contractility were independent of any alteration in Ca2+ transient (Table 3). There was also no significant difference in average cardiomyocyte size isolated from NTG and TG mouse hearts (data not shown).
FIGURE 4.

Analysis of contractile mechanics and Ca2+ transients of mouse ventricular cardiomyocytes. Shown are representative recordings of unloaded cell shortening (A), kinetics of twitch contraction and relaxation (B), and intracellular Ca2+ transients (see Table 3 for parameters) (C) of single isolated cardiomyocytes from 4-month-old NTG and TG animals. D, quantification of the contractile parameters (percentage of shortening and maximal rates of contraction and relaxation). Five NTG animals (15 cells) and four TG (11 cells) mouse hearts were studied. * and #, p < 0.05 versus NTG. Error bars, S.E.
TABLE 3.
Ca2+ transients of cardiomyocytes from 4-month-old NTG and TG-α-TM-D137L mice
Representative recordings are shown in Fig. 4C. n indicates the number of cells from 5 NTG and 4 TG mice hearts.
| NTG 4 months (n = 15) | TG 4 months (n = 11) | |
|---|---|---|
| Base line | 0.37 ± 0.01 | 0.36 ± .010 |
| Amplitude (ratio units) | 0.49 ± 0.01 | 0.44 ± 0.02 |
| Tau (ms) | 257.64 ± 9.52 | 255.43 ± 8.54 |
Myofilament Ca2+ Sensitivity Was Decreased in TG Mouse Hearts
To further elucidate the mechanisms responsible for the functional alterations observed in the hearts of TG mice, we focused on the sarcomere level and studied Ca2+-dependent tension development of skinned fiber bundles from NTG and TG mouse hearts. Results depicted a significant decrease in Ca2+ sensitivity of myofilaments from 4-month-old TG mice without any change in the maximum developed tension (Fig. 5A). No age-dependent alteration in the decreased state of Ca2+ sensitivity was detected at fibers from 8-month-old TG mouse hearts (supplemental Table S2). A significant increase in the Hill coefficient of TG myofilaments at both ages was also present (Table 4 and supplemental Table S2).
FIGURE 5.
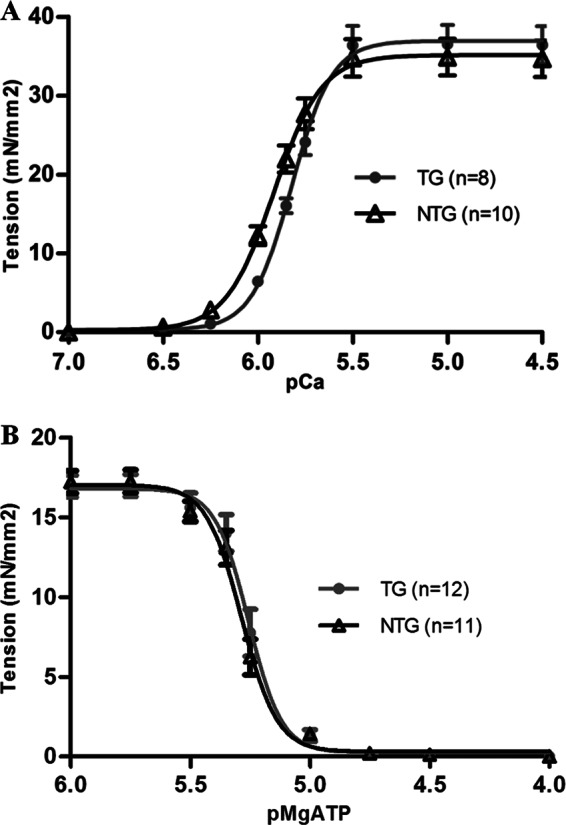
A, pCa-tension relation of skinned fiber bundles from 4-month-old NTG (black) and TG (gray) mouse hearts. Measurements were carried out at sarcomere length = 2.2 μm at 25 °C. (see Table 4 for parameters). B, pMgATP-tension relation of skinned fiber bundles from 4-month-old NTG (black) and TG (dark gray) mouse hearts. Sarcomere length = 2.2 μm at 25 °C. NTG [MgATP]50 = 5.28 ± 0.01, TG [MgATP]50 = 5.26 ± 0.01. Error bars, S.E.
TABLE 4.
Parameters from Fig. 5A describing Ca2+-dependent tension generation in skinned fiber bundles from 4-month-old NTG and TG mouse hearts
n indicates the number of fibers from five NTG and four TG mice hearts.
| NTG 4 months (n = 10) | TG 4 months (n = 8) | |
|---|---|---|
| pCa50 | 5.92 ± 0.01 | 5.82 ± 0.02a |
| nH | 3.50 ± 0.08 | 4.12 ± 0.06a |
| Maximum force (millinewtons/mm2) | 35.12 ± 2.34 | 36.93 ± 2.45 |
a p < 0.05 versus NTG.
Rigor Cross-bridge-dependent Activation of Myofilaments from TG Mouse Hearts Was Not Altered
To probe for modification in strong cross-bridge-dependent activation in the presence of α-TM-D137L, we determined the relation between [MgATP] and skinned fiber tension generation. There was no difference in the ability of strong cross-bridges to activate thin filaments from 4-month-old NTG and TG mouse hearts in the absence of Ca2+ at various ATP concentrations (Fig. 5B).
The D137L Variant Did Not Alter PTM Status of Major Myofilament Proteins
PTMs of sarcomeric proteins are one of the main modes of the heart to meet changing hemodynamic demands. It is possible that compensatory responses may occur in response to the altered contractile dynamics in α-TM-D137L TG mouse hearts. Therefore, to elucidate whether α-TM-D137L expression alters the modification status of other myofilament proteins in TG mouse hearts, we employed the 2D-DIGE approach. Results indicated no significant difference between modification levels of major myofibrillar proteins (cTnI, cTnT, MLC, and MyBP-C) from 4-month-old NTG and TG mice (Fig. 6 and Table 5). The modification state of myofilament proteins remained unchanged in samples from 8-month-old NTG and TG mouse hearts (supplemental Table S3).
FIGURE 6.

Comparison of PTM status and expression profile of myofibrillar proteins from 4-month-old NTG and TG mouse hearts by 2D-DIGE. Representative 2D-DIGE images (merged) show the region of cTnI (A), MLC (B), cTnT (C), and MyBP-C (D). In A–C, U indicates unmodified. pH values indicate the pH range of the strip used for the first dimension. Samples were labeled with different Cy dyes and equally mixed to run in the same two-dimensional gel. Quantification of protein spots is shown in Table 5.
Slightly Increased β-MyHC Expression Was Detected in TG Mouse Hearts
In addition to PTMs, variable expression of myofibrillar protein isoforms also regulates heart function. One important protein that is known to undergo isoform switching with cardiac stress is MyHC (44). There are two cardiac MyHC isoforms: α- and β-MyHC. Adult mouse hearts predominantly express α-MyHC in ventricles. We measured the relative levels of α- and β-MyHC expression in ventricular tissue from 4- and 8-month-old NTG and TG mice. Results demonstrated that β-MyHC expression was slightly higher in myofibrillar preparations from 4-month-old TG mice compared with controls (Fig. 7). However, there was no age-dependent, statistically significant increase in β-MyHC expression in 8-month-old TG mouse hearts (supplemental Fig. S2).
FIGURE 7.

SDS-polyacrylamide gel of myofibrillar proteins from 4-month-old NTG and TG mouse hearts. Only the MyHC area is shown. First lane, myofibrillar protein sample from 1-day-old neonatal mouse heart (2.5 μg/μl). NTG and TG mice samples were loaded at 9 μg/μl concentration. A percentage relative quantification for α- and β-MyHC bands in each lane is shown below the gel.
DISCUSSION
This is the first study to show that the flexibility of TM structure provided by the residue Asp-137 plays a physiologically important role in ejecting mouse hearts. Our findings advance understanding of functionally important structural characteristics of TM and suggest a possible link between altered structural flexibility of TM and cardiac disorders.
Our results from DSC measurements extend earlier findings investigating the flexibility of α-TM. The conserved, non-canonical Asp-137 residue is located at a particularly unstable region of α-TM that is susceptible to cleavage by trypsin. Sumida et al. (11) reported that substitution of Asp-137 with a canonical Leu reduced proteolytic susceptibility of the variant that was associated with reduced TM flexibility. In agreement with this study, our DSC results suggested a decrease in flexibility of the α-TM-D137L variant, which was detected in both N- and C-terminal thermal transition domains. This overall impact observed in α-TM-D137L structure is in agreement with our NMR observations. Our 1H-13C-edited HSQC measurements took advantage of reductive methylation of TM samples and revealed long range structural rearrangements in TM structure due to the stabilizing Leu substitution at position 137. These long range effects in TM structure caused by a single amino acid substitution may provide unique insight into how hypertrophic cardiomyopathy- or dilated cardiomyopathy-linked single point mutations mapped to the TM gene can significantly alter dynamic properties of TM and hence affect regulatory functions of TM in the heart.
In this study, we significantly extended in vitro work and employed transgenesis to study the naturally incorporated α-TM-D137L variant in the whole heart, single cardiomyocytes, and myofilaments. This highly integrative approach allowed us to investigate how altered TM flexibility due to D137L conversion influenced the function of regulated cardiac thin filaments. Cardiac specific expression of α-TM-D137L led to a phenotype with prominent systolic dysfunction along with diastolic dysfunction. There was a robust reduction in systolic parameters of the heart, and in isolated cardiomyocyte measurements, cell shortening was diminished. Along with decreased myofilament Ca2+ sensitivity and mild dilation of ventricles, the physiological profile of α-TM-D137L TG mice is similar to dilated cardiomyopathy. Previously, the dilated cardiomyopathy-linked E54K mutation was also reported to increase the temperature stability of α-TM (45). In contrast, increased flexibility appears to be a characteristic of hypertrophic cardiomyopathy-linked TM mutations (E180G, D175N, K70T, and A63V) (17, 46, 47). These findings indicate a possible association between TM flexibility and the complex mechanisms leading to cardiac disorders.
In 2D-DIGE experiments, we demonstrated that α-TM-D137L incorporation into myofilaments did not affect expression and PTM profiles of major myofilament proteins: TM, cTnI, cTnT, MLC, and MyBP-C. These myofilament proteins are often modified in the case of altered cardiac function due to pathological conditions resulting from impaired cardiac function (42, 43, 48–51). Our results suggested that the level of functional changes in the heart and reduced Ca2+ sensitivity of myofilaments regulated by α-TM-D137L did not provoke activation of compensatory signal transduction mechanisms or alter major myofilament proteins as substrates for kinases or phosphatases.
Another factor besides myofilament Ca2+ sensitivity and covalent modification of myofibrillar proteins that may affect the power-generating capacity of the heart is variable expression of MyHC. Despite the detected increase in β-MyHC expression in TG mouse hearts, the lack of the hypertrophic phenotype suggested that this slight increase may not be functionally important. Although it remains possible that increased β-MyHC expression might have contributed to reduced contractile function detected in TG mouse hearts (52), studies comparing the relative expression of β-MyHC with loaded shortening velocity and power demonstrated no significant effects of the presence of up to 10% β-MyHC (53).
Regulation of cardiac muscle contraction and relaxation requires complex interactions between Ca2+, thin filament proteins, and myosin cross-bridges. TM molecules undergo regulatory relocations over the surface of actin filaments in response to Ca2+ binding to Tn and myosin binding to actin. We believe that the structural flexibility of TM plays a key role in regulating the position of TM (blocked-closed-open) on the actin surface, and perturbation of the TM position affects physiological responses of cardiac muscle. Although we observed decreased Ca2+ sensitivity of myofilaments from TG mouse hearts, [MgATP]-tension measurements demonstrated no change in the ability of strong cross-bridges to activate thin filaments in the absence of Ca2+. These findings suggest that decreased TM flexibility may impede Ca2+-dependent relocation of TM between blocked and closed states, therefore resulting in a delay in time-sensitive activation and relaxation processes in cardiac muscle. The myosin-dependent closed to open state relocation of TM in thin filaments might be also affected; however, we could not detect a difference in strong cross-bridge-dependent activation in myofilaments regulated by α-TM-D137L compared with α-TM. As expected, expression of a stiffer TM resulted in an increase of the Hill n values in Ca2+-dependent myofilament activation. This indicates that the alterations in TM flexibility may be significant in a mechanism of cooperative activation that is intrinsic to the filaments (54, 55).
It is also possible that α-TM-D137L incorporation into myofilaments alters interactions of TM with thin filament proteins. It was previously reported that flexibility of the middle region of TM, where residue 137 lies, is not crucial for actin binding and that D137L substitution does not alter the actin binding properties of TM (11, 12). However, a direct interaction between the C-terminal mobile domain of cTnI and residue 146 of TM was previously proposed by Mudalige et al. (56). Long range structural perturbations in TM structure due to D137L conversion potentially affect this interaction with cTnI and alter the Tn-dependent blocked-closed state equilibrium. This possibility would also help in explaining decreased Ca2+ sensitivity of myofilaments in the absence of any change in Ca2+ transients of isolated cardiomyocytes observed in this study. Altered signal transmission between thin filament components due to expression of a decreased flexibility variant of TM may modify Ca2+ affinity of cTnC.
To conclude, our findings advance understanding of the functional importance of unconventional structural features of TM (e.g. negatively charged Asp residue at the hydrophobic core of the coiled-coil molecule). The results from our study demonstrated that a marked decrease in the structural flexibility of TM due to substitution of Asp-137 with Leu affects systolic and diastolic parameters of cardiac contraction, ultimately leading to a phenotype similar to dilated cardiomyopathy in α-TM-D137L TG mouse hearts. These investigations shed light on a structural characteristic of TM that is essential for its regulatory functions in cardiac muscle dynamics in the heart and suggest a possible association between flexibility of TM and cardiac disorders. We think that these findings will be highly relevant to interpretation of in vitro data as well as to perspectives on the clinical and physiological significance of TM flexibility.
Acknowledgments
Dr. Larry Tobacman graciously provided WT tropomyosin plasmid. We thank Chad Warren for technical help with two-dimensional gels. We also thank Dr. Ozgur Ogut for careful reading of the manuscript and valuable comments.
This work was supported, in whole or in part, by National Institutes of Health Grants PO1 HL62426 (Project 1) (to R. J. S.), R01 CA135341 (to V. G.), and RO1 HL081680 (to D. F. W.). This work was also supported by American Heart Association Predoctoral Fellowship 12PRE9260034 (to S. Y.).

This article contains supplemental Figs. S1 and S2 and Tables S1–S3.
- TM
- tropomyosin
- TG
- transgenic
- NTG
- non-transgenic
- MyHC
- myosin heavy chain
- Tn
- troponin
- MyBP-C
- myosin binding protein-C
- MLC
- myosin light chain
- cTnI
- cardiac troponin-I
- cTnT
- cardiac troponin-T
- cTnC
- cardiac troponin-C
- HSQC
- heteronuclear single quantum coherence
- DSC
- differential scanning calorimetry
- 2D-DIGE
- two-dimensional difference gel electrophoresis
- PTM
- post-translational modification
- BES
- 2-[bis(2-hydroxyethyl)amino]ethanesulfonic acid.
REFERENCES
- 1. Parry D. A., Squire J. M. (1973) Structural role of tropomyosin in muscle regulation. Analysis of the x-ray diffraction patterns from relaxed and contracting muscles. J. Mol. Biol. 75, 33–55 [DOI] [PubMed] [Google Scholar]
- 2. Haselgrove J. C. (1973) X-ray evidence for a conformational change in the actin-containing filaments of vertebrate striated muscle. Cold Spring Harb. Symp. Quant. Biol. 37, 341–352 [Google Scholar]
- 3. Lehman W., Hatch V., Korman V., Rosol M., Thomas L., Maytum R., Geeves M. A., Van Eyk J. E., Tobacman L. S., Craig R. (2000) Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J. Mol. Biol. 302, 593–606 [DOI] [PubMed] [Google Scholar]
- 4. Vibert P., Craig R., Lehman W. (1997) Steric-model for activation of muscle thin filaments. J. Mol. Biol. 266, 8–14 [DOI] [PubMed] [Google Scholar]
- 5. McKillop D. F., Geeves M. A. (1993) Regulation of the interaction between actin and myosin subfragment-1. Evidence for 3 states of the thin filament. Biophys. J. 65, 693–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Straussman R., Ben-Ya'acov A., Woolfson D. N., Ravid S. (2007) Kinking the coiled coil-negatively charged residues at the coiled-coil interface. J. Mol. Biol. 366, 1232–1242 [DOI] [PubMed] [Google Scholar]
- 7. Chen Y., Lehrer S. S. (2004) Distances between tropomyosin sites across the muscle thin filament using luminescence resonance energy transfer. Evidence for tropomyosin flexibility. Biochemistry 43, 11491–11499 [DOI] [PubMed] [Google Scholar]
- 8. Singh A., Hitchcock-DeGregori S. E. (2003) Local destabilization of the tropomyosin coiled coil gives the molecular flexibility required for actin binding. Biochemistry 42, 14114–14121 [DOI] [PubMed] [Google Scholar]
- 9. Singh A., Hitchcock-DeGregori S. E. (2006) Dual requirement for flexibility and specificity for binding of the coiled-coil tropomyosin to its target, actin. Structure 14, 43–50 [DOI] [PubMed] [Google Scholar]
- 10. Nitanai Y., Minakata S., Maéda K., Oda N., Maeda Y. (2007) Crystal structures of tropomyosin. Flexible coiled-coil. Adv. Exp. Med. Biol. 592, 137–151 [DOI] [PubMed] [Google Scholar]
- 11. Sumida J. P., Wu E., Lehrer S. S. (2008) Conserved Asp-137 imparts flexibility to tropomyosin and affects function. J. Biol. Chem. 283, 6728–6734 [DOI] [PubMed] [Google Scholar]
- 12. Nevzorov I. A., Nikolaeva O. P., Kainov Y. A., Redwood C. S., Levitsky D. I. (2011) Conserved noncanonical residue Gly-126 confers instability to the middle part of the tropomyosin molecule. J. Biol. Chem. 286, 15766–15772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Monteiro P. B., Lataro R. C., Ferro J. A., Reinach Fde C. (1994) Functional α-tropomyosin produced in Escherichia coli. A dipeptide extension can substitute the amino-terminal acetyl group. J. Biol. Chem. 269, 10461–10466 [PubMed] [Google Scholar]
- 14. Gill S. C., von Hippel P. H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 15. Means G. E., Feeney R. E. (1968) Reductive alkylation of amino groups in proteins. Biochemistry 7, 2192–2201 [DOI] [PubMed] [Google Scholar]
- 16. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 17. Kremneva E., Boussouf S., Nikolaeva O., Maytum R., Geeves M. A., Levitsky D. I. (2004) Effects of two familial hypertrophic cardiomyopathy mutations in α-tropomyosin, Asp175Asn and Glu180Gly, on the thermal unfolding of actin-bound tropomyosin. Biophys. J. 87, 3922–3933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Subramaniam A., Jones W. K., Gulick J., Wert S., Neumann J., Robbins J. (1991) Tissue-specific regulation of the α-myosin heavy chain gene promoter in transgenic mice. J. Biol. Chem. 266, 24613–24620 [PubMed] [Google Scholar]
- 19. Muthuchamy M., Grupp I. L., Grupp G., O'Toole B. A., Kier A. B., Boivin G. P., Neumann J., Wieczorek D. F. (1995) Molecular and physiological effects of overexpressing striated muscle β-tropomyosin in the adult murine heart. J. Biol. Chem. 270, 30593–30603 [DOI] [PubMed] [Google Scholar]
- 20. Gaffin R. D., Peña J. R., Alves M. S., Dias F. A., Chowdhury S. A., Heinrich L. S., Goldspink P. H., Kranias E. G., Wieczorek D. F., Wolska B. M. (2011) Long-term rescue of a familial hypertrophic cardiomyopathy caused by a mutation in the thin filament protein, tropomyosin, via modulation of a calcium cycling protein. J. Mol. Cell Cardiol. 51, 812–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lang R. M., Bierig M., Devereux R. B., Flachskampf F. A., Foster E., Pellikka P. A., Picard M. H., Roman M. J., Seward J., Shanewise J. S., Solomon S. D., Spencer K. T., Sutton M. S., Stewart W. J. (2005) Recommendations for chamber quantification. A report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J. Am. Soc. Echocardiogr. 18, 1440–1463 [DOI] [PubMed] [Google Scholar]
- 22. Nagueh S. F., Appleton C. P., Gillebert T. C., Marino P. N., Oh J. K., Smiseth O. A., Waggoner A. D., Flachskampf F. A., Pellikka P. A., Evangelisa A. (2009) Recommendations for the evaluation of left ventricular diastolic function by echocardiography. Eur. J. Echocardiogr. 10, 165–193 [DOI] [PubMed] [Google Scholar]
- 23. Wolska B. M., Solaro R. J. (1996) Method for isolation of adult mouse cardiac myocytes for studies of contraction and microfluorimetry. Am. J. Physiol. 271, H1250–H1255 [DOI] [PubMed] [Google Scholar]
- 24. Louch W. E., Sheehan K. A., Wolska B. M. (2011) Methods in cardiomyocyte isolation, culture, and gene transfer. J. Mol. Cell Cardiol. 51, 288–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wolska B. M., Keller R. S., Evans C. C., Palmiter K. A., Mutuchamy M., Wieczorek D. F., de Tombe P. P., Solaro R. J. (1998) Correlation between myofilament response to Ca2+ and altered dynamics of contraction and relaxation in transgenic cardiac cells expressing β-tropomyosin. Circulation 98, 465–465 [DOI] [PubMed] [Google Scholar]
- 26. Brandt P. W., Roemer D., Schachat F. H. (1990) Co-operative activation of skeletal muscle thin filaments by rigor crossbridges. The effect of troponin C extraction. J. Mol. Biol. 212, 473–480 [DOI] [PubMed] [Google Scholar]
- 27. Fabiato A. (1988) Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 157, 378–417 [DOI] [PubMed] [Google Scholar]
- 28. Godt R. E., Lindley B. D. (1982) Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle-fibers of the frog. J. Gen. Physiol. 80, 279–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scruggs S. B., Hinken A. C., Thawornkaiwong A., Robbins J., Walker L. A., de Tombe P. P., Geenen D. L., Buttrick P. M., Solaro R. J. (2009) Ablation of ventricular myosin regulatory light chain phosphorylation in mice causes cardiac dysfunction in situ and affects neighboring myofilament protein phosphorylation. J. Biol. Chem. 284, 5097–5106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Warren C. M., Arteaga G. M., Rajan S., Ahmed R. P., Wieczorek D. F., Solaro R. J. (2008) Use of 2-D DIGE analysis reveals altered phosphorylation in a tropomyosin mutant (Glu54Lys) linked to dilated cardiomyopathy. Proteomics 8, 100–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yuan C., Sheng Q., Tang H., Li Y., Zeng R., Solaro R. J. (2008) Quantitative comparison of sarcomeric phosphoproteomes of neonatal and adult rat hearts. Am. J. Physiol. Heart Circ. Physiol. 295, H647–H656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kirk J. A., MacGowan G. A., Evans C., Smith S. H., Warren C. M., Mamidi R., Chandra M., Stewart A. F., Solaro R. J., Shroff S. G. (2009) Left ventricular and myocardial function in mice expressing constitutively pseudophosphorylated cardiac troponin I. Circ. Res. 105, 1232–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Warren C. M., Greaser M. L. (2003) Method for cardiac myosin heavy chain separation by sodium dodecyl sulfate gel electrophoresis. Anal. Biochem. 320, 149–151 [DOI] [PubMed] [Google Scholar]
- 34. Kobayashi T., Patrick S. E., Kobayashi M. (2009) Ala Scanning of the inhibitory region of cardiac troponin I. J. Biol. Chem. 284, 20052–20060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wider G., Wüthrich K. (1999) NMR spectroscopy of large molecules and multimolecular assemblies in solution. Curr. Opin. Struct. Biol. 9, 594–601 [DOI] [PubMed] [Google Scholar]
- 36. Gerken T. A., Jentoft J. E., Jentoft N., Dearborn D. G. (1982) Intramolecular interactions of amino groups in 13C reductively methylated hen egg-white lysozyme. J. Biol. Chem. 257, 2894–2900 [PubMed] [Google Scholar]
- 37. Kurinov I. V., Mao C., Irvin J. D., Uckun F. M. (2000) X-ray crystallographic analysis of pokeweed antiviral protein-II after reductive methylation of lysine residues. Biochem. Biophys. Res. Commun. 275, 549–552 [DOI] [PubMed] [Google Scholar]
- 38. Rayment I. (1997) Reductive alkylation of lysine residues to alter crystallization properties of proteins. Methods Enzymol. 276, 171–179 [PubMed] [Google Scholar]
- 39. Potekhin S. A., Privalov P. L. (1982) Co-operative blocks in tropomyosin. J. Mol. Biol. 159, 519–535 [DOI] [PubMed] [Google Scholar]
- 40. Sturtevant J. M., Holtzer M. E., Holtzer A. (1991) A scanning calorimetric study of the thermally induced unfolding of various forms of tropomyosin. Biopolymers 31, 489–495 [DOI] [PubMed] [Google Scholar]
- 41. Williams D. L., Jr., Swenson C. A. (1981) Tropomyosin stability. Assignment of thermally induced conformational transitions to separate regions of the molecule. Biochemistry 20, 3856–3864 [DOI] [PubMed] [Google Scholar]
- 42. Nixon B. R., Liu B., Scellini B., Tesi C., Piroddi N., Ogut O., John Solaro R., Ziolo M. T., Janssen P. M., Davis J. P., Poggesi C., Biesiadecki B. J. (2012) Tropomyosin Ser-283 pseudo-phosphorylation slows myofibril relaxation. Arch. Biochem. Biophys., doi: 10.1016/j.abb.2012.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schulz E. M., Correll R. N., Sheikh H. N., Lofrano-Alves M. S., Engel P. L., Newman G., Schultz Jel J., Molkentin J. D., Wolska B. M., Solaro R. J., Wieczorek D. F. (2012) Tropomyosin dephosphorylation results in compensated cardiac hypertrophy. J. Biol. Chem. 287, 44478–44489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nadal-Ginard B., Mahdavi V. (1989) Molecular basis of cardiac performance. Plasticity of the myocardium generated through protein isoform switches. J. Clin. Invest. 84, 1693–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rajan S., Ahmed R. P., Jagatheesan G., Petrashevskaya N., Boivin G. P., Urboniene D., Arteaga G. M., Wolska B. M., Solaro R. J., Liggett S. B., Wieczorek D. F. (2007) Dilated cardiomyopathy mutant tropomyosin mice develop cardiac dysfunction with significantly decreased fractional shortening and myofilament calcium sensitivity. Circ. Res. 101, 205–214 [DOI] [PubMed] [Google Scholar]
- 46. Loong C. K., Zhou H. X., Chase P. B. (2012) Familial hypertrophic cardiomyopathy related E180G mutation increases flexibility of human cardiac α-tropomyosin. FEBS Lett. 586, 3503–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heller M. J., Nili M., Homsher E., Tobacman L. S. (2003) Cardiomyopathic tropomyosin mutations that increase thin filament Ca2+ sensitivity and tropomyosin N-domain flexibility. J. Biol. Chem. 278, 41742–41748 [DOI] [PubMed] [Google Scholar]
- 48. Solaro R. J., Henze M., Kobayashi T. (2013) Integration of troponin I phosphorylation with cardiac regulatory networks. Circ. Res. 112, 355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dubois E., Richard V., Mulder P., Lamblin N., Drobecq H., Henry J. P., Amouyel P., Thuillez C., Bauters C., Pinet F. (2011) Decreased serine 207 phosphorylation of troponin T as a biomarker for left ventricular remodelling after myocardial infarction. Eur. Heart J. 32, 115–123 [DOI] [PubMed] [Google Scholar]
- 50. Scruggs S. B., Solaro R. J. (2011) The significance of regulatory light chain phosphorylation in cardiac physiology. Arch. Biochem. Biophys. 510, 129–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. James J., Robbins J. (2011) Signaling and myosin-binding protein C. J. Biol. Chem. 286, 9913–9919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Krenz M., Robbins J. (2004) Impact of β-myosin heavy chain expression on cardiac function during stress. J. Am. Coll. Cardiol. 44, 2390–2397 [DOI] [PubMed] [Google Scholar]
- 53. Korte F. S., Herron T. J., Rovetto M. J., McDonald K. S. (2005) Power output is linearly related to MyHC content in rat skinned myocytes and isolated working hearts. Am. J. Physiol. Heart Circ. Physiol. 289, H801–H812 [DOI] [PubMed] [Google Scholar]
- 54. Sun Y. B., Irving M. (2010) The molecular basis of the steep force-calcium relation in heart muscle. J. Mol. Cell Cardiol. 48, 859–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Solaro R. J. (2009) Maintaining cooperation among cardiac myofilament proteins through thick and thin. J. Physiol. 587, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mudalige W. A., Tao T. C., Lehrer S. S. (2009) Ca2+-dependent photocrosslinking of tropomyosin residue 146 to residues 157–163 in the C-terminal domain of troponin I in reconstituted skeletal muscle thin filaments. J. Mol. Biol. 389, 575–583 [DOI] [PMC free article] [PubMed] [Google Scholar]