

Background: TTF-1 inhibits lung cancer progression via HMGA2 down-regulation.
Results: TTF-1 up-regulates miR-33a, which in turn directly represses HMGA2.
Conclusion: The signaling axis of TTF-1 to HMGA2, important in controlling lung cancer metastasis, is mediated by miR-33a.
Significance: This study explains the mechanism of HMGA2 suppression by TTF-1.
Keywords: Cholesterol Regulation, Gene Regulation, Lung Cancer, Metastasis, MicroRNA, HMGA2, SREBP2, TTF-1, miR-32, miR-33a
Abstract
In lung cancers, TTF-1 displays seemingly paradoxical activities. Although TTF-1 is amplified in primary human lung cancers, it inhibits primary lung tumors from metastasizing in a mouse model system. It was reported that the oncogenic proepithelial mesenchymal transition (EMT) high mobility group AT-hook 2 gene (HMGA2) mediates the antimetastatic function of TTF-1. To gain mechanistic insight into the metastasis-critical signaling axis of TTF-1 to HMGA2, we used both reverse and forward strategies and discovered that microRNA-33a (miR-33a) is under direct positive regulation of TTF-1. By chromatin immunoprecipitation, we determined that TTF-1 binds to the promoter of SREBF2, the host gene of miR-33a. The 3′-untranslated region (UTR) of HMGA2 contains three predicted binding sites of miR-33a. We showed that the first two highly conserved sites are conducive to HMGA2 repression by miR-33a, establishing HMGA2 as a genuine target of miR-33a. Functional studies revealed that enforced expression of miR-33a inhibits the motility of lung cancer cells, and this inhibition can be rescued by overexpression of the form of HMGA2 without the 3′-UTR, suggesting that TTF-1 keeps the prometastasis gene HMGA2 in check via up-regulating miR-33a. This study reports the first miRNAs directly regulated by TTF-1 and clarifies how TTF-1 controls HMGA2 expression. Moreover, the documented importance of SREBF2 and miR-33a in regulating cholesterol metabolism suggests that TTF-1 may be a modulator of cholesterol homeostasis in the lung. Future studies will be dedicated to understanding how miRNAs influence the oncogenic activity of TTF-1 and the role of TTF-1 in cholesterol metabolism.
Introduction
MicroRNAs (miRNAs)2 are small RNAs that do not encode proteins (1–4). The biological consequences of microRNA expression perturbation are manifested by a wide range of cellular or organismal phenotypes, including cancers (2). In the lung, miRNAs play critical roles in both development and tumorigenesis (5–8). Over 100 miRNAs are dynamically regulated during organogenesis of a normal murine lung (9). Selected examples of miRNAs influencing lung development include miR-17∼92, as evidenced by the lung developmental defect in the knock-out mice (10), and miR-302/367, which regulates lung endoderm (11). The better characterized let-7 miRNA family also shows decreased expression in advanced lung cancers (12). Let-7s target a number of oncogenes, including HMGA2 (13–15). The let-7-directed repression of HMGA2 is robust, probably due to the fact that the 3-kb HMGA2 3′-UTR contains seven let-7 binding sites. Human tumor-associated gene translocation of HMGA2 eliminates its 3′-UTR, thus liberating HMGA2 from the let-7-directed repression (13, 14).
Despite advancements in our understanding of the miRNA biology in lung cancer (16), the extent of the interconnection between miRNA-based networks and critical lung cancer genes remains poorly characterized. In this regard, we focus on a master regulator of the lung developmental transcription program termed thyroid transcription factor 1 (TTF-1 or NKX2–1). In addition to being indispensable to fetal lung organogenesis and morphogenesis (17), TTF-1 also contributes to adult lung tumorigenesis based on the genetic evidence that TTF-1 is part of a recurrent multigenic amplicon in lung cancers (18–21). Subsequent studies have identified ROR1 and LMO3 as indispensable downstream mediators of TTF-1 in lung adenocarcinomas (22, 23). Seemingly at odds with the observation that TTF-1 is a lung oncogene, Ttf-1 was also found to prevent primary lung adenocarcinomas from metastasizing in a mouse model system (24). Moreover, a loss of the Ttf-1 allele cooperates with oncogenic KrasG12D, causing pulmonary tumors in transgenic mice that were phenotypically similar to human mucinous adenocarcinomas (25, 26). In view of the multifaceted activities of TTF-1 in lung biology, we believe that mapping the connections between the miRNA network and the TTF-1-directed transcriptional program would provide novel entry points to investigate the lung biology orchestrated by TTF-1. To this end, we have recently reported the discovery of the first miRNA (i.e. miR-365) that directly regulates TTF-1 expression via binding to the TTF-1 3′-UTR (27). In this study, we concentrate on searching for the miRNAs acting downstream to TTF-1 and have uncovered multiple microRNAs that are directly regulated by TTF-1. One such miRNA, miR-33a, was chosen for a comprehensive characterization in view of the fact that it scored in both reverse and forward screens. The results unambiguously place miR-33a under the positive transcriptional control of TTF-1. Moreover, we discovered that the HMGA2 oncogene, known to be repressed by TTF-1 (24), is a direct target repressed by miR-33a. Loss- and gain-of-function analyses validate miR-33a as a mediator of the HMGA2 repression by TTF-1 (TTF-1 → miR-33a ⊣ HMGA2). In light of our observations, we believe that TTF-1 utilizes miR-33a as a means to abate HMGA2 expression. Considering the known activities of miR-33a outside of cancer biology (e.g. cholesterol metabolism (28)), the results of this study are expected to carve out novel directions for future research on TTF-1-orchestrated lung biology.
EXPERIMENTAL PROCEDURES
Cell Culture and Expression Vectors
The human lung cancer cell lines NCI-H358, NCI-H441, A549, NCI-H1299, and BEAS-2B were acquired from the American Type Culture Collection (ATCC), and maintained as described previously (29). Mouse 394T4-bc37 (shLuc) and 394T4-E1 (shTtf-1) cells were provided by Dr. Monte Winslow (24) and maintained in DMEM supplemented with 10% fetal bovine serum, penicillin, and streptomycin. The pGL4.10 SPB promoter reporter construct and pcDNA3.1 TTF-1 and TTF-1 homeodomain deletion mutant expression vectors were constructed previously (29). The SREBF2 (−998 to −3, relative to the transcription start site) and C9ORF5 (−1000 to −5) promoters were PCR-amplified from human genomic DNA using primers listed in Table 1 and cloned into the promoterless luciferase vector pGL4.10 Basic (Promega). Deletion mutants of miR-33a binding sites were derived from a psiCHECK2 vector containing the 3′-UTR of HMGA2 fused to the 3′-end of a Renilla luciferase gene, kindly provided by Dr. Marcus Peter (30). Mutation constructs were created using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol; the primers are listed in Table 1. The human HMGA2 cDNA expression plasmid was obtained from Dr. Jian-Jun Wei (31), and the murine Hmga2 cDNA was from Addgene (Cambridge, MA).
TABLE 1.
List of PCR primers used in this study
| Name | Direction | Sequence |
|---|---|---|
| Mouse RT-QPCR primers | ||
| Gapdh | Forward | 5′-GCTTGTCATCAACGGGAAGC-3′ |
| Reverse | 5′-TTTGATGTTAGTGGGGTCTCGC-3′ | |
| Ttf-1 | Forward | 5′-AAAACTGCGGGGATCTGAG-3′ |
| Reverse | 5′-TGCTTTGGACTCATCGACAT-3′ | |
| Srebf1 | Forward | 5′-CCAGAGGGTGAGCCTGACA-3′ |
| Reverse | 5′-AGCCTCTGCAATTTCCAGATCT-3′ | |
| Srebf2 | Forward | 5′-TGTGAACCTGGCCGAGTGT-3′ |
| Reverse | 5′-CGCTGTCAGGTGGATCTCAA-3′ | |
| Hmga2 | Forward | 5′-GGGCGCCGACATTCAAT-3′ |
| Reverse | 5′-ACTGCAGTGTCTTCTCCCTTCAA-3′ | |
| Human RT-QPCR primers | ||
| GAPDH | Forward | 5′-GGAGTCAACGGATTTGGTCGTA-3′ |
| Reverse | 5′-GGCAACAATATCCACTTTACCAGAGT-3′ | |
| TTF1 | Forward | 5′-CGTTCTCAGTGTCTGACATCTTGA-3′ |
| Reverse | 5′-CCTCCATGCCCACTTTCTTG-3′ | |
| SREBF1 | Forward | 5′-GCTCCTCCATCAATGACAAAATC-3′ |
| Reverse | 5′-TGCAGAAAGCGAATGTAGTCGA-3′ | |
| SREBF2 | Forward | 5′-CGAATTGAAAGACCTGGTCATG-3′ |
| Reverse | 5′-TCCTCAGAACGCCAGACTTGT-3′ | |
| HMGA2 | Forward | 5′-CCCAAAGGCAGCAAAAACAA-3′ |
| Reverse | 5′-GCCTCTTGGCCGTTTTTCTC-3′ | |
| ChIP QPCR primers | ||
| C9Orf5 (distal) | Forward | 5′-TGTCTGCCTTAAAAGCTTGTGTTC-3′ |
| Reverse | 5′-TTTGCTCCAGTACTTTGCACACTT-3′ | |
| C9Orf5 (proximal) | Forward | 5′-GGTATTCTAGTATGCAGCTTGGGTTT-3′ |
| Reverse | 5′-GAAGCTCCTTTCACTCATTCGTTAA-3′ | |
| SREBF2 (distal) | Forward | 5′-GAAGCGTGAATTGGCCTTATG-3′ |
| Reverse | 5′-TCTGACCCTTTCCCGTTGAT-3′ | |
| SREBF2 (proximal) | Forward | 5′-GCAAAGCAGAAGACGTAAAATCC-3′ |
| Reverse | 5′-CACGGAGGCTTGACAAGGTT-3′ | |
| SPB (distal) | Forward | 5′-CCGAAGGCTGTAGGGAAGAA-3′ |
| Reverse | 5′-GCTCAGGAACACTAGGGATTGC-3′ | |
| SPB (proximal) | Forward | 5′-TCCACCCACAAAAGCACTGTAG-3′ |
| Reverse | 5′-TCTCCCTAGTGGAGGCTCACA-3′ | |
| HMGA2 3′-UTR psiCHECK2 site-directed mutagenesis primersa | ||
| Δ1 | Forward | 5′-CAGGGGACACAGCTTAAGGTACCCTTTTAATTACTG-3′ |
| Reverse | 5′-CAGTAATTAAAAGGGTACCTTAAGCTGTGTCCCCTG-3′ | |
| Δ2 | Forward | 5′-GCTGCTATACACAAGGGTACCGAAAAAAACTTACTG-3′ |
| Reverse | 5′-CAGTAAGTTTTTTTCGGTACCCTTGTGTATAGCAGC-3′ | |
| Δ3 | Forward | 5′-GCCCTGCTTTTGCATGGTACCTCAAAAATATGTG-3′ |
| Reverse | 5′-CACATATTTTTGAGGTACCATGCAAAAGCAGGGC-3′ | |
a The KpnI cut site is underlined.
RNA Oligonucleotide Reagents and Transfection
All siRNAs, miRNA mimics, and inhibitors were purchased from Dharmacon. Cells were transiently transfected with plasmid DNA, siRNA (non-targeting negative control, D-001210-01; TTF-1 A/B/C, D019105–03/04/17), antisense oligonucleotide miRNA inhibitors (non-targeting negative control, IN-001001–01; hsa-miR-33a inhibitor, IH-300509–08), or miRNA mimics (non-targeting control, CN-001005–01; hsa-miR-33a (C-300509–07) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Transfection efficiency was monitored with pMAXGFP plasmid or BLOCK-iT fluorescently labeled negative control oligonucleotides (Invitrogen) and judged to be >90% for each cell line.
RNA Isolation and Reverse Transcription (RT)-Quantitative Real-time PCR (QPCR) Analysis
Total RNA (miRNA and mRNA) was isolated from cells using TRIzol (Invitrogen). For mRNA quantification, RNA was reverse transcribed using the High Capacity cDNA synthesis kit (Applied Biosystems). The resultant cDNA was evaluated by real-time PCR using the primers listed in Table 1 and Brilliant II SYBR Green master mix (Stratagene) with a StepOne Plus real-time PCR system (Applied Biosystems). MicroRNAs were quantified using miRCURYTM (Exiqon) or qScript (Quanta Biosciences) miRNA cDNA synthesis kits followed by real-time PCR analysis using locked nucleic acid (LNA) miRNA-specific PCR primers and miRCURYTM SYBR Green master mix (Exiqon) or PerfeCta miRNA assays and PerfeCta SYBR Green master mix (Quanta Biosciences), respectively.
MicroRNA Array Profiling
Total RNA was harvested for profiling using the Cell and Plant miRCURYTM RNA isolation kit (Exiqon). The quality of the total RNA was verified by an Agilent 2100 bioanalyzer profile. Total RNA (700 ng) from sample and reference was labeled with Hy3TM and Hy5TM fluorescent label, respectively, using the miRCURYTM LNA Array power labeling kit (Exiqon) following the procedure described by the manufacturer. The Hy3TM-labeled samples and a Hy5TM-labeled reference RNA sample were mixed pairwise and hybridized to the miRCURYTM LNA Array version 5th Generation (Exiqon), which contains capture probes targeting all miRNAs for humans, mice, or rats registered in miRBASE version 16.0 at the Sanger Institute. The hybridization was performed according to the miRCURYTM LNA array manual using a Tecan HS4800 hybridization station. After hybridization, the microarray slides were scanned and stored in an ozone-free environment (ozone level below 2.0 ppb) in order to prevent potential bleaching of the fluorescent dyes. The miRCURYTM LNA array microarray slides were scanned using the Agilent G2565BA microarray scanner system (Agilent Technologies), and the image analysis was carried out using ImaGene version 9.0 software (BioDiscovery). The quantified signals were background-corrected (Normexp with offset value 10 (32)) and normalized using quantile normalization.
Western Blotting
Proteins were harvested from cells as described previously (29). Cell lysates (10–20 μg) were fractionated by SDS-PAGE and electrophoretically transferred to nitrocellulose membranes. Membranes were blocked in nonfat dry milk (5%) and incubated overnight with primary antibody against TTF-1 (clone 8G7G3/1, Santa Cruz Biotechnology, Inc. (Santa Cruz, CA)), HMGA2 (Biocheck), GAPDH (Cell Signaling), or HSP90 (BD Transduction Laboratories). Proteins were detected with appropriate HRP-conjugated secondary antibody (Thermo Scientific) and chemiluminescent substrates.
Luciferase Reporter Assays
Promoter reporter assays were carried out in 96-well plates as described previously (29). Briefly, cells were co-transfected with a firefly luciferase reporter construct and a Renilla luciferase control vector pGL4.73 (Promega). Twenty-four hours after transfection, firefly and Renilla luciferase values were quantified using Dual-Glo luciferase assay (Promega) on a GloMax-96 plate reader (Promega). Firefly luciferase values were normalized to Renilla luciferase values and expressed as relative values. For 3′-UTR-based reporter studies, cells were seeded onto 24-well plates and co-transfected with psiCHECK2 reporter constructs (300 ng) and RNA oligonucleotides on the following day. Firefly and Renilla luciferase values were measured 48 h after transfection, and Renilla luciferase signals were normalized to firefly luciferase signals.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed on NCI-H441 cells as described previously (29), using either a TTF-1 antibody (H190) or a normal rabbit IgG (Santa Cruz Biotechnology, Inc.). Target sequences were detected by QPCR using Brilliant II SYBR Green master mix (Stratagene). QPCR signals of the immunoprecipitated chromatins were normalized to the signal from the total lysate (input) for each primer pair. Primers used for ChIP analyses are listed in Table 1.
Transwell Migration and Invasion Assays
For Boyden chamber migration assays, 5 × 104 transfected cells were seeded in triplicate onto upper chamber of a migration insert with 8-μm pore size (catalog no. 354578, BD Biosciences) in serum-free media; media supplemented with 5% fetal bovine serum, as a chemoattractant, was added to the lower well. After 24 h, cells from the top of the chamber membrane were removed, and the remaining cells on the bottom of the membrane were fixed with methanol and stained with hematoxylin. Average nuclei were determined in five ×100 fields using a Nikon Eclipse microscope and NIS Elements D software (Nikon), with nuclei counted blind and manually. Invasion assays were performed in the same manner as the migration assay but utilized Matrigel-coated inserts with 8-μm pore size (catalog no. 354480, BD Biosciences).
Statistical Analysis
GraphPad Prism version 5 software was used to perform statistical analyses, including Student's t test, when comparing two groups (control and experimental). One-way analysis of variance with Tukey's post-test was used to compare more than two groups, and a two-way analysis of variance test was used to analyze time course experiments. Representative experiments repeated at least twice are shown as mean ± S.D. Data were considered statistically significant when p was <0.05 (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
RESULTS
TTF-1 Represses HMGA2 in Human Lung Cancer Cells
Winslow et al. (24) reported a surprising finding, that Ttf-1 suppresses metastasis formation of primary lung adenocarcinomas driven by mutant Kras and p53 loss (KrasG12D/+;p53Δ/Δ) in a mouse model system. By gene expression profiling and functional analyses, it was determined that a pro-EMT oncogene, Hmga2, is a downstream target of Ttf-1 and that repression of Hmga2 expression by Ttf-1 is a basis to the antimetastatic activity of Ttf-1 (24). To test if the Ttf-1/Hmga2 relationship is conserved in human lung cancer cells, we used two independent small interfering RNAs (siRNAs) against TTF-1 to knock down the endogenous expression of TTF-1 in a human lung adenocarcinoma cell line with endogenous HMGA2 protein expression (NCI-H358). Interestingly, both siTTF-1s led to a greater than 2-fold derepression in HMGA2 protein expression (Fig. 1A). This observation suggests that the Ttf-1 ⊣ Hmga2 axis, originally discovered in the murine lung adenocarcinoma cells derived from tumors initiated by somatic activation of oncogenic Kras and p53 deletion (24), holds true in human lung cancer cells as well.
FIGURE 1.

TTF-1 negatively regulates HMGA2 in NCI-H358 cells. A, NCI-H358 cells were transfected with non-targeting RNA oligonucleotide (negative control; NC) or two separate siRNAs targeting TTF-1 (siTTF-1 A and B), and proteins were harvested after 72 h. Protein levels of TTF-1, HMGA2, and GAPDH (loading control) were determined by immunoblotting. B, NCI-H358 cells were transfected with a negative control or siTTF-1-targeting siRNAs, along with a psiCHECK2 vector containing the HMGA2 3′-UTR fused to the 3′-end of a Renilla luciferase gene. Luciferase assays were performed after 48 h (n = 3). RLU, relative luminescence units. Error bars, S.D. ***, p < 0.001.
Although it is known that HMGA2 is subject to regulation by the let-7 family of miRNAs (33), let-7s do not appear to play a role in mediating the repression of Hmga2 by Ttf-1 in murine lung cancer cells (24). Thus, we hypothesized that there are other miRNAs mediating the Ttf-1/Hmga2 regulation. To this end, we utilized a reporter plasmid where the 3′-UTR of human HMGA2 was fused to the 3′-end of the Renilla luciferase gene in the psiCHECK2 vector backbone (30). This reporter plasmid was transfected into the NCI-H358 human lung cancer cell line in which the endogenous TTF-1 expression was knocked down using two independent siTTF-1s. The results demonstrate that a decrease of TTF-1 expression leads to a concomitant increase in the luciferase activity of Luc-HMGA2/3′-UTR (Fig. 1B). This derepression of HMGA2 3′-UTR upon TTF-1 knockdown implicates the involvement of HMGA2 3′-UTR in the TTF-1-induced silencing of HMGA2.
Discovery of miRNAs That Mediate the Ttf-1 ⊣ Hmga2 Relationship in Murine Lung Cancer Cells
Our working hypothesis was that TTF-1 may directly up-regulate certain miRNAs, which in turn repress HMGA2 expression via direct binding to the 3′-UTR of HMGA2. To identify these putative miRNA regulators, we utilized the non-metastatic (TnonMet) murine primary lung tumor cells (394T4) generated and modified by Winslow et al. (24) to stably express a small hairpin RNA (shRNA) against Ttf-1 (394T4-shTtf-1). The Ttf-1 knockdown and the corresponding Hmga2 up-regulation in 394T4-shTtf-1 cells elicited a higher metastatic phenotype, whereas the control cells, 394T4-shLuc carrying an shRNA against luciferase (shLuc), were non-metastatic and thus similar to the parental 394T4 cells (24). We reasoned that the putative miRNA(s) responsible for suppressing Hmga2 in a Ttf-1-dependent manner in the 394T4-shLuc (Ttf-1high) cells would be down-regulated in 394T4-shTtf-1 (Ttf-1low) cells due to Ttf-1 knockdown. To identify these miRNA regulators that are present in both mice and humans, we created a custom QPCR array that contains 44 LNA-based QPCR probes in duplicates (Exiqon). Each of the LNA probes detects an miRNA predicted by TargetScan version 5.2 (34) to bind to human HMGA2 3′-UTR (Table 2). Approximately 57% (i.e. 25) of the 44 human miRNA probes are able to detect the mouse counterparts based on sequence conservation. This QPCR array was employed to quantify the murine miRNA expression differences between 394T4-shLuc (Ttf-1high) and 394T4-shTtf-1 (Ttf-1low) cells. Twelve murine miRNAs were scored in the QPCR array assay, and the results are presented as ratios of miRNA expression levels (shLuc/shTtf-1; Fig. 2A). Interestingly, two human miRNA probes (hsa-miR-33a and hsa-miR-495) detected high expression ratios, suggesting that the two corresponding murine miRNAs may be positively regulated by Ttf-1. Indeed, secondary confirmational studies utilizing an independent miRNA detection system (Quanta Biosciences) validated the QPCR array data for mmu-miR-33 (i.e. Ttf-1 knockdown repressing mmu-miR-33 expression) (Fig. 2B). (Note that mmu-miR-33 is the mouse homolog of human hsa-miR-33a. The two miRNAs are 100% identical in sequence. Hereafter, hsa-miR-33a and mmu-miR-33 are referred to as miR-33a and miR-33, respectively.) We did not pursue the second positive hit of the QPCR array screen (mmu-miR-495) because this miRNA, unlike miR-33, was not predicted to target the mouse Hmga2 3′-UTR by TargetScan; therefore, mmu-miR-495 is not likely to be involved in the Ttf-1-mediated repression of Hmga2 in the murine lung cancer cells.
TABLE 2.
Probe list of the custom HMGA2-3′-UTR-targeting QPCR array
| miRNA | Comment | |
|---|---|---|
| 1 | hsa-let-7b | Included in analysis |
| 2 | hsa-let-7c | Included in analysis |
| 3 | hsa-let-7d | Included in analysis |
| 4 | hsa-let-7e | Included in analysis |
| 5 | hsa-let-7f | Included in analysis |
| 6 | hsa-let-7i | Included in analysis |
| 7 | hsa-miR-186 | Included in analysis |
| 8 | hsa-miR-26b | Included in analysis |
| 9 | hsa-miR-33a | Included in analysis |
| 10 | hsa-miR-495 | Included in analysis |
| 11 | hsa-miR-532-3p | Included in analysis |
| 12 | hsa-miR-9 | Included in analysis |
| 13 | hsa-miR-137 | Not detected |
| 14 | hsa-miR-196a | Not detected |
| 15 | hsa-miR-204 | Not detected |
| 16 | hsa-miR-361-5p | Not detected |
| 17 | hsa-miR-370 | Not detected |
| 18 | hsa-miR-539 | Not detected |
| 19 | hsa-let-7a | Excluded, replicate Ct >1.5 cycles |
| 20 | hsa-let-7g | Excluded, replicate Ct >1.5 cycles |
| 21 | hsa-miR-129-5p | Excluded, replicate Ct >1.5 cycles |
| 22 | hsa-miR-196b | Excluded, replicate Ct >1.5 cycles |
| 23 | hsa-miR-26a | Excluded, replicate Ct >1.5 cycles |
| 24 | hsa-miR-760 | Excluded, replicate Ct >1.5 cycles |
| 25 | hsa-miR-98 | Excluded, replicate Ct >1.5 cycles |
| 26 | hsa-miR-202 | Excluded, human-specific miRNA |
| 27 | hsa-miR-211 | Excluded, human-specific miRNA |
| 28 | hsa-miR-337-3p | Excluded, human-specific miRNA |
| 29 | hsa-miR-33b | Excluded, human-specific miRNA |
| 30 | hsa-miR-376c | Excluded, human-specific miRNA |
| 31 | hsa-miR-450b-5p | Excluded, human-specific miRNA |
| 32 | hsa-miR-509-3-5p | Excluded, human-specific miRNA |
| 33 | hsa-miR-520d-5p | Excluded, human-specific miRNA |
| 34 | hsa-miR-522 | Excluded, human-specific miRNA |
| 35 | hsa-miR-524-5p | Excluded, human-specific miRNA |
| 36 | hsa-miR-548c-3p | Excluded, human-specific miRNA |
| 37 | hsa-miR-554 | Excluded, human-specific miRNA |
| 38 | hsa-miR-556-5p | Excluded, human-specific miRNA |
| 39 | hsa-miR-570 | Excluded, human-specific miRNA |
| 40 | hsa-miR-573 | Excluded, human-specific miRNA |
| 41 | hsa-miR-578 | Excluded, human-specific miRNA |
| 42 | hsa-miR-579 | Excluded, human-specific miRNA |
| 43 | hsa-miR-588 | Excluded, human-specific miRNA |
| 44 | hsa-miR-608 | Excluded, human-specific miRNA |
FIGURE 2.
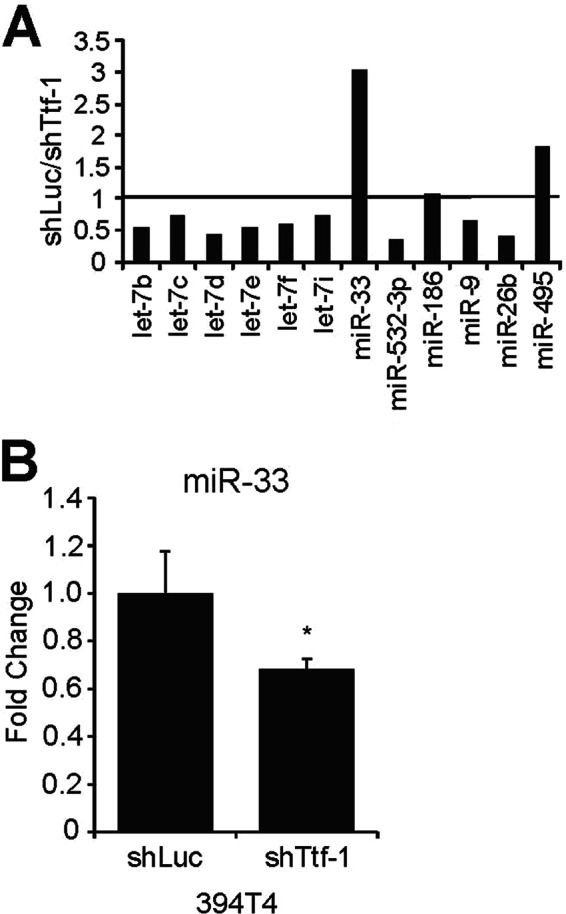
Screening murine lung cancer cells for Ttf-1-regulated miRNAs that target HMGA2 using an LNA-QPCR array. A, total RNA isolated from 394T4-shLuc (Ttf-1high) and 394T4-shTtf-1 (Ttf-1low) cells was quantified in duplicate using an LNA-QPCR array designed to include miRNAs predicted to target the 3′-UTR of human HMGA2 gene. Normalized expression values between the two cell lines are expressed as -fold change of shLuc over shTtf-1 cells. B, down-regulation of mmu-miR-33 in 394T4-shTtf-1 cells was confirmed by RT-QPCR using an miRNA detection system and PCR primers (Quanta Biosciences) (n = 3). Error bars, S.D. *, p < 0.05.
Searching for TTF-1-regulated miRNAs in a Human Lung Epithelial System with Inducible TTF-1 Expression
To substantiate the candidacy of miR-33a as a TTF-1-regulated miRNA and to explore TTF-1-regulated miRNAs globally in human cells, we took on a gain-of-function strategy using a human lung epithelial cell system in which the expression of a human TTF-1 transgene could be turned on by doxycycline (i.e. a doxycycline (dox)-on system) (29). The host cells of this inducible system are the premalignant, viral oncogene-immortalized human lung epithelial cells, BEAS-2B (35). We chose this cell strain due to the fact that it lacks endogenous TTF-1 expression (36), thus maximizing the miRNA expression perturbation in response to the dox-induced expression of the TTF-1 transgene. As shown by immunoblotting, the expression of TTF-1 protein was turned on following a 24-h dox treatment (Fig. 3A), with little leaky expression of the TTF-1 transgene without dox in the culture media. Concomitantly, HMGA2 mRNA decreased by 40% upon dox induction (data not shown). For global miRNA profiling, we collected total RNA from these four samples in duplicates: (i) BEAS-2B-rtTA3-TTF-1 in the presence of dox (TTF-1 + Dox), (ii) BEAS-2B-rtTA3-TTF-1 in the absence of dox (TTF-1 − Dox), (iii) BEAS-2B-rtTA3-empty vector in the presence of dox (EV + Dox), and (iv) BEAS-2B-rtTA3-empty vector in the absence of dox (EV − Dox). The TTF-1 transgene was turned on only under the (i) TTF-1 + Dox condition. These RNAs were analyzed for global miRNA expression using a commercial microarray platform (Exiqon miRCURYTM LNA microRNA Array 5th Generation; the number of miRNA species the array targeted was as follows: 1250 human, 1115 mouse, and 692 rat). Two outliers, hsa-miR-33a and hsa-miR-32, were up-regulated in the TTF-1 + Dox group relative to either the TTF-1 − Dox or EV + Dox group (Fig. 3B; see supplemental Table S1 for the complete list of differentially expressed miRNAs), suggesting that the observed expression changes were dependent on TTF-1 expression but independent of dox treatment. For validation, LNA-based probes to miR-33a and miR-32 were used in RT-QPCR to confirm the array profiling data. The results, corroborating the array profiling observations, indicate a 1.7–2-fold increase of miR-33a and miR-32 upon TTF-1 induction (Fig. 3C).
FIGURE 3.

A global screen for TTF-1-regulated miRNAs using a microarray platform. A, immortalized human lung epithelial cells with Dox-inducible TTF-1 elements (BEAS-2B-rtTA3-TTF1) were treated with or without dox (0.8 μg/ml) for 24 h prior to protein harvest. Western blot analysis confirmed the induction of TTF-1, with HSP90 protein as the loading control. B, RNA isolated from BEAS-2B-rtTA3-EV or TTF-1 cells treated with or without dox (0.8 μg/ml) for 24 h was evaluated in duplicates for changes in miRNA expression using an LNA-miRNA microarray (Exiqon). Expression of miRNAs between the different groups was compared by log median scores (LMS), and the average miRNA expression changes are graphed as indicated. Lines on the graph indicate the 1st and 99th percentile of the data. C, up-regulation of miR-32 and miR-33a in BEAS-2B-rtTA3-TTF-1 cells was confirmed using RT-QPCR (n = 3). Error bars, S.D. *, p < 0.05; **, p < 0.01.
Identification of miR-33a and miR-32 as Direct Transcriptional Targets of TTF-1
In view of the biochemical property of TTF-1 as a transcription factor (17, 37, 38), we surmised that TTF-1 would activate the promoter of the respective genes hosting miR-33a and miR-32. To approach this issue, we cloned the promoter region (∼1 kb) of SREBF2 (NM_004599, the host gene of hsa-miR-33a) and C9ORF5 (NM_032012, the host gene of hsa-miR-32) into the 5′-end of a promoterless luciferase reporter vector (Fig. 4A). The luciferase activity readout of the resultant reporter plasmids was then used to assess the responsiveness of each promoter to TTF-1 or a transcriptionally inactive mutant of TTF-1 lacking the homeodomain (the homeodomain deletion mutant (29)) in A549 cells, a human lung cancer cell line commonly used to study transcriptional activity of exogenous TTF-1 (39). The data in Fig. 4B show that both promoters respond to TTF-1 transactivation, whereas the homeodomain deletion mutant of TTF-1 failed to activate the promoters, consistent with the notion that the homeodomain-dependent DNA binding activity of TTF-1 is essential for this function (40). The positive luciferase data are in line with the thesis that SREBF2 is under TTF-1 transcriptional control. To further test this relationship, we used two siTTF-1s to knock down endogenous TTF-1 in the NCI-H441 human lung adenocarcinoma cell line, which has high endogenous TTF-1 expression (29). RT-QPCR analyses show that TTF-1 knockdown induced a concomitant 40–50% decrease in SREBF2 RNA (Fig. 4C).
FIGURE 4.

TTF-1 binds to and activates transcription from the promoters of the miR-32 and miR-33a host genes. A, diagram of the promoter regions of the miR-32 and miR-33a host genes, C9Orf5 and SREBF2, respectively. Locations for negative control (NC) and promoter PCR primer pairs used in the ChIP analyses are shown relative to the respective gene's predicted transcriptional start site (TSS). B, A549 cells were transfected with the indicated expression vector (empty vector (EV), TTF-1, or a TTF-1 homeodomain deletion mutant, HDD) and a luciferase promoter reporter construct for C9Orf5 (miR-32) or SREBF2 (miR-33a). Luciferase assays were performed 24 h after transfection (n = 4). y axis, -fold change in relative luminescence units (RLU). C, TTF-1 knockdown resulted in a decrease of SREBF2 RNA. NCI-H441 cells were transfected with mock or a negative control oligonucleotide or an individual siTTF-1. After 48 h, RT-QPCR was conducted to quantify the expression level of SREBF2 RNA (n = 3). SiTTF-1B and SiTTF-1C were from Dharmacon/Thermo Scientific (catalog nos. D019105-04 and D019105-17, respectively). D, chromatin immunoprecipitation of endogenous TTF-1 in NCI-H441 cells. Sheared chromatins were precipitated with either rabbit immunoglobulin (Rb IgG) or anti-TTF-1 antibody and subsequently analyzed using QPCR location probes shown in A (n = 3). Error bars, S.D. ***, p < 0.001.
To determine whether TTF-1 directly bound to the promoters of the respective host genes of miR-33a and miR-32, we conducted ChIP using an anti-TTF-1 antibody. The immunoprecipitated chromatins from NCI-H441 cells were analyzed using two genomic location probes: a proximal probe at <1 kb and a distal probe at 5 kb upstream to the transcription start site (Fig. 4A, TSS). In both cases, the proximal QPCR probes detected quantitative recovery of promoter DNA sequences for both host genes in the TTF-1-precipitated chromatins immunoprecipitated (Fig. 4D), implying a direct interaction between the promoter region of both host genes with TTF-1. In view of the positive results for miR-33a in both loss- and gain-of-function analyses using mouse and human cells, miR-33a stood out as a strong candidate miRNA under direct TTF-1 control.
TTF-1-induced Down-regulation of HMGA2 Is Mediated by miR-33a
Both human HMGA2 and mouse Hmga2 were ranked by TargetScan (34) as potential target genes of hsa-miR-33a and mmu-miR-33, respectively (Table 3). In addition, there are three putative binding sites for miR-33a and one binding site for miR-32 in the HMGA2 3′-UTR (Fig. 5A). In view of these data, we initiated studies to validate HMGA2 as an authentic target gene of miR-33a and miR-32. An miR-33a mimetic oligonucleotide or an miRNA known to target HMGA2 (let-7d) was cotransfected with a luciferase reporter of HMGA2 3′-UTR into a human lung cancer cell line, NCI-H1299. We chose NCI-H1299 because the endogenous HMGA2 expression in NCI-H1299 is responsive to let-7 regulation (13), implicating the existence of a functional miRNA-dependent surveillance of HMGA2 in NCI-H1299 cells. Chemiluminescence measurement revealed a 50% reduction of reporter activity in the miR-33a transfectants (Fig. 5B); the positive control let-7d induced a stronger inhibition of HMGA2 3′-UTR reporter (∼80%), which is probably due to the fact that there are seven let-7 binding sites in the 3′-UTR of HMGA2 (Fig. 5A). Interestingly, although the miR-32 mimic elicited a slight response in the reporter assay, this response appeared to be independent of the predicted miR-32 site (Fig. 5C). Thus, we conclude that the predicted binding site of miR-32 in the 3′-UTR of HMGA2 may be nonfunctional. We then proceeded to measure the endogenous RNA and protein expression of HMGA2 following miR-33a or let-7d transfection in NCI-H1299 cells (Fig. 5, D and E). In addition, we also measured the HMGA2 protein level following miR-33a transfection in the murine lung cancer cells (394T4-shTtf-1; Fig. 5F). Overall the results corroborate the notion that HMGA2 is a target gene of miR-33a.
TABLE 3.
Top five predicted targets for hsa-miR-33a and mmu-miR-33 by TargetScan 5.2 and 6.2
| Rank | Target gene | Gene name | Total context score |
|---|---|---|---|
| hsa-miR-33a TargetScan 5.2 | |||
| 1 | ABCA1 | ATP-binding cassette, subfamilyA (ABC1), member 1 | −1.25 |
| 2 | CROT | Carnitine O-octanoyltransferase | −0.84 |
| 3 | CDK6 | Cyclin-dependent kinase 6 | −0.8 |
| 4 | HMGA2 | High mobility group AT-hook 2 | −0.75 |
| 5 | LIMCH1 | LIM and calponin homology domains 1 | −0.71 |
| mmu-miR-33 TargetScan 5.2 | |||
| 1 | Abca1 | ATP-binding cassette, subfamilyA (ABC1), member 1 | −0.86 |
| 2 | Hmga2 | High mobility group AT-hook 2 | −0.83 |
| 3 | Slc12a5 | Solute carrier family 12, member 5 | −0.74 |
| 4 | Zfp281 | Zinc finger protein 281 | −0.72 |
| 5 | Rgs7bp | Regulator of G-protein signaling 7-binding protein | −0.67 |
| hsa-miR-33a TargetScan 6.2 | |||
| 1 | ABCA1 | ATP-binding cassette, subfamilyA (ABC1), member 1 | −0.88 |
| 2 | CROT | Carnitine O-octanoyltransferase | −0.65 |
| 3 | CCNYL1 | Cyclin Y-like 1 | −0.54 |
| 4 | PCDH18 | Protocadherin 18 | −0.5 |
| 5 | AKAP2 | A kinase (PRKA) anchor protein 2 | −0.48 |
| 32 | HMGA2 | High mobility group AT-hook 2 | −0.36 |
| mmu-miR-33 TargetScan 6.2 | |||
| 1 | Abca1 | ATP-binding cassette, subfamilyA (ABC1), member 1 | −0.72 |
| 2 | Slc12aA5 | Solute carrier family 12 (potassium/chloride transporter), member 5 | −0.65 |
| 3 | Hmga2 | High mobility group AT-hook 2 | −0.52 |
| 4 | Znf281 | Zinc finger protein 281 | −0.52 |
| 5 | Dcun1d5 | DCN1, defective in cullin neddylation 1, domain-containing 5 | −0.47 |
FIGURE 5.

HMGA2 is a novel target of miR-33a in mouse and human lung cancer cells. A, diagram depicting the 3′-UTR of the human HMGA2 gene. Locations of predicted miRNA binding sites for let-7 (black), miR-33a (gray), and miR-32 (white) are marked with arrows, and seed site locations are listed below the arrows. B, NCI-H1299 cells were transfected with the HMGA2 3′-UTR reporter and either a scrambled control oligonucleotide (Scr) or an miR-33a mimic (miR-33a). Luciferase activity was read 48 h post-transfection (n = 3). C, HMGA2 3′-UTR is not a target for miR-32. NCI-H1299 cells were cotransfected with an miR-32 mimic or a negative control oligonucleotide (NC). At the same time, the transfections include a wild-type HMGA2 3′-UTR reporter construct (Wt) or an HMGA2 3′-UTR reporter with a deleted miR-32 binding site (ΔmiR-32). After 48 h, Renilla and firefly luciferase activities were assayed. Although miR-32 resulted in a slight inhibition of the HMGA2 reporter, deletion of the sole predicted miR-32 binding site did not cause a derepression of the HMGA2 reporter. RLU, relative luminescence units. D, quantification of endogenous HMGA2 mRNA after transfection of NCI-H1299 cells with a scrambled control oligonucleotide or an miR-33a mimic. HGMA2 expression levels were normalized to GAPDH (n = 3). E, Western blot analysis confirmed a reduced expression of HMGA2 in the human NCI-H1299 cells transfected with miR-33a mimic. F, the Hmga2 expression was knocked down in the murine 394T4-shTtf-1 cells transfected with an miR-33a mimic. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To substantiate that the Ttf-1-induced up-regulation of mmu-miR-33 in the 394T4 murine cells was responsible for repressing Hmga2, we first treated 394T4-shLuc (Ttf-1high) cells with an miR-33a miRNA inhibitor (anti-miR-33a, which targets both hsa-miR-33a and mmu-miR-33) and found that the Hmga2 protein level increased by ∼2-fold (Fig. 6A). To extend this observation to human lung cancer cells, we next treated NCI-H358 cells with anti-miR-33a. Inhibition of miR-33a in these cells again resulted in a ∼3-fold increase in HMGA2 protein (Fig. 6B), corroborating the results seen with the murine 394T4-shLuc cells. Finally, we analyzed the impact of anti-miR-33a on the Hmga2 repression imposed by exogenous TTF-1. To this end, we transfected human TTF-1 cDNA into a murine Ttf-1 knockdown background (394T4-shTtf-1 cells). The stably expressed shRNA against the mouse Ttf-1 gene in the 394T4-shTtf-1 cells does not down-regulate the human TTF-1 gene due to the fact the particular shRNA targeting sequence (CGCCATGTCTTGTTCTACCTT) is unique to mouse Ttf-1. The expression of human TTF-1 clearly conferred repression of Hmga2 (Fig. 6C). Importantly, anti-miR-33a abolished the Hmga2 protein repression imposed by the exogenous human TTF-1 (Fig. 6C), proving that miR-33a is a critical mediator of the TTF-1-induced HMGA2 repression.
FIGURE 6.
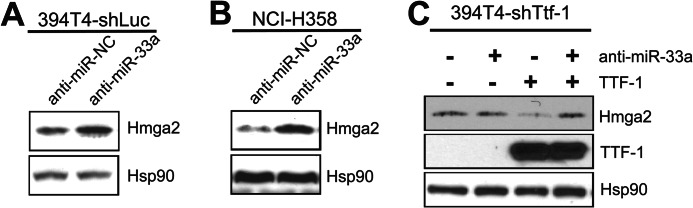
TTF-1 up-regulation of miR-33a suppresses HMGA2 expression in human and mouse lung epithelial cells. A, Western blot analysis of Hmga2, Ttf-1, and Hsp90 expression in murine 394T4-shLuc cells transfected with either a negative control (anti-miR-NC) or an miR-33a inhibitor (anti-miR-33a). B, Western blot analysis of HMGA2, TTF-1, and Hsp90 expression in the human NCI-H358 cells transfected with either a negative control or an miR-33a inhibitor. C, 394T4-shTtf-1 cells were transfected as indicated, and the corresponding cell lysates were analyzed by immunoblotting for the expression of Ttf-1, Hmga2, and Hsp90.
Analysis of miR-33a Binding Sites in the 3′-UTR of HMGA2
To analyze the relative contribution of the three miR-33a binding sites to HMGA2 repression by miR-33a, we mutated the three TargetScan-predicted miR-33a binding sites individually and in all possible pairwise combinations, affording a total of seven mutant reporter plasmids of HMGA2 3′-UTR for analysis (Fig. 7A). The luciferase reporter activities of individual mutant plasmids were evaluated following cotransfection into NCI-H1299 cells along with either an exogenous miR-33a mimetic oligonucleotide or a scrambled control oligonucleotide. Of the three single-site mutants (Δ1, Δ2, and Δ3), Δ1 showed the strongest derepression (Fig. 7B), whereas Δ3 was essentially identical to the wild-type reporter plasmid. This suggests that the relative contribution of the three miR-33a binding sites to repression of HMGA2 3′UTR is in the following order: site 1 > site 2 > site 3. Data obtained with the three double mutants (Δ1/2, Δ1/3, and Δ2/3) generally agreed with the observation gathered from single mutants (Fig. 7B). It is intriguing to note that the derepression of losing both sites 1 and 2 (Δ1/2) appeared to be larger than the sum of the two single mutants (Δ1 + Δ2), indicating potential synergy between sites 1 and 2. Although miRNA binding sites are known to work synergistically in repressing target genes, the two miRNA binding sites would generally have to be close in distance for optimal cooperativity (34, 41). Therefore, in view of the long distance (>1 kb) between miR-33a sites 1 and 2 in the 3′-UTR of HMGA2, it is surprising that these two sites might work cooperatively.
FIGURE 7.

Characterization of the three predicted miR-33a binding sites located within the HMGA2 3′-UTR. A, diagram depicting the HMGA2 3′-UTR reporter construct mutants used to characterize functional miR-33a sites. The miR-33a seed sequences were mutated by replacement with a KpnI restriction enzyme recognition sequence (GGTACC). B, NCI-H1299 cells, transfected with individual HMGA2 3′-UTR reporter constructs from A and an miR-33a mimetic oligonucleotide or a scrambled control oligonucleotide (each at 20 nm), were assayed for luciferase activities 48 h post-transfection. Relative luminescence units (RLU) were normalized to the corresponding scrambled control for each reporter construct (n = 3). Error bars, S.D.
miR-33a Impedes Motility of Human and Murine Lung Cancer Cells
Because Ttf-1 was shown to regulate metastatic dissemination (24), we chose to examine the migratory and invasive properties of human and murine lung cancer cells following modulation of TTF-1/miR-33a/HMGA2 levels. Initially, we compared the migration and invasiveness of the two murine lung cancer cell lines, 394T4-shLuc (Ttf-1high) and 394T4-shTtf-1 (Ttf-1low), using transwell migration (uncoated control inserts) and invasion (Matrigel-coated inserts) assays. Although the motility of the 394T4-shTtf-1 cells was greater than the shLuc cells, there was no difference in invasiveness between the treatment groups (Fig. 8, A and B). Transfection of the human NCI-H1299 cells with an exogenous miR-33a oligonucleotide impeded migration compared with a scrambled control oligonucleotide in the transwell migration assay. However, the invasiveness of cells as determined by the transwell assay was not altered by miR-33a transfection (Fig. 8, C and D). Therefore, we focused the subsequent studies on the relevance of the TTF-1 → miR-33a ⊣ HMGA2 axis to cellular motility. The assumption was that miR-33a could functionally replace TTF-1 in slowing migration due to repression of HMGA2, which promotes cell motility (31). The murine 394T4-shLuc cells (Ttf-1high), transfected with a mmu-miR-33 inhibitor (anti-miR-33a) or a negative control, were evaluated for migratory potential using the transwell assays. The anti-miR-33a conferred an increase in migration by 1.8-fold (Fig. 9A), indicating that derepression of Hmga2 by anti-miR-33a enhances cellular migration. Conversely, enforced overexpression of Hmga2 in the 394T4-shLuc cells increased its motility (Fig. 9B, lane 1 versus lane 2), phenocopying the cellular responses to anti-miR-33a. Transfection of the miR-33a mimic into 394T4-shLuc cells slowed down cellular motility, as shown in Fig. 9B (lane 1 versus lane 3). Importantly, stable overexpression of Hmga2 lacking 3′-UTR overcame the inhibitory effect of miR-33a on cellular migration (Fig. 9B, lane 3 versus lane 4).
FIGURE 8.
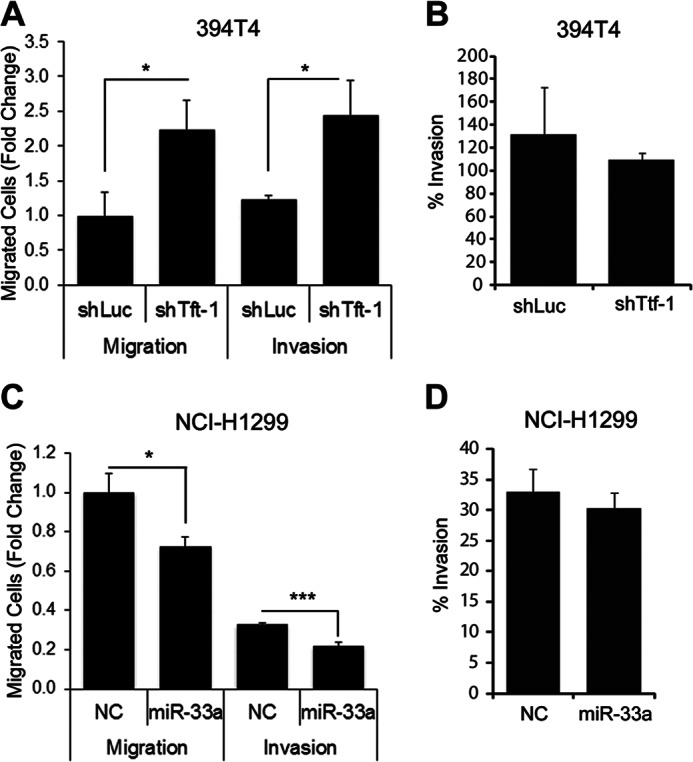
Knockdown of endogenous Ttf-1 and exogenous miR-33a slow migration but do not alter invasiveness. A, murine 394T4-shLuc and shTtf-1 cells were allowed to migrate through a membrane with 8-μm pores (Migration) or a membrane with 8-μm pores coated with Matrigel (Invasion) for 22 h. B, data from A were used to calculate the percentage invasion (% invasion = (mean number of cells invaded through the Matrigel-coated membrane)/(mean number of cells invaded through the uncoated membrane)). C and D, experiments here were conducted as in A and B except that NCI-H1299 cells transfected with a negative control oligonucleotide (NC) or an miR-33a mimic oligonucleotide were analyzed. Error bars, S.D. *, p < 0.05; ***, p < 0.001.
FIGURE 9.

The TTF-1 → miR-33a ⊣ HMGA2 signaling axis inhibits motility of lung cancer cells. A, the murine 394T4-shLuc (Ttf-1high) cells were transfected with a negative control oligonucleotide (NC) or an miR-33 inhibitor (anti-miR-33a) and allowed to migrate through uncoated transwell inserts for 22 h. Migrated cells were counted and normalized to negative control (n = 3). B, the murine 394T4-Ttf-1 cells stably expressing a transgene Hmga2 lacking 3′-UTR were transfected with a scrambled control oligonucleotide (Scr Oligo) or an miR-33a mimetic oligonucleotide (miR-33a mimic). A transwell migration assay was performed as in A. C, RT-QPCR analysis of HMGA2 RNA expression of NCI-H1299 transfectant cells. Human HMGA2 was stably expressed via retrovirus-mediated gene transfer. Subsequently, a scrambled control oligonucleotide or an miR-33a mimetic oligonucleotide was transfected, and the RNA of total HMGA2 (endogenous plus exogenous) was quantified by RT-QPCR. D, the human NCI-H1299 cells stably expressing a transgene HMGA2 lacking 3′-UTR were transfected with a scrambled control oligonucleotide or an miR-33a mimetic oligonucleotide (miR-33a mimic). Transwell migration assay was performed as in A. Error bars, S.D. **, p < 0.01; ***, p < 0.001.
To complement these observations derived from the mouse lung cancer cells, we turned to the human NCI-H1299 cells, which are TTF-1low. A human HMGA2 cDNA lacking 3′-UTR and thus non-targetable by miR-33a was retrovirally transduced into NCI-H1299 cells for stable expression. Subsequently, either an miR-33a mimetic oligonucleotide or a scrambled control RNA oligonucleotide was introduced via transfection. RT-QPCR analysis showed that miR-33a mimic reduced the endogenous HMGA2 RNA by 64% in NCI-H1299 cells (Fig. 9C). By the transwell assay, the miR-33a mimic transfection conferred a 40% reduction in motility of NCI-H1299 cells compared with a scrambled oligonucleotide (Fig. 9D, lane 1 versus lane 3). Importantly, this miR-33a-induced decrease in motility was rescued by the stable expression of the HMGA2 transgene (Fig. 9D, lane 3 versus lane 4). These data implicate HMGA2 as a major mediator of miR-33a-directed impediment of lung cancer cell migration.
DISCUSSION
The study of Winslow et al. reported that TTF-1-dependent suppression of HMGA2 is critical to the antimetastatic function of TTF-1 (24). In that study, the potential involvement of miRNAs in Ttf-1-driven suppression of Hmga2 was focused on the let-7 family of miRNAs because of the known target/miRNA relationship between HMGA2 and let-7s (13–15). Using a reporter plasmid that reads out let-7 activity, the primary and metastatic KrasG12D/+;p53Δ/Δ mouse lung cancer cells of different Ttf-1 expression status exhibited equivalent let-7 activity, suggesting that the let-7 miRNAs do not intervene in the Ttf-1-directed Hmga2 repression. However, considering the long 3′-UTR (∼3 kb) of HMGA2/Hmga2, we hypothesized that other miRNAs may be dispatched by Ttf-1 to repress Hmga2. Our earlier investigation identified the first miRNA (i.e. miR-365) that directly represses TTF-1 expression (27). This finding prompted us to initiate the present work to identify the miRNAs that are downstream to and directly regulated by TTF-1 using both loss- and gain-of-TTF-1-function strategies. A motivating factor was that the TTF-1-controlled miRNAs may target HMGA2, thus shedding mechanistic light on the TTF-1 ⊣ HMGA2 signaling axis. Therefore, our initial reverse (loss-of-function) screen via a QPCR array was HMGA2 3′-UTR-centric in that we compared the expression level of a series of miRNAs predicted to bind to HMGA2 3′-UTR in the Ttf-1low and Ttf-1high mouse lung cancer cell lines created by Winslow et al. (24). For the forward (gain-of-function) miRNA screen, we employed a TTF-1-inducible human lung cell system in which a TTF-1-transgene is under dox control (29). In this system, we conducted an unbiased global screen for miRNAs whose expression was altered from the TTF-1off to TTF-1on state using a commercial microarray bearing probes to all known human, mouse, and rat miRNAs. Interestingly, hsa-miR-33a/mmu-miR-33 was scored in both types of screens as an miRNA positively regulated by TTF-1. Curiously, miR-32 was also identified in the global forward screen as an miRNA that is up-regulated by TTF-1. Indeed, by chromatin immunoprecipitation, TTF-1 was demonstrated to bind to the promoter region of the host genes of both miR-33a and miR-32. However, only miR-33a was validated as a genuine miRNA that targets HMGA2. Clearly, the functional consequences of the TTF-1 → miR-32 regulation remain to be determined. Considering the documented expression alterations of miR-32 in lung cancer (down-regulation (42)), multiple myeloma (up-regulation (43)), and prostate cancer (up-regulation (44)), it is likely that miR-32 may also relay functionally significant signaling from TTF-1 in lung cancer.
The regulation of HMGA2 is complex. For example, loss of the gastrointestinal transcription factor Hnf4a causes derepression of Hmga2 in the Ttf-1-negative murine lung tumors (26). The data presented in this study unequivocally establish that TTF-1 relies on miR-33a to hold HMGA2 in check. This mode of restraining the HMGA2 oncogene in lung cells appears conserved from mice to humans per our observations. In our experimental systems, it appears that miR-33a represents an important mediator of TTF-1-induced HMGA2 repression in view of our “add-back” experiment (Fig. 6C) in which the endogenous Hmga2 suppression imposed by the exogenous human TTF-1 in the background of Ttf-1low mouse cells (394T4-shTtf-1) was fully abolished by anti-miR-33a. However, it remains to be investigated whether TTF-1 may directly or indirectly influence HMGA2 expression via an miR-33a-independent manner, as suggested by Winslow et al. (24). Together with our recent finding of miR-365 directly targeting TTF-1 (27), we believe that TTF-1 can be unambiguously placed in the context of an miRNA-based network, with both up- and downstream microRNA signaling partners (miR-365 ⊣ TTF-1 → miR-33a ⊣ HMGA2). Given the fact that TTF-1 is crucial to lung and thyroid development (45, 46), the miRNA network linked to TTF-1 will undoubtedly be shown to play a critical role in the development of these organs in the future.
TTF-1 joins an expanding list of cancer genes, such as MDM2 (47), NOTCH (48), and WT1 (49), displaying both pro- and antitumorigenic activities. This functional dimorphism is perhaps not surprising from the viewpoint that gene activities vary with genetic context. Established oncogenes like AKT1 and MYC have also been shown to inhibit cancer invasion and metastasis (50, 51). Recently, we uncovered that TTF-1 directly transactivates epithelial tight junction genes, OCCLUDIN and CLAUDIN-1, impeding lung cancer cell motility and inducing anoikis (29). This suggests that the antimetastatic activity of TTF-1 may be multipronged in several molecules. Consistent with this thesis, TTF-1 has been shown to reduce cell motility via transactivating MYBPH (52) and antagonizing TGFβ-induced EMT (39). In this study, the prominent phenotype of miR-33a overexpression is also on a metastasis-related phenotype, cell motility. It is curious that TTF-1 would invoke both transcriptional and post-transcriptional (miRNA) mechanisms to restrain lung cell migratory capacity. On the flip side, our findings beg the question of whether miRNAs also manifest the pro-oncogenic function of TTF-1.
In humans, there are two highly conserved miR-33 species (hsa-miR-33a and hsa-miR-33b). These two mature miRNAs differ by two nucleotides. However, in mice, there is a single species, mmu-miR-33, which is identical to hsa-miR-33a. In 2010, it was discovered that hsa-miR-33a is embedded in the 16th intron of SREBF2, and hsa-miR-33b is embedded in the 17th intron of SREBP1 (53, 54). This new metabolism regulator, hsa-miR-33a, acts in concert with SREBP2 (the protein product of SREBF2) to supervise cholesterol homeostasis (53, 54), whereas SREBP2 has long been known as a master regulator of cholesterol metabolism, directly controlling the expression of many key enzymes along the cholesterol biosynthetic pathway (55, 56). In view of the data herein, one must ask if TTF-1 influences cholesterol homeostasis regionally in the lung. Furthermore, because ABCA1, a cholesterol exporter and a direct target of miR-33a (53, 54), displays anti-cancer activity (57), we postulate that cholesterol metabolism may be an integral and indispensable component of the pro- and/or antitumorigenic activities of TTF-1. Motivated by the finding that miR-33a targets the PIM-1 oncogene (58), Ibrahim et al. (59) obtained positive data for an miR-33a replacement therapy in a model of colon carcinoma. Seeing the increasing cancer target multiplicity of miR-33a (HMGA2 (this study), PIM-1 (58), CDK6 (60), CCND1 (60), PTHrP (61), and ABCA1 (53, 54)), we suggest that miR-33a acts as a liaison interfacing cholesterol homeostasis and tumorigenesis. In the future, it will be exciting to define the roles of the miRNA network in coupling the two processes.
Acknowledgments
We thank Dr. Jian-Jun Wei for the HMGA2 cDNA and Dr. Marcus Peter for the HMGA2 3′-UTR reporter construct.
This work was supported, in whole or in part, by National Institutes of Health, NCI, Grant CA127547 (to D. M.).

This article contains supplemental Table 1.
- miRNA
- microRNA
- QPCR
- quantitative real-time PCR
- LNA
- locked nucleic acid
- dox and Dox
- doxycycline.
REFERENCES
- 1. Bartel D. P. (2004) MicroRNAs. Genomics, Biogenesis, Mechanism, and Function. Cell 116, 281–297 [DOI] [PubMed] [Google Scholar]
- 2. Croce C. M. (2009) Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 10, 704–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Esquela-Kerscher A., Slack F. J. (2006) Oncomirs. MicroRNAs with a role in cancer. Nat. Rev. Cancer 6, 259–269 [DOI] [PubMed] [Google Scholar]
- 4. He L., Hannon G. J. (2004) MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 5, 522–531 [DOI] [PubMed] [Google Scholar]
- 5. Bhaskaran M., Wang Y., Zhang H., Weng T., Baviskar P., Guo Y., Gou D., Liu L. (2009) MicroRNA-127 modulates fetal lung development. Physiol. Genomics 37, 268–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lizé M., Herr C., Klimke A., Bals R., Dobbelstein M. (2010) MicroRNA-449a levels increase by several orders of magnitude during mucociliary differentiation of airway epithelia. Cell Cycle 9, 4579–4583 [DOI] [PubMed] [Google Scholar]
- 7. Sozzi G., Pastorino U., Croce C. M. (2011) MicroRNAs and lung cancer. From markers to targets. Cell Cycle 10, 2045–2046 [DOI] [PubMed] [Google Scholar]
- 8. Yu S. L., Chen H. Y., Chang G. C., Chen C. Y., Chen H. W., Singh S., Cheng C. L., Yu C. J., Lee Y. C., Chen H. S., Su T. J., Chiang C. C., Li H. N., Hong Q. S., Su H. Y., Chen C. C., Chen W. J., Liu C. C., Chan W. K., Chen W. J., Li K. C., Chen J. J., Yang P. C. (2008) MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell 13, 48–57 [DOI] [PubMed] [Google Scholar]
- 9. Dong J., Jiang G., Asmann Y. W., Tomaszek S., Jen J., Kislinger T., Wigle D. A. (2010) MicroRNA networks in mouse lung organogenesis. PLoS One 5, e10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ventura A., Young A. G., Winslow M. M., Lintault L., Meissner A., Erkeland S. J., Newman J., Bronson R. T., Crowley D., Stone J. R., Jaenisch R., Sharp P. A., Jacks T. (2008) Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132, 875–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tian Y., Zhang Y., Hurd L., Hannenhalli S., Liu F., Lu M. M., Morrisey E. E. (2011) Regulation of lung endoderm progenitor cell behavior by miR302/367. Development 138, 1235–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boyerinas B., Park S. M., Hau A., Murmann A. E., Peter M. E. (2010) The role of let-7 in cell differentiation and cancer. Endocr. Relat. Cancer 17, F19–F36 [DOI] [PubMed] [Google Scholar]
- 13. Lee Y. S., Dutta A. (2007) The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 21, 1025–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mayr C., Hemann M. T., Bartel D. P. (2007) Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 315, 1576–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park S.-M., Shell S., Radjabi A. R., Schickel R., Feig C., Boyerinas B., Dinulescu D. M., Lengyel E., Peter M. E. (2007) Let-7 prevents early cancer progression by suppressing expression of the embryonic gene HMGA2. Cell Cycle 6, 2585–2590 [DOI] [PubMed] [Google Scholar]
- 16. Qi J., Mu D. (2012) MicroRNAs and lung cancers. From pathogenesis to clinical implications. Front. Med. 6, 134–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maeda Y., Davé V., Whitsett J. A. (2007) Transcriptional control of lung morphogenesis. Physiol. Rev. 87, 219–244 [DOI] [PubMed] [Google Scholar]
- 18. Kendall J., Liu Q., Bakleh A., Krasnitz A., Nguyen K. C., Lakshmi B., Gerald W. L., Powers S., Mu D. (2007) Oncogenic cooperation and coamplification of developmental transcription factor genes in lung cancer. Proc. Natl. Acad. Sci. U.S.A. 104, 16663–16668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kwei K. A., Kim Y. H., Girard L., Kao J., Pacyna-Gengelbach M., Salari K., Lee J., Choi Y. L., Sato M., Wang P., Hernandez-Boussard T., Gazdar A. F., Petersen I., Minna J. D., Pollack J. R. (2008) Genomic profiling identifies TITF1 as a lineage-specific oncogene amplified in lung cancer. Oncogene 27, 3635–3640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tanaka H., Yanagisawa K., Shinjo K., Taguchi A., Maeno K., Tomida S., Shimada Y., Osada H., Kosaka T., Matsubara H., Mitsudomi T., Sekido Y., Tanimoto M., Yatabe Y., Takahashi T. (2007) Lineage-specific dependency of lung adenocarcinomas on the lung development regulator TTF-1. Cancer Res. 67, 6007–6011 [DOI] [PubMed] [Google Scholar]
- 21. Weir B. A., Woo M. S., Getz G., Perner S., Ding L., Beroukhim R., Lin W. M., Province M. A., Kraja A., Johnson L. A., Shah K., Sato M., Thomas R. K., Barletta J. A., Borecki I. B., Broderick S., Chang A. C., Chiang D. Y., Chirieac L. R., Cho J., Fujii Y., Gazdar A. F., Giordano T., Greulich H., Hanna M., Johnson B. E., Kris M. G., Lash A., Lin L., Lindeman N., Mardis E. R., McPherson J. D., Minna J. D., Morgan M. B., Nadel M., Orringer M. B., Osborne J. R., Ozenberger B., Ramos A. H., Robinson J., Roth J. A., Rusch V., Sasaki H., Shepherd F., Sougnez C., Spitz M. R., Tsao M. S., Twomey D., Verhaak R. G., Weinstock G. M., Wheeler D. A., Winckler W., Yoshizawa A., Yu S., Zakowski M. F., Zhang Q., Beer D. G., Wistuba I. I., Watson M. A., Garraway L. A., Ladanyi M., Travis W. D., Pao W., Rubin M. A., Gabriel S. B., Gibbs R. A., Varmus H. E., Wilson R. K., Lander E. S., Meyerson M. (2007) Characterizing the cancer genome in lung adenocarcinoma. Nature 450, 893–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Watanabe H., Francis J. M., Woo M. S., Etemad B., Lin W., Fries D. F., Peng S., Snyder E. L., Tata P. R., Izzo F., Schinzel A. C., Cho J., Hammerman P. S., Verhaak R. G., Hahn W. C., Rajagopal J., Jacks T., Meyerson M. (2013) Integrated cistromic and expression analysis of amplified NKX2–1 in lung adenocarcinoma identifies LMO3 as a functional transcriptional target. Genes Dev. 27, 197–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamaguchi T., Yanagisawa K., Sugiyama R., Hosono Y., Shimada Y., Arima C., Kato S., Tomida S., Suzuki M., Osada H., Takahashi T. (2012) NKX2–1/TITF1/TTF-1-induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell 21, 348–361 [DOI] [PubMed] [Google Scholar]
- 24. Winslow M. M., Dayton T. L., Verhaak R. G., Kim-Kiselak C., Snyder E. L., Feldser D. M., Hubbard D. D., DuPage M. J., Whittaker C. A., Hoersch S., Yoon S., Crowley D., Bronson R. T., Chiang D. Y., Meyerson M., Jacks T. (2011) Suppression of lung adenocarcinoma progression by Nkx2–1. Nature 473, 101–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maeda Y., Tsuchiya T., Hao H., Tompkins D. H., Xu Y., Mucenski M. L., Du L., Keiser A. R., Fukazawa T., Naomoto Y., Nagayasu T., Whitsett J. A. (2012) Kras(G12D) and Nkx2–1 haploinsufficiency induce mucinous adenocarcinoma of the lung. J. Clin. Invest. 122, 4388–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Snyder E. L., Watanabe H., Magendantz M., Hoersch S., Chen T. A., Wang D. G., Crowley D., Whittaker C. A., Meyerson M., Kimura S., Jacks T. (2013) Nkx2–1 represses a latent gastric differentiation program in lung adenocarcinoma. Mol. Cell 50, 185–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qi J., Rice S. J., Salzberg A. C., Runkle E. A., Liao J., Zander D. S., Mu D. (2012) MiR-365 regulates lung cancer and developmental gene thyroid transcription factor 1. Cell Cycle 11, 177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bommer G. T., MacDougald O. A. (2011) Regulation of lipid homeostasis by the bifunctional SREBF2-miR33a locus. Cell Metab. 13, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Runkle E. A., Rice S. J., Qi J., Masser D., Antonetti D. A., Winslow M. M., Mu D. (2012) Occludin is a direct target of thyroid transcription factor-1 (TTF-1/NKX2–1). J. Biol. Chem. 287, 28790–28801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shell S., Park S. M., Radjabi A. R., Schickel R., Kistner E. O., Jewell D. A., Feig C., Lengyel E., Peter M. E. (2007) Let-7 expression defines two differentiation stages of cancer. Proc. Natl. Acad. Sci. U.S.A. 104, 11400–11405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu J., Liu Z., Shao C., Gong Y., Hernando E., Lee P., Narita M., Muller W., Liu J., Wei J.-J. (2011) HMGA2 overexpression-induced ovarian surface epithelial transformation is mediated through regulation of EMT genes. Cancer Res. 71, 349–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ritchie M. E., Silver J., Oshlack A., Holmes M., Diyagama D., Holloway A., Smyth G. K. (2007) A comparison of background correction methods for two-colour microarrays. Bioinformatics 23, 2700–2707 [DOI] [PubMed] [Google Scholar]
- 33. Fusco A., Fedele M. (2007) Roles of HMGA proteins in cancer. Nat. Rev. Cancer 7, 899–910 [DOI] [PubMed] [Google Scholar]
- 34. Grimson A., Farh K. K., Johnston W. K., Garrett-Engele P., Lim L. P., Bartel D. P. (2007) MicroRNA targeting specificity in mammals. Determinants beyond seed pairing. Mol. Cell 27, 91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reddel R. R., Ke Y., Gerwin B. I., McMenamin M. G., Lechner J. F., Su R. T., Brash D. E., Park J. B., Rhim J. S., Harris C. C. (1988) Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Res. 48, 1904–1909 [PubMed] [Google Scholar]
- 36. Hsu D. S., Acharya C. R., Balakumaran B. S., Riedel R. F., Kim M. K., Stevenson M., Tuchman S., Mukherjee S., Barry W., Dressman H. K., Nevins J. R., Powers S., Mu D., Potti A. (2009) Characterizing the developmental pathways TTF-1, NKX2–8, and PAX9 in lung cancer. Proc. Natl. Acad. Sci. U.S.A. 106, 5312–5317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boggaram V. (2009) Thyroid transcription factor-1 (TTF-1/Nkx2.1/TITF1) gene regulation in the lung. Clin. Sci. 116, 27–35 [DOI] [PubMed] [Google Scholar]
- 38. Damante G., Fabbro D., Pellizzari L., Civitareale D., Guazzi S., Polycarpou-Schwartz M., Cauci S., Quadrifoglio F., Formisano S., Di Lauro R. (1994) Sequence-specific DNA recognition by the thyroid transcription factor-1 homeodomain. Nucleic Acids Res. 22, 3075–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saito R. A., Watabe T., Horiguchi K., Kohyama T., Saitoh M., Nagase T., Miyazono K. (2009) Thyroid transcription factor-1 inhibits transforming growth factor-β-mediated epithelial-to-mesenchymal transition in lung adenocarcinoma cells. Cancer Res. 69, 2783–2791 [DOI] [PubMed] [Google Scholar]
- 40. Guazzi S., Price M., De Felice M., Damante G., Mattei M. G., Di Lauro R. (1990) Thyroid nuclear factor 1 (TTF-1) contains a homeodomain and displays a novel DNA binding specificity. EMBO J. 9, 3631–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saetrom P., Heale B. S., Snøve O., Jr., Aagaard L., Alluin J., Rossi J. J. (2007) Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res. 35, 2333–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yanaihara N., Caplen N., Bowman E., Seike M., Kumamoto K., Yi M., Stephens R. M., Okamoto A., Yokota J., Tanaka T., Calin G. A., Liu C. G., Croce C. M., Harris C. C. (2006) Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9, 189–198 [DOI] [PubMed] [Google Scholar]
- 43. Pichiorri F., Suh S. S., Ladetto M., Kuehl M., Palumbo T., Drandi D., Taccioli C., Zanesi N., Alder H., Hagan J. P., Munker R., Volinia S., Boccadoro M., Garzon R., Palumbo A., Aqeilan R. I., Croce C. M. (2008) MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 12885–12890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ambs S., Prueitt R. L., Yi M., Hudson R. S., Howe T. M., Petrocca F., Wallace T. A., Liu C. G., Volinia S., Calin G. A., Yfantis H. G., Stephens R. M., Croce C. M. (2008) Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res. 68, 6162–6170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Antonica F., Kasprzyk D. F., Opitz R., Iacovino M., Liao X. H., Dumitrescu A. M., Refetoff S., Peremans K., Manto M., Kyba M., Costagliola S. (2012) Generation of functional thyroid from embryonic stem cells. Nature 491, 66–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kimura S., Hara Y., Pineau T., Fernandez-Salguero P., Fox C. H., Ward J. M., Gonzalez F. J. (1996) The T/ebp null mouse. Thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 10, 60–69 [DOI] [PubMed] [Google Scholar]
- 47. Manfredi J. J. (2010) The Mdm2-p53 relationship evolves. Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 24, 1580–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Radtke F., Raj K. (2003) The role of Notch in tumorigenesis. Oncogene or tumour suppressor? Nat. Rev. Cancer 3, 756–767 [DOI] [PubMed] [Google Scholar]
- 49. Yang L., Han Y., Suarez Saiz F., Minden M. D. (2007) A tumor suppressor and oncogene. The WT1 story. Leukemia 21, 868–876 [DOI] [PubMed] [Google Scholar]
- 50. Liu H., Radisky D. C., Nelson C. M., Zhang H., Fata J. E., Roth R. A., Bissell M. J. (2006) Mechanism of Akt1 inhibition of breast cancer cell invasion reveals a protumorigenic role for TSC2. Proc. Natl. Acad. Sci. U.S.A. 103, 4134–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu H., Radisky D. C., Yang D., Xu R., Radisky E. S., Bissell M. J., Bishop J. M. (2012) MYC suppresses cancer metastasis by direct transcriptional silencing of αv and β3 integrin subunits. Nat. Cell Biol. 14, 567–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hosono Y., Yamaguchi T., Mizutani E., Yanagisawa K., Arima C., Tomida S., Shimada Y., Hiraoka M., Kato S., Yokoi K., Suzuki M., Takahashi T. (2012) MYBPH, a transcriptional target of TTF-1, inhibits ROCK1, and reduces cell motility and metastasis. EMBO J. 31, 481–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moore K. J., Rayner K. J., Suárez Y., Fernández-Hernando C. (2011) The role of microRNAs in cholesterol efflux and hepatic lipid metabolism. Annu. Rev. Nutr. 31, 49–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rottiers V., Näär A. M. (2012) MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 13, 239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brown M. S., Goldstein J. L. (1997) The SREBP pathway. Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340 [DOI] [PubMed] [Google Scholar]
- 56. Horton J. D., Goldstein J. L., Brown M. S. (2002) SREBPs. Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Smith B., Land H. (2012) Anticancer activity of the cholesterol exporter ABCA1 gene. Cell Rep. 2, 580–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thomas M., Lange-Grünweller K., Weirauch U., Gutsch D., Aigner A., Grünweller A., Hartmann R. K. (2012) The proto-oncogene Pim-1 is a target of miR-33a. Oncogene 31, 918–928 [DOI] [PubMed] [Google Scholar]
- 59. Ibrahim A. F., Weirauch U., Thomas M., Grünweller A., Hartmann R. K., Aigner A. (2011) MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 71, 5214–5224 [DOI] [PubMed] [Google Scholar]
- 60. Cirera-Salinas D., Pauta M., Allen R. M., Salerno A. G., Ramírez C. M., Chamorro-Jorganes A., Wanschel A. C., Lasuncion M. A., Morales-Ruiz M., Suarez Y., Baldan Á., Esplugues E., Fernández-Hernando C. (2012) Mir-33 regulates cell proliferation and cell cycle progression. Cell Cycle 11, 922–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kuo P. L., Liao S. H., Hung J. Y., Huang M. S., Hsu Y. L. (2013) MicroRNA-33a functions as a bone metastasis suppressor in lung cancer by targeting parathyroid hormone related protein. Biochim. Biophys. Acta 1830, 3756–3766 [DOI] [PubMed] [Google Scholar]