

Background: Regulation of insulin receptor substrate (IRS) 2 protein levels is crucial for glucose homeostasis.
Results: Elevated cAMP levels increase phosphorylation of serine 1137/1138 residues on IRS2 protein, which mediates its binding to 14-3-3 proteins and enhances its stability.
Conclusion: Serine 1137/1138 are novel cAMP-dependent phosphorylation sites on IRS2 that regulate its protein degradation via interaction with 14-3-3 proteins.
Significance: Novel cAMP-dependent mechanism to control IRS2 protein levels.
Keywords: Cyclic AMP (cAMP), Insulin, Phosphorylation, Protein Degradation, Protein Kinase A (PKA), 14-3-3, IRS2
Abstract
Insulin receptor substrate (IRS) 2 as intermediate docking platform transduces the insulin/IGF-1 (insulin like growth factor 1) signal to intracellular effector molecules that regulate glucose homeostasis, β-cell growth, and survival. Previously, IRS2 has been identified as a 14-3-3 interaction protein. 14-3-3 proteins can bind their target proteins via phosphorylated serine/threonine residues located within distinct motifs. In this study the binding of 14-3-3 to IRS2 upon stimulation with forskolin or the cAMP analog 8-(4-chlorophenylthio)-cAMP was demonstrated in HEK293 cells. Binding was reduced with PKA inhibitors H89 or Rp-8-Br-cAMPS. Phosphorylation of IRS2 on PKA consensus motifs was induced by forskolin and the PKA activator N6-Phe-cAMP and prevented by both PKA inhibitors. The amino acid region after position 952 on IRS2 was identified as the 14-3-3 binding region by GST-14-3-3 pulldown assays. Mass spectrometric analysis revealed serine 1137 and serine 1138 as cAMP-dependent, potential PKA phosphorylation sites. Mutation of serine 1137/1138 to alanine strongly reduced the cAMP-dependent 14-3-3 binding. Application of cycloheximide revealed that forskolin enhanced IRS2 protein stability in HEK293 cells stably expressing IRS2 as well as in primary hepatocytes. Stimulation with forskolin did not increase protein stability either in the presence of a 14-3-3 antagonist or in the double 1137/1138 alanine mutant. Thus the reduced IRS2 protein degradation was dependent on the interaction with 14-3-3 proteins and the presence of serine 1137/1138. We present serine 1137/1138 as novel cAMP-dependent phosphorylation sites on IRS2 and show their importance in 14-3-3 binding and IRS2 protein stability.
Introduction
Insulin receptor substrate (IRS)2 2 belongs to the family of insulin receptor substrates and serves as an intermediate docking platform to transmit the insulin and IGF-1 signal upon ligand binding to intracellular effector molecules for the regulation of glucose and lipid metabolism. Knock-out models have shown the physiological importance of IRS2 for glucose homeostasis, β-cell growth, and survival (1–3). The dynamic regulation of IRS2 protein levels in liver is important for the adaptation of the glucose metabolism to fasting and refeeding, whereas in pancreatic β-cells sufficient IRS2 protein levels are crucial for growth and survival (4). β-Cell-specific expression of IRS2 in IRS2 knock-out, obese, and streptozotocin-treated mice prevented diabetes in these mice (5). Ligand-stimulated autophosphorylation of tyrosine residues on insulin/IGF-1 receptors leads to their interaction with IRS proteins and other Src homology domain 2 containing proteins such as the p85 regulatory subunit of PI 3-kinase or Grb2. Serine and threonine residues on IRS2 can be phosphorylated by several kinases including ERK, JNK, or glycogen synthase kinase 3 (6–9) and this has been associated with a negative feedback control of insulin signaling (10, 11). The serine/threonine phosphorylation patterns of IRS proteins may also have stimulatory consequences for insulin/IGF-1 signal transduction (12–14). An important mechanism to control insulin signaling at the level of IRS2 is the regulation of IRS2 mRNA and total protein amounts. They are tightly regulated by fasting and refeeding (2, 7, 15). Elevated cAMP levels are a major stimulus for increased expression of IRS2 in the liver and pancreatic β-cell. IRS2 gene activation is initiated by activation of CREB (cAMP response element-binding protein) (16) through phosphorylation by PKA (protein kinase A) with TORC2 (transducer of regulated CREB activity 2) as an important co-activator for CREB-dependent hepatic IRS2 expression (15). Insulin suppresses IRS2 protein expression by reducing the rate of transcription of the IRS2 gene (17) and induces post-translational modifications on the IRS2 molecule itself, thus leading to proteasomal degradation (18).
14-3-3 proteins are versatile regulators of a variety of intracellular processes and participate in neuronal development and control of cell cycle, cell growth, gene transcription, and apoptosis. In mammals, 7 genes encode for the 7 14-3-3 isoforms that are entitled with the Greek letters β, γ, ϵ, η, σ, τ, and ζ (19). 14-3-3 proteins can also be found in varying numbers in yeast, plants, and other eukaryotes. A special feature is the high sequence homology of all 14-3-3 proteins and the finding that 14-3-3 proteins from different species are interchangeable and functionally redundant. Being synthesized as ∼30 kDa proteins, 14-3-3 proteins form homo- and heterodimers with the exception of 14-3-3σ, which preferentially forms homodimers (20). Each monomer shows a cup-like shape with a central groove wherein the amino acids are strictly conserved (21). In most of the reported cases, phosphorylation of a serine/threonine residue within a 14-3-3 binding motif on a binding partner is crucial for interaction with 14-3-3 proteins. Muslin et al. (22) identified a putative motif for 14-3-3 binding (RSXpSXP, pS indicates phosphorylated serine, X denotes any amino acid) and showed that phosphorylated serine is crucial for recognition by 14-3-3. Further studies led to the nomination of two 14-3-3 binding motifs, namely mode I (RSXpSXP) and mode II (RX(Y/F)pSXP) (23, 24). Nevertheless, in a few publications phosphorylation-independent interaction of 14-3-3 proteins with binding partners has been described (25–27). This indicates additional structural features of target proteins that can be important for interaction with 14-3-3 proteins in certain cases. 14-3-3 proteins can bind one or two simultaneously phosphorylated residues within one protein and binding can have several consequences: obscuring a nearby binding/interaction motif, thus modulating interaction of the target protein with other binding partners (28), regulation of nuclear import/export upon binding (29, 30), a conformational change that influences the activity of the target protein (31), and acting as scaffold protein to connect signaling pathways (32).
IRS2 and 14-3-3 have been shown to interact with each other upon insulin and IGF-1 stimulation (33, 34). Here we identified cAMP as a novel stimulus that triggered IRS2 and 14-3-3 interaction. Given the crucial role of IRS2 for glucose homeostasis and β-cell survival and the manifold of regulatory functions that can be exerted by 14-3-3 proteins we set out to characterize this interaction and its consequences further.
EXPERIMENTAL PROCEDURES
Materials
PKA substrate antibody (catalog number 9621), β-actin antibody (4970), p-Thr-308 Akt/PKB antibody (9275), p-Thr-202/p-Tyr-204 ERK antibody (9101), ERK1/2 protein antibody (9102), and PD98059 (9900) were from Cell Signaling Technology (Frankfurt, Germany) and p-Ser-157 VASP antibody (ab58555) was from Abcam (Cambridge, UK). GFP (green fluorescent protein) antibody (sc-8334) was from Santa Cruz (Santa Cruz, CA) and Protein A-Sepharose (17-5280-04), Protein G-Sepharose (17-0618-01), glutathione-Sepharose 4B (17-0756-01), and GST (glutathione S-transferase) antibody (27-4577-01) from GE Healthcare Europe (Munich, Germany). Recombinant human 14-3-3ζ-HRP (HRP-2669) was from R&D Systems (Minneapolis, MN) and IRS2 antibody (06-506 and MABS15) from Millipore (Schwalbach, Germany). InSolutionTM Akt Inhibitor VIII Akti-1/2 (124017), 14-3-3 antagonist I (100081), and H89 (371963) were from Calbiochem (Schwalbach, Germany) and forskolin (A2165) from Applichem (Darmstadt, Germany). IGF-1 (I3769), EGF (E9644), 8-(4-chlorophenylthio) (CPT)-cAMP (C3912), and phosphatase inhibitors (sodium fluoride, sodium pyrophosphate, sodium orthovanadate, and β-glycerophosphate) were from Sigma. Polyethylenimine (24765) was from Polysciences (Eppelheim, Germany). Akt/PKB protein antibody (610861) was from BD Transduction Laboratories (Erembodegem, Belgium). N6-Phe-cAMP (P006), 8-pCPT-2′-O-Me-cAMP (C041), and Rp-8-Br-cAMPS (B001) were from BioLog (Bremen, Germany). Flp-In HEK293 cells and 4–12% BisTris mini gels (NP0321) were obtained from Invitrogen (Karlsruhe, Germany) and GFP-Trap (gta-20) was from Chromotek (Martinsried, Germany). HEK293 cells were from The European Collection of Cell Cultures (Salisbury, UK) and Myc antibody and Myc-14-3-3γ expression vector was provided by Reiner Lammers (University Hospital Tuebingen, Tuebingen, Germany).
Plasmid Constructs
The coding region for mouse IRS2 (NM_001081212.1) was amplified from a pRK5 IRS2 vector (kindly provided by M. F. White, Boston, MA) using primers 5′-gaggatccatggctagcgcgcccctgcctg-3′ and 5′-ctgcggccgctcgagtcactctttcacgactgtggcttccttc-3′, cloned into vector pSC-B (Stratagene) and sequenced. IRS2 was subcloned from this plasmid into pcDNA5/FRT/TO-GFP as a BamHI/NotI insert to give vector pcDNA5/FRT/TO-GFP-IRS2 in which IRS2 is tagged with GFP at the N terminus. For truncated IRS2 versions containing the IRS2 sequence from amino acids 1–300 and 1–600 a stop codon was created after position 300 (sense, 5′-gagttccggcctcgctgaaagagtcagtcgtcc-3′; antisense, 5′-ggacgactgactctttcagcgaggccggaactc-3′) or 600 (sense, 5′-ctcatgagggccacctgatctggtagttcaggtc-3′; antisense, 5′-gacctgaactaccagatcaggtggccctcatgag-3′), respectively. For the constructs 301–1321 and 601–1321, a BamHI restriction site was created prior to positions 301 and 601 and the obtained inserts were ligated into pcDNA5/FRT/TO-GFP. The constructs mentioned so far were generated at the University of Dundee (Dundee, UK). Construct GFP-IRS2-601-952 was generated by creating a stop codon at position 952 and pcDNA5/FRT/TO-GFP-IRS2-601-1321 served as template. The following constructs were generated with the Stratagene QuikChange XL Site-directed Mutagenesis method (200522-5, Stratagene, La Jolla, CA): GFP-IRS2-601-952, S1137A, S1138A, S1163A, and S1137/S1138A, and verified by sequencing.
Cell Culture and Transfections
All cells were kept at 37 °C with 5% CO2 and 95% humidity. Flp-In HEK293 and HEK293 cells were cultivated in DMEM containing 4.5 g/liter of glucose (BE12–741F, Lonza, Cologne, Germany) supplemented with 10% FBS (SV30160.03, HyClone, Thermo Fisher Scientific, Schwerte, Germany), 1% glutamine, 100 units/ml of penicillin, and 100 units/ml of streptomycin (DE17–602E, Lonza, Cologne, Germany). Flp-In HEK293 cells were stably transfected by the calcium phosphate method (35) and positive clones were selected with hygromycin B, whereas HEK293 cells were transiently transfected using polyethylenimine in 25 mm HEPES. 45 h after transfection cells were serum starved for 3 h and incubated with various substances, as described in detail under “Results” and in the figure legends.
Cell Lysis, GFP Pulldown, Myc Co-immunoprecipitation, and Western Blotting
Cell lysis took place in lysis buffer consisting of 50 mm HEPES, 150 mm NaCl, 1.5 mm MgCl2, 1 mm EDTA, 10% glycerol, 1% Triton X-100, set to pH 7.5, containing phosphatase inhibitors (1 mm sodium fluoride, 0.5 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 1 mm β-glycerophosphate). Centrifugation of lysates at 16,000 × g for 5 min at 4 °C was followed by protein determination (Bio-Rad Protein Assay). To minimize unspecific binding lysates were precleared with Protein A-Sepharose for 30 min before incubation with GFP-Trap for 2 h at 4 °C. Beads were washed twice with high salt buffer (50 mm Tris, pH 7.5, 150 mm NaCl) and twice with no salt buffer (50 mm Tris, pH 7.5, 1 mm EGTA, 0.1% mercaptoethanol) before denaturation. If not indicated otherwise, all the samples were separated on 7.5% SDS gels and proteins were transferred onto nitrocellulose membranes by semi-dry Western blot. For co-immunoprecipitation experiments, lysates were incubated with Myc antibody and Protein G-Sepharose for 2 h at 4 °C, washed two times with lysis buffer and separated on 5–15% SDS gels followed by Western blotting. Western blotting and visualization of proteins was performed using enhanced chemiluminescence (ECL) as described in Ref. 36.
Overlay Assay
Overlay assay was used to visualize interaction of 14-3-3 with target proteins. Membranes were blocked with 5% milk in TBS-T (25 mm Tris, pH 7.4, 0.15 m NaCl, 0.1% Tween 20) for 1 h at room temperature. Overnight incubation at 4 °C with recombinant 14-3-3ζ-HRP was followed by washing 5 times with TBS-T and detection using ECL.
Isolation and Maintenance of Primary Mouse Hepatocytes
14-Week-old male C57Bl/6 mice were anesthetized and the portal vein was catheterized. Liver was perfused with Krebs-Henseleit buffer I (115 mm NaCl, 25 mm NaHCO3, 5.9 mm KCl, 1.18 mm MgCl2, 1.23 mm NaH2PO4, 1.195 mm Na2SO4, 0.5 mm EGTA, 20 mm HEPES, pH 7.4) and subsequently with Krebs-Henseleit buffer II (115 mm NaCl, 25 mm NaHCO3, 5.9 mm KCl, 1.18 mm MgCl2, 1.23 mm NaH2PO4, 1.195 mm Na2SO4, 2.5 mm CaCl2, 20 mm HEPES, pH 7.2) containing 1 mg/ml of Collagenase II (Biochrom, Berlin, Germany). Isolated hepatocytes were washed and counted with Trypan Blue. 5 × 105 cells were cultivated in hepatocyte maintenance medium with supplements (Provitro, Berlin, Germany) and seeded on 6-well plates precoated with Collagen R (Serva, Heidelberg, Germany). 24 h after isolation cells were serum starved overnight and stimulated as indicated under “Results” and in the figure legends. All procedures were approved by the local Animal Care and Use committee.
GST-14-3-3 Fusion Protein Expression and GST Pulldown
GST-14-3-3β and -ϵ were a gift from Dr. Angelika Hausser (University of Stuttgart) and propagated in Escherichia coli. Expression was induced by adding 1 mm isopropyl β-d-1-thiogalactopyranoside for 5 h and cells were harvested by centrifugation for 15 min at 4500 × g at 4 °C. 40 strokes with a probe-type sonicater were followed by addition of Triton X-100 to a final concentration of 1% and centrifugation for 20 min at 4500 × g at 4 °C. The GST-14-3-3 fusion proteins were coupled to glutathione-Sepharose 4B and the amount of GST-14-3-3 was determined by SDS-PAGE and subsequent Coomassie staining. Lysates were incubated with 2 μg of GST-14-3-3 for 2 h in lysis buffer at 4 °C and washed three times with lysis buffer before denaturation. Centrifugation between washes was carried out at 1100 × g for 1 min at 4 °C.
Sample Preparation for Mass Spectrometry
HEK293 cells were seeded at a density of 7 × 106 cells onto 15-cm diameter cell culture dishes and transfected 24 h later with 10 μg of DNA/plate using 80 μg of polyethylenimine/plate. 45 h later, cells were serum starved and incubated for 30 min with 20 μm forskolin alone or after preincubation with 30 μm H89 for 30 min. Cells were lysed with 900 μl of lysis buffer, after which GFP-IRS2 was purified from 5 mg of precleared total protein using GFP-Trap. Prior to LC-MS/MS analysis, samples were separated on 4–12% BisTris gels and after Coomassie staining bands corresponding to IRS2 were cut and subjected to tryptic proteolysis as described by Gloeckner et al. (37). Resulting peptides were separated using the UltiMate 3000 nano-HPLC system. They were eluted from the trap column onto the analytical column (Acclaim PepMap RSLC 75 μl × 25-cm C18 2-μm 100 Å) by applying an acetonitrile gradient of 2 to 35% of eluent (80% acetonitrile, 0.08% formic acid) for 33 min. The eluted peptides were analyzed by mass spectrometry (LTQ Orbitrap Velos, Thermo Fisher Scientific). Mass spectra were extracted by Mascot Daemon without charge state deconvolution and de-isotoping and the results were analyzed using Mascot (Matrix Science version 2.3.0, Boston, MA). Because mouse IRS2 was transfected, Mascot was set up to search the Mus musculus subset of the SwissProt database (release 2011_12, 533657 entries) assuming trypsin as the digestion enzyme. Mascot was searched with a fragment ion mass tolerance of 1 Da and a parent ion tolerance of 10 ppm. Oxidation of methionine, deamidation of asparagines and glutamine, and phosphorylation of serine, threonine, and tyrosine were specified in Mascot as variable modifications. Carbamidomethylation of cysteine was specified as a fixed modification. The Mascot result files were loaded into Scaffold software (Scaffold version 3.3.1, Proteome Software Inc., Portland, OR) to validate MS/MS-based peptide identifications. Peptide identifications were accepted if they could be established at a probability greater than 80%, as specified by the Mascot Daemon. Further analysis of the MS/MS data with regard to post-translational modifications was performed using the Proteome Discoverer software (version 1.3, Thermo Fisher Scientific) and Skyline (version 1.2) (38).
Statistical Analysis
Statistical analysis was performed with JMP 10.0.0 (SAS Institute Inc., NC) and data are presented as mean ± S.E. t test was employed to test for significant differences between groups and a result was considered significant if p < 0.05.
RESULTS
Elevated cAMP Levels Enable 14-3-3 Binding to IRS2 in a PKA-dependent Manner
Dubois et al. (34) reported an interaction of IRS2 with 14-3-3 proteins upon activation of the PI 3-kinase pathway by IGF-1 stimulation in HEK293 cells. We tested if other intracellular pathways can mediate 14-3-3 binding to IRS2 in HEK293 cells expressing GFP-IRS2. The adenylyl cyclase activator forskolin increased binding of 14-3-3 to IRS2 as shown by an overlay assay (Fig. 1A). Dependence of the IRS2/14-3-3 interaction on elevated cAMP levels was also shown with the cell-permeable cAMP analog CPT-cAMP (Fig. 1B). The interaction was completely abrogated with the PKA inhibitor H89 (Fig. 1, A and B) or reduced to basal levels with the specific PKA inhibitor Rp-8-Br-cAMPS (Fig. 1C). These data suggest the involvement of a PKA-mediated phosphorylation of IRS2 for its interaction with 14-3-3 proteins. PKA-dependent phosphorylation of IRS2 was studied with a PKA substrate antibody in HEK293 cells expressing GFP-IRS2 after GFP pulldown. Forskolin induced the phosphorylation of IRS2 (Fig. 1, D–F). Successful stimulation with forskolin was shown by phosphorylation of Ser-157 of vasodilator-stimulated protein (VASP) (Fig. 1, D and E). Elevated cAMP levels cannot only enhance phosphorylation of IRS2 via PKA, but can also involve exchange proteins directly activated by cAMP (EPAC) and downstream kinases ERK, Akt/PKB, and p90 ribosomal S6 kinase (RSK). Moreover, the PKA substrate antibody used recognizes a consensus site that partially overlaps with other arginine-directed kinases such as Akt/PKB and RSK. Therefore, the putative involvement of these kinases was studied. Stimulation with EGF (epidermal growth factor) as an activator of the ERK pathway did not lead to phosphorylation of any of the sites recognized by the PKA substrate antibody (Fig. 1D). Successful EGF stimulation was confirmed by checking the phosphorylation status of ERK. Pretreatment with the MEK1 inhibitor PD98059 before forskolin treatment did not influence cAMP-dependent phosphorylation of PKA sites on IRS2 (Fig. 1D). Next, cells were treated with IGF-1 and the Akt/PKB inhibitor Akti. Successful IGF-1 stimulation was confirmed by checking the phosphorylation of threonine 308 of Akt/PKB. However, the stimulation by IGF-1 did not lead to phosphorylation of any of the sites recognized by the PKA substrate antibody (Fig. 1E). Furthermore, we tested the direct PKA activator N6-Phe-cAMP. Stimulation of the cells with this compound increased the phosphorylation of IRS2, whereas the EPAC activator 8-pCPT-2′-O-Me-cAMP did not increase the phosphorylation to a similar extend (Fig. 1F). The use of the specific PKA inhibitor Rp-8-Br-cAMPS prevented the phosphorylation (Fig. 1, F and G). These results indicate that elevated cAMP levels via activation of PKA lead to phosphorylation of IRS2 and interaction between IRS2 and 14-3-3. Furthermore, phosphorylation of IRS2 on PKA sites was detected in primary hepatocytes after forskolin stimulation, which was prevented by H89 pretreatment (Fig. 1H).
FIGURE 1.

14-3-3 interacts with IRS2 upon elevated cAMP levels and PKA phosphorylates IRS2. A, HEK293 cells were transiently transfected with GFP-IRS2. Cells were incubated for 30 min with 20 μm forskolin alone or after preincubation with 30 μm H89 for 30 min. 100 μg of protein was used for GFP pulldown. 14-3-3 binding was visualized by performing an overlay assay and the membrane was stripped and reprobed for GFP-IRS2 as expression and loading control. Densitometric analysis of 14-3-3 binding is shown as band intensities normalized for GFP-IRS2. GFP-IRS2 treated with forskolin was set as 1 (mean ± S.E., n = 3, *, p < 0.05, forskolin versus unstimulated; #, <0.05, forskolin versus H89). B, cells were stimulated with 100 μm CPT-cAMP for 30 min or after preincubation with 30 μm H89 for 30 min. 200 μg of protein was used for GFP pulldown. GFP-IRS2 treated with CPT-cAMP was set as 1 (mean ± S.E., n = 3, *, p < 0.05, CPT-cAMP versus unstimulated; #, <0.05, CPT-cAMP versus H89). C, cells were incubated for 30 min with 20 μm forskolin alone or after preincubation with 100 μm Rp-8-Br-cAMPS for 30 min. 400 μg of protein was used for GFP pulldown. GFP-IRS2 treated with forskolin was set as 1 (mean ± S.E., n = 3, *, p < 0.05, forskolin versus unstimulated; #, <0.05, forskolin versus Rp-8-Br-cAMPS). D and E, GFP-IRS2 was transiently expressed in HEK293 cells and cells were incubated for 30 min with 1 μm Akt inhibitor VIII Akti 1/2 (Akti), 20 μm forskolin, 50 ng/ml of IGF-1, 50 μm PD98059, and 100 ng/ml of EGF as indicated. 200 μg of total protein was used for GFP pulldown. Membrane was incubated with PKA substrate antibody. Phosphorylation of serine 157 of VASP was assessed as forskolin stimulation control, phosphorylation of threonine 308 of Akt/PKB as IGF-1 stimulation control, whereas phosphorylation of threonine 202/tyrosine 204 of ERK was checked as control for successful EGF stimulation. Corresponding IRS2, Akt/PKB, and ERK reblots are also shown (n = 3). F and G, GFP-IRS2 was expressed transiently in HEK293 cells. Cells were incubated with the following reagents for 30 min: 100 μm N6-Phe-cAMP, 100 μm 8-pCPT-2′-O-Me-cAMP, 100 μm Rp-8-Br-cAMPS or 20 μm forskolin. 400 μg of protein was used for GFP pulldown. Membrane was incubated with PKA substrate antibody, after which it was stripped and reprobed with IRS2 antibody (n = 3). H, primary hepatocytes were incubated with 20 μm forskolin alone or after preincubation with 30 μm H89 for 30 min, respectively. 100 μg of protein was separated on SDS gel and membranes were incubated with PKA substrate antibody, phosphoserine 157 VASP antibody, or β-actin antibody. PKA substrate antibody membrane was stripped and reprobed with IRS2 antibody (n = 3).
Identification of the cAMP-dependent 14-3-3 Binding Region on IRS2
Fragments spanning different regions of the IRS2 protein were generated. The two fragments GFP-IRS2-(1–300) and GFP-IRS2-(1–600) encompass the front part of the IRS2 molecule including the pleckstrin homology and the phosphotyrosine binding domains. The two fragments GFP-IRS2-(301–1321) and GFP-IRS2-(601–1321) comprise the rear part of IRS2 with the kinase regulatory loop binding domain (Fig. 2A). HEK293 cells were transiently transfected with wild type IRS2 and IRS2 fragments and treated with forskolin and H89. Phosphorylation of PKA sites was tested after GFP pulldown (Fig. 2B). The antibody detected PKA phosphorylation sites on IRS2 wild type and fragments GFP-IRS2-(301–1321) and GFP-IRS2-(601–1321) after forskolin stimulation that were not detectable when cells were pretreated with H89. No signal was detected on fragments GFP-IRS2-(1–300) and GFP-IRS2-(1–600). Fragment GFP-IRS2-(601–952) was generated to further narrow down the area for forskolin-dependent phosphorylation of IRS2. Because the PKA substrate antibody did not detect any signals from this fragment we assumed that the PKA phosphorylation sites on IRS2 were located after amino acid position 952 (Fig. 2C). To test if the fragments containing PKA phosphorylation sites were also able to interact with 14-3-3 proteins, pulldown experiments with a GST-14-3-3 fusion protein were performed (Fig. 2D). IRS2 wild type protein interacted only in the forskolin-stimulated sample with GST-14-3-3β. GFP-IRS2-(301–1321) and GFP-IRS2-(601–1321) also showed strong interaction with GST-14-3-3β in the forskolin-stimulated condition, and only minimal interaction in the unstimulated or H89 pretreated condition. No interaction between fragment GFP-IRS2-(601–952) and GST-14-3-3β could be detected. Incubation with the PKA substrate antibody confirmed the presence of PKA phosphorylation sites on wild type IRS2 and IRS2 fragments 301–1321 and 601–1321. Therefore we concluded that PKA phosphorylation sites on IRS2 that mediate interaction with 14-3-3 were located after amino acid position 952.
FIGURE 2.

Site(s) for PKA phosphorylation on IRS2 are located after amino acid position 952. A, schematic illustration of IRS2 and IRS2 fragments. Five fragments spanning different regions of the IRS2 protein were generated according to mouse numbering: GFP-IRS2-(1–300), GFP-IRS2-(1–600), GFP-IRS2-(301–1321), GFP-IRS2-(601–1321), and GFP-IRS2-(601–952). All constructs were GFP tagged at the N terminus. B and C, HEK293 cells were transiently transfected with GFP-IRS2, GFP-IRS2-(1–300), GFP-IRS2-(1–600), GFP-IRS2-(301–1321), GFP-IRS2-(601–1321), or GFP-IRS2-(601–952). Cells were incubated for 30 min with 20 μm forskolin alone or after preincubation with 30 μm H89 for 30 min. 100–200 μg of protein was used for GFP pulldown and after separation on 5–15% SDS gels and Western blotting, the membrane was incubated with PKA substrate antibody. Stripping and reprobing with GFP antibody as expression and loading control is also shown (n = 3). D, GFP-IRS2, GFP-IRS2-(301–1321), GFP-IRS2-(601–1321), and GFP-IRS2-(601–952) were transiently expressed in HEK293 cells. Stimulation was carried out with 20 μm forskolin for 30 min or 30 μm H89 for 30 min prior forskolin stimulation. Lysate containing 250 μg of total protein was incubated with 2 μg of GST-14-3-3β for 2 h and samples were separated by SDS-PAGE. Transfer onto nitrocellulose membranes followed by incubation with GFP antibody to visualize the amount of protein that interacted with GST-14-3-3 and with GST to ensure equal pulldown and loading. Incubation with PKA substrate antibody was carried out to visualize phosphorylation of PKA motifs (n = 3).
Mass Spectrometric Identification of Potential PKA Phosphorylation and 14-3-3 Binding Sites
We applied mass spectrometry to identify possible candidates for PKA phosphorylation of IRS2. HEK293 cells were transiently transfected with GFP-IRS2 and treated with forskolin and H89. GFP-IRS2 was captured from cell lysates and after Coomassie staining, bands corresponding to GFP-IRS2 were cut and prepared for mass spectrometric analysis. This experiment was performed three times and only sites that matched the following criteria were considered for further testing: (i) present in all three replicates in the forskolin-stimulated sample and not present in the H89 pretreated sample, (ii) matched the consensus sequence for PKA (RXXpS), and (iii) matched a 14-3-3 binding motif (mode I, RSXpS/pTXP; mode II, RX(F/Y)XpS/pTXP). From that evaluation three candidates emerged: serine 1137, serine 1138, and serine 1163, depicted with their surrounding amino acids in Fig. 3A. Of note, phosphorylated serine 1137 and serine 1138 could be found in tryptic peptides as single phosphorylated residues but also doubly phosphorylated peptides were detectable as shown in Fig. 3B. Serine 1137 (GRRRHS1137SETFSS) and serine 1138 (GRRRHSS1138ETFSS) could probably compensate for each other, because in the case of substitution of either residue with an alanine there would still be the same PKA recognition motif present. Therefore, not only were the single residues mutated to alanine but an additional double mutant S1137A/S1138A was also generated. Transient expression of GFP, GFP-IRS2, GFP-IRS2-S1137A, GFP-IRS2-S1138A, GFP-IRS2-S1163A, or GFP-IRS2-S1137A/S1138A in HEK293 cells was followed by stimulation with forskolin and H89 and subsequent GFP pulldown. The PKA substrate antibody only detected signals in the forskolin-treated samples of GFP-IRS2 and GFP-IRS2-S1163A (Fig. 3C). This result argued against serine 1163 as a PKA phosphorylation site and argued for serine 1137 and serine 1138 as PKA phosphorylation sites on IRS2. Due to the detection of peptides phosphorylated on both serine residues 1137 and 1138 and the PKA consensus site still present in the single mutants, the double mutant GFP-IRS2-S1137A/S1138A was used for further studies. Sequence alignments of IRS2 protein sequences from human, mouse, rat, and frog revealed sequence conservation of these potential PKA phosphorylation sites (Fig. 3D).
FIGURE 3.

Mass spectrometry identifies three candidate residues in IRS2 for phosphorylation by PKA. A, sequences surrounding the potential PKA phosphorylation sites serine 1137, serine 1138, and serine 1163 are shown according to IRS2 mouse numbering. B, MS/MS analysis of a tryptic peptide with the parent mass (M+3H) = 951.4145 revealed a mass spectrum consisting of single- and double-charged fragment masses (preferably ions most relevant to phosphorylation sites are annotated) matching the doubly phosphorylated IRS2-peptide sequence RHpSpSETFSSTTTVTPVSPSFAHNSK. C, HEK293 cells were transiently transfected with GFP, GFP-IRS2, GFP-IRS2-S1137A, GFP-IRS2-S1138A, GFP-IRS2-S1163A, or GFP-IRS2-S1137A/S1138A. Cells were stimulated with 20 μm forskolin for 30 min alone or after pretreatment with 30 μm H89 for 30 min. GFP pulldown assay was performed with 200 μg of total protein and after SDS-PAGE and Western blotting the membrane was incubated with PKA substrate antibody. The corresponding blot reprobed with IRS2 antibody is also shown (n = 3). D, amino acid areas surrounding serine 1137 and serine 1138 from IRS2 in different species are shown: Homo sapiens (NP_003740.2), Mus musculus (NP_001074681.1), Rattus norvegicus (NP_001162104.1), and Xenopus laevis (AAH72768.1).
Mutation of Serine 1137/1138 on IRS2 Reduces Interaction with 14-3-3
If serine 1137 and serine 1138 are important for cAMP-mediated interaction with 14-3-3 proteins, mutation of these sites should reduce this interaction. This hypothesis was tested by stimulating cells that transiently expressed GFP-IRS2 and GFP-IRS2-S1137A/S1138A with forskolin. GST pulldown experiments revealed that wild type IRS2 interacted slightly with both GST-14-3-3β and GST-14-3-3ϵ fusion proteins in the serum-starved condition, which might be due to the detected weak basal phosphorylation on PKA motifs or other sites (Fig. 4A). Forskolin stimulation increased the interaction, and treatment of cells with H89 prior to forskolin stimulation completely abolished it. The double mutant showed no interaction with GST-14-3-3ϵ in the basal condition and a minor interaction with GST-14-3-3β. There was also an interaction detectable in the forskolin-treated samples, but the extent was significantly less in comparison to wild type IRS2 (Fig. 4B). To further confirm these results we performed co-immunoprecipitation assays, where HEK293 cells were transiently transfected with GFP-IRS2 or GFP-IRS2-S1137A/S1138A in addition with Myc-tagged 14-3-3γ. Similar to the GST pulldown assays, the immunoprecipitation of Myc-14-3-3γ revealed a clear reduction of the binding of the double mutant when compared with GFP-IRS2 (Fig. 4, C and D). The regulation of binding by forskolin and H89 was less pronounced when compared with the GST pulldown experiments. Taken together, the data show that serine 1137/1138 are important for the PKA-mediated interaction between IRS2 and 14-3-3.
FIGURE 4.

Mutation of serine 1137/1138 of IRS2 significantly reduces interaction with 14-3-3. A, GFP-IRS2 and GFP-IRS2-S1137A/S1138A were transiently expressed in HEK293 cells and stimulated for 30 min with 20 μm forskolin alone or after preincubation with 30 μm H89 for 30 min. Lysates containing 250 μg of total protein were incubated with 2 μg of GST-14-3-3ϵ or GST-14-3-3β for 2 h, after which they were separated by SDS-PAGE. Transfer onto nitrocellulose membranes was followed by incubation with GFP antibody to visualize the amount of protein that interacted with GST-14-3-3, with PKA substrate antibody and with GST antibody to ensure equal pulldown and loading. B, densitometric analysis of pulldown with GST-14-3-3ϵ. GFP signal intensities were normalized to GST and GFP-IRS2 stimulated with forskolin was set as 1 (mean ± S.E., n = 3, *, p < 0.05 GFP-IRS2 forskolin versus GFP-IRS2-S1137A/S1138A forskolin-treated. C, HEK293 cells were transiently co-transfected with GFP-IRS2 and Myc-14-3-3γ or GFP-IRS2-S1137A/S1138A and Myc-14-3-3γ, and incubated for 30 min with 20 μm forskolin alone or after pretreatment with 30 μm H89 for 30 min. Lysates containing 200 μg of protein were incubated with Myc antibody and Protein G-Sepharose for 3 h at 4 °C. Samples were separated on 5–15% SDS gels and after Western blotting, the membranes were incubated with GFP antibody, PKA substrate antibody and Myc antibody. D, densitometric analysis of co-immunoprecipitated GFP. Band intensities were normalized for Myc and GFP-IRS2 treated with forskolin was set as 1 (mean ± S.E., n = 3, *, p < 0.05, GFP-IRS2 forskolin versus GFP-IRS2-S1137A/S1138A forskolin-treated).
Stimulation with Forskolin Prevents Protein Degradation of IRS2
A strong effect of elevated cAMP levels on the IRS2 protein amount via transcriptional activation has already been established (16). Based on our results we hypothesized additional effects of cAMP, namely on IRS2 protein stability. HEK293 cells stably expressing IRS2 were generated, because stable transfection provides the advantage of a continuous expression of the desired protein at considerably lower levels than in transiently transfected cells, thus preventing an obscuring of IRS2 protein half-life due to permanently high expression levels. To visualize IRS2 protein degradation cells were incubated with cycloheximide to inhibit protein translation. After 3 and 6 h of incubation, IRS2 protein content was reduced by 80%. Concomitant incubation with forskolin reduced the IRS2 protein content only by about 25% (Fig. 5, A and B). Comparable results were also obtained in primary hepatocytes (Fig. 5C).
FIGURE 5.

Forskolin prevents protein degradation of IRS2. A, Flp-In HEK293 cells stably expressing IRS2 were incubated with 25 μg/ml of cycloheximide (CHX) alone or in combination with 20 μm forskolin (FSK) for 1, 3, and 6 h. 10 μg of protein was separated and membranes were incubated with antibodies against IRS2, phosphorylated serine 157 of VASP as stimulation control and β-actin as loading control. B, densitometric analysis of IRS2 protein bands. IRS2 content was normalized for β-actin and IRS2 from untreated cells was set as 100% (mean ± S.E., n = 8, *, p < 0.05 cycloheximide (open squares) versus cycloheximide/forskolin-treated (filled squares)). C, hepatocytes were isolated from male C57Bl/6 mice, plated onto 6-well plates, treated as described in A and 100 μg of protein was analyzed (n = 3).
14-3-3 and Serine 1137/1138 Are Important for IRS2 Protein Stability
For clarification of whether 14-3-3 binding to IRS2 is important for its stability, a dipeptidyl-phosphoserine compound was used (39). It binds to endogenous 14-3-3 proteins, thereby disrupting its binding to target proteins. Treatment of cells with cycloheximide, forskolin, and the 14-3-3 antagonist in parallel abolished the increased IRS2 protein stability that was observed after incubation with cycloheximide and forskolin alone (Fig. 6, A and B). The involvement of the identified PKA phosphorylation sites of serine 1137/1138 in IRS2 protein stability was also tested. GFP-IRS2 stably expressed in Flp-In HEK293 cells showed increased protein stability upon stimulation with forskolin compared with the S1137A/S1138A mutant (Fig. 6, C and D). Thus, the presence of phosphorylated serine 1137/1138 and the interaction with 14-3-3 proteins is necessary for cAMP-dependent IRS2 protein stability.
FIGURE 6.
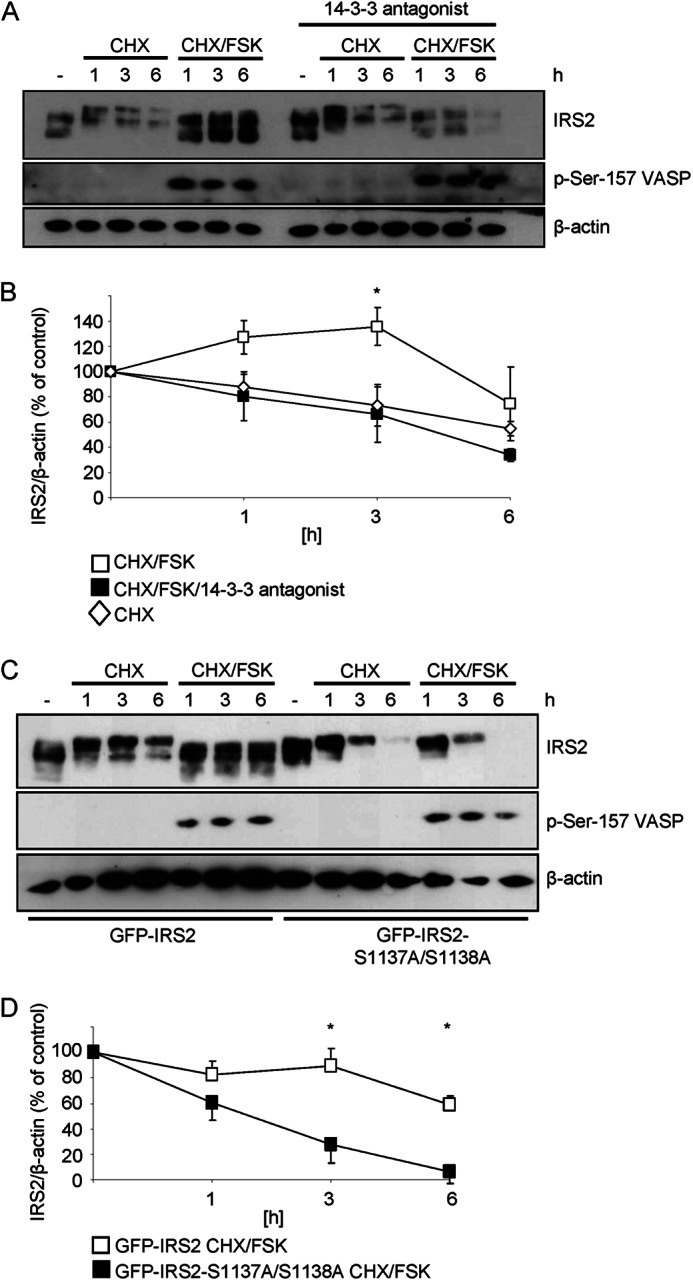
14-3-3 proteins and serine 1137/1138 are important for IRS2 protein stability. A, Flp-In HEK293 cells stably expressing GFP-IRS2 were treated for the indicated time points with 25 μg/ml of cycloheximide (CHX) alone or in combination with 20 μm forskolin (FSK). Another set of cells was treated with 20 μm 14-3-3 antagonist overnight and during the incubation times. 25 μg of protein was separated by SDS-PAGE and membranes were incubated with following antibodies: IRS2, phosphoserine 157 of VASP and β-actin. B, densitometric analysis of IRS2 protein degradation. IRS2 content was normalized for β-actin and IRS2 from untreated cells was set as 100% (mean ± S.E., n = 4, *, p < 0.05 cycloheximide/forskolin (open squares) versus cycloheximide/forskolin/14-3-3 antagonist-treated (filled squares) at 3 h). C, Flp-In HEK293 cells stably expressing GFP-IRS2 or GFP-IRS2-S1137A/S1138A were incubated with 25 μg/ml of CHX and 20 μm FSK for 1, 3, and 6 h. 25 μg of protein was separated by SDS-PAGE and membranes were incubated with IRS2 antibody, phosphoserine 157 of VASP and β-actin antibody. D, the extent of IRS2 protein degradation was quantified by scanning immunoblots and normalization of IRS2 signal intensities for β-actin. Untreated cells expressing GFP-IRS2 or GFP-IRS2-S1137A/S1138A were set to 100% (mean ± S.E., n = 4, *, p < 0.05, GFP-IRS2 (open squares) versus GFP-IRS2-S1137A/S1138A (filled squares) at 3 and 6 h).
DISCUSSION
An increasing number of studies have shown the importance of 14-3-3 proteins in controlling cellular processes like cell cycle, cell growth, gene transcription, and apoptosis as reviewed in Refs. 40–43. IRS proteins and IRS1 and IRS2 in particular serve as intracellular docking and adapter molecules that integrate stimuli from different cellular pathways. Our results provide clear evidence that IRS2 and 14-3-3 are binding partners under conditions of increased cAMP levels. The binding is at least partially mediated via PKA-dependent phosphorylation of IRS2 on serine 1137/1138 and enhances IRS2 protein stability. The binding of 14-3-3 proteins to IRS2 has been shown by Ogihara et al. (33) in Sf9 cells and can also be regulated by an IGF-1/insulin-dependent pathway (34, 44). Now we could demonstrate a second, IGF-1/insulin-independent regulatory mechanism induced by elevated cAMP levels.
Our results indicate that the cAMP-activated phosphorylation of IRS2 involves PKA. Elevated cAMP levels can also activate EPAC (45, 46) and as downstream kinases ERK, RSK, and Akt/PKB (47). Application of different activators and inhibitors of the above mentioned targets revealed that PKA is the major kinase responsible for the phosphorylation of IRS2 upon cAMP stimulation in our system. The activators of EPAC, ERK, and Akt/PKB did not lead to phosphorylation of IRS2 on PKA sites, whereas the PKA activator did. Inhibitors of Akt/PKB or ERK had no effect on forskolin-stimulated IRS2 phosphorylation, but H89 and the PKA inhibitor Rp-8-Br-cAMPS prevented phosphorylation and the increased binding of IRS2 to 14-3-3 proteins. Taken together, these results provide clear indications of involvement of the cAMP-PKA pathway as mediator of IRS2 phosphorylation and binding of 14-3-3 proteins. The inhibitor H89 is very effective in reducing the interaction of IRS2 and 14-3-3 below basal levels. It does not solely inhibit PKA, but can also inhibit MSK1, S6K1, or ROCK-II with a similar potency (48). Therefore it is possible that other pathways regulated by H89 can modulate the IRS2/14-3-3 interaction.
Mass spectrometric analysis and serine to alanine mutants led to the identification of serine 1137 and serine 1138 as PKA phosphorylation sites. Because peptides phosphorylated on both residues 1137 and 1138 at the same time were detected and the single mutation of either serine 1137 (RRHA1137SETF) and serine 1138 (RRHSA1138ETF) still resulted in a PKA phosphorylation motif (RXXpS) we performed the subsequent experiments with the double GFP-IRS2-S1137A/S1138A mutant. Yi et al. (49) found in a previous report that the homologue positions serine 1100 and 1101 in human IRS1 are phosphorylated by PKA in an in vitro kinase assay using expressed GST-IRS1 fusion proteins followed by HPLC-ESI-MS/MS. Due to the lack of additional definitive spectra the authors could not clearly state if only serine 1100 or serine 1101 was phosphorylated or if a doubly phosphorylated peptide was present. These earlier findings of Yi et al. (49) further support the idea of PKA as a kinase to phosphorylate these residues in IRS1 and IRS2.
According to our data serine 1137/1138 seem to be the predominant cAMP-dependent phosphorylation sites on the IRS2 molecule that are located in a PKA motif and can mediate cAMP-induced 14-3-3 binding to IRS2. The importance of serine 1137/1138 was verified by three different experimental approaches: overlay assay, GST pulldown, and co-immunoprecipitation of the transfected Myc-14-3-3 protein. In some experiments interaction between GFP-IRS2 wild type and 14-3-3 fusion proteins could also be found at basal conditions, whereas pretreatment with H89 always reduced the interaction below basal levels. This indicates the existence of further 14-3-3 binding sites on IRS2 that are phosphorylated at basal conditions. These sites could be phosphorylated for example, by other arginine-directed kinases. This would also explain the weak interaction that could be observed between 14-3-3 fusion proteins and the double mutant S1137A/S1138A.
We could show in Flp-In HEK293 cells stably expressing either IRS2 or GFP-IRS2 and also in primary hepatocytes that the half-life of the IRS2 protein was increased when cells were stimulated with forskolin. Serine 1137/1138 proved to be crucial for this increased stability, because a double GFP-IRS2-S1137A/S1138A mutant did not show increased protein stability during forskolin stimulation. Based on the present data we hypothesize that phosphorylation on serine 1137/1138 is necessary for both, the cAMP-enhanced binding to 14-3-3 proteins and the stabilization of the protein. The use of a 14-3-3 antagonist supports the proposed molecular mechanism of cAMP/PKA-dependent IRS2 protein stabilization. Cells stably expressing GFP-IRS2 showed no increased IRS2 protein stability when treated with a 14-3-3 antagonist prior to and during the forskolin stimulation. Other groups also reported increased protein stability associated with 14-3-3 binding. Short et al. (50) showed an AMP-activated protein kinase-dependent phosphorylation of threonine 197 in p27, which increased its 14-3-3 binding and protein half-life. The histone deacetylase HDAC7 was associated with 14-3-3 proteins upon phosphorylation by calcium/calmodulin-dependent kinases, thus preventing its ubiquitination and subsequent degradation via the 26 S proteasome (51). Finally, Yang et al. (52) provided evidence for 14-3-3σ-dependent stabilization of the cell cycle checkpoint protein p53. 14-3-3 binding blocked Mdm2-dependent ubiquitination and nuclear export of p53.
The amount of IRS2 protein present in cells is dependent on mRNA expression and protein stability (2). The stability of the IRS2 protein is regulated by post-translational modifications that mark the molecule for ubiquitination and subsequent proteasomal degradation (18, 53). To mark a protein for proteasomal degradation three classes of enzymes are needed: ubiquitin-activating enzyme, ubiquitin-conjugating enzyme, and ubiquitin ligase (54). In addition, post-translational modifications of IRS2 can lead to a pronounced reduction in the electrophoretic mobility of IRS2 suggesting an increased molecular weight. For example, in refed mice reduced electrophoretic mobility of hepatic IRS2 has been observed by our group (7) and others (2), which are at least partially due to hyperphosphorylation of the IRS2 protein, but ubiquitination might also be involved. In our experiments, cells were incubated with cycloheximide and a shift of IRS2 was observable. It is possible that this shift is caused by a time-dependent increase in the ubiquitination of the protein, which is prevented by the concomitant incubation with forskolin.
We present data suggesting a protection from proteasomal degradation upon cAMP-mediated 14-3-3 binding to IRS2. It is imaginable that this protein/protein interaction results in a conformational change that obscures the recognition motif for an E3 ubiquitin ligase, thus protecting IRS2 from proteasomal degradation. This hypothesis is supported by GlobPlot® analysis that predicted the C-terminal part of the IRS2 molecule to be unstructured. Unstructured regions can often be found in proteins that serve as regulatory and signaling proteins (55), because these intrinsically unstructured regions are suitable for restructuring upon interaction with a binding partner. 14-3-3 proteins display a very rigid structure and therefore can provide themselves as an anvil whereupon the binding partner can be restructured (56). Conformational change upon 14-3-3 binding has been shown in several examples: the phosphatidylinositol 4-kinase IIIβ is protected from dephosphorylation in the active site by 14-3-3 binding (57), 14-3-3 binding to BAD (Bcl-2-associated death promoter) enhances accessibility of serine 155 for phosphorylation by prosurvival kinases (58), and AANAT (serotonin N-acetyltransferase) shows an increased affinity for its substrates upon 14-3-3 binding (31).
Our data indicate an additional mechanism for the up-regulation of IRS2 protein levels in physiological conditions with elevated cAMP concentrations. During fasting, blood glucagon levels are increased and are known to induce IRS2 mRNA expression through glucagon-dependent G protein-coupled increases in cAMP levels, activation of PKA, and subsequent activation of the transcription factor CREB and its coactivator TORC2 (15, 16). Of note, IRS2 expression in the fasting state is important to negatively regulate glucose output from the liver to avoid hyperglycemia (15). Increased IRS2 protein levels in hepatocytes during the fasting state ensure fast and effective signaling upon insulin stimulation in the transition from the fasted to the fed state. Notably, we were able to show phosphorylation of IRS2 on PKA sites in primary mouse hepatocytes after forskolin stimulation as well as increased IRS2 protein stability. The pronounced up-regulation of IRS2 protein levels observed already after 30 min of forskolin stimulation suggests that the primary cell culture model in particular, is responsive to the effects of cAMP on the regulation of IRS2 protein amount.
In pancreatic β-cells IRS2 expression is crucial for growth and survival (5). Increased blood glucose levels in the refed state are followed by insulin secretion of the β-cell. Glucose itself can induce IRS2 mRNA and protein expression in β-cells by a calcium-dependent mechanism (59). In addition, glucagon-like peptide 1 is released into the systemic circulation from enteroendocrine cells during feeding and potentiates insulin secretion. Glucagon-like peptide 1 increases cAMP levels in β-cells, thus promoting β-cell viability via increased IRS2 expression (16). Our finding of increased IRS2 protein stability due to elevated cAMP levels could provide an additional mechanism for the β-cell to protect itself from reduced IRS2 protein levels. Van de Velde et al. (60) observed rapid gene expression of CREB target genes by forskolin stimulation that decreased to baseline levels after 4 h, whereas elevated IRS2 protein expression was measurable by Western blot up to 16 h in INS-1 cells. This discrepancy in decreased in IRS2 gene activation but elevated IRS2 protein levels could be explained with our finding of increased IRS2 protein stability due to cAMP-induced 14-3-3 binding to IRS2. Of course, our considerations are based on the mechanistic investigation of cAMP-dependent interaction between IRS2 and 14-3-3 in our study and have to be confirmed in β-cells and subsequently in vivo.
To conclude, we identified serine 1137/1138 as PKA phosphorylation sites on IRS2 that mediate 14-3-3 binding and regulate IRS2 protein stability. The increased IRS2 protein stability upon elevated cAMP levels provides an additional mechanism to cAMP-induced IRS2 mRNA and subsequent protein expression to ensure sufficient amounts of IRS2 protein.
Acknowledgments
We are grateful to Prof. Carol MacKintosh and Dr. Rachel Toth from the University of Dundee for their help in generation of some of the expression vectors used in this work and the initial experiments that led to this study. Furthermore, we thank Prof. Reiner Lammers for providing the Myc-14-3-3γ construct and the Myc antibody, Angelika Hausser for GST-14-3-3 expression vectors, and Morris White for the pRK5 IRS2 expression vector. In addition we thank Heike Runge and Ann Kathrin Pohl for their outstanding technical assistance and Madhura Panse for proofreading the manuscript.
This work was supported by Grant GRK 1302/2 from the Deutsche Forschungsgemeinschaft (to H. U. H., E. S., and C. W.) in part by a grant from the German Federal Ministry of Education and Research (BMBF) to the German Center for Diabetes Research (DZD e.V.), and by European Community's Seventh Framework Programme FP7/2009 Grant 241955.
- IRS
- insulin receptor substrate
- Akti
- Akt inhibitor
- CPT-cAMP
- 8-(4-chlorophenylthio) cAMP
- 8-pCPT-2′-O-Me-cAMP
- 8-(4-chlorophenylthio)-2′-O-methyl-cAMP
- CREB
- cAMP response element-binding protein
- EPAC
- exchange factor directly activated by cAMP
- Grb2
- growth factor receptor-bound protein 2
- GSK3
- glycogen synthase kinase 3
- IGF-1
- insulin-like growth factor 1
- MSK
- mitogen and stress activated kinase
- 6-Phe-cAMP
- N6-phenyladenosine-cAMP
- PKA
- protein kinase A
- S6K
- protein S6 kinase
- Rp-8-Br-cAMPS
- 8-bromoadenosine-3′,5′-cyclic monophosphorothioate, Rp-isomer
- ROCK
- Rho-associated kinase
- VASP
- vasodilator-stimulated protein
- EGF
- epidermal growth factor
- BisTris
- BisTris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Kubota N., Tobe K., Terauchi Y., Eto K., Yamauchi T., Suzuki R., Tsubamoto Y., Komeda K., Nakano R., Miki H., Satoh S., Sekihara H., Sciacchitano S., Lesniak M., Aizawa S., Nagai R., Kimura S., Akanuma Y., Taylor S. I., Kadowaki T. (2000) Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory beta-cell hyperplasia. Diabetes 49, 1880–1889 [DOI] [PubMed] [Google Scholar]
- 2. Kubota N., Kubota T., Itoh S., Kumagai H., Kozono H., Takamoto I., Mineyama T., Ogata H., Tokuyama K., Ohsugi M., Sasako T., Moroi M., Sugi K., Kakuta S., Iwakura Y., Noda T., Ohnishi S., Nagai R., Tobe K., Terauchi Y., Ueki K., Kadowaki T. (2008) Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metab. 8, 49–64 [DOI] [PubMed] [Google Scholar]
- 3. Withers D. J., Gutierrez J. S., Towery H., Burks D. J., Ren J. M., Previs S., Zhang Y., Bernal D., Pons S., Shulman G. I., Bonner-Weir S., White M. F. (1998) Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391, 900–904 [DOI] [PubMed] [Google Scholar]
- 4. Schuppin G. T., Pons S., Hügl S., Aiello L. P., King G. L., White M., Rhodes C. J. (1998) A specific increased expression of insulin receptor substrate 2 in pancreatic beta-cell lines is involved in mediating serum-stimulated beta-cell growth. Diabetes 47, 1074–1085 [DOI] [PubMed] [Google Scholar]
- 5. Hennige A. M., Burks D. J., Ozcan U., Kulkarni R. N., Ye J., Park S., Schubert M., Fisher T. L., Dow M. A., Leshan R., Zakaria M., Mossa-Basha M., White M. F. (2003) Up-regulation of insulin receptor substrate-2 in pancreatic beta cells prevents diabetes. J. Clin. Invest. 112, 1521–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sharfi H., Eldar-Finkelman H. (2008) Sequential phosphorylation of insulin receptor substrate-2 by glycogen synthase kinase-3 and c-Jun NH2-terminal kinase plays a role in hepatic insulin signaling. Am. J. Physiol. Endocrinol. Metab. 294, E307-E315 [DOI] [PubMed] [Google Scholar]
- 7. Fritsche L., Neukamm S. S., Lehmann R., Kremmer E., Hennige A. M., Hunder-Gugel A., Schenk M., Häring H.U., Schleicher E. D., Weigert C. (2011) Insulin-induced serine phosphorylation of IRS-2 via ERK1/2 and mTOR. Studies on the function of Ser-675 and Ser-907. Am. J. Physiol. Endocrinol. Metab. 300, E824-E836 [DOI] [PubMed] [Google Scholar]
- 8. Solinas G., Naugler W., Galimi F., Lee M. S., Karin M. (2006) Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc. Natl. Acad. Sci. U.S.A. 103, 16454–16459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oriente F., Formisano P., Miele C., Fiory F., Maitan M. A., Vigliotta G., Trencia A., Santopietro S., Caruso M., Van Obberghen E., Beguinot F. (2001) Insulin receptor substrate-2 phosphorylation is necessary for protein kinase Cζ activation by insulin in L6hIR cells. J. Biol. Chem. 276, 37109–37119 [DOI] [PubMed] [Google Scholar]
- 10. Greene M. W., Garofalo R. S. (2002) Positive and negative regulatory role of insulin receptor substrate 1 and 2 (IRS-1 and IRS-2) serine/threonine phosphorylation. Biochemistry 41, 7082–7091 [DOI] [PubMed] [Google Scholar]
- 11. Gurevitch D., Boura-Halfon S., Isaac R., Shahaf G., Alberstein M., Ronen D., Lewis E. C., Zick Y. (2010) Elimination of negative feedback control mechanisms along the insulin signaling pathway improves beta-cell function under stress. Diabetes 59, 2188–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weigert C., Kron M., Kalbacher H., Pohl A. K., Runge H., Häring H. U., Schleicher E., Lehmann R. (2008) Interplay and effects of temporal changes in the phosphorylation state of serine-302, -307, and -318 of insulin receptor substrate-1 on insulin action in skeletal muscle cells. Mol. Endocrinol. 22, 2729–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Danielsson A., Ost A., Nystrom F. H., Strålfors P. (2005) Attenuation of insulin-stimulated insulin receptor substrate-1 serine 307 phosphorylation in insulin resistance of type 2 diabetes. J. Biol. Chem. 280, 34389–34392 [DOI] [PubMed] [Google Scholar]
- 14. Copps K. D., Hancer N. J., Opare-Ado L., Qiu W., Walsh C., White M. F. (2010) Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab. 11, 84–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Canettieri G., Koo S. H., Berdeaux R., Heredia J., Hedrick S., Zhang X., Montminy M. (2005) Dual role of the coactivator TORC2 in modulating hepatic glucose output and insulin signaling. Cell Metab. 2, 331–338 [DOI] [PubMed] [Google Scholar]
- 16. Jhala U. S., Canettieri G., Screaton R. A., Kulkarni R. N., Krajewski S., Reed J., Walker J., Lin X., White M., Montminy M. (2003) cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev. 17, 1575–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hirashima Y., Tsuruzoe K., Kodama S., Igata M., Toyonaga T., Ueki K., Kahn C.R., Araki E. (2003) Insulin down-regulates insulin receptor substrate-2 expression through the phosphatidylinositol 3-kinase/Akt pathway. J. Endocrinol. 179, 253–266 [DOI] [PubMed] [Google Scholar]
- 18. Rui L., Fisher T. L., Thomas J., White M.F. (2001) Regulation of insulin/insulin-like growth factor-1 signaling by proteasome-mediated degradation of insulin receptor substrate-2. J. Biol. Chem. 276, 40362–40367 [DOI] [PubMed] [Google Scholar]
- 19. Kleppe R., Martinez A., Døskeland S. O., Haavik J. (2011) The 14-3-3 proteins in regulation of cellular metabolism. Semin. Cell Dev. Biol. 22, 713–719 [DOI] [PubMed] [Google Scholar]
- 20. Wilker E. W., Grant R. A., Artim S. C., Yaffe M. B. (2005) A structural basis for 14-3-3σ functional specificity. J. Biol. Chem. 280, 18891–18898 [DOI] [PubMed] [Google Scholar]
- 21. Liu D., Bienkowska J., Petosa C., Collier R. J., Fu H., Liddington R. (1995) Crystal structure of the ζ isoform of the 14-3-3 protein. Nature 376, 191–194 [DOI] [PubMed] [Google Scholar]
- 22. Muslin A. J., Tanner J. W., Allen P. M., Shaw A. S. (1996) Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84, 889–897 [DOI] [PubMed] [Google Scholar]
- 23. Yaffe M. B., Rittinger K., Volinia S., Caron P. R., Aitken A., Leffers H., Gamblin S. J., Smerdon S. J., Cantley L. C. (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91, 961–971 [DOI] [PubMed] [Google Scholar]
- 24. Rittinger K., Budman J., Xu J., Volinia S., Cantley L. C., Smerdon S.J., Gamblin S. J., Yaffe M. B. (1999) Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol. Cell. 4, 153–166 [DOI] [PubMed] [Google Scholar]
- 25. Masters S. C., Pederson K. J., Zhang L., Barbieri J. T., Fu H. (1999) Interaction of 14-3-3 with a nonphosphorylated protein ligand, exoenzyme S of Pseudomonas aeruginosa. Biochemistry 38, 5216–5221 [DOI] [PubMed] [Google Scholar]
- 26. Sumioka A., Nagaishi S., Yoshida T., Lin A., Miura M., Suzuki T. (2005) Role of 14-3-3γ in FE65-dependent gene transactivation mediated by the amyloid β-protein precursor cytoplasmic fragment. J. Biol. Chem. 280, 42364–42374 [DOI] [PubMed] [Google Scholar]
- 27. Seimiya H., Sawada H., Muramatsu Y., Shimizu M., Ohko K., Yamane K., Tsuruo T. (2000) Involvement of 14-3-3 proteins in nuclear localization of telomerase. EMBO J. 19, 2652–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O'Kelly I., Butler M. H., Zilberberg N., Goldstein S. A. (2002) Forward transport. 14-3-3 binding overcomes retention in endoplasmic reticulum by dibasic signals. Cell 111, 577–588 [DOI] [PubMed] [Google Scholar]
- 29. Zhao X., Gan L., Pan H., Kan D., Majeski M., Adam S. A., Unterman T. G. (2004) Multiple elements regulate nuclear/cytoplasmic shuttling of FOXO1. Characterization of phosphorylation- and 14-3-3-dependent and -independent mechanisms. Biochem. J. 378, 839–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sakiyama H., Wynn R. M., Lee W.R., Fukasawa M., Mizuguchi H., Gardner K. H., Repa J. J., Uyeda K. (2008) Regulation of nuclear import/export of carbohydrate response element-binding protein (ChREBP). Interaction of an α-helix of ChREBP with the 14-3-3 proteins and regulation by phosphorylation. J. Biol. Chem. 283, 24899–24908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Obsil T., Ghirlando R., Klein D. C., Ganguly S., Dyda F. (2001) Crystal structure of the 14-3-3ζ:serotonin N-acetyltransferase complex. A role for scaffolding in enzyme regulation. Cell 105, 257–267 [DOI] [PubMed] [Google Scholar]
- 32. Agarwal-Mawal A., Qureshi H. Y., Cafferty P. W., Yuan Z., Han D., Lin R., Paudel H. K. (2003) 14-3-3 connects glycogen synthase kinase-3β to Tau within a brain microtubule-associated tau phosphorylation complex. J. Biol. Chem. 278, 12722–12728 [DOI] [PubMed] [Google Scholar]
- 33. Ogihara T., Isobe T., Ichimura T., Taoka M., Funaki M., Sakoda H., Onishi Y., Inukai K., Anai M., Fukushima Y., Kikuchi M., Yazaki Y., Oka Y., Asano T. (1997) 14-3-3 protein binds to insulin receptor substrate-1, one of the binding sites of which is in the phosphotyrosine binding domain. J. Biol. Chem. 272, 25267–25274 [DOI] [PubMed] [Google Scholar]
- 34. Dubois F., Vandermoere F., Gernez A., Murphy J., Toth R., Chen S., Geraghty K.M., Morrice N.A., MacKintosh C. (2009) Differential 14-3-3 affinity capture reveals new downstream targets of phosphatidylinositol 3-kinase signaling. Mol. Cell. Proteomics 8, 2487–2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen C., Okayama H. (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7, 2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weigert C., Brodbeck K., Staiger H., Kausch C., Machicao F., Häring H. U., Schleicher E. D. (2004) Palmitate, but not unsaturated fatty acids, induces the expression of interleukin-6 in human myotubes through proteasome-dependent activation of nuclear factor-κB. J. Biol. Chem. 279, 23942–23952 [DOI] [PubMed] [Google Scholar]
- 37. Gloeckner C. J., Boldt K., Schumacher A., Roepman R., Ueffing M. (2007) A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics 7, 4228–4234 [DOI] [PubMed] [Google Scholar]
- 38. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline. An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu H., Ge J., Yao S. Q. (2010) Microarray-assisted high-throughput identification of a cell-permeable small-molecule binder of 14-3-3 proteins. Angew. Chem. Int. Ed. Engl. 49, 6528–6532 [DOI] [PubMed] [Google Scholar]
- 40. Obsilová V., Silhan J., Boura E., Teisinger J., Obsil T. (2008) 14-3-3 proteins. A family of versatile molecular regulators. Physiol. Res. 57, S11-S21 [DOI] [PubMed] [Google Scholar]
- 41. Mackintosh C. (2004) Dynamic interactions between 14-3-3 proteins and phosphoproteins regulate diverse cellular processes. Biochem. J. 381, 329–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ferl R. J. (2004) 14-3-3 proteins. Regulation of signal-induced events. Physiol Plant. 120, 173–178 [DOI] [PubMed] [Google Scholar]
- 43. Fu H., Subramanian R. R., Masters S. C. (2000) 14-3-3 proteins. Structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40, 617–647 [DOI] [PubMed] [Google Scholar]
- 44. Neukamm S. S., Toth R., Morrice N., Campbell D. G., Mackintosh C., Lehmann R., Haering H. U., Schleicher E. D., Weigert C. (2012) Identification of the amino acids 300–600 of IRS-2 as 14-3-3 binding region with the importance of IGF-1/insulin-regulated phosphorylation of Ser-573. PLoS One 7, e43296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kawasaki H., Springett G. M., Mochizuki N., Toki S., Nakaya M., Matsuda M., Housman D. E., Graybiel A. M. (1998) A family of cAMP-binding proteins that directly activate Rap1. Science 282, 2275–2279 [DOI] [PubMed] [Google Scholar]
- 46. de Rooij J., Zwartkruis F. J., Verheijen M. H., Cool R. H., Nijman S. M., Wittinghofer A., Bos J. L. (1998) Epac is a Rap1 guanine-nucleotide exchange factor directly activated by cyclic AMP. Nature 396, 474–477 [DOI] [PubMed] [Google Scholar]
- 47. Roscioni S. S., Elzinga C. R., Schmidt M. (2008) Epac. Effectors and biological functions. Naunyn Schmiedebergs Arch. Pharmacol. 377, 345–357 [DOI] [PubMed] [Google Scholar]
- 48. Davies S. P., Reddy H., Caivano M., Cohen P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yi Z., Luo M., Carroll C. A., Weintraub S. T., Mandarino L. J. (2005) Identification of phosphorylation sites in insulin receptor substrate-1 by hypothesis-driven high-performance liquid chromatography-electrospray ionization tandem mass spectrometry. Anal. Chem. 77, 5693–5699 [DOI] [PubMed] [Google Scholar]
- 50. Short J. D., Dere R., Houston K. D., Cai S. L., Kim J., Bergeron J. M., Shen J., Liang J., Bedford M. T., Mills G. B., Walker C. L. (2010) AMPK-mediated phosphorylation of murine p27 at T197 promotes binding of 14-3-3 proteins and increases p27 stability. Mol. Carcinog. 49, 429–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li X., Song S., Liu Y., Ko S. H., Kao H.Y. (2004) Phosphorylation of the histone deacetylase 7 modulates its stability and association with 14-3-3 proteins. J. Biol. Chem. 279, 34201–34208 [DOI] [PubMed] [Google Scholar]
- 52. Yang H. Y., Wen Y. Y., Chen C. H., Lozano G., Lee M. H. (2003) 14-3-3σ positively regulates p53 and suppresses tumor growth. Mol. Cell. Biol. 23, 7096–7107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rui L., Yuan M., Frantz D., Shoelson S., White M. F. (2002) SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J. Biol. Chem. 277, 42394–42398 [DOI] [PubMed] [Google Scholar]
- 54. Hershko A., Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 55. Iakoucheva L. M., Brown C. J., Lawson J. D., Obradović Z., Dunker A. K. (2002) Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 323, 573–584 [DOI] [PubMed] [Google Scholar]
- 56. Yaffe M. B. (2002) How do 14-3-3 proteins work? Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 513, 53–57 [DOI] [PubMed] [Google Scholar]
- 57. Hausser A., Link G., Hoene M., Russo C., Selchow O., Pfizenmaier K. (2006) Phospho-specific binding of 14-3-3 proteins to phosphatidylinositol 4-kinase IIIβ protects from dephosphorylation and stabilizes lipid kinase activity. J. Cell Sci. 119, 3613–3621 [DOI] [PubMed] [Google Scholar]
- 58. Datta S. R., Katsov A., Hu L., Petros A., Fesik S. W., Yaffe M. B., Greenberg M.E. (2000) 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol. Cell 6, 41–51 [PubMed] [Google Scholar]
- 59. Lingohr M. K., Briaud I., Dickson L. M., McCuaig J. F., Alárcon C., Wicksteed B. L., Rhodes C. J. (2006) Specific regulation of IRS-2 expression by glucose in rat primary pancreatic islet beta-cells. J. Biol. Chem. 281, 15884–15892 [DOI] [PubMed] [Google Scholar]
- 60. Van de Velde S., Hogan M. F., Montminy M. (2011) mTOR links incretin signaling to HIF induction in pancreatic beta cells. Proc. Natl. Acad. Sci. U.S.A. 108, 16876–16882 [DOI] [PMC free article] [PubMed] [Google Scholar]