

Background: Polyketide synthases (PKSs) synthesize complex lipid components of the cell wall in M. tuberculosis.
Results: We determined the crystal structure of M. tuberculosis PKS11 and identified its substrate, intermediates, and product.
Conclusion: M. tuberculosis PKS11 synthesizes a unique cyclic methyl-branched alkylpyrone.
Significance: Our identification of alkylpyrones as the product of this PKS suggests a previously unknown component of the mycobacterial cell wall.
Keywords: Crystal Structure, Crystallography, Lipid Synthesis, Polyketides, Tuberculosis, Mycobacterium tuberculosis, PKS11, Alkylpyrones, Lipids
Abstract
PKS11 is one of three type III polyketide synthases (PKSs) identified in Mycobacterium tuberculosis. Although many PKSs in M. tuberculosis have been implicated in producing complex cell wall glycolipids, the biological function of PKS11 is unknown. PKS11 has previously been proposed to synthesize alkylpyrones from fatty acid substrates. We solved the crystal structure of M. tuberculosis PKS11 and found the overall fold to be similar to other type III PKSs. PKS11 has a deep hydrophobic tunnel proximal to the active site Cys-138 to accommodate substrates. We observed electron density in this tunnel from a co-purified molecule that was identified by mass spectrometry to be palmitate. Co-crystallization with malonyl-CoA (MCoA) or methylmalonyl-CoA (MMCoA) led to partial turnover of the substrate, resulting in trapped intermediates. Reconstitution of the reaction in solution confirmed that both co-factors are required for optimal activity, and kinetic analysis shows that MMCoA is incorporated first, then MCoA, followed by lactonization to produce methyl-branched alkylpyrones.
Introduction
Mycobacterium tuberculosis polyketide synthase 11 (PKS11)2 (Rv1665) is one of three type III polyketide synthases found in the M. tuberculosis genome. Although its biological function is currently unknown, PKS11 has been implicated in the biosynthesis of long chain fatty acid-derived lipids (1). PKS11 has been shown to be nonessential in vitro (2) and in vivo in a mouse model of infection by transposon mutagenesis (3). Transposon insertion mutants of PKS11 were found to be defective in the biosynthesis of phthiocerol dimycocersate (4), although these defects might have been due to secondary mutations in the phthiocerol dimycocersate locus, which are frequently observed in laboratory settings (5).
In other organisms, PKSs produce a wide range of secondary metabolites, from flavonoids and phytoalexins in plants, to complex cell wall lipids in prokaryotes, as well as many antibacterial and antifungal natural products (e.g. anthracyclines, macrolides). PKSs carry out repeated two-carbon chain extensions of their substrates, through condensation with malonyl-CoA (MCoA) and thioesterification of intermediates. In type I PKSs (modular), each catalytic step is carried out by a different domain. Type II PKSs are complexes that bring multiple functional domains together. Type III PKSs utilize an iterative mechanism to extend a substrate multiple times with a single domain.
Although several of the products of the PKSs in M. tuberculosis that have been identified are all linear ketides (for example, mycocerosic acid, mycoceranic acid, mycolepenic acid, and mycoketide, synthesized by mas, pks2, pks3/4, and pks12, respectively), the final products of PKSs are often cyclized in other organisms (e.g. chalcone, stilbene, and resveratrol (6)). In fact, PKS11 has 57% amino acid identity to Streptomyces griseus SrsA, which produces cyclic alkyl-resorcinols (1,3-dihydroxyphenols), which are phenolic lipids that serve as a membrane permeability barrier and prevent the entry of antibacterial agents (7). The cyclization step of some PKSs, whereby an aromatic ring is formed from the linear ketide, has been shown to follow one of three different mechanistic routes. Medicago sativa (alfalfa) chalcone synthase (CHS) and Streptomyces coelicolor tetrahydroxynaphthylene synthase utilize a Claisen condensation (nucleophilic attack of C6 on C1, using the conventional numbering of carbons in the acyl chain starting from the esterified carbon) (8, 9); Pinus sylvestris (pine) stilbene synthase and Neurospora crassa 2′-oxoresorcylic acid synthase (ORAS) use an aldol condensation of C2 with C7 (10, 11). Finally, pyrone rings may be formed by lactonization via attack of the C5 keto oxygen on the thioesterified C1. Whereas the latter is considered a “derailment” product for most enzymes, it is the primary mechanism used to synthesize the antifungal gerberin by 2-pyrone synthase in Gerbera hydrida (12). The Claisen condensation and lactonization mechanisms autonomously cleave the protein-bound thioester, whereas the aldol reaction requires a subsequent hydrolysis step to release the product from the enzyme.
M. tuberculosis PKS11 has 26% amino acid identity to PKS18, another type III PKS in the M. tuberculosis genome, which catalyzes the formation of alkylpyrones from C6 to C20 substrates (13). Similarly, PKS11 has also been shown to produce alkylpyrones from hexanoyl- and lauroyl-CoA when incubated with MCoA (1), although it is not clear whether these shorter chain fatty acids represent cognate substrates. PKS18 bears structural similarity to N. crassa ORAS, which also has a deep hydrophobic substrate-binding channel for binding long chain fatty acid substrates (up to C24) (11). However, N. crassa ORAS has been shown to produce alkyl-resorcinols from longer chain substrates through aldol condensation (14). Although mutations of residues lining the substrate-binding channel of PKS18 have been shown to modulate substrate specificity (via length), the reasons leading to pyrone formation are unknown in this enzyme (15).
In this paper, we report the crystal structure of M. tuberculosis PKS11, along with several complexes. Although the overall fold is similar to other type III PKSs, unexpected density was observed in the PKS11 active site for compounds that were identified as palmitate that co-purified with the protein. Co-crystallization of PKS11 with MCoA and, separately, with methylmalonyl-CoA (MMCoA) led to partial turnover of this substrate. Thus, in contrast to previous studies, we show that one of the elongation steps is preformed with MMCoA, resulting in a novel product, a methyl-branched alkyl-pyrone, from a longer starter molecule (C16). The reaction was reconstituted in solution, and mass spectrometry and burst-phase kinetic analysis were used to resolve the order of co-factor incorporation.
EXPERIMENTAL PROCEDURES
Cloning and Expression of M. tuberculosis PKS11
A 1,062-bp DNA fragment containing the PKS11 (Rv1665) gene was amplified by PCR, using M. tuberculosis H37Rv genomic DNA as a template, by the following oligonucleotide primers 5′-AGA TGA AGC ATA TGA ACG AGG CGC TCG ACG AT-3′ and 5′-AGA GTA AGC TTA TGG TAT ATG CTG CCT ATC GC-3′. The amplified DNA fragment was digested with the NdeI and HindIII restriction enzymes and subcloned into the corresponding restriction sites in the M. smegmatis vector P1602-dest17. The ligated plasmid was transformed into M. smegmatis mc2 155 by the electroporation method. The transformed cells were grown to exponential phase at 37 °C in 7H9 medium containing kanamycin (20 μg/ml) and hygromycin (80 μg/ml). Cells were induced with acetamide (final concentration 0.2%), and the cells were harvested after growth for 8 h at 37 °C. Se-Met protein was prepared by the inhibition method (16). For production of Se-Met-labeled protein, the cells were grown in minimal medium, supplemented with selected amino acids and selenomethionine. Expression of PKS11 was then induced using acetamide (final concentration 0.2%), and the cells were harvested after growth for 14 h at 37 °C.
Protein Purification
The harvested cells were resuspended in buffer A (20 mm Tris-HCl, pH 7.5, 500 mm NaCl, and 1% Triton X-100) containing 1 mm PMSF, DNase, and complete EDTA-free protease inhibitors (Roche Applied Science). The cells were lysed at 25,000 p.s.i. using a French press (M-110P; Microfluid Corporation), and the cell suspension was centrifuged at 15,000 rpm for 1 h. The clear supernatant was used for ammonium sulfate precipitation, and PKS11 precipitated between 20–40% ammonium sulfate. The above pellet was dissolved and dialyzed against 20 mm Tris, pH 8.0, containing protease inhibitors, 1 mm EDTA, 2 mm DTT, and 5% glycerol. PKS11 was purified to near homogeneity using Q-Sepharose ion exchange (GE Healthcare) and a gel filtration column (Sephacryl S-300). In the gel filtration column PKS11 eluted as a homodimer (70 kDa). The protein was >95% pure, as observed by SDS-PAGE. The PKS11-C138S mutant protein was expressed and purified using the same protocol.
Mutagenesis
The C138S mutant of PKS11 was constructed using the QuikChange site-directed mutagenesis kit (Stratagene). Mutagenesis was carried out in accordance with the manufacturer's instruction. The presence of the mutation was confirmed by DNA sequencing. Mutant plasmids were transformed into M. smegmatis, and protein expression was verified. All mutants expressed protein at 37 °C, however the level of expression was low compared with the wild-type protein. The forward primer used was TTC GGT CTG GGC TCC GTG GCA GGG, and the reverse primer used was AAG CCA GAC CCG AGG CAC CGT CCC.
Crystallization
Crystallization of PKS11 was carried out in sitting drops consisting of 1 μl of purified selenomethionine-labeled protein (16) (10 mg of protein) and 1 μl of crystallization buffer from the well solution. The crystallization buffer contained 0.1 m sodium acetate trihydrate, pH 5.2–6.0, 5.6% PEG 4000, and 10% isopropyl alcohol as a precipitant. Crystals were grown at 16 °C, and diffraction-quality crystals were obtained within 2–5 days and optimized by the hanging drop method. Crystals were soaked in mother solution containing 30% ethylene glycol, and they were flash cooled at 100 K in a nitrogen gas stream. PKS11 was preincubated with MCoA (1:10 and 1:75 molar ratio) for 12 h at 4 °C. Crystallization of the protein complex was carried out using the sitting drop method by mixing of 1 μl of protein with 1 μl of well solution. The PKS11 and MCoA complex crystallized in well solution containing 0.2 m ammonium acetate and 29% PEG 3350 at pH 7.1. Diffraction quality crystals were obtained within 8 days. Crystallization of PKS11 with MMCoA and MCoA, as well as PKS11-MCoA or PKS11-MMCoA complex, was carried out using the sitting drop method. PKS11 was preincubated with the respective ligands for 12 h at 4 °C. Optimization and diffracting quality crystals were obtained by the sitting drop method.
PKS11 C138S crystals were obtained by the hanging drop method. The crystallization buffer contained sodium citrate buffer, pH 5.2–6.0, 5.6% PEG 4000 and 10% isopropyl alcohol as the precipitant. These crystals were grown at 16 °C, and diffraction quality crystals were obtained within 7 days.
Data Collection and Processing
Highly redundant and complete selenium K-edge multiple anomalous dispersion diffraction data from a single SeMet PKS11 crystal were collected at two wavelengths (peak and inflection), using an ADSC CCD detector on beam line 23-ID at the Advanced Photon Source (APS) of the Argonne National Laboratory (ANL). Crystals mounted in cryoloops were flash cooled in a liquid N2 stream (120 K) after brief soaks in mother liquid containing 20% ethylene glycol. The diffraction data were reduced using DENZO (17) and intensities were scaled with SCALEPACK (17). The reflections were indexed as primitive monoclinic. Examination of the integrated and scaled data indicated the primitive monoclinic space group P21. Solvent content calculations (18) indicated the presence of four molecules (VM 2.68, VS 54%) in the asymmetric unit.
Determination of PKS11 Structure
Experimental phases for M. tuberculosis PKS11 were obtained by multiple anomalous dispersion phasing (19) (Table 1). Sharp located the 16 selenium sites in the asymmetric unit, consistent with four PKS11 molecules in the asymmetric unit (20). Further phase improvement with solvent flattening in AUTOSHARP (21) followed by DM (21) resulted in density-modified maps of high quality showing clear electron density for all four molecules of protein in the asymmetric unit. Inspection of electron density maps and subsequent model building were performed in Xfit and Coot. Water molecules were added manually during iterative cycles of model building and refinement using a Fourier difference map. Several cycles of manual model building, and NCS-restrained maximum likelihood refinement in REFMAC5 (22), yielded a high quality model with an R-factor of 21.0% and R-free of 26.0% (Table 1) for the PKS11 dimer complex. Coordinate and parameter files for the ligands were generated using the PRODRG website.
TABLE 1.
Data collection, refinement, and geometry statistics
| Parameters | Crystal 1: PKS11 | Crystal 2: PKS11+MCoA | Crystal 3: PKS11+MMCoA | Crystal 4: PKS11+MCoA+ MMCoA |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P21 | P21 | P21 | P21 |
| Wavelength (Å) | 0.97 | 0.97 | 0.97 | 0.97 |
| a (Å) | 71.12 | 72.00 | 72.01 | 72.59 |
| b (Å) | 48.42 | 48.71 | 48.62 | 48.53 |
| c (Å) | 189.69 | 194.29 | 193.65 | 195.23 |
| α | 90.00 | 90.00 | 90.00 | 90.00 |
| β | 97.25 | 97.89 | 97.57 | 98.40 |
| γ | 90.00 | 90.00 | 90.00 | 90.00 |
| Resolution (Å) | 50-2.05 | 50-1.73 | 50-1.83 | 50-1.98 |
| Highest resolution bin (Å) | 2.05-2.15 | 1.73-1.77 | 1.83-1.87 | 1.97-2.04 |
| Observed reflectionsa | 81,856 | 140,395 | 117,929 | 95,038 |
| Unique reflectionsa | 63,039 | 125,862 | 90,986 | 66,722 |
| % Completeness | 96 (92.2) | 89 (74.3) | 80.5 (33.1) | 70.9 (23.6) |
| Rmergea | 0.054 | 0.058 | 0.080 | 0.0712 |
| <I/σ(I)>a | 11.66 (6.39) | 12.64 (1.65) | 6.34 (0.22) | 7.42 (0.81) |
| Refinement | ||||
| Free R value,a random, 5% | 27.04 | 22.06 | 28.71 | 31.41 |
| R valuea | 20.79 | 17.34 | 22.93 | 26.8 |
| Protein residues | 1,408 | 1,408 | 1,408 | 1,408 |
| Water molecules | 389 | 822 | 61 | |
| Ligands | Palmitate (subunits A, D); thioester of 2-methylhepta decanoate (in B, C) | Triketide thioester (subunits A, D); free diketide (B, C) | Thioester of 2-methyldiketide (A, B); palmitate (C, D) | |
| Co-factors built | CoA (in B and C) | CoA (in B and C) | ||
| R.m.s.d. bond length (Å)b | 0.01 | 0.02 | 0.02 | 0.01 |
| R.m.s.d. bond angle (Å)b | 2.038 | 2.483 | 2.293 | 1.761 |
| Overall coordinate error (Å)c | 0.26 | 0.12 | 0.20 | 0.36 |
| RSCC (REFMAC5)d | 0.95 | 0.96 | 0.96 | |
| Most favored (%) | 1,313 (94.05)% | 1,351 (97.26)% | 1,310 (94.04)% | 1,257 (89.72)% |
| Generously allowed (%) | 66 (4.73)% | 33 (2.38)% | 64 (4.59)% | 117 (8.35)% |
| Disallowed (%) | 17 (1.22)% | 5 (0.36)% | 19 (1.36)% | 27 (1.93)% |
Structures of PKS11-MCoA, PKS11-MMCoA, and PKS11-MCoA-MMCoA complexes were solved by molecular replacement using EPMR (23) using PKS11-SeMet structure as a search model. These crystals belonged to the P21 space group and contained two functional dimers in the asymmetric unit. These models were further manually built using Coot and refined in REFMAC5 (22). Water molecules were added manually during iterative cycles of manual model building and refined with REFMAC5 against the high resolution data set. The crystallographic data collection, statistics, and refinement parameters are summarized in Table 1.
Isolation and Identification of the Ligand Co-purified with PKS11
The bound substrate molecule was extracted from purified PKS11 by the acidification method. The standard reaction mixture containing 200 μl of 100 mm sodium acetate buffer, pH 4.2, 1 mm EDTA, and 200 μl of PKS11 C138S (10 mg/ml) was mixed to precipitate the proteins. After the precipitation the sample was centrifuged at 15,000 rpm for 10 min at 4 °C. The supernatant was separated from the pellet and mixed with an equal volume of ethyl acetate. The organic layer was separated from the mixture, and the procedure was repeated at least three times. The organic extracts were pooled and evaporated to dryness under nitrogen. The residual material was dissolved in 50 μl of methanol and analyzed by mass spectrometry.
Enzyme Assay
The standard reaction mixture contained 200 μm starter unit, 100 μm MCoA, or 100 μm MCoA and 100 μm MMCoA, 100 m Tris-HCl, pH 8.0, 1 mm EDTA, 1 mm 2-mercaptoethanol, and 20 μm PKS11 or PKS11 C138S in a total volume of 400 μl. The reactions were incubated at 30 °C for 2 h and then quenched with 5% acetic acid. The reaction products were extracted with 2 × 300 μl of ethyl acetate, and the organic layer was evaporated to dryness under nitrogen. The residual material was dissolved in methanol and analyzed by negative mode ESI FT-ICR or ESI TOF (Applied BioSystems Q-Star) mass spectrometry to detect the formation of fatty acyl or polyketide products.
For single turnover studies, the enzyme (∼0.5 mm) was incubated with a single equivalent of the appropriate CoA substrate or substrates in 0.1 m potassium phosphate buffer, pH 8.0. When reactions were carried out under “burst” conditions, the enzyme concentration remained at ∼0.5 mm, but the CoA substrates were present at 2–5 mm. The palmitoyl-CoA hydrolase and MMCoA decarboxylase activities were monitored by reverse-phase HPLC with detection at 254 nm. The stationary phase was a Supelcosil LC-18-T octadecylsilyl column (150 × 4.5 mm, 3-μm particles). Analysis was performed on an Agilent 1200 series LC system. The mobile phase consisted of a gradient of methanol in 0.01 or 0.1 m potassium phosphate buffer, pH 6.6. Prior to LC analysis, protein was removed from the sample by centrifugal ultrafiltration (10-kDa MWCO filters).
Fourier Transform Ion Cyclotron Resonance Mass Spectrometry
All ESI mass spectra were acquired on a SolariX 9.4 T FT-ICR mass spectrometer (Bruker Daltronics, Billerica, MA) equipped with an ESI source. MS instrumentation was externally calibrated prior to sample analysis. Spray voltage and sample flow rate were optimized for each sample with typical values of −4500 V and 1.5 μl/min, respectively. Full scan mass spectra were typically acquired over a mass range of m/z 100–1000, with a 0.10-s ion accumulation time and data acquisition of 1 million values. Each combined spectrum was the average of four scans.
RESULTS
Three-dimensional Structure of PKS11
Recombinant PKS11 crystallized in the P21 space group with four PKS11 molecules in the asymmetric unit. The structure of PKS11 was solved by the multiwavelength anomalous dispersion method. The electron density map was interpretable for the entire polypeptide chain in all four subunits (except for the N-terminal methionine in one of the subunits). After several cycles of manual model building and NCS refinement in REFMAC5 (22), a high quality model was obtained (Table 1). Each subunit of PKS11 exhibits an αβαβα thiolase fold (Fig. 1), which is a highly conserved fold among structurally determined type III PKSs (8, 9, 15). In the unit cell, the four subunits are organized as two pairs of subunits related by a 2-fold symmetry, with each pair tightly packed together (supplemental Fig. S1). The solvent-accessible surface area for an individual PKS11 subunit is 15,703 Å2, of which 2,323 Å2 (15%) is buried by the interface with the other subunit in the pair, well above the threshold for typical physiological dimers (24). In contrast, the buried surface area between subunits of adjacent pairs in the tetramer is only 3%, which likely represent nonphysiological crystal contacts.
FIGURE 1.

Tertiary structure of M. tuberculosis PKS11, represented as a ribbon diagram of the protein backbone, colored continuously from N terminus (blue) to C terminus (red). This figure was made with Chimera.
The backbone Cα r.m.s.d. of PKS11 to plant CHS (9) is 1.56 Å over 343 residues, the r.m.s.d. to bacterial stilbene synthase (10) is 1.56 Å over 343 residues, and the r.m.s.d. to N. crassa 2′-ORAS (11) is 1.63 Å over 328 residues. Superposition of PKS11 on M. tuberculosis PKS18 shows the backbone Cα r.m.s.d. between the M. tuberculosis PKS11 and PKS18 structures is 2.1 Å (over 329 residues). However, the Cα backbone diverges between these two structures. Notably, the PKS11 Cα backbone deviates significantly in four regions in the vicinity of the substrate-binding tunnel, including residues 18–49, 52–71, 172–181, and 228–241 (Cα r.m.s.d. 5.9, 4.5, 5.0, and 2.5 Å, for the four regions, respectively) (supplemental Fig. S2). Residues from these regions line the active site cavity and participate in substrate binding and hence might contribute to differences in substrate specificity.
The active site contains a catalytic triad consisting of Cys-138, His-277, and Asn-310 (contact distances: 3.4 Å from Cys-138 to His-277, 3.3 Å from His-277 to Asn-310), which are required for catalysis in other PKSs (12). By analogy to other enzymes like 2-pyrone synthase (12), His-277 serves as a general base to deprotonate Cys-138, facilitating transesterification of acyl substrates between CoA thioesters and the cysteine thiolate. Asn-310 contributes a stabilizing hydrogen bond to the thioester oxygen. In addition, residue 341 is a proline, which is conserved in all other type III PKSs (including CHS and stilbene synthase) but replaced by a large, hydrophobic side chain such as Phe or Leu in more distantly related KAS II (β-ketoacyl carrier protein synthase) enzymes (25). This residue is situated in a β-hairpin turn, and the rigidity afforded by the proline has been postulated to promote cyclization of products (26).
One of the distinctive features of the active site of PKS11, compared with other type III PKSs, is the presence of a deep hydrophobic substrate-binding pocket (Fig. 2). The active site lies at the confluence of two channels: a binding pocket for CoA that extends to the surface, and a buried substrate-binding pocket. The substrate-binding pocket is entirely enclosed (no opening to the surface). In contrast to CHS, which has a smaller pocket volume for accommodating a shorter fatty acid substrate and the cyclized chalcone product, the substrate-binding pocket of PKS11 is a long hydrophobic tunnel, suggesting that it has the ability to bind substrates such as long-chain fatty acids. The cavity has dimensions of ∼20 × 5 Å and is lined primarily with hydrophobic residues. The hydrophobic tunnel is similar to other type III PKSs that bind long chain fatty acids (e.g. M. tuberculosis PKS18, and ORAS, from N. crassa, which binds eicosanoic acid; (14), as opposed to those family members that bind shorter substrates like acetoacetyl-CoA or coumaroyl-CoA. In the superposition of PKS11 with CHS (Protein Data Bank code 1CGZ, complexed with resveratrol), a large residue (Trp-230, substituted by Gly-256 in CHS; Phe-252 in ORAS) protrudes into the position occupied by one of the aromatic rings of the ligand (resveratrol), and Cys-168 conflicts with the other ring, but Phe-188 and Thr-171 in PKS11 rotate to open up a portal to the long internal cavity. A similar opening is observed in the ORAS structure.
FIGURE 2.

Active site architecture (wall-eyed stereo). The catalytic residues consist of Cys-138, His-277, and Asn-310. Phe-188 and Trp-230 lie at the mouth of a long hydrophobic channel (light blue region) in which the tail of the long chain fatty acid ligand resides. The entry of the CoA-binding channel is from the left (peach-colored region).
Palmitate Co-purified from Whole Cell Expression Is Observed Bound in the Active Site of PKS11
Although no exogenous substrates were added to the crystallization buffer, significant electron density was observed in the Fo − Fc Fourier difference electron density map in the substrate-binding channel. The density was observed in all four subunits consistent with a long chain fatty acid bound in the internal hydrophobic tunnel. Electron density was clearly continuous with Cys-138 in subunits B and C, and discontinuous with Cys-138 in subunits A and D. These observations suggested that a long chain fatty acid had been co-purified from the cell extract and was crystallized covalently attached in two of the subunits. To determine the identity of this ligand, the purified enzyme was treated with acetic acid, and the fatty acid was isolated and analyzed by ESI-TOF mass spectrometry (Fig. 3). The mass spectrum showed a peak at 255.2632 Da ([M-H]−), consistent with palmitate (C16). These data suggest that, during expression in M. smegmatis, the enzyme bound to palmitate (or palmitoyl-CoA, which subsequently hydrolyzed), which is a major product of the FAS-I biosynthetic pathway in mycobacteria.
FIGURE 3.

The co-purified ligand was extracted from PKS11 by acid hydrolysis, and its mass was analyzed by ESI mass spectrometry. The m/z of the major peak was 255.2632 atomic mass units in negative mode, which corresponds to palmitic acid.
Palmitate was initially modeled into all four subunits (as free palmitate in A and D, and palmitoyl thioester in B and C) (Fig. 4). The alkyl chains of these ligands make multiple hydrophobic contacts with the residues lining the substrate-binding channel. The tail of the fatty acyl ligands (distal to Cys-138) is enclosed in a hydrophobic vestibule that defines the bottom of the substrate-binding tunnel, defined by residues Tyr-172, Tyr-59, Phe-25, Phe-28, Ile-21, Leu-56, Leu-52, and Arg-50 (Fig. 5). One oxygen of the carboxylate head group of the free palmitate hydrogen bonds with both the backbone N and sulfydryl Sγ of Cys-138 (∼3 Å to each). The superposition of PKS11 with ORAS (1.63 Å backbone r.m.s.d.) shows a striking similarity between the conformations of the bound substrates (palmitate for PKS11, and eicosanoic acid for ORAS), which both occupy a similar hydrophobic tunnel. The carboxylate head group of palmitate is in a position similar to the carboxylate of eicosanoic acid in the superposition with ORAS (Protein Data Bank code 3EUT), shifted by only 1.8 Å. The alkyl tail is 4 carbons longer in eicosanoic acid (C20) than in palmitate, and the space to accommodate the longer substrate at the distal end of the pocket of ORAS is filled in by the side chain of Tyr-59 in PKS11, which is in a loop that is shortened by a deletion of 4 amino acids compared with ORAS.
FIGURE 4.

Co-purified ligands observed in PKS11 crystal. a, free palmitate observed in subunit A of crystal 1. b, monoketide with methyl branch observed in subunit B of crystal 1, covalently attached to Cys-138. Trp-230 and Phe-188 are shown for reference. Electron density is 2Fo − Fc map contoured at 1.5σ. This figure was made with PyMOL.
FIGURE 5.

Substrate- and co-factor-binding channels. Palmitoyl thioester is shown covalently attached to Cys-138 (at the top center). The alkyl chain occupies a deep, internal channel, surround by Phe-188 and Trp-230 (cyan) at the mouth and terminating in a hydrophobic cavity enclosed by residues primarily from two loops (residues Phe-25, Phe-28, Tyr-59, and Tyr-172 are shown in yellow). The (M)MCoA-binding channel can be seen entering from left and converging at the catalytic Cys-138.
Examination of the Fo − Fc difference density map after refinement with ligands (as well as an omit map) indicated that there was additional density at the α-position relative to the thioester for the covalently attached ligand in subunits B and C, presumed to be a methyl branch (Fig. 4b). The conformation of the branch could clearly be distinguished as the (S) stereoisomer. The methyl group makes hydrophobic contact with Pro-341 (4.2 Å). Furthermore, the density for this ligand extends deeper into the pocket by 1 atom. Thus, we infer that the covalently attached ligand is (S)-2-methylheptadecanoic acid. The model was re-refined with this methyl-branched ligand in subunits B and C.
Co-crystallization with Malonyl-CoA and Methylmalonyl-CoA
Based on what is known for other polyketide synthases, it was anticipated that MCoA might be utilized as a co-factor to react with and extend the substrate by two carbon units at a time. Purified PKS11 was co-crystallized with 10 times molar excess of MCoA. The crystal structure was determined to a resolution of 1.73 Å (in the same space group, with a similar unit cell) and was again found to form a tetramer in the asymmetric unit. Very little conformational difference was observed in the overall structure, having a Cα r.m.s.d. of 0.45 Å when superimposed on the initial structure (structure 1, nothing added). When the active sites of PKS11 in this crystal were examined, the Fo − Fc difference density was observed consistent with the presence of an acyl ligand covalently esterified to Cys-138 in subunits A and D. The density was consistent with a triketide, with three planar branch points representing ketone groups at C1 (the thioester) and C3 and C5 (implying two rounds of elongation). The ligands in subunits B and C appeared to be de-esterified diketides, as electron density for these ligands was discontiguous with Cys-138.
In this crystal structure, the CoA-binding cavity was occupied in both subunits B and C; however, the same cavity was vacant in subunits A and D. The adenine is sandwiched between Pro-243 and Val-179 near the surface of the protein. The phosphates are solvent-exposed and make salt bridges with several lysine and arginine residues on the protein surface. The CoA is probably derived from a reaction product of the added MCoA. Even soaking with 75-fold molar excess of MCoA did not reveal significant occupancy of the CoA-binding channel in subunits A and D.
Because some polyketide synthases use MMCoA as an extender unit, purified PKS11 was co-crystallized with a 10-fold molar excess of MMCoA. The crystal structure was determined to a resolution of 1.83 Å and found to form a tetramer that superimposes with an r.m.s.d. of 0.47 Å to crystal structure 1. Difference electron density was observed in the active site, in this case consistent with a covalent acyl ligand with a methyl branch at the α position (C2) and a ketone oxygen on C3 in subunits A and B. De-esterified palmitate molecules were observed in the active sites of chains C and D. The acyl chain appears to be 18 carbons in length, consistent with a methyl-branched diketide, the product of one round of elongation of palmitate with MMCoA. This crystal structure suggests that methylmalonyl-CoA can also act as an extender substrate.
Finally, we co-crystallized PKS11 in the presence of both malonyl-CoA and methylmalonyl-CoA (1:10:10 molar ratio). When the enzyme is co-crystallized with both of these extender CoA thioesters, both active sites are devoid of any polyketide intermediates or products (crystal 4), suggesting progression of the reaction and release of a product. The enzyme thus appears to require both extender units to synthesize polyketide products efficiently. The fact that only intermediates were detected in the crystal when incubated with MCoA or MMCoA alone implies that both substrates are required for complete product formation.
Biochemical Characterization of PKS11 in Solution Demonstrates Synthesis of Methyl-branched Alkylpyrones
Given the crystallographic evidence that PKS11 can bind palmitoyl-CoA and react with MCoA and MMCoA, we sought to reconstitute the reaction in solution and to identify the product. We incubated PKS11 in 1:10 molar ratios each with palmitoyl-, methylmalonyl-, and malonyl-CoA for 2 h under steady-state turnover conditions and analyzed the resulting reaction mixture by ESI FT-ICR mass spectrometry (negative ion mode). A product is observed that is consistent with a cyclic alkylpyrone (Fig. 6) resulting from two rounds of elongation, once by MMCoA, and once by MCoA, corresponding to a peak at 335.261 Da (Fig. 7A). This mass is distinct from the mass that would be expected for a linear (noncyclized) polyketide product after two rounds of elongation (353 Da), where the mass difference of 18 Da is due to loss of a water molecule on cyclization. When incubated with MCoA alone the analysis revealed a peak at 321.245 Da (Fig. 7B), consistent with formation of 4-hydroxy-6-alkyl-2-pyrone. The peak for MCoA+MMCoA is higher abundance than with MCoA alone, suggesting that both substrates are required. We infer from the methyl-branched palmitoyl-diketide observed in the co-crystals of PKS11 and MMCoA that reaction with MMCoA occurs first, placing the branch in the final cyclic product at the 5 position: 4-hydroxy-5-methyl-6-alkyl-2-pyrone (overall proposed reaction shown in Fig. 8). This is also supported by biochemical evidence of preference for reaction with MMCoA over MCoA in competition studies discussed below. The mass is the same as would be expected from a potential Claisen condensation product, as seen in other polyketide synthases (9). However, analysis by tandem mass spectrometry shows a major fragmentation peak at m/z = 291.270, indicating loss of carbon dioxide (mass difference of 44 Da; Fig. 7A, inset), which is characteristic of alkylpyrones, rather than a phenolic Claisen product. Incubation of stearoyl-CoA (C18) with MCoA and MMCoA produced a peak at 363.293 Da, which also showed a loss of 44 Da (m/z = 319.301 Da, Fig. 7C). Thus, PKS11 catalyzes the formation of methylated pyrone products in vitro.
FIGURE 6.
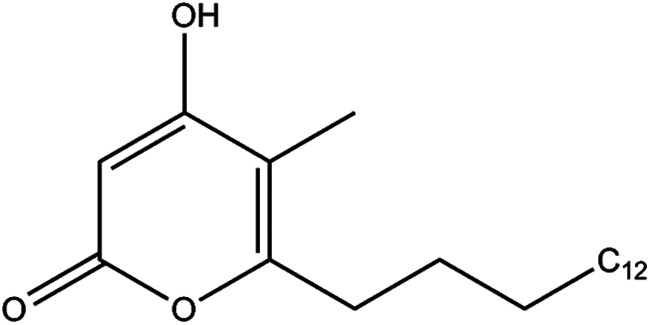
Structure of the methyl-branched alkylpyrone product of PKS11.
FIGURE 7.

Formation of alkylpyrones from fatty acyl-CoAs and (methyl)malonyl-CoA, catalyzed by PKS11. The major peak at 255.234 m/z (or 283.266) represents the fatty acid hydrolysis product of the substrate, palmitoyl-CoA (or stearyl-CoA). The arrows indicate products with masses consistent with alkylpyrones. A, incubation with palmitoyl-, methylmalonyl-, and malonyl-CoA produces a peak at 335.261 representing 4-hydroxy-5-methyl-6-pentadecyl-2-pyrone. Inset, tandem MS spectrum derived from fragmentation of 335.261, showing loss of a 44-Da fragment characteristic of alkylpyrones. B, incubation with palmitoyl- and malonyl-CoA. 321.245, 4-hydroxy-6-pentadecyl-2-pyrone (the peak at 295.265 represents extension of palmitate by MCoA without subsequent cyclization. C, incubation with stearyl-, methylmalonyl-, and malonyl-CoA. 363.293, 4-hydroxy-5-methyl-6-heptadecyl-2-pyrones. D, no pyrone product observed when the C138S active site mutant enzyme is used. This mutant can catalyze the hydrolysis of palmitoyl-CoA to palmitate (255.234) and the decarboxylation of (methyl)malonyl-CoA, but unlike the wild-type enzyme it cannot couple the two reactions to catalyze polyketide formation.
FIGURE 8.
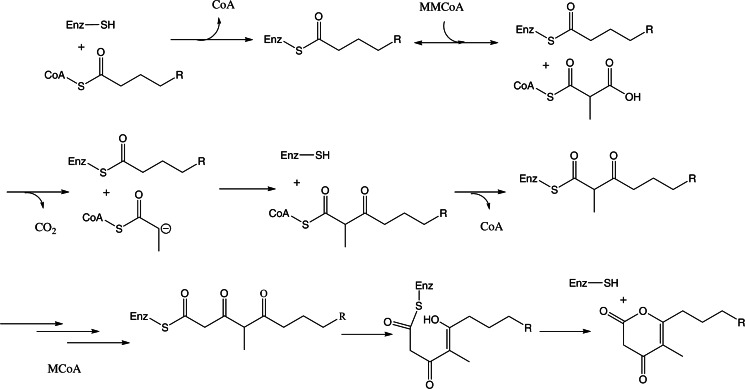
Proposed mechanism for PKS11-mediated pyrone formation. Thioesterification of a fatty acid from CoA onto Cys-138, followed by elongation, which involves binding of MMCoA, decarboxylation, formation of the enolate intermediate, and nucleophilic attack on substrate, followed by acyl transfer of the C2-lengthened chain back to Cys-138. The cycle is repeated a second time with MCoA, and then the product is proposed to undergo cyclization and release as an alkylpyrone.
To further characterize the enzymatic properties of PKS11, we investigated the palmitoyl transferase and (methyl)malonyl-CoA decarboxylase activities of purified PKS11 using reverse-phase HPLC. The HPLC assay detected the formation of CoA from palmitoyl-CoA and from coupled decarboxylation-elongation with (methyl)malonyl-CoA. Polyketide products were not readily detected by this assay, probably due to a combination of low abundance and the peak broadening imparted by their considerable hydrophobicity.
Wild-type PKS11 catalyzes the hydrolysis of palmitoyl-CoA in the absence of (methyl)malonyl-CoA. This is consistent with the observation of both covalent palmitoylation and the noncovalently bound palmitic acid in distinct active sites of the crystal 1. The enzyme catalyzed similar hydrolysis reactions with comparable efficiency for lauroyl- and stearyl-CoA (data not shown), suggesting that it is capable of efficiently processing longer chain fatty acyl-CoAs. However, the hydrolysis reaction was considerably less efficient for hexanoyl-CoA, indicating that the enzyme can distinguish between primer substrates based on their hydrocarbon chain length.
In the presence of malonyl- or methylmalonyl-CoA, or both, the enzyme still catalyzed the formation of CoA from palmitoyl-CoA, whereas the principal products of decarboxylation of the extenders were the abortively protonated ones, i.e. acetyl-CoA and propionyl-CoA (Fig. 9). The extent of formation of CoA in the presence of the extender CoAs was not detectably greater than in their absence, suggesting that coupling of decarboxylation to chain extension is inefficient. When only malonyl-CoA was included as an extender CoA, palmitoyl triketide could be detected by mass spectrometry, as described above. No such product was observed with methylmalonyl-CoA. Activity assays carried out under burst conditions ([S] ∼ 5×Eo) indicated that malonyl and methylmalonyl-CoA compete for the PKS11 active site; they can mutually modulate the extent of their respective uncoupled decarboxylation reactions.
FIGURE 9.

Malonyl- and methylmalonyl-CoA compete for decarboxylation at the PKS11 active site during polyketide synthesis. Reverse-phase HPLC analysis of the reaction of malonyl and/or methylmalonyl-CoA catalyzed by PKS11 in the presence of palmitoyl-CoA is shown. The reactions were carried out under burst conditions (substrate concentrations 5–10×Eo). a, analysis of reaction with PKS11 C138S mutant. The enzyme can efficiently catalyze the decarboxylation of MCoA (retention time 9.1 min) to produce acetyl-CoA (ACoA, 10.0 min, red trace) when the decarboxylation is not coupled to polyketide chain extension. The enzyme can catalyze a similar decarboxylation of MMCoA (9.4 min), producing propionyl-CoA (PrCoA, 10.5 min, green trace). When both CoA thioesters are present in the reaction mixture, the decarboxylation reactions occur at almost the same rates as for the individual substrates. Coenzyme-A (CoASH, 9.5 min) is mainly from hydrolysis of palmitoyl-CoA and also to a small extent from (methyl)malonyl-CoA decarboxylation which becomes coupled to chain extension. b, analysis of reaction with wild-type PKS11. The enzyme can catalyze the decarboxylation of both malonyl and methylmalonyl-CoA, but is less efficient than the C138S mutant at catalyzing the malonyl-CoA decarboxylation. When treated with a mixture of both extender substrates, the efficiency of decarboxylation of malonyl-CoA increases whereas that of methylmalonyl-CoA is unchanged or decreases slightly. In this case, and in contrast to the C138S mutant, the black, red, and green chromatograms have different peak heights.
Interestingly, the C138S mutant of PKS11 could also catalyze the hydrolysis of palmitoyl-CoA with efficiency equal to or greater than that of the wild-type enzyme (also shown in Fig. 9). This mutant could also efficiently catalyze the decarboxylation of extender CoAs. However, no cyclic polyketide products could be detected with this enzyme (Fig. 7D), confirming that chain extension does not occur. Surprisingly, the individual extender CoAs did not modulate the other's rate of uncoupled decarboxylation, as for the wild-type enzyme, suggesting that this competition is kinetically linked to the functional state of Cys-138 and the active site occupancy. This is consistent with the requirement that the enzyme select for differing extender substrates at different points in the chain extension process.
DISCUSSION
The M. tuberculosis cell wall is composed of a thick lipid coat; ∼50–60% (by mass) of these lipids are unique to mycobacteria, and many of these are synthesized by polyketide synthases. The macromolecular composition of the M. tuberculosis cell wall changes in response to the external environment, including adaptation for survival during the bacterial dormant state associated with latent tuberculosis infection in humans (27). The thick lipid coat has been proposed to function as an impermeable barrier that protects the organism from antimicrobial agents, making the mycobacterial cell wall a focus for drug discovery. It has been postulated that these polyketide cell wall components might regulate cell wall permeability (28). Many of them are also immunogenic (29, 30) and potentially contribute to virulence (31).
We have shown that PKS11 can acquire palmitate directly from the cellular environment and bind it with high enough affinity to be retained during purification. Given the assumed similarity of the intracellular environment of M. smegmatis (used for expression) and M. tuberculosis, we conclude that palmitoyl-CoA or a similar long chain fatty acid derivative is the likely substrate of PKS11 in vivo. Palmitate is expected to be abundant in M. tuberculosis, as it is the primary product of the FAS-I system in mycobacteria (32). Similar to other PKSs, this substrate is then extended by two rounds of condensation with MCoA and MMCoA to form a triketide thioester. This intermediate then undergoes lactonization to form a methyl-branched alkylpyrone (Fig. 6). The complexes with both covalently and noncovalently attached long chain fatty acids, along with the products formed by reaction with (M)MCoA, are consistent with the typical chain-elongation cycle carried out by other type III PKSs, including a substrate loading phase (acyl transfer from palmitoyl-CoA to Cys-138), binding of an extender unit (MCoA or MMCoA), decarboxylation and enolate formation, nucleophilic attack on the thioester to form fatty acyl-CoA extended by two carbons, and finally reloading (acyl transfer back to Cys-138).
The reaction appears to require both MCoA and MMCoA. This is supported both by the apparently lower yield observed when PKS11 and palmitoyl-CoA are incubated with MCoA alone versus a higher yield when co-incubated with MCoA and MMCoA, as well as the crystallographic data showing that intermediates are trapped in the crystals when crystallized with either MCoA or MMCoA alone, but the active sites are empty when crystallized with both, suggesting complete substrate turnover.
Although for many PKSs, pyrones are considered to be derailment products that fail to convert into polyphenolic flavonoids, they are the principal product for 2-pyrone synthase, which produces gerberin as an antifungal/insecticide natural product in daisies (12). Furthermore, the closely related PKS18 from M. tuberculosis was shown previously also to produce fatty acid-derived alkylpyrones in vitro (13). Our results extend previous studies of PKS11 by showing that the polyketide product has a methyl branch resulting from elongation by MMCoA and utilizes a longer starting molecule (palmitate).
Alkylpyrones have not yet been detected in extracts from M. tuberculosis cultures. The production of cyclic ketides by PKS11 contrasts with all other previously characterized PKSs in M. tuberculosis (with the exception of PKS18), which produce linear polyketide products as components of the cell wall. For example, mycocerosic acid (synthesized by mycocerosic acid synthase, mas) and phthiocerol (synthesized by ppsABCDE) are both (linear) components of phthiocerol dimycocerosate. PKS2 produces (linear) mycoceranic acid as part of sulfolipid I (33–35), PKS12 produces (linear) mycoketide in mannosyl-β-1-phosphomycoketide (36), PKS1/15 produces (linear) phenolphthiocerol in phenolglyoclipid (30), and PKS3/4 produces (linear) mycolipenic acid incorporated in polyacyl trehaloses (37). All of these mycobacterial PKSs do not undergo cyclization in the final step, but instead rely on a thioesterase or acyl transferase to release the product (38). Nonetheless, the data presented in this paper demonstrate that PKS11 is capable of producing cyclic methyl-branched alkylpyrones, warranting further investigation of the biology surrounding these metabolites in mycobacteria.
Acknowledgment
We thank Neha Patel for help on some of the experiments.
This work was supported, in whole or in part, by National Institutes of Health Grant P01AI095208 (to J. C. S.). This work was also supported by Robert A. Welch Foundation Grants A-0034 (to T. P. B.) and A-0015 to (J. C. S.). Use of the Advanced Photon Source (beamline 23ID) was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357.

This article contains supplemental Figs. S1 and S2.
The atomic coordinates and structure factors (codes 4JAO, 4JAQ, 4JAP, and 4JAR) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PKS11
- polyyketide synthase 11
- ESI
- electrospray ionization
- MCoA
- malonyl-CoA
- MMCoA
- methylmalonyl-CoA
- ORAS
- 2′-oxoresorcylic acid synthase
- r.m.s.d.
- root mean square deviation
- CHS
- chalcone synthase.
REFERENCES
- 1. Saxena P., Yadav G., Mohanty D., Gokhale R. S. (2003) A new family of type III polyketide synthases in Mycobacterium tuberculosis. J. Biol. Chem. 278, 44780–44790 [DOI] [PubMed] [Google Scholar]
- 2. Griffin J. E., Gawronski J. D., Dejesus M. A., Ioerger T. R., Akerley B. J., Sassetti C. M. (2011) High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7, e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sassetti C. M., Rubin E. J. (2003) Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U.S.A. 100, 12989–12994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Waddell S. J., Chung G. A., Gibson K. J., Everett M. J., Minnikin D. E., Besra G. S., Butcher P. D. (2005) Inactivation of polyketide synthase and related genes results in the loss of complex lipids in Mycobacterium tuberculosis H37Rv. Lett. Appl. Microbiol. 40, 201–206 [DOI] [PubMed] [Google Scholar]
- 5. Domenech P., Reed M. B. (2009) Rapid and spontaneous loss of phthiocerol dimycocerosate (PDIM) from Mycobacterium tuberculosis grown in vitro: implications for virulence studies. Microbiology 155, 3532–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Austin M. B., Noel J. P. (2003) The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 20, 79–110 [DOI] [PubMed] [Google Scholar]
- 7. Funabashi M., Funa N., Horinouchi S. (2008) Phenolic lipids synthesized by type III polyketide synthase confer penicillin resistance on Streptomyces griseus. J. Biol. Chem. 283, 13983–13991 [DOI] [PubMed] [Google Scholar]
- 8. Ferrer J. L., Jez J. M., Bowman M. E., Dixon R. A., Noel J. P. (1999) Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nat. Struct. Biol. 6, 775–784 [DOI] [PubMed] [Google Scholar]
- 9. Austin M. B., Izumikawa M., Bowman M. E., Udwary D. W., Ferrer J. L., Moore B. S., Noel J. P. (2004) Crystal structure of a bacterial type III polyketide synthase and enzymatic control of reactive polyketide intermediates. J. Biol. Chem. 279, 45162–45174 [DOI] [PubMed] [Google Scholar]
- 10. Austin M. B., Bowman M. E., Ferrer J. L., Schröder J., Noel J. P. (2004) An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chem. Biol. 11, 1179–1194 [DOI] [PubMed] [Google Scholar]
- 11. Rubin-Pitel S. B., Zhang H., Vu T., Brunzelle J. S., Zhao H., Nair S. K. (2008) Distinct structural elements dictate the specificity of the type III pentaketide synthase from Neurospora crassa. Chem. Biol. 15, 1079–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jez J. M., Austin M. B., Ferrer J., Bowman M. E., Schröder J., Noel J. P. (2000) Structural control of polyketide formation in plant-specific polyketide synthases. Chem. Biol. 7, 919–930 [DOI] [PubMed] [Google Scholar]
- 13. Sankaranarayanan R., Saxena P., Marathe U. B., Gokhale R. S., Shanmugam V. M., Rukmini R. (2004) A novel tunnel in mycobacterial type III polyketide synthase reveals the structural basis for generating diverse metabolites. Nat. Struct. Mol. Biol. 11, 894–900 [DOI] [PubMed] [Google Scholar]
- 14. Funa N., Awakawa T., Horinouchi S. (2007) Pentaketide resorcylic acid synthesis by type III polyketide synthase from Neurospora crassa. J. Biol. Chem. 282, 14476–14481 [DOI] [PubMed] [Google Scholar]
- 15. Goyal A., Saxena P., Rahman A., Singh P. K., Kasbekar D. P., Gokhale R. S., Sankaranarayanan R. (2008) Structural insights into biosynthesis of resorcinolic lipids by a type III polyketide synthase in Neurospora crassa. J. Struct. Biol. 162, 411–421 [DOI] [PubMed] [Google Scholar]
- 16. Van Duyne G. D., Standaert R. F., Karplus P. A., Schreiber S. L., Clardy J. (1993) Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229, 105–124 [DOI] [PubMed] [Google Scholar]
- 17. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 18. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 19. Hendrickson W. A., Ogata C. M. (1997) Phase determination from multiwavelength anomalous diffraction measurements. Methods Enzymol. 276, 494–523 [DOI] [PubMed] [Google Scholar]
- 20. Sheldrick G. M., Gould R.O. (1995) Structure solution by iterative peaklist optimization and tangent expansion in space group P1. Acta Crystallogr. B 51, 423–431 [Google Scholar]
- 21. Cowtan K. D., Main P. (1996) Phase combination and cross-validation in iterated density-modification calculations. Acta Crystallogr. D Biol. Crystallogr. 52, 43–48 [DOI] [PubMed] [Google Scholar]
- 22. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 23. Kissinger C. R., Gehlhaar D. K., Fogel D. B. (1999) Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D Biol. Crystallogr. 55, 484–491 [DOI] [PubMed] [Google Scholar]
- 24. Janin J. (1997) Specific versus non-specific contacts in protein crystals. Nat. Struct. Biol. 4, 973–974 [DOI] [PubMed] [Google Scholar]
- 25. Huang W., Jia J., Edwards P., Dehesh K., Schneider G., Lindqvist Y. (1998) Crystal structure of β-ketoacyl-acyl carrier protein synthase II from E. coli reveals the molecular architecture of condensing enzymes. EMBO J. 17, 1183–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suh D. Y., Fukuma K., Kagami J., Yamazaki Y., Shibuya M., Ebizuka Y., Sankawa U. (2000) Identification of amino acid residues important in the cyclization reactions of chalcone and stilbene synthases. Biochem. J. 350, 229–235 [PMC free article] [PubMed] [Google Scholar]
- 27. Seiler P., Ulrichs T., Bandermann S., Pradl L., Jörg S., Krenn V., Morawietz L., Kaufmann S. H., Aichele P. (2003) Cell-wall alterations as an attribute of Mycobacterium tuberculosis in latent infection. J. Infect. Dis. 188, 1326–1331 [DOI] [PubMed] [Google Scholar]
- 28. Camacho L. R., Constant P., Raynaud C., Laneelle M. A., Triccas J. A., Gicquel B., Daffe M., Guilhot C. (2001) Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis: evidence that this lipid is involved in the cell wall permeability barrier. J. Biol. Chem. 276, 19845–19854 [DOI] [PubMed] [Google Scholar]
- 29. de Jong A., Arce E. C., Cheng T. Y., van Summeren R. P., Feringa B. L., Dudkin V., Crich D., Matsunaga I., Minnaard A. J., Moody D. B. (2007) CD1c presentation of synthetic glycolipid antigens with foreign alkyl branching motifs. Chem. Biol. 14, 1232–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reed M. B., Domenech P., Manca C., Su H., Barczak A. K., Kreiswirth B. N., Kaplan G., Barry C. E., 3rd (2004) A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature 431, 84–87 [DOI] [PubMed] [Google Scholar]
- 31. Jackson M., Stadthagen G., Gicquel B. (2007) Long-chain multiple methyl-branched fatty acid-containing lipids of Mycobacterium tuberculosis: biosynthesis, transport, regulation and biological activities. Tuberculosis 87, 78–86 [DOI] [PubMed] [Google Scholar]
- 32. Bloch K. (1977) Control mechanisms for fatty acid synthesis in Mycobacterium smegmatis. Adv. Enzymol. Relat. Areas Mol. Biol. 45, 1–84 [DOI] [PubMed] [Google Scholar]
- 33. Sirakova T. D., Thirumala A. K., Dubey V. S., Sprecher H., Kolattukudy P. E. (2001) The Mycobacterium tuberculosis pks2 gene encodes the synthase for the hepta- and octamethyl-branched fatty acids required for sulfolipid synthesis. J. Biol. Chem. 276, 16833–16839 [DOI] [PubMed] [Google Scholar]
- 34. Bhatt K., Gurcha S. S., Bhatt A., Besra G. S., Jacobs W. R., Jr. (2007) Two polyketide synthase-associated acyltransferases are required for sulfolipid biosynthesis in Mycobacterium tuberculosis. Microbiology 153, 513–520 [DOI] [PubMed] [Google Scholar]
- 35. Kumar P., Schelle M. W., Jain M., Lin F. L., Petzold C. J., Leavell M. D., Leary J. A., Cox J. S., Bertozzi C. R. (2007) PapA1 and PapA2 are acyltransferases essential for the biosynthesis of the Mycobacterium tuberculosis virulence factor sulfolipid-1. Proc. Natl. Acad. Sci. U.S.A. 104, 11221–11226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matsunaga I., Bhatt A., Young D. C., Cheng T. Y., Eyles S. J., Besra G. S., Briken V., Porcelli S. A., Costello C. E., Jacobs W. R., Jr., Moody D. B. (2004) Mycobacterium tuberculosis pks12 produces a novel polyketide presented by CD1c to T cells. J. Exp. Med. 200, 1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hatzios S. K., Schelle M. W., Holsclaw C. M., Behrens C. R., Botyanszki Z., Lin F. L., Carlson B. L., Kumar P., Leary J. A., Bertozzi C. R. (2009) PapA3 is an acyltransferase required for polyacyltrehalose biosynthesis in Mycobacterium tuberculosis. J. Biol. Chem. 284, 12745–12751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gokhale R. S., Sankaranarayanan R., Mohanty D. (2007) Versatility of polyketide synthases in generating metabolic diversity. Curr. Opin. Struct. Biol. 17, 736–743 [DOI] [PubMed] [Google Scholar]
- 39. Cruickshank D. W. (1999) Remarks about protein structure precision. Acta Crystallogr. D Biol. Crystallogr. 55, 583–601 [DOI] [PubMed] [Google Scholar]