

Background: LKB1S activates AMPK despite its lack of Ser-428/431, previously reported as essential for LKB1-mediated activation of AMPK.
Results: Ser-399, a novel phosphorylation site in LKB1S, is essential for AMPK activation.
Conclusion: Ser-399 is a PKCζ-dependent phosphorylation site similar to Ser-428/431 and likely plays a compensatory role.
Significance: This study clarifies paradoxical findings regarding the role of the C terminus in LKB1 signaling.
Keywords: AMP-activated Kinase (AMPK), LKB, Phosphorylation, Protein Kinase C (PKC), Signal Transduction, Peroxynitrite, Metformin
Abstract
Two splice variants of LKB1 exist: LKB1 long form (LKB1L) and LKB1 short form (LKB1S). In a previous study, we demonstrated that phosphorylation of Ser-428/431 (in LKB1L) by protein kinase Cζ (PKCζ) was essential for LKB1-mediated activation of AMP-activated protein kinase (AMPK) in response to oxidants or metformin. Paradoxically, LKB1S also activates AMPK although it lacks Ser-428/431. Thus, we hypothesized that LKB1S contained additional phosphorylation sites important in AMPK activation. Truncation analysis and site-directed mutagenesis were used to identify putative PKCζ phosphorylation sites in LKB1S. Substitution of Ser-399 to alanine did not alter the activity of LKB1S, but abolished peroxynitrite- and metformin-induced activation of AMPK. Furthermore, the phosphomimetic mutation (S399D) increased the phosphorylation of AMPK and its downstream target phospho-acetyl-coenzyme A carboxylase (ACC). PKCζ-dependent phosphorylation of Ser-399 triggered nucleocytoplasmic translocation of LKB1S in response to metformin or peroxynitrite treatment. This effect was ablated by pharmacological and genetic inhibition of PKCζ, by inhibition of CRM1 activity and by substituting Ser-399 with alanine (S399A). Overexpression of PKCζ up-regulated metformin-mediated phosphorylation of both AMPK (Thr-172) and ACC (Ser-79), but the effect was ablated in the S399A mutant. We conclude that, similar to Ser-428/431 (in LKB1L), Ser-399 (in LKB1S) is a PKCζ-dependent phosphorylation site essential for nucleocytoplasmic export of LKB1S and consequent AMPK activation.
Introduction
Liver kinase B1 (LKB1) is a tumor suppressor gene whose inactivating mutations are implicated in various sporadic cancers and are responsible for the majority of Peutz-Jeghers syndrome cases (1–5). Expression of LKB1 is ubiquitous in many cell types and tissues, and its high expression in fetal tissues is consistent with the critical role of LKB1 in embryonic development (6–9). The functions of LKB1 are mediated through activation of various downstream targets including AMPK2 and at least 13 AMPK-related kinases (2, 5, 10, 11). It plays a fundamental regulatory role in cell polarity, cell growth, and cellular metabolism (5, 11–15). The function of LKB1 in cellular metabolism is largely mediated by phosphorylation and subsequent activation of AMPK (13, 16, 17)
Human and mouse LKB1 consist of 433 and 436 amino acid residues, respectively (2). Residues 49–309 (human) span the catalytic domain flanked by regulatory N-and C-terminal domains (2, 16). The N terminus contains a nuclear localization signal (residues 38–43) and regulates activity by sequestering LKB1 in the nucleus (2, 18). However, kinase activity requires LKB1 to exit the nucleus and localize in the cytoplasm, a process regulated by STRAD and MO25 (19, 20). The regulatory role of the LKB1 C terminus is still unclear, but mutations in this domain are present in Peutz-Jeghers syndrome and sporadic tumors (21). The C-terminal domain contains a farnesylation site and phosphorylation sites, which implies that post-translational modifications play a regulatory role in LKB1 signaling (21).
There are two splice variants of LKB1 that differ in their C terminus, the LKB1 long form (LKB1L) and the LKB1 short form (LKB1s) (22–24). In the alternatively spliced LKB1 short form, 34 amino acids replace the last 63 amino acids present in the C terminus of human LKB1L (25). This substitution leads to loss of the farnesylation site and the Ser-431 (human Ser-428) phosphorylation site in the LKB1 C-terminal domain (22, 24). Ser-431 has been the focus of various studies seeking to understand the role of C-terminal phosphorylation in regulation of LKB1 activity. These studies have yielded conflicting results, particularly regarding the role of Ser-431 in the subcellular distribution of LKB1 and its ability to activate downstream targets (22, 26–29). Therefore, further investigation for the role of the C-terminal domain in regulating LKB1 signaling is needed before we can fully understand the various functions of LKB1. For instance, the upstream kinases responsible for phosphorylation of LKB1 and the associated downstream signaling that potentially lead to differential LKB1 functions remain unclear.
Because LKB1S has a modified C terminus (22), it affords a unique opportunity to investigate the role of the C terminus in functional regulation of LKB1. To date, studies (22, 26) using LKB1S have demonstrated that phosphorylation at Ser-431, previously reported as essential for LKB1 activation (29), is dispensable for subcellular localization and downstream signaling of LKB1. Hence, we sought to examine this seeming dissonance and to determine whether an additional phosphorylation site compensates for the absence of Ser-431/Ser-428 in LKB1S. We have identified Ser-399 as a novel phosphorylation site in LKB1 short form (LKB1S) and propose that its function may be analogous to that of Ser-428/431 in LKB1 (LKB1L). Phosphorylation of Ser-399 by protein kinase Cζ (PKCζ), following peroxynitrite (ONOO−) or metformin treatment, induces translocation of LKB1S from the nucleus to the cytoplasm and results in activation of AMPK.
EXPERIMENTAL PROCEDURES
Materials
Recombinant human PKCζ and myristoylated pseudosubstrate peptides for PKC (Myr-Ser-Ile-Tyr-Arg-Arg-Gly-Ala-Arg-Arg-Trp-Arg-Lys-Leu-OH) were obtained from BIOSOURCE International, Inc. (Camarillo, CA). SAMS peptide was purchased from EMD Millipore (Billerica, MA). PKCζ substrate peptide was purchased from Enzo Life Sciences (Farmingdale, NY). Protein A-Sepharose CL-4B beads were from GE Healthcare. ONOO− was obtained from Calbiochem. Plasmid and siRNA delivery agent Lipofectamine TM 2000 and RNAiMAX were from Invitrogen. Other chemicals and organic solvents of the highest grade available were obtained from Sigma-Aldrich.
Antibodies
Rabbit antibodies against phospho-AMPK (Thr-172), AMPKα1/α2, lactate dehydrogenase, H2AX, phospho-acetyl-coenzyme A carboxylase (ACC) (Ser-79), phospho-PKCζ (Thr-410/403), PKCζ, and CRM1 were obtained from Cell Signaling, Inc. (Beverly, MA). Mouse monoclonal antibody recognizing both LKB1L and LKB1S (Ley37D/G6), anti-STRAD (STRAD N13), anti-glyceraldehyde-3-phosphate dehydrogenase, and anti-actin were from Santa Cruz Biotechnology (Santa Cruz, CA). FLAG (M2) was from Sigma.
Cell Culture
HeLa-S3 and A549 cells (ATCC) were grown in Dulbecco's modified Eagle's medium containing 10% heat-inactivated fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm l-glutamine, at 37 °C in a humidified atmosphere of 5% CO2. Cells were maintained in serum-free medium for 2 h prior to treatment with either 5 mm metformin for 2 h or 100 μm ONOO− for 15 min. Following the treatment, cells were washed briefly with ice-cold phosphate-buffered saline (PBS) and prepared for immunoprecipitation or Western blot analysis.
Preparation of cDNA Expression Constructs and Site-directed Mutagenesis
cDNA encoding LKB1 truncates were amplified from full-length human cDNA clone (purchased from Invitrogen, clone 3689780) with LKB1-specific oligonucleotide primers using High-Fidelity DNA polymerase (Invitrogen). The primers used were: forward primer (including FLAG tag and EcoRI restriction site) 5′-GGC GGA ATT CAT GGA TTA TAA AGA TGA TGA CGA TAA AAT GGA GGT GGT GGA CCC GCA GCA G-3′, and reverse primer (including SalI restriction site) 5′-AAT GTC GAC TCA CTG CTG CTT GCA GGC CGA-3′, and the C-terminal truncation of LKB1 reverse primers (including SalI restriction site) (LKB1 1–309) 5′-AAA TGT CGA CTC AGA ACC AGC TGT GCT GCC GGA T-3′, (LKB1 1–343) 5′-AAT GTC GAC TCA GTC CTC CAA GTA CGG CAC CAC-3′, (LKB1 1–371) 5′-AAT GTC GAC TCA TCC GGG CAC CGT GAA GTC-3′, (LKB1 1–309) 5′-AAA TGT CGA CTC AGA ACC AGC TGT GCT GCC GGA T-3′, (LKB1 1–343) 5′-AAT GTC GAC TCA GTC CTC CAA GTA CGG CAC CAC-3′, and (LKB1 1–371) 5′-AAT GTC GAC TCA TCC GGG CAC CGT GAA GTC-3′.
The products were purified by agarose gel electrophoresis, digested with EcoRI and SalI, and cloned into pCI-neo mammalian expression vector (Promega, catalog number A1360). The authenticity of the inserts was confirmed by DNA sequencing.
Point mutations encoding the substitutions in LKB1S, i.e. S375A, S388A, S391A, S394A, S399A, S399E, and S399D, were generated using the QuikChange II site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The mutations were verified by DNA Sequencing.
Cell Transfection and Adenoviral Infection
Plasmid DNA was prepared using a Qiagen midiprep kit according to the manufacturer's instructions. Cells cultured overnight were transfected using Lipofectamine TM 2000 (Invitrogen) following the manufacturer's protocol. After 24 h, the cells were treated as described under “Cell Culture” above. A replication-defective adenoviral vector expressing PKCζ wild type (WT), dominant-negative PKCζ (PKCζ-DN), or green fluorescent protein (GFP) as negative control was used to infect A549 or HeLa-S3 for 24 h. Moiety of infection was more than 50 in these experiments.
Preparation of Subcellular Fractions
Transfected A549 or HeLa-S3 cells were harvested to isolate cellular cytosolic and nuclear fractions according to the manufacturer's instructions (Thermo Scientific).
Determination of PKCζ Phosphorylation Site
To analyze phosphorylation of LKB1S by PKCζ in vitro, we incubated the substrate, a specific peptide of LKB1S containing Ser-399 (GEEASRPAPQ) or mutant (GEEAARPAPQ), with rPKCζ for 15 min at 37 °C in kinase buffer with [32P]ATP. PKCζ substrate peptide was used as a positive control. Incorporation of 32P into LKB1s peptide was monitored by a γ counter.
To determine the PKCζ phosphorylation site in the cells, lysates of LKB1-deficient HeLa-S3 cells (transfected with empty vector or plasmid encoding LKB1S S399E, S399D, and S399A mutants for 24 h) were immunoprecipitated. Thereafter, immunoprecipitates were used as a substrate for incubation with recombinant PKCζ in the presence of [32P]ATP in kinase buffer for 15 min at 37 °C. The reaction mixture was then supplemented with 20 μl of 3× sample buffer to terminate the reaction, boiled for 5 min at 95 °C, and separated by 12% SDS-PAGE as described by Xie et al. (25). The dried gels were subjected to autoradiography to analyze incorporation of 32P into LKB1S.
Western Blotting and Immunoprecipitation
Cells were lysed with ice-cold buffer from Santa Cruz Biotechnology containing 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mm Na3VO4, and 10 mm NaF. Lysates were centrifuged at 16,000 × g for 20 min at 4 °C. Protein concentration was measured using the BCA protein assay (Pierce Biotechnology). Samples containing 20–50 μg of proteins were separated on polyacrylamide gel with Tris-glycine-SDS running buffer (Bio-Rad) and transferred to a polyvinylidene difluoride (PVDF) membrane. The membranes were blocked for 1 h in 5% milk in Tris-buffered saline-Tween 20 (0.1%) and incubated overnight at 4 °C with the specific primary antibodies. Thereafter, membranes were washed and incubated with horseradish peroxidase-linked secondary antibodies, and the reactive bands were detected by ECLTM Western blotting detection reagents (Amersham Biosciences).
For immunoprecipitation, cells were lysed by the addition of 500 μl of ice cold lysis buffer (50 mm Hepes, pH 7.4, 5 mm sodium pyrophosphate, 50 mm NaF, 1 mm EDTA, 10% (v/v) glycerol, 1% (v/v) Triton X-100, 1 mm dithiothreitol, 4 μg/ml trypsin inhibitor, 0.1 mm phenylmethylsulfonyl fluoride, 1 mm benzamidine). The lysates were centrifuged at 16,000 × g for 15 min at 4 °C and used for subsequent assays.
LKB1 Activity Assays
Cell lysates from HeLa-S3 cells expressing LKB1 and LKB1 truncates were incubated with protein A-Sepharose beads for 1 h at 4 °C. The ectopic protein was immunoprecipitated by incubation with anti-FLAG antibody, and LKB1 activity in the supernatants was measured by analyzing incorporation of 32P into LKBtide as described previously (30).
AMPK Activity Assay
Total AMPK was immunoprecipitated from 500 μg of protein using an antibody against AMPKα, and AMPK activity was assessed by determining the incorporation of 32P into the synthetic SAMS peptide as described previously (31). Briefly, immunoprecipitates were incubated at 37 °C for 15 min in the presence of [32P]ATP (1 μCi) and the SAMS peptide (200 μm), with or without AMP (200 μm). Aliquots of the reaction mixture were then subjected to scintillation counting to determine AMPK activity (31).
Immunofluorescence Staining
A549 cells and HeLa-S3 cultured overnight on coverslips were transfected and treated accordingly (ONOO− or metformin). Subsequently, the cells were washed twice with PBS, fixed in 4% paraformaldehyde, permeabilized with PBS containing 0.1% (v/v) Triton X-100, and blocked with PBS containing 2% (v/v) donkey serum and 5% (w/v) bovine serum albumin before incubation with the appropriate primary antibodies. After thorough washing with TBS, the cells were incubated with the appropriate secondary antibody conjugated to Alexa Fluor® 488 or Alexa Fluor® 594 (Invitrogen). Cells were mounted in SHUR/MountTM with DAPI and visualized on Leica TCS SP2 confocal microscope using a 100× or 63× objective.
RESULTS
C-terminal Truncate (1–371aa) Does Not Affect the Activity of LKB1 in Cells
To gain insight into the function of the LKB1 C-terminal domain and identify putative phosphorylation sites, we developed a series of constructs (1–309aa, 1–343aa, 1–371aa) encoding LKB1 truncates lacking various C-terminal residues (Fig. 1A). Each of these LKB1 constructs, including wild type LKB1L (full length) and the short splice variant, LKB1S (kindly provided by Dr. Angela Woods from Cell Stress Group, Medical Research Council (MRC) Clinical Sciences Centre), were tagged with a FLAG epitope on the N-terminal domain. HeLa-S3 cells, which lack endogenous LKB1, were transiently transfected with 1–309aa, 1–343aa, 1–371aa, LKB1S, or LKB1L. Western blot analysis indicated a similar expression level of LKB1 and the LKB1 C-terminal truncates (data not shown). Transfected cells were treated with ONOO−, which activates AMPK via a PKCζ-dependent pathway (30). Cell lysates were immunoprecipitated using anti-FLAG to isolate expressed LKB1 and LKB1 truncates for subsequent activity assays. The activity of LKB1, under basal and stimulated conditions, was determined by measuring the incorporation of [32P]ATP into the LKBtide. As expected, the LKB1 truncate (1–309) lacking the C-terminal domain showed diminished LKB1 activity as compared with 1–343 (minimum required for STRAD and MO25 binding). Other constructs containing additional amino acid residues at the C terminus had greater activity in comparison with the LKB1 truncate 1–309 (Fig. 1B), suggesting that the PKC-ζ-dependent C-terminal phosphorylation of LKB1 is essential for maintaining LKB1 activity. Interestingly, despite the presence of serine 307 in 1–309 of LKB1S, a site that we previously reported to be important in PKC-ζ-dependent LKB1L nucleus export (25), LKB1 activity in 1–309 LKB1S mutants was very low, suggesting that the function by serine 307 is likely dependent on the presence of C-terminal of LKB1. Similar to previous studies (22, 26), our results did not show any difference between the activity of LKB1S and LKB1L (Fig. 1B). Furthermore, our C-terminal truncate (1–371aa) containing the first 371 residues of LKB1 revealed LKB1 activity similar to that of LKB1S and LKB1L. In addition, ONOO− treatment had no effect on LKB1 activity (Fig. 1B).
FIGURE 1.

Effect of LKB1 C-terminal truncation on AMPK activation. A, schematic representation LKB1L and LKB1s as well as the three truncates. B, immunoprecipitated lysates of LKB1 deficient HeLa-S3 cells, transfected with empty vector or plasmids expressing LKB1 and its truncates and treated with or without 100 μm peroxynitrite. Con, control. C, cell lysates from B were immunoprecipitated with anti-AMPKα antibody to measure AMPK activity. D, as B, but the lysates of HeLa-S3 were used to determine the phosphorylation of AMPK (P-AMPK) at Thr-172 and the phosphorylation of Ser-79 on ACC (P-ACC) through Western blotting. AU, arbitrary units. E, as D, but transfected HeLa-S3 cells were incubated with 5 mm metformin (Met). FL, full length. Error bars indicate S.D.
C-terminal Truncate (1–371) Affects ONOO−/Metformin-dependent Activity of AMPK in Cells
We next determined whether LKB1 C-terminal truncations affect the activation of AMPK, a well characterized downstream target of LKB1. We treated HeLa-S3 cells ectopically expressing LKB1S, LKB1L, or truncates with ONOO− and used the SAMS peptide assay to determine the activity of the AMPK immunoprecipitates. Under basal conditions, AMPK activity was similar in all LKB1 constructs containing the C-terminal domain (Fig. 1C). In addition, stimulation with ONOO− resulted in similar AMPK activity for both LKB1S and LKB1L. However, in comparison with LKB1S, the LKB1 truncate 1–371 had significantly (p < 0.05) reduced AMPK activation in response to ONOO− treatment. Similar results were obtained using Western blot analysis (Fig. 1D). These data indicate that ONOO−-mediated AMPK activation is dependent upon the LKB1 C-terminal region, specifically the residues spanning from amino acid 371 to the end of the LKB1S C terminus.
We previously reported that the activation of AMPK by the antidiabetic drug metformin requires phosphorylation of Ser-428/431 in the LKB1 C terminus (29) and that activation of AMPK by metformin is mediated by ONOO− (31). We next evaluated the effects of metformin on AMPK activation in HeLa-S3 cells transiently expressing LKB1 and LKB1 C-terminal truncates. Metformin-mediated phosphorylation of AMPK (Thr-172) was reduced in cells expressing the 1–371 LKB1 truncate as compared with those expressing LKB1S (Fig. 1E). In addition, the 1–371 truncate led to diminished phosphorylation of ACC (Ser-79), a downstream target of AMPK (Fig. 1E). These results further suggest the presence of a regulatory site in the LKB1S C-terminal region that is required for metformin-induced AMPK activation.
LKB1S C-terminal Mutations Reduce AMPK Activation
Our results prompted us to investigate the site in the LKB1S C terminus that regulates ONOO− or metformin-induced AMPK activation. Based on the sequence of LKB1S, there are five serines between amino acid 371 and the end of the LKB1S C terminus. We mutated these serines to alanine, generating five LKB1S mutants, S375A, S388A, S391A, S394A, and S399A. The sequence of each mutant was verified by DNA sequencing. HeLa-S3 cells transfected with the plasmids encoding LKB1S or the mutants were treated with ONOO− or metformin. Cell lysates were immunoprecipitated for use in LKB1 and AMPK activity assays or prepared for Western blot analysis. Our results demonstrate that LKB1S is constitutively active and that its activity is not affected by mutation of the C terminus (Fig. 2A). However, mutation of LKB1S at Ser-399 diminished ONOO−-enhanced AMPK activation in the cells (Fig. 2B). Western blot results also revealed that the LKB1 C-terminal mutation at Ser-399 impaired ONOO−/metformin-induced phosphorylation of AMPK and its downstream target ACC (Fig. 2, C and D). These results demonstrate that Ser-399 in LKB1S plays an important role in ONOO−/metformin-mediated activation of AMPK.
FIGURE 2.

LKB1S serine 399 is associated with AMPK activation by ONOO− and metformin. A, the lysates of HeLa-S3 transiently transfected with plasmid encoding LKB1S and various mutants were immunoprecipitated with anti-FLAG antibody following exposure of the transfected HeLa-S3 to 100 μm peroxynitrite. The activity of LKB1S in immunoprecipitates was analyzed by measuring incorporation of 32P into LKBtide. Con, control. B, as A, but lysates were immunoprecipitated with anti-AMPKα antibody to measure the activity of AMPK. C and E, as A, but lysates used for Western blot analysis of phospho-ACC (Ser-79) (P-ACC) and phospho-AMPK (Thr-172) (P-AMPK). D and F, as C and E, but prior to lysis, transfected HeLa-S3 were exposed to 5 mm metformin for 2 h. FL, full length. Error bars indicate S.D. *, p < 0.05.
To confirm these results, we constructed constitutively activated LKB1S mutants (S399D and S399E) through site-directed mutagenesis. Substituting Ser-399 with aspartic acid (S399D), to mimic phosphorylation in LKB1S, resulted in increased phosphorylation of AMPK (Thr-172) and ACC (Ser-79) in the presence or absence of ONOO− or metformin (Fig. 2, E and F). These results confirm that phosphorylation of Ser-399 in the C terminus of LKB1S is important for ONOO−- or metformin-mediated AMPK activation. Unexpectedly, we found that unlike S399D gain-of-the-function mutant, S399E mutants, in which Ser-399 was substituted with glutamate (S399E) to mimic phosphorylation in LKB1S, caused only a modest increase of phospho-AMPK and phospho-ACC in resting states or in response to ONOO− (Fig. 2E). This result indicated that unlike aspartic acid, glutamate replacement did not mimic serine phosphorylation. Indeed, other investigators (32) also found that “aspartate is a better substitute for phosphoserine than glutamate.”
PKCζ-mediated Phosphorylation of Ser-399 in LKB1s Is Required for Metformin-induced AMPK Activation
Our group had previously shown that PKCζ is an upstream kinase that phosphorylates LKB1 at Ser-428/431 and Ser-307 and that this phosphorylation is required for metformin/ONOO− enhanced activation of AMPK (25, 29). To investigate whether PKCζ also phosphorylates Ser-399 in the LKB1S C terminus, we utilized an in vitro assay to determine the effect of PKCζ on phosphorylation of the LKB1S peptide (GEEASRPAPQ). The peptide was incubated with recombinant PKCζ for 15 min at 37 °C in kinase buffer in the presence of [32P]ATP. Fig. 3A shows a significantly (p < 0.01) higher incorporation of 32P in the wild type LKB1S peptide as compared with the LKB1S peptide (GEEAARPAPQ) with an alanine substitution at Ser-399 (S399A). These results demonstrate that Ser-399 in the LKB1S C-terminal domain is targeted for phosphorylation by PKCζ.
FIGURE 3.
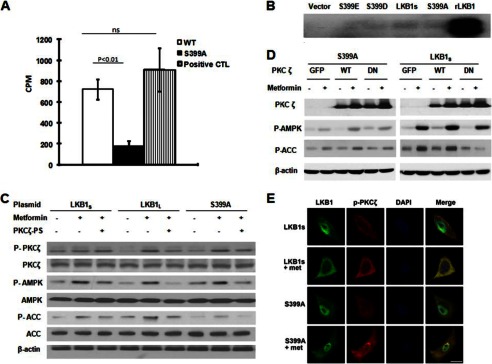
PKCζ phosphorylation of LKB1S Ser-399 is required for metformin activation of AMPK. A, in vitro analysis of phosphorylation of LKB1S peptide by PKCζ. A specific peptide of LKB1S containing Ser-399 (as a substrate) was incubated with rPKCζ for 15 min at 37 °C in kinase buffer with [32P]ATP. 32P incorporation into LKB1S peptide was monitored by γ counter. Error bars indicate S.D. ns, not significant. B, autoradiography of LKB1S and Ser-399 mutants. Immunoprecipitates from HeLa-S3 expressing LKB1S and mutants were incubated with rPKCζ in kinase buffer with [32P]ATP for 15 min at 37 °C, and incorporation of 32P into LKB1s was monitored by autoradiography. The blot is a representative of three blots from three independent experiments (n = 3). C, LKB1-deficient HeLa-S3 cells transfected with plasmids encoding LKB1L, LKB1S, and S399A for 24 h were preincubated with PKCζ-PS for 30 min before being exposed to metformin. After treatment, the cells were lysed and prepared for Western blotting. The blot is a representative of three blots obtained from three individual experiments. P-PKCζ, phospho-PKCζ; P-AMPK, phospho-AMPK; P-ACC, phospho-ACC. D, HeLa-S3 cells were co-transfected by plasmids encoding LKB1S or S399A with adenovirus encoding PKCζ WT, PKCζ-DN (DN), or GFP for 24 h, then treated with metformin for 2 h. The lysates of treated HeLa-S3 were used to examine the phosphorylation of AMPK at Thr-172 and the phosphorylation of Ser-79 on ACC through Western blotting. The blot is a representative of four blots from four individual experiments. E, immunocytochemical staining of metformin (met)-enhanced phosphorylation of PKCζ and LKB1S translocation in A549 cells. LKB1-deficient A549 cells transfected with plasmids encoding LKB1S and S399A for 24 h were incubated with metformin for 2 h before fixation of cells. Mouse anti-LKB1 and rabbit anti-phospho-PKCζ Thr-410/403 were used as the primary antibodies, whereas Alexa Fluor® 488-conjugated goat anti-mouse IgG (ab′) 2 and Alexa Fluor® 594-conjugated donkey anti-rabbit IgG(ab′) 2 were used as secondary antibodies. Images were visualized using a Leica TCS SP2 confocal microscope. Scale bar is 8 μm.
To further confirm that PKCζ directly phosphorylates LKB1S at Ser-399, plasmids encoding LKB1S and the site-directed mutants S399A, S399D, and S399E were transfected separately into LKB1-deficient HeLa-S3 cells. After 24 h, cell lysates were immunoprecipitated with an anti-FLAG antibody, and the immunoprecipitates were incubated with recombinant PKCζ for 15 min at 37 °C in kinase buffer containing [32P]ATP. We identified that PKCζ led to phosphorylation of wild type LKB1S but had minimal effects on Ser-399 mutants of LKB1S (Fig. 3B). This confirms that PKCζ directly phosphorylates LKB1S at Ser-399 in the cells.
Our previous results determined that PKCζ is the upstream kinase required for LKB1L-induced AMPK activation in response to ONOO−and metformin (29, 30). The next studies focused on confirming this role for PKCζ in metformin-mediated LKB1S activation and concomitant AMPK phosphorylation. We evaluated the effects of pharmacological (PKCζ-PS) and genetic (PKCζ-DN) inhibitors of PKCζ in HeLa-S3 cells expressing LKB1S or its mutant S399A. PKCζ-specific pseudosubstrate peptide (PKCζ-PS), a synthetic peptide that selectively inhibits PKCζ (30), suppressed metformin-enhanced phosphorylation of both AMPK (Thr-172) and ACC (Ser-79) in LKB1L, LKB1S, and the LKB1S mutant S399A (Fig. 3C). In addition, adenoviral expression of PKCζ-WT significantly increased metformin-mediated phosphorylation of AMPK (Fig. 3D) in cells expressing LKB1S but not in the S399A-expressing cells. In contrast, overexpression of a dominant-negative PKCζ mutant (PKCζ-DN) eliminated the effects of metformin on AMPK (Fig. 3D) in cells expressing LKB1S. These data imply that metformin-mediated phosphorylation of AMPK is dependent on Ser-399 and PKCζ. Also in agreement with our previous findings (29), metformin induced translocation of LKB1S from the nucleus, an effect that was reduced in the cells expressing the mutant S399A (Fig. 3E). We used confocal microscopy to demonstrate that phospho-PKCζ colocalizes with wild-type LKB1S in response to metformin treatment but that this effect is ablated with the S399A mutant (Fig. 3E). Taken together, our results show that PKCζ is a mediator of metformin-induced AMPK activation and that these effects are dependent upon Ser-399 in LKB1S.
Metformin Increases the Translocation of LKB1S from the Nucleus to Cytosol
When inactive, LKB1 is localized to the nucleus and is able to translocate to the cytosol and become active when it associates with STRAD and MO25 (20, 33). Therefore, we investigated the effect of metformin on subcellular localization of LKB1S. Previous studies by our group demonstrated that nucleocytoplasmic translocation of LKB1L, which requires phosphorylation of Ser-428/431 and Ser-307 in the LKB1L C-terminal domain, is needed for metformin-mediated activation of AMPK (25, 29). To investigate the distribution of LKB1S in the cells, we transfected LKB1-deficient HeLa-S3 cells with plasmids encoding LKB1S or its mutant (S399A). Following transfection, we subjected cells to subcellular fractionations and subsequent Western blotting using a mouse anti-LKB1 antibody. The purity of the subcellular fractions was determined by probing for lactate dehydrogenase, an enzyme located in the cytosol, and nuclear histone H2AX (25). Our results demonstrated that LKB1S is predominantly localized in the nuclei of HeLa-S3 cells transfected with either LKB1S or the S399A mutant (Fig. 4A). Notably, metformin increased (p < 0.05) LKB1S in the cytosol of HeLa-S3 transfected with LKB1S but not in the cells transfected with the S399A mutant (Fig. 4A). These results were further confirmed by confocal microscopy. As shown in Fig. 4B, mutation of Ser-399 restricted LKB1S to the nucleus, whereas metformin treatment increased cytoplasmic translocation of LKB1S but had no effect on the Ser-399 mutant. These data suggest that Ser-399 phosphorylation is required for metformin-induced nuclear export of LKB1S. Notably, the effects of a Ser-399 mutation on nuclear localization of LKB1S were also evident in the presence of hypoxia (Fig. 4C), suggesting that the importance of Ser-399 phosphorylation might extend beyond the metformin-LKB1-AMPK pathway.
FIGURE 4.

Metformin/ONOO− increases the translocation of LKB1S from nucleus to cytosol. A, HeLa-S3 cells were transfected with LKB1S or S399A mutant for 24 h followed by treatment with 5 mm metformin for 2 h. Subcellular fractions were prepared, and the protein level of LKB1S was detected in each fraction by Western blot. Lactate dehydrogenase was used as the loading control for the cytoplasmic fraction (C), and histone H2AX was used as the loading control for the nuclear fraction (N). The blot is a representative of three blots from three individual experiments (n = 3). B, as A, but treated HeLa S3 were fixed in 4% paraformaldehyde and probed with a mouse monoclonal anti-LKB1 (Ley37D/6G) that recognizes both LKB1L and LKB1S. Alexa Fluor® 488-conjugated rabbit anti-mouse IgG was used as the secondary antibody, and images were visualized using a Leica TCS SP2 confocal microscope. Scale bar is 16 μm. C, A549 cells were transfected with LKB1S or S399A mutant for 24 h before exposure to hypoxia (1% O2 for 4 h). LKB1S and S399A distribution was examined by immunostaining of treated A549. Scale bar is 14 μm. D, A549 cells were transfected with LKB1S for 24 h and then pretreated with leptomycin B (LMB) (200 nm) for 1 h followed by treatment with ONOO− (100 μm) for 15 min. Subcellular fractions were prepared as described under “Experimental Procedures.” The protein level of LKB1S and CRM1 in each fraction was analyzed by Western blot. β-Actin was used as the loading control for the cytoplasmic fraction, and histone H2AX was used as the loading control for the nuclear fraction. The blot is a representative of three blots from three individual experiments (n = 3). Error bars in A and D indicate S.D. *, p < 0.05. E, A549 cells were treated as D, and then immunostaining was used to examine LKB1S and S399A distribution. The images are a representative of three individual experiments (n = 3). Scale bar is 14 μm.
Inhibition of CRM1 Abolishes Nucleocytoplasmic Shuttling of CRM1 and LKB1s
We next wanted to gain more insight into the mechanism of ONOO−/metformin-mediated translocation of LKB1S from the nucleus. We chose to assess the role of CRM1, a nuclear export protein that binds LKB1 via the adaptor protein, STRADα, resulting in exportation of LKB1 from the nucleus (20). As described previously (25), we used leptomycin B, a specific inhibitor of CRM1 (20), to determine the effect of CRM1 on ONOO−-induced nuclear export of LKB1S in A549 cells transfected with LKB1S or the S399A mutant. We used both Western blot analysis of fractionated cellular components and immunocytochemical analysis of cells to evaluate the localization of LKB1S. We determined that treatment with ONOO− (15 min) significantly (p < 0.05) increased the cytoplasmic portion of LKB1S and reduced the nuclear content of LKB1S (Fig. 4D). This effect was reversed by treatment with the CRM1 inhibitor, leptomycin B (Sigma). Using confocal microscopy, we demonstrated that ONOO− caused LKB1S to translocate from the nucleus and that this interaction was suppressed by leptomycin B, as well as mutation of LKB1S Ser-399 to S399A (Fig. 4E). Taken together, these results suggest that ONOO−-induced nucleocytoplasmic translocation of LKB1S is dependent upon both CRM1 and phosphorylation of Ser-399.
LKB1S Ser-399 Is Not Required for Trimeric Complex Formation
Because the formation of the LKB1-STRADα-MO25α heterotrimeric complex is crucial for nuclear export and cytoplasmic stability of LKB1 (20, 33), we sought to determine whether Ser-399 mutation affects the ability of LKB1S to form the heterotrimeric complex. Plasmids encoding LKB1L, LKB1S, and mutants S399A and S399D were co-transfected with or without STRADα and MO25α into HeLa-S3 LKB1-deficient cells. Cells were harvested after 24 h and subjected to nuclear/cytosolic fractionation. Western blot analysis and immunofluorescence staining were used to determine the distribution of LKB1L, LKB1S, and its mutants in the transfected cells. Co-expression of LKB1 with STRADα and MO25α resulted in the translocation of LKB1L, LKB1S, and its mutants from the nucleus to the cytoplasm (Fig. 5, A and B). These results are in agreement with the findings of others (22, 26) that LKB1S, like LKB1L, is able to form the heterotrimeric complex with STRADα and MO25α. In addition, mutation of Ser-399 in the C terminus of LKB1S did not hinder formation of the LKB1S-STRADα-MO25α complex. However, all forms of LKB1 remained sequestered in the nucleus without co-expression with STRADα-MO25α. These results imply that a stimulus for heterotrimeric complex formation is required for LKB1-dependent pathways, including ONOO−/metformin-mediated AMPK activation.
FIGURE 5.

LKB1S Ser-399 mutant does not impair its ability to bind MO25α and STRADα. A, HeLa-S3 cells were transiently transfected with LKB1S/mutants alone (left panel) or co-transfected with MO25α and STRADα constructs (right panel) for 24 h. Subcellular fractions were prepared as described under “Experimental Procedures.” The protein level of LKB1S was detected in each fraction by Western blot. Lactate dehydrogenase was used as the loading control for the cytoplasmic fraction, and histone H2AX was used as the loading control for nuclear fraction. The blot is a representative of three blots from three individual experiments (n = 3). B, transfected or co-transfected HeLa-S3 cells were fixed to determine distribution of LKB1S or mutants in HeLa-S3 cells through immunocytochemical staining. Scale bar is 10 μm.
DISCUSSION
In this study, we have identified Ser-399 as a novel phosphorylation site in the C terminus of LKB1S. Mutation of this site to alanine suppresses ONOO−- or metformin-induced AMPK activation, but does not affect LKB1 activity or base-line AMPK activation. Furthermore, we identified that Ser-399 is phosphorylated by PKCζ and that PKCζ was critical for metformin-induced AMPK activation in cells transfected with LKB1S. Furthermore, we identified that metformin and ONOO− were able to induce nuclear export of LKB1S and that these effects were dependent upon Ser-399 in the LKB1S C terminus. Finally, we found that ONOO−-induced translocation of LKB1S is mediated by CRM1.
Our results suggest that Ser-399 in the C terminus of LKB1S is analogous to Ser-431/428 in LKB1 (LKB1L). In this case, metformin treatment leads to activation of PKCζ, which phosphorylates Ser-399 in LKB1S (or Ser-428/431 in LKB1L) (29), thereby increasing LKB1S association with CRM1. The consequence of this interaction results in the nuclear export of LKB1 and subsequent AMPK activation. This novel mechanism provides a logical explanation of the previous findings (22, 26) that LKB1L and LKB1S have similar activity and downstream activation of base-line AMPK. Moreover, LKB1L and LKB1S have comparable responses to ONOO−/metformin treatment, suggesting that Ser-399 in LKB1S mimics the functionality of Ser-431/428 in LKB1L. This rectifies previous, seemingly paradoxical findings (22, 26, 28, 29, 35, 36) regarding the necessity of the Ser-431/428 site in LKB1L by establishing that the Ser-399 site plays a compensatory role in LKB1S.
These findings might provide insight into regulation of LKB1 signaling, particularly the role of the C-terminal domain in regulation of LKB1 function. As pointed out by a recent review (34), “regulation of the LKB1 signaling network is highly context-dependent.” The upstream kinases that phosphorylate LKB1 might depend upon different cell types or stimuli. In the context of metformin-induced AMPK activation, our previous (25, 29) and present studies suggest that phosphorylation of Ser-428/431/Ser-307 in LKB1L and Ser-399 in LKB1S by PKCζ is required for this pathway. Xie et al. (25) demonstrated that Ser-307, present in both LKB1S and LKB1L, is necessary for the association of LKB1 with STRAD and CRM1 and subsequent export of LKB1 from the nucleus to the cytoplasm, consequently activating AMPK. Ser-307 is essential for regulation of LKB1L by PKCζ (25) and would likely play a major role in metformin-induced AMPK activation. Because LKB1 activity in 1–309 truncate was very low (Fig. 1B), the function by serine 307 might require the presence of the LKB1 C terminus. However, the S307A mutant of LKB1L exhibits low activity, underscoring the importance of Ser-307 even in the presence of the C terminus (25). A previous study from our group (29) determined that the two other upstream kinases, protein kinase A (PKA) and p90 ribosomal S6 kinase (RSK), known to phosphorylate LKB1 at the Ser-428 site (16), are not involved in metformin-enhanced phosphorylation of AMPK (Thr-172) in bovine aortic endothelial cells (29). Moreover the study of Dension et al. (22) showed that treatment with forskolin, an activator of the PKA pathway, did not enhance LKB1-dependent activation of AMPK as evidenced by increased AMPK activity in cells expressing catalytically inactive LKB1. This implies that alternative pathway(s) is (are) implicated in PKA-induced AMPK activation, which further supports the notion that regulation and activity of LKB1 are context-dependent (34). Other studies (35, 36) have suggested an important regulatory role played by post-translational modifications in LKB1. It is probable that LKB1 is differentially phosphorylated by different stimuli in different cell types (25), warranting further investigation of the LKB1 regulatory C-terminal domain.
Evidence from our research has revealed that PKCζ phosphorylates LKB1 at various sites in its C terminus in response to stimulation by oxidants (ONOO−) and that the effects of the antidiabetic drug, metformin, are mediated through ONOO− (25, 28–31). This suggests that PKCζ-dependent LKB1 regulation might be oxidant (metformin)-specific. It is plausible that oxidant-induced activation of PKCζ, leading to phosphorylation and translocation of LKB1 from the nucleus and subsequent AMPK activation, is a preconditioning that would serve to sensitize cells to redox sensing. In this case, activation of AMPK triggers a negative feedback loop resulting in cell defense mechanisms to protect cells from further oxidative damage as we have proposed before (31). Our studies offer insight into a unique pathway that would lead to preferential regulation of LKB1 function in response to oxidative stress. PKCζ is implicated in regulation of various cellular functions (37), including metabolism (38, 39) and polarity (40, 41), which are similar to the functions of LKB1. Because PKCζ can phosphorylate both LKB1L and LKB1S, this pathway might play an important physiological role in various cell defense mechanisms and pathophysiological conditions characterized by oxidative stress.
In summary, we have identified Ser-399, a novel phosphorylation site in the C-terminal domain of the LKB1 short form, that is likely analogous to Ser-428/431 in full-length LKB1. Phosphorylation of Ser-399 by PKCζ does not affect LKB1S kinase activity, but it is essential for ONOO−/metformin-induced nucleocytoplasmic translocation of the LKB1 short form and subsequent AMPK activation. Our findings enhance the understanding of the role of the C-terminal domain in regulating LKB1 functions and provide insight into the mechanism for metformin-stimulated AMPK activation. These results underscore the importance of PKCζ as an upstream LKB1 kinase that is critical in the metformin/oxidant-LKB1-AMPK pathway.
This work was supported, in whole or in part, by National Institutes of Health Grants HL079584, HL080499, HL074399, HL089920, HL096032, HL105157, and HL110488) (to M. H. Z.). This work was also supported by a Research Award from the American Diabetes Association and funds from the Warren Chair of the University of Oklahoma Health Sciences Center (to M. H. Z.).
- AMPK
- AMP-activated protein kinase
- PKCζ-PS
- PKCζ pseudosubstrate peptides
- rPKCζ
- recombinant PKCζ
- PKCζ-DN
- dominant-negative PKCζ mutant
- ONOO−
- peroxynitrite
- STRAD
- Ste20-related adaptor)
- ACC
- phospho-acetyl-coenzyme A carboxylase
- aa
- amino acid.
REFERENCES
- 1. Hemminki A., Markie D., Tomlinson I., Avizienyte E., Roth S., Loukola A., Bignell G., Warren W., Aminoff M., Höglund P., Järvinen H., Kristo P., Pelin K., Ridanpää M., Salovaara R., Toro T., Bodmer W., Olschwang S., Olsen A. S., Stratton M. R., de la Chapelle A., Aaltonen L. A. (1998) A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391, 184–187 [DOI] [PubMed] [Google Scholar]
- 2. Hezel A. F., Bardeesy N. (2008) LKB1; linking cell structure and tumor suppression. Oncogene 27, 6908–6919 [DOI] [PubMed] [Google Scholar]
- 3. Sanchez-Cespedes M. (2007) A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene 26, 7825–7832 [DOI] [PubMed] [Google Scholar]
- 4. Sanchez-Cespedes M., Parrella P., Esteller M., Nomoto S., Trink B., Engles J. M., Westra W. H., Herman J. G., Sidransky D. (2002) Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 62, 3659–3662 [PubMed] [Google Scholar]
- 5. Shackelford D. B., Shaw R. J. (2009) The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9, 563–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Collins S. P., Reoma J. L., Gamm D. M., Uhler M. D. (2000) LKB1, a novel serine/threonine protein kinase and potential tumour suppressor, is phosphorylated by cAMP-dependent protein kinase (PKA) and prenylated in vivo. Biochem. J. 345, 673–680 [PMC free article] [PubMed] [Google Scholar]
- 7. Jishage K.-i., Nezu J.-i., Kawase Y., Iwata T., Watanabe M., Miyoshi A., Ose A., Habu K., Kake T., Kamada N., Ueda O., Kinoshita M., Jenne D. E., Shimane M., Suzuki H. (2002) Role of Lkb1, the causative gene of Peutz-Jegher's syndrome, in embryogenesis and polyposis. Proc. Natl. Acad. Sci. U.S.A. 99, 8903–8908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rowan A., Churchman M., Jefferey R., Hanby A., Poulsom R., Tomlinson I. (2000) In situ analysis of LKB1/STK11 mRNA expression in human normal tissues and tumours. J. Pathol. 192, 203–206 [DOI] [PubMed] [Google Scholar]
- 9. Ylikorkala A., Rossi D. J., Korsisaari N., Luukko K., Alitalo K., Henkemeyer M., Mäkelä T. P. (2001) Vascular abnormalities and deregulation of VEGF in Lkb1-deficient mice. Science 293, 1323–1326 [DOI] [PubMed] [Google Scholar]
- 10. Lizcano J. M., Göransson O., Toth R., Deak M., Morrice N. A., Boudeau J., Hawley S. A., Udd L., Mäkelä T. P., Hardie D. G., Alessi D. R. (2004) LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 23, 833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mirouse V., Billaud M. (2011) The LKB1/AMPK polarity pathway. FEBS Lett. 585, 981–985 [DOI] [PubMed] [Google Scholar]
- 12. Boudeau J., Sapkota G., Alessi D. R. (2003) LKB1, a protein kinase regulating cell proliferation and polarity. FEBS Lett. 546, 159–165 [DOI] [PubMed] [Google Scholar]
- 13. Jansen M., Ten Klooster J. P., Offerhaus G. J., Clevers H. (2009) LKB1 and AMPK family signaling: the intimate link between cell polarity and energy metabolism. Physiol. Rev. 89, 777–798 [DOI] [PubMed] [Google Scholar]
- 14. Shaw R. J. (2008) LKB1: cancer, polarity, metabolism, and now fertility. Biochem. J. 416, e1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tiainen M., Vaahtomeri K., Ylikorkala A., Mäkelä T. P. (2002) Growth arrest by the LKB1 tumor suppressor: induction of p21WAF1/CIP1. Hum. Mol. Genet. 11, 1497–1504 [DOI] [PubMed] [Google Scholar]
- 16. Alessi D. R., Sakamoto K., Bayascas J. R. (2006) LKB1-dependent signaling pathways. Annu. Rev. Biochem. 75, 137–163 [DOI] [PubMed] [Google Scholar]
- 17. Woods A., Johnstone S. R., Dickerson K., Leiper F. C., Fryer L. G., Neumann D., Schlattner U., Wallimann T., Carlson M., Carling D. (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008 [DOI] [PubMed] [Google Scholar]
- 18. Smith D. P., Spicer J., Smith A., Swift S., Ashworth A. (1999) The mouse Peutz-Jeghers syndrome gene Lkb1 encodes a nuclear protein kinase. Hum. Mol. Genet. 8, 1479–1485 [DOI] [PubMed] [Google Scholar]
- 19. Baas A. F., Boudeau J., Sapkota G. P., Smit L., Medema R., Morrice N. A., Alessi D. R., Clevers H. C. (2003) Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J. 22, 3062–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dorfman J., Macara I. G. (2008) STRADα regulates LKB1 localization by blocking access to importin-α, and by association with Crm1 and exportin-7. Mol. Biol. Cell 19, 1614–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Forcet C., Etienne-Manneville S., Gaude H., Fournier L., Debilly S., Salmi M., Baas A., Olschwang S., Clevers H., Billaud M. (2005) Functional analysis of Peutz-Jeghers mutations reveals that the LKB1 C-terminal region exerts a crucial role in regulating both the AMPK pathway and the cell polarity. Hum. Mol. Genet. 14, 1283–1292 [DOI] [PubMed] [Google Scholar]
- 22. Denison F. C., Hiscock N. J., Carling D., Woods A. (2009) Characterization of an alternative splice variant of LKB1. J. Biol. Chem. 284, 67–76 [DOI] [PubMed] [Google Scholar]
- 23. Denison F. C., Smith L. B., Muckett P. J., O'Hara L., Carling D., Woods A. (2011) LKB1 is an essential regulator of spermatozoa release during spermiation in the mammalian testis. PLoS ONE 6, e28306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Towler M. C., Fogarty S., Hawley S. A., Pan D. A., Martin D. M., Morrice N. A., McCarthy A., Galardo M. N., Meroni S. B., Cigorraga S. B., Ashworth A., Sakamoto K., Hardie D. G. (2008) A novel short splice variant of the tumour suppressor LKB1 is required for spermiogenesis. Biochem. J. 416, 1–14 [DOI] [PubMed] [Google Scholar]
- 25. Xie Z., Dong Y., Zhang J., Scholz R., Neumann D., Zou M.-H. (2009) Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Mol. Cell. Biol. 29, 3582–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fogarty S., Hardie D. G. (2009) C-terminal phosphorylation of LKB1 is not required for regulation of AMP-activated protein kinase, BRSK1, BRSK2, or cell cycle arrest. J. Biol. Chem. 284, 77–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sapkota G. P., Kieloch A., Lizcano J. M., Lain S., Arthur J. S., Williams M. R., Morrice N., Deak M., Alessi D. R. (2001) Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90RSK and cAMP-dependent protein kinase, but not its farnesylation at Cys433, is essential for LKB1 to suppress cell growth. J. Biol. Chem. 276, 19469–19482 [DOI] [PubMed] [Google Scholar]
- 28. Song P., Xie Z., Wu Y., Xu J., Dong Y., Zou M. H. (2008) Protein kinase Cζ-dependent LKB1 serine 428 phosphorylation increases LKB1 nucleus export and apoptosis in endothelial cells. J. Biol. Chem. 283, 12446–12455 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29. Xie Z., Dong Y., Scholz R., Neumann D., Zou M. H. (2008) Phosphorylation of LKB1 at serine 428 by protein kinase C-ζ is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation 117, 952–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xie Z., Dong Y., Zhang M., Cui M. Z., Cohen R. A., Riek U., Neumann D., Schlattner U., Zou M. H. (2006) Activation of protein kinase Cζ by peroxynitrite regulates LKB1-dependent AMP-activated protein kinase in cultured endothelial cells. J. Biol. Chem. 281, 6366–6375 [DOI] [PubMed] [Google Scholar]
- 31. Zou M. H., Kirkpatrick S. S., Davis B. J., Nelson J. S., Wiles W. G., 4th, Schlattner U., Neumann D., Brownlee M., Freeman M. B., Goldman M. H. (2004) Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo: role of mitochondrial reactive nitrogen species. J. Biol. Chem. 279, 43940–43951 [DOI] [PubMed] [Google Scholar]
- 32. Huang W., Erikson R. L. (1994) Constitutive activation of Mek1 by mutation of serine phosphorylation sites. Proc. Natl. Acad. Sci. U.S.A. 91, 8960–8963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boudeau J., Baas A. F., Deak M., Morrice N. A., Kieloch A., Schutkowski M., Prescott A. R., Clevers H. C., Alessi D. R. (2003) MO25α/β interact with STRADα/β enhancing their ability to bind, activate, and localize LKB1 in the cytoplasm. EMBO J. 22, 5102–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vaahtomeri K., Mäkelä T. P. (2011) Molecular mechanisms of tumor suppression by LKB1. FEBS Lett. 585, 944–951 [DOI] [PubMed] [Google Scholar]
- 35. Barnes A. P., Lilley B. N., Pan Y. A., Plummer L. J., Powell A. W., Raines A. N., Sanes J. R., Polleux F. (2007) LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell 129, 549–563 [DOI] [PubMed] [Google Scholar]
- 36. Shelly M., Cancedda L., Heilshorn S., Sumbre G., Poo M. M. (2007) LKB1/STRAD promotes axon initiation during neuronal polarization. Cell 129, 565–577 [DOI] [PubMed] [Google Scholar]
- 37. Hirai T., Chida K. (2003) Protein kinase Cζ (PKCζ): activation mechanisms and cellular functions. J. Biochem. 133, 1–7 [DOI] [PubMed] [Google Scholar]
- 38. Cantó C., Suárez E., Lizcano J. M., Griñó E., Shepherd P. R., Fryer L. G., Carling D., Bertran J., Palacín M., Zorzano A., Gumà A. (2004) Neuregulin signaling on glucose transport in muscle cells. J. Biol. Chem. 279, 12260–12268 [DOI] [PubMed] [Google Scholar]
- 39. Liu L. Z., Zhao H. L., Zuo J., Ho S. K., Chan J. C., Meng Y., Fang F. D., Tong P. C. (2006) Protein kinase Cζ mediates insulin-induced glucose transport through actin remodeling in L6 muscle cells. Mol. Biol. Cell 17, 2322–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grifoni D., Garoia F., Bellosta P., Parisi F., De Biase D., Collina G., Strand D., Cavicchi S., Pession A. (2007) aPKCζ cortical loading is associated with Lgl cytoplasmic release and tumor growth in Drosophila and human epithelia. Oncogene 26, 5960–5965 [DOI] [PubMed] [Google Scholar]
- 41. Real E., Faure S., Donnadieu E., Delon J. (2007) Cutting edge: Atypical PKCs regulate T lymphocyte polarity and scanning behavior. J. Immunol. 179, 5649–5652 [DOI] [PubMed] [Google Scholar]