

Background: GlgE is involved in the biosynthesis of α-glucans and is a genetically validated antituberculosis target.
Results: Phosphorylation of GlgE by the Ser/Thr protein kinase PknB lowers enzyme activity in vitro and in vivo.
Conclusion: Phosphorylation of GlgE will negatively regulate flux through the GlgE pathway in actinomycetes.
Significance: This study provides new opportunities to target the GlgE pathway therapeutically.
Keywords: Bacterial pathogenesis, Bacterial protein kinases, Microbiology, Mycobacterium tuberculosis, phosphorylation, Serine threonine protein kinase, glucan, maltosyltransferase
Abstract
GlgE is a maltosyltransferase involved in the biosynthesis of α-glucans that has been genetically validated as a potential therapeutic target against Mycobacterium tuberculosis. Despite also making α-glucan, the GlgC/GlgA glycogen pathway is distinct and allosterically regulated. We have used a combination of genetics and biochemistry to establish how the GlgE pathway is regulated. M. tuberculosis GlgE was phosphorylated specifically by the Ser/Thr protein kinase PknB in vitro on one serine and six threonine residues. Furthermore, GlgE was phosphorylated in vivo when expressed in Mycobacterium bovis bacillus Calmette–Guérin (BCG) but not when all seven phosphorylation sites were replaced by Ala residues. The GlgE orthologues from Mycobacterium smegmatis and Streptomyces coelicolor were phosphorylated by the corresponding PknB orthologues in vitro, implying that the phosphorylation of GlgE is widespread among actinomycetes. PknB-dependent phosphorylation of GlgE led to a 2 orders of magnitude reduction in catalytic efficiency in vitro. The activities of phosphoablative and phosphomimetic GlgE derivatives, where each phosphorylation site was substituted with either Ala or Asp residues, respectively, correlated with negative phosphoregulation. Complementation studies of a M. smegmatis glgE mutant strain with these GlgE derivatives, together with both classical and chemical forward genetics, were consistent with flux through the GlgE pathway being correlated with GlgE activity. We conclude that the GlgE pathway appears to be negatively regulated in actinomycetes through the phosphorylation of GlgE by PknB, a mechanism distinct from that known in the classical glycogen pathway. Thus, these findings open new opportunities to target the GlgE pathway therapeutically.
Introduction
Tuberculosis is caused by Mycobacterium tuberculosis and remains a major threat to global health, claiming the life of 2 million individuals annually (1). The ability to control the tuberculosis pandemic is limited by a lack of new therapeutic agents and the rapid emergence of multi-resistant and extensively drug-resistant M. tuberculosis strains, which are essentially untreatable at this time. The development of new drugs targeting resistant strains is clearly now a priority.
In searching for new vulnerable processes in M. tuberculosis to enable the development of more efficient antituberculosis chemotherapies, a novel antimycobacterial drug target has recently been discovered. GlgE is an essential maltosyltransferase that elongates α-glucans. Kalscheuer et al. (2) characterized this novel carbohydrate-active enzyme and defined its role within a new nonclassical primary metabolic pathway for α-glucan biosynthesis. This four-step pathway comprises the TreS, Pep2, GlgE, and GlgB enzymes for the conversion of trehalose into a branched α-glucan that resembles glycogen (3). A key enzyme in this pathway is the essential GlgE maltosyltransferase that uses maltose 1-phosphate (M1P)4 as a donor substrate to generate the linear glucan backbone that is further branched by the GlgB enzyme. Moreover, the GlgE pathway is of particular interest because it is feasible that it is associated with virulence and persistence of M. tuberculosis as it might participate in the formation of capsular α-glucan, an extracellular pericellular wall component potentially involved in immune evasion (4–7). Surprisingly, the lethality associated with targeting GlgE is not due to the absence of product formation but is correlated with the hyper-accumulation to a toxic level of the GlgE donor substrate, M1P, which leads directly or indirectly to pleiotropic effects, toxicity, DNA damage, and cell death (2). This novel mode of killing by self-poisoning, together with the lack of GlgE in humans, makes it an attractive candidate for future antitubercular drugs (8).
The primary metabolic GlgE pathway combines gene essentiality with a synthetic lethal interaction with another α-glucan pathway defined by the glycosyl transferase Rv3032 (2). In addition, there is a complex interplay between the GlgE and Rv3032 pathways with the classical GlgA-GlgC glycogen pathway in mycobacteria such that they appear collectively to produce three types of α-glucans: cytosolic glycogen, capsular α-glucan, and methylglucose lipopolysaccharide (3, 7, 9). Therefore, each pathway is likely to be tightly regulated. To date, only the allosteric regulation of GlgC by metabolites has been reported (10).
To overcome the stressful conditions imposed by a host, pathogens have evolved various protective and offensive responses generally achieved through cascades of phosphorylation reactions. Many of the stimuli encountered by M. tuberculosis are transduced via sensor kinases in the membrane, allowing the pathogen to adapt for survival in hostile environments. In addition to the classical two-component systems, M. tuberculosis contains 11 eukaryotic-like serine/threonine protein kinases (STPKs) (11, 12). There is now an increasing body of evidence suggesting that many STPKs in M. tuberculosis are involved in regulating metabolic processes, transport of metabolites, cell division, and virulence (13, 14). Signaling through Ser/Thr phosphorylation has recently emerged as a key regulatory mechanism in pathogenic mycobacteria (13, 14). This idea is supported by recent studies elucidating the network of post-translational modifications of complex metabolic pathway such as mycolic acid biosynthesis, where most enzymes are regulated by Ser/Thr phosphorylation (15–20).
This study was undertaken to establish whether GlgE is regulated by phosphorylation in M. tuberculosis, with the potential to provide new approaches to target it therapeutically. To this end, we have identified GlgE as a new substrate of M. tuberculosis STPKs and identified its phosphorylation sites. This allowed us to address the role and contribution of phosphorylation in regulating the maltosyltransferase activity of GlgE. Importantly, with both in vitro and in vivo approaches, we provide for the first time evidence that phosphorylation will negatively regulate flux through the GlgE pathway.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth Conditions
Strains used for cloning and expression of recombinant proteins were Escherichia coli 10G (Lucigen) and E. coli BL21(DE3)Star (Novagen) as detailed in supplemental Table S1. They were grown in LB medium at 37 °C. Media were supplemented with ampicillin (100 μg/ml), hygromycin (200 μg/ml), kanamycin (25 μg/ml), or spectinomycin (100 μg/ml) when required. Mycobacteria strains were grown aerobically on Middlebrook 7H10 agar plates with oleic acid/albumin/dextrose/catalase enrichment (Difco) or in Middlebrook 7H9 medium supplemented with 10% (v/v) oleic acid/albumin/dextrose/catalase enrichment, 0.5% (v/v) glycerol and 0.05% (v/v) tyloxapol. Hygromycin (50 μg/ml) and kanamycin (25 μg/ml) were added for the selection of appropriate strains. Validamycin A (Research Products International Corp., Mount Prospect, IL) was used at 10 mm for inhibiting TreS activity when required.
Cloning, Expression, and Purification of M. tuberculosis GlgE and Mutant Proteins
The glgE gene was amplified by PCR using M. tuberculosis H37Rv chromosomal DNA as the template and the forward and reverse primers listed in supplemental Table S2 containing NdeI and BamHI restriction sites, respectively. The amplified product was digested with NdeI and BamHI and ligated into the pETPhos plasmid (21), a variant of pET15b (Novagen) that includes a tobacco etch virus protease site instead of the thrombin site and an N-terminal His tag free of Ser/Thr/Tyr residues, thus generating pETPhos_glgE. The glgE genes containing either the seven mutations T10A, T59A, S85A, T148A, T191A, T193A, and T370A or the mutations T10D, T59D, S85D, T191D, T193D, and T370D were both synthesized by GenScript with 5′-NdeI and 3′-BamHI restriction sites. These two glgE mutants were cloned into the pETPhos vector, generating pETPhos_glgE_T10A/T59A/S85A/T148A/T191A/T193A/T370A and pETPhos_ glgE_T10D/T59D/S85D/T148D/T191D/T193D/T370D named pEThos_glgE_Ala and pETPhos_glgE_Asp, respectively. A duet strategy was used to generate hyper-phosphorylated GlgE protein as described previously (22). The glgE gene was cloned into the pCDFDuet-1 vector already carrying the sequence encoding the PknB kinase domain using primers listed in supplemental Table S2, thus generating plasmid pDuet_glgE, which was used to transform E. coli BL21(DE3)Star cells to overexpress His-tagged GlgE. All constructs were verified by DNA sequencing. Recombinant strains harboring the different constructs were used to inoculate 200 ml of LB medium supplemented with ampicillin or spectinomycin, and the resulting cultures were incubated at 37 °C with shaking until the optical density of the culture reached an A600 of 0.5. Isopropyl-1-thio-β-d-galactopyranoside (1 mm final) was added to induce the overexpression, and growth was continued for 3 h at 37 °C. Purification of the His-tagged recombinant proteins was performed as described by the manufacturer (Qiagen).
Cloning, Expression, and Purification of Mycobacterium smegmatis GlgEMs and PknBMs and Streptomyces coelicolor GlgESc and AfsK
The glgE gene from M. smegmatis was amplified by PCR using M. smegmatis mc2155 chromosomal DNA as the template and the forward and reverse primers containing NdeI and BamHI restriction sites (supplemental Table S2), respectively. The amplified product was digested with NdeI and BamHI and ligated into the pETPhos plasmid, yielding pETPhos_glgEMs, which was used as a source of His-tagged GlgEMs. The pknB kinase domain open reading frame from M. smegmatis was amplified in the same way except that the reverse primer contained a BglII restriction site (supplemental Table S2). The amplified product was digested with NdeI and BglII and ligated into the pETPhos plasmid that was previously digested with NdeI and BamHI, resulting in pETPhos_pknBMs, which was used to prepare His-tagged PknBMs. The glgE isoform I gene from S. coelicolor (Sco5443) was amplified using S. coelicolor M145 chromosomal DNA as the template with the forward and reverse primers containing NdeI and BamHI restriction sites (supplemental Table S2), respectively. The amplified product was digested with NdeI and BamHI and ligated into pETPhos, thus generating pETPhos_glgESc, which was used to prepare His-tagged GlgESc. The afsK kinase domain open reading frame from S. coelicolor was similarly amplified with the forward and reverse primers (supplemental Table S2) containing BamHI and HindIII restriction sites, respectively. The amplified product was digested with BamHI and HindIII, and ligated into the pGEX(M) vector, yielding pGEX_afsK, which was used for the production of GST-tagged AfsK, as described previously (23).
In Vitro Kinase Assay
In vitro phosphorylation was performed with 4 μg of wild-type GlgE or GlgE derivatives in 20 μl of buffer P (25 mm Tris-HCl, pH 7.0, containing 1 mm DTT, 5 mm MgCl2, and 1 mm EDTA) with 200 μCi/ml [γ-33P]ATP (PerkinElmer Life Sciences, 3000 Ci/mmol) and 2–4 μg of kinase to obtain the optimal autophosphorylation activity for each mycobacterial kinase for 30 min at 37 °C. For in vitro phosphorylation with PknBMs or AfsK, 3 μg of kinase were used. Each reaction mixture was stopped by the addition of an equal volume of 5× Laemmli buffer, and the mixture was heated at 100 °C for 5 min. After electrophoresis, gels were soaked in 16% TCA for 10 min at 90 °C and dried. Radioactive proteins were visualized by autoradiography using direct exposure to films.
Mass Spectrometry Analysis
Purified His-tagged hyper-phosphorylated GlgE (GlgE-P) from the E. coli strain carrying pDuet_glgE and co-expressing the PknB kinase domain was subjected to mass spectrometry without further treatment. Subsequent mass spectrometric analyses were performed as reported previously (16, 20).
Overexpression of GlgE and a Derivative in Mycobacterium bovis BCG and Their Purification
M. tuberculosis glgE and glgE_Ala genes were amplified from the corresponding pETPhos vector constructs and cloned into the shuttle vector pVV16 (24) using the primers listed in supplemental Table S2. The resulting constructs pVV16_glgE and pVV16_glgE_Ala were electroporated into M. bovis BCG. Transformants were grown and used for the purification of the His-tagged GlgE proteins as described above. The purified recombinant proteins were used for immunoblotting using anti-phosphothreonine, anti-phosphoserine, and anti-phosphotyrosine antibodies according to the manufacturer's instructions (Invitrogen) and revealed with secondary antibodies labeled with IRDye infrared dyes (Odyssey Classic) to increase the detection sensitivity of phosphorylated proteins.
Enzymatic Assays
Before assaying for enzyme activity, proteins were further purified using size exclusion chromatography using an Superdex S200 16/60 column (Amersham Biosciences) with 20 mm Tris buffer, pH 8.5, containing 100 mm NaCl at 1 ml/min. Fractions containing GlgE were pooled, dialyzed against 20 mm Tris, pH 8.5, concentrated to ∼2.3 mg/ml, and stored in aliquots at −80 °C. GlgE specific activity was monitored at 21 °C using an end-point assay involving inorganic phosphate release detection with malachite green (2). Reaction mixtures of 25 μl comprised 1 mm maltohexaose, 0.25 mm M1P, 100 mm Bis-Tris propane, pH 7.0, and 50 mm NaCl. Enzyme concentrations were such to allow reactions to progress linearly for 30 min with total donor consumption being <40%. Reactions were quenched with 175 μl of malachite green and incubated for 20 min at 21 °C, and the absorbance at 630 nm was measured on a SpectraMax Plus microplate spectrophotometer using SoftMax Pro 3.1.1 software. The affinity for maltohexaose was determined using this method with a 0–10 mm maltohexaose concentration range and 0.25 mm M1P. The affinity for M1P was determined at 37 °C with a 0.25–5 mm M1P concentration range and 1 mm maltohexaose using a modified method allowing the monitoring of initial rates as described previously (2).
Circular Dichroism Spectroscopy
Circular dichroism spectra were collected on a JASCO J-710 spectropolarimeter. Samples comprised 10 mm sodium phosphate buffer, pH 7.0, and 0.5 mg/ml protein. Far-ultraviolet (180–260 nm) spectra were collected using a 0.02 mm quartz cuvette. Spectra were recorded with a sensitivity of 100 millidegrees, 0.2-nm data pitch, 4-s response time, and a 1.0-nm bandwidth at a scan speed of 20 nm/min. Triplicate spectra were averaged followed by the subtraction of buffer-only control spectra. Far-ultraviolet spectra were analyzed with Dichroweb (25) using the CDSSTR algorithm and reference set 1 (26).
Complementation Studies
The M. smegmatis mc27124 (ΔglgE) strain (2) was transformed with pVV16, pVV16_glgE, pVV16_glgE_Ala, or pVV16_glgE_Asp (supplemental Table S1) that were constructed as follows. The glgE derivatives (lacking the stop codon) were amplified using the corresponding pETPhos constructs as the template with the primers listed in supplemental Table S2 and cloned into the pVV16 vector digested with NdeI/HindIII. The resulting constructs were electroporated into M. smegmatis mc27124. Clones selected on kanamycin were grown in 7H9 medium at 37 °C to mid-log phase and plated at 37 °C for 3–5 days on Middlebrook 7H10 agar plates with oleic acid/albumin/dextrose/catalase enrichment plus 1 mm of trehalose and/or 10 mm validamycin A (Research Products International Corp.) when required. Moreover, transformants were grown in similar growth conditions, and His-tagged GlgE proteins were purified as described above.
RESULTS
GlgE Is Phosphorylated in Vitro by the Mycobacterial Ser/Thr Kinase PknB
The M. tuberculosis genome encodes 11 STPKs (11, 12). Although these mycobacterial kinases appear to be involved in different key pathways such as cell wall metabolism, antibiotic susceptibility, and virulence (13, 14, 27, 28), little is known about the nature of the substrates that are phosphorylated. Due to our interest in Ser/Thr kinase regulation and central metabolism in M. tuberculosis, we investigated whether the newly discovered GlgE metabolic pathway for nonclassical glucan biosynthesis is regulated by Ser/Thr phosphorylation. This was first investigated in vitro in the presence of purified STPKs. The soluble kinase domains of several transmembrane kinases from M. tuberculosis were expressed as His-tagged fusion proteins and purified from E. coli as reported earlier (19). The kinase enzymes were incubated with M. tuberculosis GlgE and [γ-33P]ATP, the proteins were resolved by SDS-PAGE, and the protein phosphorylation status was analyzed by autoradiography. The presence of an intense radioactive signal indicated that GlgE was phosphorylated by PknB (Fig. 1A). No signal was observed in the presence of PknA, PknD, PknE, or PknF, all of which displayed autokinase activities as reported earlier (19). As expected, no radioactive band was observed in the absence of kinase. These results clearly indicate that GlgE interacts with and is specifically phosphorylated by PknB, at least in vitro, suggesting that this key enzyme of glucan biosynthesis could be regulated by phosphorylation in mycobacteria.
FIGURE 1.

A, in vitro phosphorylation of M. tuberculosis GlgE by PknB. The soluble domains of five recombinant M. tuberculosis STPKs (PknA to PknF) were expressed and purified as His-tagged fusions and incubated with purified His-tagged GlgE and [γ-33P]ATP. The amount of the STPKs used varied from 2 to 4 μg to obtain the optimal autophosphorylation activity for each kinase. Samples were separated by SDS-PAGE, stained with Coomassie Blue (upper panel), and visualized by autoradiography after overnight exposure to a film (lower panel). Upper bands reflect the phosphorylation signal of GlgE, and the lower bands correspond to the autophosphorylation activity of each kinase. M, molecular mass markers. B, in vitro phosphorylation of the GlgE_Ala mutant. Purified GlgE and phosphoablative GlgE (GlgE_Ala) were incubated with PknB and [γ-33P]ATP. Samples were separated by SDS-PAGE, stained with Coomassie Blue (upper panel), and visualized by autoradiography (lower panel) after overnight exposure to a film.
GlgE Is Phosphorylated on Serine and Threonine Residues
Mass spectrometry was used to identify the nature and location of the phosphorylation site(s) on M. tuberculosis GlgE. Such a method has been successfully used to elucidate the phosphorylation sites in a sequence-specific fashion for several M. tuberculosis STPK substrates (15–17, 20, 29, 30). Phosphorylated GlgE-P was purified from E. coli co-expressing PknB and GlgE (pDuet_glgE) and subjected to mass spectrometric analysis after tryptic digestion. A sequence coverage of 95% that included all Ser and Thr residues was obtained. Spectra were analyzed with the paragon algorithm from the ProteinPilot® 2.0 database-searching software (Applied Biosystems) using the phosphorylation emphasis criterion against a homemade database that included the sequences of GlgE and its derivatives. The phosphopeptides identified by the software were then validated by manual examination of the corresponding MS/MS spectra. Manual validations were performed based on neutral loss of H3PO4 from the precursor ions and the assignment of major fragment ions to b- and y-ion series or to the corresponding neutral loss of H3PO4 from these fragment ions. The MS/MS spectra unambiguously identified the presence of seven phosphate groups on peptides (Table 1 and supplemental Fig. S1), thus indicating that GlgE is phosphorylated on six threonines, Thr-10, Thr-59, Thr-148, Thr-191, Thr-193, and Thr-370, and on a single serine, Ser-85.
TABLE 1.
Phosphoacceptors identified after purification of M. tuberculosis GlgE from the E. coli strain co-expressing M. tuberculosis PknB
Sequences of the phosphorylated peptides identified in GlgE as determined by mass spectrometry following tryptic digestion are indicated, and phosphorylated residues (pT or pS) are shown in bold.
| Phosphorylated tryptic peptide sequence of GlgE purified from pCDFDuet co-expressing PknB | Number of detected phosphate groups (LC/MS/MS) | Phosphorylated residue(s) |
|---|---|---|
| [5–17] AIGTEpTEWWVPGR | 1 | Thr-10 |
| [51–63] EGHEAVAApTLVVR | 1 | Thr-59 |
| [80–90] VLPTPpSEPQQR | 1 | Ser-85 |
| [141–163] LDAGQGEpTELSNDLLVGAVLLER | 1 | Thr-148 |
| [185–192] TPGDPVpTR | 1 | Thr-191 |
| [185–211] TPGDPVTRpTALALTPEIEELLADYPLR | 1 | Thr-193 |
| [361–379] QWFTELPDGpTIAYAENPPK | 1 | Thr-370 |
Then, to prevent in vitro phosphorylation, these residues were mutated to alanine. The corresponding phosphoablative GlgE_T10A/T59A/S85A/T148A/T191A/T193A/T370A mutant (GlgE_Ala) was expressed as a His-tagged protein in E. coli BL21(DE3)Star harboring pETPhos_glgE_Ala. The resulting GlgE_Ala mutant protein was purified, analyzed by circular dichroism to confirm its proper folding (see below and supplemental Table S3), and incubated with PknB in the presence of [γ-33P]ATP. Following separation by SDS-PAGE and analysis by autoradiography, total abrogation of the phosphorylation signal occurred when compared with the wild-type GlgE (Fig. 1B). As expected, GlgE_Ala was phosphorylated by neither PknA, PknD, PknE, nor PknF (supplemental Fig. S2). These results suggest that specific phosphorylation of these seven residues by PknB may play a role in the regulation of M. tuberculosis GlgE activity.
GlgE Is Phosphorylated in Mycobacteria
To address the relevance of in vitro phosphorylation, we next investigated the in vivo phosphorylation status of M. tuberculosis GlgE in GlgE-overexpressing strains of M. bovis BCG, a species frequently used as a nonvirulent surrogate strain closely related to M. tuberculosis, by Western blotting using anti-phosphothreonine, anti-phosphoserine, or anti-phosphotyrosine antibodies (20). First, the specificity of the antibodies was determined using the GlgE isoforms purified from either E. coli or E. coli co-expressing PknB, based on the strategy described by us (22). Only the phosphorylated GlgE isoform derived from pDuet_glgE (GlgE-P) reacted with the anti-phosphothreonine and anti-phosphoserine antibodies as expected, whereas the unphosphorylated GlgE isoform derived from the pETPhos_glgE construct (GlgE) failed to generate a signal with these antibodies (Fig. 2). To address the phosphorylation state of GlgE in mycobacteria, the genes encoding GlgE or GlgE_Ala (harboring Ser/Thr to Ala substitutions at all seven positions) as C-terminal His tag fusions were cloned into the shuttle vector pVV16, and the resulting constructs, designated pVV16_glgE and pVV16_glgE_Ala, respectively, were introduced into M. bovis BCG. The His tag-purified proteins derived from these M. bovis BCG recombinant strains were subjected to Western blotting using anti-phosphothreonine, anti-phosphoserine, and anti-phosphotyrosine antibodies. As shown in Fig. 2, a clear signal for the wild-type GlgE was detected with anti-phosphothreonine and anti-phosphoserine antibodies, whereas no signal was observed with the GlgE_Ala (Fig. 2). No signal could be detected when probing the membrane with anti-phosphotyrosine antibodies (data not shown). Overall, these results not only confirm the presence of phosphorylated Ser and Thr residues only, but also demonstrate that GlgE is phosphorylated in mycobacteria.
FIGURE 2.

Phosphorylation of GlgE in mycobacteria. E. coli harboring pETPhos_glgE was used as a source of nonphosphorylated GlgE (GlgE), and the strain harboring pDuet_glgE coexpressing PknB and GlgE provided the phosphorylated GlgE isoform (GlgE-P). GlgE and GlgE_Ala were produced in M. bovis BCG strains harboring pVV16_glgE or pVV16_glgE_Ala, respectively. Three μg of purified His-tagged GlgE derivatives were analyzed by SDS-PAGE after staining with Coomassie Blue (upper panel), detected on independent SDS-PAGE gels by immunoblotting using anti-phosphothreonine (middle panel) or anti-phosphoserine (lower panel) antibodies according to the manufacturer's instructions (Invitrogen), and revealed with secondary antibodies labeled with IRDye infrared dyes (Odyssey Classic). M, molecular mass markers.
Phosphorylation of GlgE Is Conserved in Other Actinomycetes
There is a high degree of sequence identity between the GlgE of M. tuberculosis and those of M. smegmatis (79%) and S. coelicolor (54%). However, not all of the phosphorylation sites identified for M. tuberculosis GlgE were fully conserved among GlgE orthologues (supplemental Fig. S3). We therefore wondered whether the GlgE proteins from these other actinomycetes are phosphorylated as well. Thus, the different GlgE orthologues were expressed in E. coli, purified, and subjected to in vitro kinase assays in the presence of their corresponding kinase counterparts. Fig. 3 clearly shows that the GlgE isoform I from S. coelicolor (GlgESc) and M. smegmatis (GlgEMs) could be phosphorylated by the S. coelicolor PknB homologue, AfsK, and the M. smegmatis PknB orthologue (PknBMs), respectively. These results indicate that GlgEMs and GlgESc are, like M. tuberculosis GlgE, kinase substrates and suggest that Ser/Thr kinase regulation of GlgE is likely to be conserved in actinomycetes despite incomplete conservation of the phosphorylation sites.
FIGURE 3.
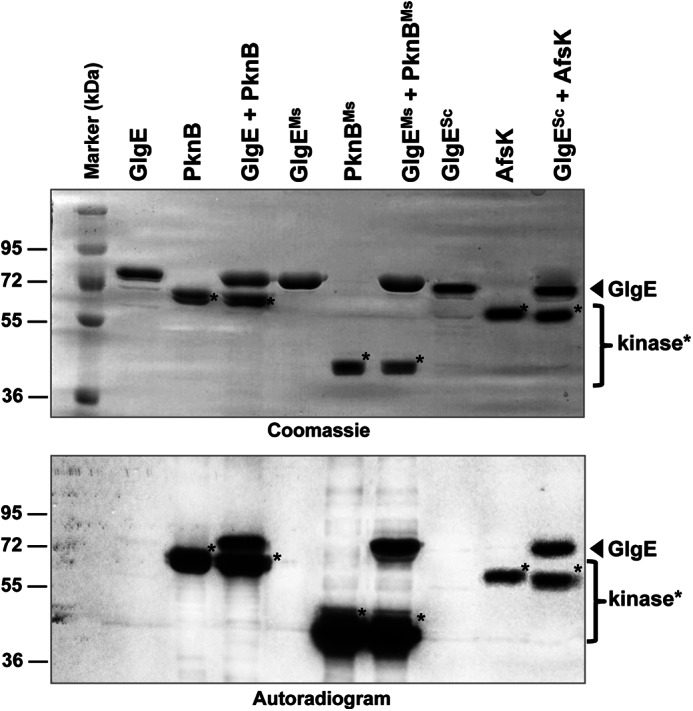
In vitro phosphorylation of the GlgE orthologues from M. smegmatis and S. coelicolor. Recombinant M. smegmatis PknB kinase (PknBMs) and S. coelicolor AfsK kinase (AfsK) were expressed as His-tagged fusion proteins in E. coli, purified, and incubated with purified His-tagged M. smegmatis GlgE (GlgEMs) and S. coelicolor GlgE (GlgESc), respectively, in the presence of [γ-33P]ATP. GlgE and PknB proteins from M. tuberculosis were included as a positive control. Samples were separated by SDS-PAGE, stained with Coomassie Blue (upper panel), and visualized by autoradiography (lower panel). Marker indicates molecular mass markers.
Phosphorylation Negatively Regulates GlgE Maltosyltransferase Activity
Phosphorylation of metabolic enzymes can be associated with either positive or negative regulation. To test whether phosphorylation affects the maltosyltransferase activity of GlgE, we compared the activity of nonphosphorylated and phosphorylated GlgE. Recombinant His-tagged M. tuberculosis GlgE from E. coli carrying pETPhos_glgE was used as a source of nonphosphorylated GlgE (designated GlgE) as confirmed by Western blot analysis (Fig. 2) and mass spectrometry (data not shown). By contrast, when co-expressed with PknB with the pDuet system, the purified protein was found to be substantially phosphorylated (designated GlgE-P; Fig. 2). Circular dichroism spectroscopy indicated that the net secondary structure of GlgE was not significantly affected by phosphorylation (supplemental Table S3) and was consistent with the known crystal structure of the S. coelicolor isoform I enzyme (31). It was then possible to determine the kinetics of the maltosyltransferase activity of GlgE and GlgE-P by monitoring the release of inorganic phosphate during the extension of maltohexaose by M1P (2). Phosphorylation of GlgE led to a 31-fold decrease in kcatapp/Kmapp for maltohexaose (Table 2), clearly demonstrating that the enzyme was negatively regulated. The dependence of GlgE-P activity on M1P concentration did not give a normal saturation curve because activity decreased markedly above 1 mm M1P, and this behavior did not adhere to normal substrate inhibition kinetics. Nevertheless, the maximum rate observed at 1 mm M1P was 63-fold lower than the corresponding kcatapp observed with the wild-type enzyme, consistent with a reduction in enzyme activity by 2 orders of magnitude as a consequence of phosphorylation.
TABLE 2.
Michaelis-Menten kinetic analysis of M. tuberculosis GlgE derivatives
Enzyme activity was monitored by detecting Pi release in triplicate and values are expressed as the mean and standard error. Quoted constants are apparent because the enzyme obeys ping-pong (substituted enzyme) kinetics (2).
| Kmapp | kcatapp | kcatapp/Kmapp | |
|---|---|---|---|
| mm | s−1 | m−1 s−1 | |
| Maltohexaosea | |||
| GlgE | 5.5 ± 0.5 | 4.3 ± 0.2 | 780 ± 80 |
| GlgE_Ala | 2.1 ± 0.2 | 0.72 ± 0.3 | 340 ± 40 |
| GlgE_Asp (dimer) | 1.6 ± 0.1 | 0.44 ± 0.01 | 280 ± 20 |
| GlgE_Asp (monomer) | 1.64 ± 0.05 | 0.216 ± 0.002 | 132 ± 4 |
| GlgE-P | 2.2 ± 0.2 | 0.053 ± 0.002 | 25 ± 2 |
| M1Pb | |||
| GlgE | 0.25 ± 0.03 | 1.25 ± 0.06 | 5,000 ± 600 |
| GlgE_Ala | 0.33 ± 0.07 | 0.59 ± 0.03 | 1,800 ± 400 |
| GlgE_Asp (dimer) | 0.45 ± 0.09 | 0.27 ± 0.01 | 600 ± 100 |
| GlgE_Asp (monomer) | 0.9 ± 0.2 | 0.15 ± 0.01 | 170 ± 40 |
| GlgE-P | ∼0.24c | 0.020 ± 0.001d | ∼80e |
a In the presence of 0.25 mm M1P.
b Maltose 1-phosphate in the presence of 1 mm maltohexaose.
c The GlgE-P enzyme was severely inhibited at concentrations of M1P > 1 mm with kinetics that did not adhere to simple substrate inhibition. Therefore, this value reflects the concentration of M1P that gave half the maximum observed rate.
d This value is the maximum observed rate that was with 1 mm M1P (see footnote c above).
e This value reflects the ratio between the maximum observed rate (footnote d) and the concentration of M1P that gave half the maximum observed rate (footnote c).
Phosphomimetic GlgE Has Reduced Enzyme Activity
To test the effect of GlgE activity in vivo, M. tuberculosis GlgE variants with constitutively altered activities were required. Earlier studies have shown that the acidic amino acids, Asp or Glu, mimic the functional effect of phosphorylation (18, 20, 32), whereas we have shown that Thr substitution by Ala prevents phosphorylation. Following a strategy successfully used to demonstrate the role of phosphorylation on Ser/Thr kinase substrates in other systems (33), we expressed and purified phosphomimetic GlgE_Asp and phosphoablative GlgE_Ala, where each phosphorylation site was mutated to mimic or ablate phosphorylation, respectively.
GlgE_Ala was almost as active as the wild-type enzyme. A 2- and 3-fold reduction in kcatapp/Kmapp for maltohexaose and M1P, respectively, was presumably due to the invasive nature of the amino acid substitutions.
Unexpectedly, the purified GlgE_Asp protein was atypical because size exclusion chromatography revealed that it was present not only as a dimer, the exclusive quaternary structure observed with all other GlgE derivatives, but also as a monomer in roughly equal proportions (supplemental Fig. S4). Circular dichroism spectroscopy showed that the distribution of protein secondary structures in both of the dimeric mutant proteins was very similar to that of the wild-type and phosphorylated proteins (supplemental Table S3). Monomeric GlgE_Asp was somewhat different because it possessed a little less α-helix (12% rather than 18% for the wild-type), presumably as a result of the loss of subunit interfaces, but was otherwise similar to wild type.
The kcatapp/Kmapp for maltohexaose and M1P with the GlgE_Asp dimer showed a 1.2 and 3-fold reduction, respectively, when compared with GlgE_Ala (Table 2). The activity of monomeric GlgE_Asp was even lower with a 2.5- and 10-fold reduction when compared with GlgE_Ala, perhaps reflecting the change in quaternary structure and slight perturbation in secondary structure. Interestingly, the activities of the monomeric and dimeric forms converged to an intermediate value over a period of days. This implied a slow equilibration between these two species consistent with the isolation of equal proportions of each species from E. coli. It therefore appears that GlgE_Asp spontaneously forms a mixture of monomeric and dimeric forms, which occurs in vivo as well as in vitro. Thus, the effective reduction in kcatapp/Kmapp between GlgE_Ala and GlgE_Asp would most probably reflect intermediate values of ∼2–7-fold. Although this reduction was not of the same magnitude as that between GlgE and GlgE-P, the strategy was nevertheless successful in terms of generating GlgE derivatives with a range of activities that could not be modified further by PknB.
With all GlgE derivatives, the reduction in kcatapp/Kmapp for maltohexaose was dominated by a reduction in kcatapp (Table 2) because the affinity for maltohexaose actually increased a little. No significant change in the affinity for the alternative acceptor, glycogen, was observed (data not shown), again consistent with a kcatapp effect. Although the reduction in kcatapp/Kmapp for the donor substrate, M1P, was also dominated by a lowering of kcatapp, there was some contribution from an increase in the Kmapp for M1P.
GlgE_Asp Does Not Complement a ΔglgE Mutant Strain
To investigate the effect of GlgE activity in vivo, we took advantage of a ΔglgE M. smegmatis (m27124) strain that had been generated previously (2). In contrast to GlgE essentiality in M. tuberculosis, null deletion of the glgE gene could be readily generated in M. smegmatis on minimal medium (2). Despite normal growth in minimal medium, the M. smegmatis ΔglgE mutant is unable to grow in complex media and is sensitive to the disaccharide trehalose (α-d-glucopyranosyl-(1→1)-α-d-glucopyranoside) (2). Thus, the M. smegmatis ΔglgE mutant is not able to grow in minimal medium in the presence of exogenous trehalose. Trehalose-induced bacteriostasis in the M. smegmatis ΔglgE mutant correlates with hyper-accumulation of the GlgE substrate, M1P, which is formed from trehalose by the successive action of the trehalose synthase TreS and the maltokinase Pep2 and which becomes toxic at very high intracellular levels (2). M. smegmatis ΔglgE was therefore transformed with either the empty pVV16 vector or the pVV16 derivatives, allowing constitutive expression of the different glgE alleles in mycobacteria together with selection for kanamycin resistance. Cultures were then plated and grown at 37 °C in the presence of 1 mm trehalose for 4–5 days. Fig. 4A shows that cells transformed with constructs carrying either the glgE or the glgE_Ala alleles could grow on trehalose, indicating that functional complementation occurred, suggesting that the GlgE wild-type and GlgE_Ala isoforms were sufficiently active to metabolize M1P, preventing the trehalose-induced toxic effect. In sharp contrast, the glgE_Asp allele failed to restore growth on trehalose, showing that the reduction in activity of the GlgE_Asp isoform was severe enough to exhibit a phenotype in vivo (Fig. 4A). These phenotypes could not be attributed to alteration in the expression levels of the various GlgE variants, as demonstrated by SDS-PAGE analysis with Coomassie Blue staining following purification of the His-tagged proteins from the complemented M. smegmatis ΔglgE strains (Fig. 4B). We also noticed that the M. smegmatis GroEL1 protein was co-purified in each preparation, presumably due to the presence of a known His-rich region capable of binding to the nickel-bearing beads, as reported earlier (34). This inadvertently provided an internal standard supporting the conclusion that there were similar expression levels of the GlgE variants. Western blots with anti-His tag antibody of crude extracts gave further support for the comparable expression levels of each GlgE variant (data not shown). The rate of proteolytic digestion of the GlgE variants was similar in each case (supplemental Fig. S5) with a small difference in the proteolytic cleavage pattern with the GlgE_Asp variant being rationalized as one of the Asp substitutions affecting one of the predicted cleavage sites. Therefore, the integrity of each GlgE variant appeared to be maintained in M. smegmatis. Attempts to measure GlgE activity in these strains were hampered by the deliberately low level of expression of each variant compounded by a significant background phosphatase activity interfering with the enzyme assay.
FIGURE 4.

A, functional complementation of M. smegmatis ΔglgE with GlgE or GlgE_Ala but not with phosphomimetic GlgE_Asp. The ΔglgE mutant of M. smegmatis, which is sensitive to trehalose, was transformed with pVV16, pVV16_glgE, pVV16_glgE_Ala, or pVV16_glgE_Asp and grown with or without trehalose (1 mm). Plates were incubated at 37 °C for 4–5 days. B, expression of the different GlgE variants in the complemented M. smegmatis ΔglgE strains. GlgE proteins from M. smegmatis ΔglgE cultures complemented with the wild-type pVV16_glgE, pVV16_glgE_Ala, or pVV16_glgE_Asp constructs were obtained by purification by nickel affinity chromatography. Equal quantities of purified His-tagged GlgE proteins per unit volume of culture were separated by SDS-PAGE, stained with Coomassie Blue, and identified using mass spectrometry. The mycobacterial GroEL1 protein was co-purified due to it harboring a region rich in histidine residues, thus providing an internal standard. Marker indicates molecular mass markers.
Finally, to correlate the trehalose-sensitive phenotype of the M. smegmatis ΔglgE mutant expressing GlgE_Asp with M1P hyper-accumulation, experiments were conducted under conditions where M1P formation was suppressed by genetic or chemical inactivation of TreS trehalose synthase activity, as demonstrated previously with the ΔglgE mutant (2). First, we used an M. smegmatis ΔglgEΔtreS mutant strain that was transformed with the different pVV16_glgE derivatives. The glgE_Asp allele could be readily introduced into the double ΔglgEΔtreS mutant in the presence of trehalose (Fig. 5A), supporting the direct link between the GlgE_Asp lethal phenotype on trehalose (Fig. 4A) and the significantly reduced maltosyltransferase activity of this isoform (Fig. 6). Second, using an alternative to the genetic approach, TreS was chemically inhibited with validamycin A to prevent M1P toxicity (Fig. 6), as demonstrated previously (2). As expected, it was possible to introduce the glgE_Asp allele into the ΔglgE mutant strain in the presence of trehalose and validamycin A (Fig. 5B). As expected, an empty vector control also allowed growth when TreS was blocked genetically or chemically, consistent with GlgE_Asp leading to lethality only through M1P toxicity (Figs. 4 and 5). Overall, these results show that introduction of the glgE_Asp GlgE allele into M. smegmatis ΔglgE cannot restore metabolic flux through the GlgE pathway in the presence of trehalose (Fig. 6), leading to mycobacterial death. Thus, both classical and chemical reverse genetic approaches are consistent with flux through the GlgE pathway correlating with GlgE activity.
FIGURE 5.
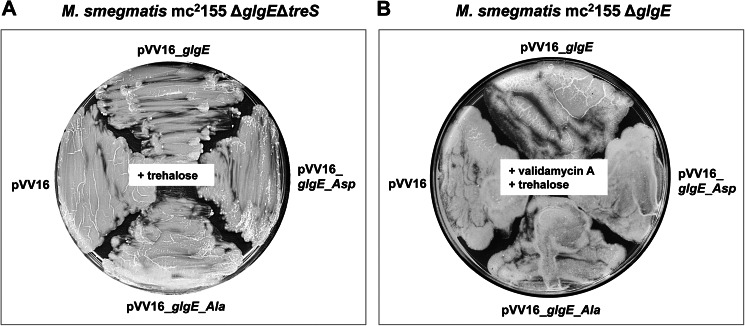
Complementation of the M. smegmatis ΔglgE single and ΔglgEΔtreS double mutants with GlgE variants. A and B, both genetic (A) and chemical (B) inactivation of TreS prevent M1P hyper-accumulation in the presence of exogenous trehalose. A, genetic inactivation of treS. The ΔglgEΔtreS mutant, which is insensitive to trehalose, was transformed with pVV16, pVV16_glgE, pVV16_glgE_Ala, or pVV16_glgE_Asp and grown with 10 mm trehalose. Plates were incubated at 37 °C for 4–5 days. B, chemical inactivation of TreS in the presence of validamycin A. The ΔglgE mutant was transformed with pVV16, pVV16_glgE, pVV16_glgE_Ala, or pVV16_glgE_Asp and grown in the presence of both validamycin A (10 mm) and trehalose (1 mm). Plates were incubated at 37 °C for 4–5 days.
FIGURE 6.

Proposed regulatory role of the phosphorylated isoform of GlgE as a molecular switch in α-glucan synthesis in M. tuberculosis. Distinct from the classic glycogen pathway, the newly identified GlgE pathway comprises four successive enzymatic steps from trehalose to an α-glucan polymer that may contribute to biosynthesis of cytosolic glycogen, capsular glucan, and/or methylglucose lipopolysaccharide-related α-glucan derivatives. In response to external stimuli, the mycobacterial Ser/Thr kinase PknB is autophosphorylated. This induces GlgE phosphorylation (P) on seven Ser/Thr residues. As a result, the maltosyltransferase activity of the Ser/Thr phosphorylated GlgE is inhibited.
DISCUSSION
We have use a combination of genetic and biochemical approaches to provide the first reported evidence that the M1P-dependent maltosyltransferase GlgE, a recently identified and genetically validated antituberculosis target (8), is negatively regulated by phosphorylation. M. tuberculosis GlgE was shown to be phosphorylated both in vitro and in vivo, showing that GlgE was missed in a recent phosphoproteomic study performed on M. tuberculosis (35). One serine (Ser-85) and six threonine (Thr-10, Thr-59, Thr-148, Thr-191, Thr-193, and Thr-370) residues were phosphorylated specifically by the Ser/Thr kinase PknB. This resulted in a reduction in enzyme activity in vitro that correlated with lethality in vivo with a GlgE phosphomimetic implying a toxic buildup of the GlgE substrate, M1P, as observed previously for a GlgE null mutant (8). Therefore, the GlgE α-glucan pathway appears to be regulated by the STPK-dependent phosphorylation of GlgE, consolidating the emerging theme of Ser/Thr phosphorylation playing a critical role in the regulation of anabolic pathways in mycobacteria (Fig. 6) (13, 14). We have also shown that the phosphorylation of GlgE occurs not only in M. tuberculosis but also in other actinomycetes such as Streptomyces. Importantly, phosphoregulation of the GlgE pathway is distinct from the regulation of the classical GlgC/GlgA-dependent glycogen biosynthetic pathway that involves allosteric regulation by metabolic intermediates (10).
The regulation of the GlgE pathway through inhibition of the GlgE-catalyzed step raises an important question because blocking this step has been shown to be lethal in M. tuberculosis (8). The implication is that there is at least one other upstream step within the glucan pathway that is co-regulated to prevent the buildup of M1P to toxic levels in M. tuberculosis. Alternatively, the extent to which GlgE is down-regulated is limited to fine-tuning, perhaps in coordination with the regulation of the GlgA-GlgC and Rv3032 pathways. Interestingly, a temperature-sensitive GlgE mutant in M. smegmatis has been reported to affect glycogen/α-glucan recycling and growth rate (36). Particularly noteworthy was the observation that the temperature-sensitive mutation was suppressed by the overexpression of GarA (36), a forkhead-associated domain protein that is known not only to regulate both carbon and nitrogen metabolism but also to be a substrate for PknB (37). Perhaps the overexpressed GarA acted as a decoy, suppressing the phosphorylation and down-regulation of an already compromised temperature-sensitive GlgE by PknB. Alternatively, GarA interacts directly with GlgE in some way.
There is no known structure of M. tuberculosis GlgE but there is one for S. coelicolor GlgE isoform I (31). To establish where the phosphorylation sites are, it was possible to map their locations on the known structure (supplemental Fig. S6) with reference to a sequence alignment (supplemental Fig. S3). All sites were solvent-exposed, as expected. Three were within the S-domain, for which there is currently no confirmed role, three were within the N-domain, a domain making subunit interfaces, and one was within the B-domain, involved in defining the active site. Although the sites within the N-domain could have been responsible for the dissociation of the normally dimeric GlgE_Asp, dissociation was not observed with GlgE-P, suggesting that the dissociation of GlgE-Asp was due to an artifact. None of the phosphorylation sites are located within known substrate binding sites. Indeed, studies on the activity of single-site phosphomimetic GlgE derivatives suggest that no single site is solely responsible for the down-regulation of activity, suggesting that a cooperative inactivation involving several sites is required.5 The phosphorylation-dependent reduction in GlgE activity appears to be primarily a result of the lowering of kcatapp (Table 2). This could therefore be a result of long range electrostatic effects on catalytic intermediates. Any contribution of a lowering of the affinity for M1P could also be a result of long range electrostatic effects on the binding of this negatively charged substrate. Why GlgE-P is inhibited at high M1P concentrations is currently unknown. More structure/function work on GlgE-P and GlgE phosphomimetic is therefore required to establish the physical and structural basis for phosphoregulation.
Our work suggests that a shift in the unphosphorylated/phosphorylated GlgE balance in favor of the phosphorylated form rapidly leads to the accumulation of toxic M1P levels, leading to mycobacterial death. Therefore, an elegant hypothesis arising from the present work is that by increasing the activity of the PknB kinase, it may be possible to alter mycobacterial growth, opening new opportunities for future antituberculosis drug development. Indeed, small molecules that modulate the activity of STPKs in general may be of great therapeutic value in inhibiting M. tuberculosis growth. Bryostatin, a natural product synthesized by a marine bacterium, which activates eukaryotic intracellular STPKs (38), is one such molecule. Interestingly, bryostatin acts directly on the Bacillus subtilis Ser/Thr kinase PrkC, which contains an extracellular domain able to bind to peptidoglycan fragments, and this signals the bacteria to exit dormancy by stimulating germination (39). PrkC, like M. tuberculosis PknB, possesses the PASTA (penicillin and Ser/Thr kinase-associated) domains, which are found in the extracellular portion of membrane-associated STPKs and which have been proposed to bind to peptidoglycan and act as signaling molecules. In this context, bryostatin or other STPK-activating molecules, along with the recent structural determination of the M. tuberculosis PknB PASTA domains (40), may provide new therapeutic strategies to be developed against tuberculosis. Alternatively, if and when a phosphatase has been identified that dephosphorylates GlgE, it could also be a potential target.
In conclusion, the present study provides a foundation for further investigation of an important functional linkage between STPKs and the GlgE α-glucan biosynthetic pathway, which may contribute to the generation of the mycobacterial capsule. An abundant surface-exposed M. tuberculosis capsular α-glucan has been identified as a novel ligand for the C-type lectin DC-SIGN (41). Binding of α-glucan to DC-SIGN stimulated the production of immunosuppressive IL-10 by LPS-activated monocyte-derived dendritic cells. This observation suggests that the α-glucan capsule may fulfill an important role in pathogenesis. Recent work has demonstrated that the surface-exposed glucan plays an important role in the virulence of these bacteria and particularly in the persistence of infection in mice (7). Although there is still much to understand about the interplay between the three α-glucan pathways and three α-glucan products in mycobacteria, the present work provides a conceptual advance in our understanding of the metabolic adaptation and regulatory mechanisms associated with the recently identified GlgE α-glucan pathway. Although challenging, future studies will help identify extracellular cues sensed by kinases leading to the phosphorylation of GlgE and other glucan pathway enzymes. This will not only allow us to understand how M. tuberculosis senses its environment and mediates its response in a coordinated manner to regulate α-glucan biosynthesis, but also establish whether there is a link between GlgE phosphorylation and the establishment of the nonreplicating persistent state.
Acknowledgment
We thank Dr. Gerhard Saalbach for technical assistance at the John Innes Centre (Norwich, UK).
This work was supported by in part grants from the French National Research Agency (Grant ANR-09-MIEN-004) (to V. M. and L. K.), the United Kingdom Biotechnology and Biological Sciences Research Council (Grant BB/I012850/1 and BB/J004561/1) and the John Innes Foundation (to S. B.), and the Jürgen Manchot Foundation (to R. K.).

This article contains supplemental Tables S1–S3 and Figs. S1–S6.
K. Syson and S. Bornemann, unpublished observations.
- M1P
- maltose 1-phosphate
- STPK
- Ser/Thr protein kinase
- Bis-Tris
- 2-(bis(2-hydroxyethyl)amino)-2-(hydroxymethyl)propane-1,3-diol
- BCG
- bacillus Calmette–Guérin.
REFERENCES
- 1. Dye C., Williams B. G. (2010) The population dynamics and control of tuberculosis. Science 328, 856–861 [DOI] [PubMed] [Google Scholar]
- 2. Kalscheuer R., Syson K., Veeraraghavan U., Weinrick B., Biermann K. E., Liu Z., Sacchettini J. C., Besra G., Bornemann S., Jacobs W. R., Jr. (2010) Self-poisoning of Mycobacterium tuberculosis by targeting GlgE in an α-glucan pathway. Nat. Chem. Biol. 6, 376–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chandra G., Chater K. F., Bornemann S. (2011) Unexpected and widespread connections between bacterial glycogen and trehalose metabolism. Microbiology 157, 1565–1572 [DOI] [PubMed] [Google Scholar]
- 4. Dinadayala P., Sambou T., Daffé M., Lemassu A. (2008) Comparative structural analyses of the α-glucan and glycogen from Mycobacterium bovis. Glycobiology 18, 502–508 [DOI] [PubMed] [Google Scholar]
- 5. Gagliardi M. C., Lemassu A., Teloni R., Mariotti S., Sargentini V., Pardini M., Daffé M., Nisini R. (2007) Cell wall-associated α-glucan is instrumental for Mycobacterium tuberculosis to block CD1 molecule expression and disable the function of dendritic cell derived from infected monocyte. Cell. Microbiol. 9, 2081–2092 [DOI] [PubMed] [Google Scholar]
- 6. Kaur D., Guerin M. E., Skovierová H., Brennan P. J., Jackson M. (2009) Chapter 2: Biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv. Appl. Microbiol. 69, 23–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sambou T., Dinadayala P., Stadthagen G., Barilone N., Bordat Y., Constant P., Levillain F., Neyrolles O., Gicquel B., Lemassu A., Daffé M., Jackson M. (2008) Capsular glucan and intracellular glycogen of Mycobacterium tuberculosis: biosynthesis and impact on the persistence in mice. Mol. Microbiol. 70, 762–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kalscheuer R., Jacobs W. R., Jr. (2010) The significance of GlgE as a new target for tuberculosis. Drug News Perspect. 23, 619–624 [DOI] [PubMed] [Google Scholar]
- 9. Mendes V., Maranha A., Alarico S., Empadinhas N. (2012) Biosynthesis of mycobacterial methylglucose lipopolysaccharides. Nat. Prod. Rep. 29, 834–844 [DOI] [PubMed] [Google Scholar]
- 10. Ballicora M. A., Iglesias A. A., Preiss J. (2003) ADP-glucose pyrophosphorylase, a regulatory enzyme for bacterial glycogen synthesis. Microbiol. Mol. Biol. Rev. 67, 213–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E., 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M. A., Rajandream M. A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J. E., Taylor K., Whitehead S., Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 12. Av-Gay Y., Everett M. (2000) The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 8, 238–244 [DOI] [PubMed] [Google Scholar]
- 13. Wehenkel A., Bellinzoni M., Graña M., Duran R., Villarino A., Fernandez P., Andre-Leroux G., England P., Takiff H., Cerveñansky C., Cole S. T., Alzari P. M. (2008) Mycobacterial Ser/Thr protein kinases and phosphatases: physiological roles and therapeutic potential. Biochim. Biophys. Acta 1784, 193–202 [DOI] [PubMed] [Google Scholar]
- 14. Molle V., Kremer L. (2010) Division and cell envelope regulation by Ser/Thr phosphorylation: Mycobacterium shows the way. Mol. Microbiol. 75, 1064–1077 [DOI] [PubMed] [Google Scholar]
- 15. Veyron-Churlet R., Zanella-Cléon I., Cohen-Gonsaud M., Molle V., Kremer L. (2010) Phosphorylation of the Mycobacterium tuberculosis β-ketoacyl-acyl carrier protein reductase MabA regulates mycolic acid biosynthesis. J. Biol. Chem. 285, 12714–12725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Slama N., Leiba J., Eynard N., Daffé M., Kremer L., Quémard A., Molle V. (2011) Negative regulation by Ser/Thr phosphorylation of HadAB and HadBC dehydratases from Mycobacterium tuberculosis type II fatty acid synthase system. Biochem. Biophys. Res. Commun. 412, 401–406 [DOI] [PubMed] [Google Scholar]
- 17. Veyron-Churlet R., Molle V., Taylor R. C., Brown A. K., Besra G. S., Zanella-Cléon I., Fütterer K., Kremer L. (2009) The Mycobacterium tuberculosis β-ketoacyl-acyl carrier protein synthase III activity is inhibited by phosphorylation on a single threonine residue. J. Biol. Chem. 284, 6414–6424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Molle V., Gulten G., Vilchèze C., Veyron-Churlet R., Zanella-Cléon I., Sacchettini J. C., Jacobs W. R., Jr., Kremer L. (2010) Phosphorylation of InhA inhibits mycolic acid biosynthesis and growth of Mycobacterium tuberculosis. Mol. Microbiol. 78, 1591–1605 [DOI] [PubMed] [Google Scholar]
- 19. Molle V., Brown A. K., Besra G. S., Cozzone A. J., Kremer L. (2006) The condensing activities of the Mycobacterium tuberculosis type II fatty acid synthase are differentially regulated by phosphorylation. J. Biol. Chem. 281, 30094–30103 [DOI] [PubMed] [Google Scholar]
- 20. Corrales R. M., Molle V., Leiba J., Mourey L., de Chastellier C., Kremer L. (2012) Phosphorylation of mycobacterial PcaA inhibits mycolic acid cyclopropanation: consequences for intracellular survival and for phagosome maturation block. J. Biol. Chem. 287, 26187–26199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Canova M. J., Kremer L., Molle V. (2008) pETPhos: a customized expression vector designed for further characterization of Ser/Thr/Tyr protein kinases and their substrates. Plasmid 60, 149–153 [DOI] [PubMed] [Google Scholar]
- 22. Molle V., Leiba J., Zanella-Cléon I., Becchi M., Kremer L. (2010) An improved method to unravel phosphoacceptors in Ser/Thr protein kinase-phosphorylated substrates. Proteomics 10, 3910–3915 [DOI] [PubMed] [Google Scholar]
- 23. Molle V., Kremer L., Girard-Blanc C., Besra G. S., Cozzone A. J., Prost J. F. (2003) An FHA phosphoprotein recognition domain mediates protein EmbR phosphorylation by PknH, a Ser/Thr protein kinase from Mycobacterium tuberculosis. Biochemistry 42, 15300–15309 [DOI] [PubMed] [Google Scholar]
- 24. Jackson M., Crick D. C., Brennan P. J. (2000) Phosphatidylinositol is an essential phospholipid of mycobacteria. J. Biol. Chem. 275, 30092–30099 [DOI] [PubMed] [Google Scholar]
- 25. Whitmore L., Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sreerama N., Woody R. W. (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287, 252–260 [DOI] [PubMed] [Google Scholar]
- 27. Walburger A., Koul A., Ferrari G., Nguyen L., Prescianotto-Baschong C., Huygen K., Klebl B., Thompson C., Bacher G., Pieters J. (2004) Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304, 1800–1804 [DOI] [PubMed] [Google Scholar]
- 28. Papavinasasundaram K. G., Chan B., Chung J. H., Colston M. J., Davis E. O., Av-Gay Y. (2005) Deletion of the Mycobacterium tuberculosis pknH gene confers a higher bacillary load during the chronic phase of infection in BALB/c mice. J. Bacteriol. 187, 5751–5760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barthe P., Roumestand C., Canova M. J., Kremer L., Hurard C., Molle V., Cohen-Gonsaud M. (2009) Dynamic and structural characterization of a bacterial FHA protein reveals a new autoinhibition mechanism. Structure 17, 568–578 [DOI] [PubMed] [Google Scholar]
- 30. Canova M. J., Kremer L., Molle V. (2009) The Mycobacterium tuberculosis GroEL1 chaperone is a substrate of Ser/Thr protein kinases. J. Bacteriol. 191, 2876–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Syson K., Stevenson C. E., Rejzek M., Fairhurst S. A., Nair A., Bruton C. J., Field R. A., Chater K. F., Lawson D. M., Bornemann S. (2011) Structure of Streptomyces maltosyltransferase GlgE, a homologue of a genetically validated anti-tuberculosis target. J. Biol. Chem. 286, 38298–38310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang C. M., Nyayapathy S., Lee J. Y., Suh J. W., Husson R. N. (2008) Wag31, a homologue of the cell division protein DivIVA, regulates growth, morphology and polar cell wall synthesis in mycobacteria. Microbiology 154, 725–735 [DOI] [PubMed] [Google Scholar]
- 33. Veyron-Churlet R., Guerrini O., Mourey L., Daffé M., Zerbib D. (2004) Protein-protein interactions within the fatty acid synthase-II system of Mycobacterium tuberculosis are essential for mycobacterial viability. Mol. Microbiol. 54, 1161–1172 [DOI] [PubMed] [Google Scholar]
- 34. Noens E. E., Williams C., Anandhakrishnan M., Poulsen C., Ehebauer M. T., Wilmanns M. (2011) Improved mycobacterial protein production using a Mycobacterium smegmatis groEL1ΔC expression strain. BMC Biotechnol. 11, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Prisic S., Dankwa S., Schwartz D., Chou M. F., Locasale J. W., Kang C. M., Bemis G., Church G. M., Steen H., Husson R. N. (2010) Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. U.S.A. 107, 7521–7526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Belanger A. E., Hatfull G. F. (1999) Exponential-phase glycogen recycling is essential for growth of Mycobacterium smegmatis. J. Bacteriol. 181, 6670–6678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. England P., Wehenkel A., Martins S., Hoos S., André-Leroux G., Villarino A., Alzari P. M. (2009) The FHA-containing protein GarA acts as a phosphorylation-dependent molecular switch in mycobacterial signaling. FEBS Lett. 583, 301–307 [DOI] [PubMed] [Google Scholar]
- 38. Hale K. J., Hummersone M. G., Manaviazar S., Frigerio M. (2002) The chemistry and biology of the bryostatin antitumour macrolides. Nat. Prod. Rep. 19, 413–453 [DOI] [PubMed] [Google Scholar]
- 39. Shah I. M., Laaberki M. H., Popham D. L., Dworkin J. (2008) A eukaryotic-like Ser/Thr kinase signals bacteria to exit dormancy in response to peptidoglycan fragments. Cell 135, 486–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Barthe P., Mukamolova G. V., Roumestand C., Cohen-Gonsaud M. (2010) The structure of PknB extracellular PASTA domain from Mycobacterium tuberculosis suggests a ligand-dependent kinase activation. Structure 18, 606–615 [DOI] [PubMed] [Google Scholar]
- 41. Geurtsen J., Chedammi S., Mesters J., Cot M., Driessen N. N., Sambou T., Kakutani R., Ummels R., Maaskant J., Takata H., Baba O., Terashima T., Bovin N., Vandenbroucke-Grauls C. M., Nigou J., Puzo G., Lemassu A., Daffé M., Appelmelk B. J. (2009) Identification of mycobacterial α-glucan as a novel ligand for DC-SIGN: involvement of mycobacterial capsular polysaccharides in host immune modulation. J. Immunol. 183, 5221–5231 [DOI] [PubMed] [Google Scholar]