

Background: Pulmonary arterial hypertension (PAH) is characterized by abnormal vascular remodeling and impaired BMP/Smad signaling.
Results: cGMP/PKGI are required for the anti-proliferative and pro-differentiation effects of BMP in pulmonary artery smooth muscle cells (PASMCs) in vivo.
Conclusion: Cooperative cGMP and BMP signaling is essential for maintaining a low proliferative, differentiated PASMC phenotype.
Significance: These data explain therapeutic cGMP effects in PAH.
Keywords: Bone Morphogenetic Protein (BMP), Cyclic GMP (cGMP), Protein Kinase G (PKG), Pulmonary Hypertension, Smad Transcription Factor
Abstract
Pulmonary arterial hypertension (PAH) is a progressive, usually fatal disease with abnormal vascular remodeling. Pulmonary artery smooth muscle cells (PASMCs) from PAH patients are hyperproliferative and apoptosis-resistant and demonstrate decreased signaling in response to bone morphogenetic proteins (BMPs). Cyclic GMP-elevating agents are beneficial in PAH, but their mechanism(s) of action are incompletely understood. Here we show that BMP signaling via Smad1/5/8 requires cGMP-dependent protein kinase isotype I (PKGI) to maintain PASMCs in a differentiated, low proliferative state. BMP cooperation with cGMP/PKGI was crucial for transcription of contractile genes and suppression of pro-proliferative and anti-apoptotic genes. Lungs from mice with low or absent PKGI (Prkg1+/− and Prkg1−/− mice) exhibited impaired BMP signaling, decreased contractile gene expression, and abnormal vascular remodeling. Conversely, cGMP stimulation of PKGI restored defective BMP signaling in rats with hypoxia-induced PAH, consistent with cGMP-elevating agents reversing vascular remodeling in this PAH model. Our results provide a mechanism for the therapeutic effects of cGMP-elevating agents in PAH and suggest that combining them with BMP mimetics may provide a novel, disease-modifying approach to PAH therapy.
Introduction
Progressive narrowing of arterial vessels in pulmonary arterial hypertension (PAH)2 increases pulmonary arterial pressure, causing right ventricular hypertrophy and ultimately heart failure (1, 2). Structural changes occur in all vessel layers, with medial thickening caused by abnormal proliferation of PASMCs (1). PASMCs can switch from a differentiated, quiescent state to a de-differentiated, proliferative state, but the mechanism is not well understood (3, 4). PAH treatment includes modulation of the NO/cGMP signaling pathway to induce vasodilatation (1), with mounting evidence indicating that cGMP-elevating agents may also ameliorate pathological vascular remodeling (5). PKGI is one of several cGMP effectors in smooth muscle cells (SMCs), involved in regulating proliferation, differentiation, and survival, albeit controversy exists about the exact functions of different cGMP targets (6).
BMPs activate BMP type 1 and 2 receptor complexes (BMPR1 and 2), with BMPR1 phosphorylating and thereby activating Smad1/5/8; the latter bind Smad4, and the complex translocates to the nucleus to regulate gene transcription (7). Most patients with heritable PAH and ∼20% of “idiopathic” cases have a heterozygous mutation in the Bmpr2 gene (1). These mutations decrease Smad signaling, and even in patients with nonmutated BMPR2, BMP receptor complexes and Smad1/5/8 signaling are reduced in vascular lesions, emphasizing the importance of BMP/Smad signaling in PAH (1, 8). However, only 10–20% of people with Bmpr2 mutations develop PAH, suggesting that other genetic and environmental factors contribute to disease penetrance (9, 10). We showed previously that PKGI facilitates Smad1/5/8 signaling in C2C12 myoblasts: PKGI binds to BMPR2 and, upon BMP stimulation, detaches from the receptor complex and associates with activated Smads; the PKGI-Smad complex translocates to the nucleus, where it recruits the transcriptional co-factor TFII-I to enhance Smad-dependent transcription from a BMP response element-containing reporter gene (11). We now demonstrate functional consequences of cGMP/BMP cross-talk in SMCs: cGMP/PKGI and BMP/Smad cooperatively regulate PASMC growth, survival, and differentiation; defective Smad phosphorylation and Smad downstream signaling are observed in mice with reduced PKG expression, and PKG activation in a rat model of PAH improves Smad signaling.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatment with Pharmacological Agents
Primary human PASMCs (Lonza) were maintained according to the manufacturer's instructions and used at passages 4–8. Spontaneously immortalized rat PASMCs (PAC1, a kind gift from A. Rothman) were cultivated in Dulbecco's modified eagle medium (DMEM) plus 10% (v/v) fetal bovine serum (FBS) and antibiotics. SMC precursors (C3H/10T1/2 cells (ATCC), passages 6–16) were cultivated in basic medium Eagle supplemented with 10% (v/v) FBS and 100 units/ml penicillin and 100 mg/ml streptomycin. Cells were serum-starved in 0.1% (v/v) FBS for 18–24 h and then treated with 3 nm BMP (R&D Systems) or 100 μm 8-pCPT-cGMP (Biolog) for 30 min, unless noted otherwise. When indicated, cells were pre-treated for 1 h with 100 μm 8-pCPT-cGMP, 50 μm Rp-pCPT-PET-cGMPS (Biolog), or 4 μm dorsomorphin (Calbiochem).
Transfections, siRNA Sequences, and Infection with Virus
Cells were transiently transfected in 12-well (or 6-well) plates at a confluence of 50–60% using 2 μl (4 μl) of Lipofectamine 2000TM (Invitrogen) and 100 pmol of siRNA in 400 μl (1 ml) of serum-containing medium. siRNA oligoribonucleotides (Qiagen) had the following siRNA target sequences or catalogue numbers: rat PKGI, 5′-CCGGACAUUUAAAGACAGCAA-3′ (targeting the common C terminus (12)); human PKGI (SI02758630), rat Smad1 (S103038070), rat Smad5, 5′-AAAGCTGAAGAAGAAGAAGGG-3′; rat Id1, 5′-AAGAGGAAAAAAAGTGCTCTC-3′; rat Id3, 5′-GCCTGAGGGGCATGGATGAGC-3′; and GFP, 5′-AAGCTGACCCTGAAGTTCATC-3′. Adenoviral vectors encoding β-galactosidase (LacZ), human PKGIα, and human Smad1 were produced using the pAd/CMV/V5-DESTTM GatewayR system (Invitrogen). 24 h after siRNA transfection, cells were infected in medium containing 0.1% FBS at a multiplicity of infection of ∼10; 18 h later cells were transferred to fresh starvation medium plus stimuli until harvest.
Quantitative RT-PCR and PCR Array
Primary human PASMCs were serum-starved and stimulated with 3 nm BMP2 for 4 h (for Id1/2/3) or 24 h (for Acta2, Tagln, and Cnn). Total RNA was isolated using TRI ReagentTM (Molecular Research Center) following the manufacturer's instructions, and 1 μg was transcribed into cDNA using iScriptTM Synthesis kit (Bio-Rad). Real-time PCR was performed using Brilliant II SYBR® Green QPCR Master Mix (Agilent), and Gapdh, Hprt, and/or β-actin served as internal references to calculate relative mRNA levels using the 2−ΔΔCT method.
Quantitative PCR Array was performed with primary human PASMCs, transfected with control or PKGI siRNA, and treated 24 h later with 3 nm BMP2 for an additional 24 h, using primers provided on Human Cell Cycle RT2 ProfilerTM PCR Array plates (Qiagen). Data were analyzed with RT2 Profiler PCR Array Data Analysis version 3.5 (Qiagen). For validation, human Quanti-Tect primers from Qiagen were used. Primer sequences and catalogue numbers are provided in the supplemental Material.
Collagen Matrix Contraction Assay
Collagen contraction assay was performed as described (13), with minor changes. Serum-deprived PASMCs were treated with 3 nm BMP2 for 48 h. Cells were trypsinized, and equal cell numbers were embedded into collagen matrices, which were incubated for another 18–20 h under full serum conditions. Collagen gel sizes were measured with ImageJ (National Institute of Health).
Immunoblotting
Cell extracts were fractionated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by Western blotting with antibodies specific for phospho-Ser463/465-Smad1/5, phospho-Ser463/465(426/428)-Smad1/5/8, Smad5, phospho-Ser239-VASP (all from Cell Signaling), PKGI (C-terminal; Abgent), Smad1, β-actin, α-tubulin (all from Santa Cruz Biotechnology), or α2-actin (clone 1A4; Sigma-Aldrich), and Western blots were scanned and analyzed using ImageJ.
Immunofluorescence
Primary human PASMCs and SMC precursors were plated on glass coverslips in 24-well plates, serum-deprived, and stimulated with 3 nm BMP2 for 30 min (Smad1 nuclear translocation) or 24 h (α2-actin expression). Cells were fixed with 3.7% paraformaldehyde and permeabilized in 0.5% Triton X-100. After blocking with 3% BSA, cells were stained using Smad1 primary antibody (Santa Cruz Biotechnology) and Alexa Fluor 555-conjugated goat anti-mouse secondary antibody (Invitrogen), or α2-actin primary antibody (clone 1A4; Sigma-Aldrich) and FITC-conjugated goat anti-mouse secondary antibody (Jackson Immunoresearch). Nuclei were stained using Hoechst 33342. For co-localization studies on lung sections, slides were boiled in 1 mm EDTA, pH 8.0, as described below, and incubated with primary antibodies followed by FITC- and TRITC-labeled secondary antibodies (Jackson Immunoresearch). Coverslips were embedded in Fluoromount-G (Southern Biotech). Pictures were taken from randomly selected fields using a confocal microscope (Olympus FV1000) and 40/0.75 or 10/0.4 objectives. Images were analyzed with Fluoview (Olympus) and Photoshop (Adobe; sizing, contrast adjustment), and identical software settings were used for image acquisition of all samples in a given experiment.
Immunohistochemical Staining and Histomorphometry
Lung tissue paraffin sections were deparaffinized and hydrated, and antigen retrieval was done by boiling (using a 1200-W microwave, full power for 3 × 15 s) in 1 mm EDTA, pH 8.0, for phospho-Smad1/5 staining, or 10 mm citrate, pH 6.0, for α2-actin staining. After blocking in TBS/0.1% Tween 20/5% goat serum, primary antibodies were incubated overnight at 4 °C in antibody diluent (Cell Signaling). After washing, slides were treated with species-specific SignalStain® Boost IHC Detection Reagent (Cell Signaling), developed with a 3,3-diaminobenzidine peroxidase substrate kit (Vector Laboratories), and dehydrated. Tissue staining was analyzed using a NanoZoomer 2.0-HT with 20/0.75 objective (Hamamatsu); image acquisition and analysis were done as described above. Histomorphometry was performed on lung sections derived from matched wild type and Prkg1+/− littermates and stained for α2-actin. Approximately 100 small pulmonary arteries (diameter <50 μm) were analyzed, and the thickness of the smooth muscle layer was measured using ImageJ.
Proliferation and Apoptosis Assays
Primary human PASMCs were seeded on glass coverslips, serum-depleted, and incubated with BMP2 (3 nm for proliferation and 10 nm for apoptosis assays) for 48 h. For BrdU uptake, cells received 200 μm BrdU for 48 h, were fixed and permeabilized as described above and incubated with 0.5 unit/μl DNase I (Sigma-Aldrich) for 30 min at 37 °C, prior to staining with anti-BrdU antibody (Sigma-Aldrich), TRITC-conjugated goat anti-mouse secondary antibody (Jackson Immunoresearch), and Hoechst 33342. Total cell counts were obtained using a hemocytometer; in some cases, trypsinized cells were incubated with 0.04% trypan blue solution prior to counting. TUNEL assays were performed according to the manufacturer's protocol (In Situ Cell Death Detection Kit; Roche Applied Science).
Prkg1 Knock-out Mice
Heterozygous Prkg1+/− mice (14) were bred to obtain wild type, homo-, and heterozygous animals. Animals from each litter were genotyped by PCR, and littermates were matched according to sex and weight. Lungs from 6- or 16-week-old mice (see figure legends) were harvested at the University of Bonn, Germany, and extracted in radioimmunoprecipitation assay buffer. Equal amounts of protein were analyzed for Smad1/5/8 phosphorylation. Quantitative RT-PCR was performed by using cDNA transcribed from 1 μg of total lung RNA. Protein and RNA samples from each litter were processed and examined at the same time.
PAH Rats
Adult male Sprague-Dawley rats were treated at the University of Giessen and Marburg Lung Center, Germany, as described (5). Briefly, the animals were injected subcutaneously with the VEGF receptor inhibitor SU5416 (20 mg/kg) and exposed to chronic hypoxia (10% oxygen) for 21 days. The animals were then randomized to be treated orally (by gavage) with vehicle or riociguat (BAY 63-2521; Bayer Healthcare) at a dose of 10 mg/kg per day for 14 days (from day 21 to 35), until euthanasia. Rats injected with saline and exposed to normoxic conditions for 35 days served as controls. Hemodynamic measurements confirming development of PAH and reversal by riociguat were performed as described (5). Equal amounts of extracted lung protein were examined by immunoblotting as described above.
Statistics
Immunoblots and photographs represent three independent experiments, and graphs show the mean ± S.E. of three independent experiments, unless noted otherwise. One-way analysis of variance followed by Bonferroni's post test was used for comparing multiple groups, and a two-tailed Student's t test for pairwise comparison (Prism; GraphPad Software). For nonparametric distribution, Wilcoxon signed-rank test (pairwise comparison) or Friedman test with Dunn's post test (multiple groups) was applied. p values < 0.05 were considered significant.
RESULTS
PKGI Facilitates BMP/Smad Signaling in SMCs
We studied interactions between BMP and cGMP signaling in primary human PASMCs, immortalized rat PASMCs, and immortalized SMC precursors (C3H/10T1/2 cells). We found that treating cells with 8-pCPT-cGMP significantly increased BMP-induced Smad phosphorylation (Fig. 1, A and B for primary human PASMCs and murine SMC precursors). Conversely, inhibiting PKG with Rp-pCPT-PET-cGMPS largely blocked BMP-mediated Smad activation (Fig. 1, C and D, and supplemental Fig. 1, A–C, for rat PASMCs). Because pharmacological inhibition of PKG could have off-target effects, we also used an siRNA approach (12) with subsequent viral reconstitution of siRNA-resistant PKGIα. We found that BMP-induced Smad phosphorylation was reduced in PKGI-depleted cells, but could be restored by re-expressing PKGIα (Fig. 1, E and F, and supplemental Fig. 1D). Moreover, inhibiting PKG blocked BMP-induced nuclear Smad1 translocation in primary human PASMCs (Fig. 1G). We previously reported similar findings in C2C12 myoblasts, where shRNA-mediated transient knockdown of PKGI diminished BMP-induced Smad phosphorylation and nuclear translocation (11).
FIGURE 1.

PKGI enhances Smad phosphorylation and downstream signaling in SMCs. A and B, primary (Prim.) human PASMCs and SMC precursors (SMC prec.) were treated with 3 nm BMP2 and/or 100 μm 8-pCPT-cGMP (cGMP) for 30 min. Smad1/5 phosphorylation was examined by immunoblotting, and three experiments were quantified, with BMP2-induced Smad phosphorylation assigned a value of 1; *, p < 0.05, two-tailed Student's t test. Error bars, S.E. C and D, cells were treated with BMP2 and/or 50 μm Rp-pCPT-PET-cGMPS (Rp), and Smad activation was analyzed as in A and B (*, p < 0.05). E and F, SMC precursors were transfected with siRNAs targeting GFP (control) or PKGI, and PKG was reconstituted by adenoviral vector encoding siRNA-resistant PKGIα. Cells were treated and analyzed as in A and B, with BMP2-induced Smad phosphorylation of GFP siRNA/LacZ virus-treated cells assigned a value of 1 (*, p < 0.05, one-way ANOVA). PKGI expression was assessed by immunoblotting. G, human PASMCs were treated as in C, and Smad1 nuclear translocation was examined by immunofluorescence labeling (red); nuclei were counterstained with Hoechst (blue). Scale bars, 5 μm. H, human PASMCs were treated with BMP2 and/or Rp for 4 h, and Id1/2/3 mRNA levels were quantified by real-time RT-PCR. After normalization to Gapdh, relative Id mRNA in untreated cells was assigned a value of 1 (*, p < 0.05; **, p < 0.01; ***, p < 0.001, one-way ANOVA).
Smads assemble transcriptional complexes at promoter regions of specific genes, including the inhibitor of differentiation (Id) gene family (7, 11). We found that inhibiting PKG decreased, although activating PKG increased Id1/2/3 mRNA expression in BMP-treated cells (Fig. 1H and supplemental Fig. 1E). Basal Id expression was not significantly affected by the PKG inhibitor. Thus, PKGI is necessary for BMP-induced Smad1/5 activation and nuclear translocation and BMP-induced Id expression in PASMCs.
cGMP and BMP Cooperate to Control PASMC Proliferation and Apoptosis
Medial hypertrophy in the vascular lesions of PAH is due mainly to abnormal PASMC proliferation and resistance to apoptosis (1), and PASMCs from PAH patients respond less to the anti-proliferative and pro-apoptotic effects of BMP compared with cells from normal subjects (15, 16). Similar to BMP, cGMP/PKGI signaling represses SMC growth (3, 17). Therefore, we examined whether PKGI mediated the anti-proliferative effects of BMP in PASMCs and found that inhibiting PKG pharmacologically or by siRNAs (12) largely prevented growth inhibition by BMP (Fig. 2, A and C, and supplemental Fig. 2, A–C). In the latter case, reconstituting siRNA-resistant PKGIα restored BMP-induced growth inhibition (Fig. 2C). As reported by others (16, 18), BMP increased cell death in PASMCs, but PKG inhibition abolished this effect (Fig. 2B and supplemental Fig. 2, D and E). In the absence of BMP, cGMP activation of PKG reduced PASMC growth and survival (Fig. 2D and supplemental Fig. 2F). Smad1/5-depleted cells were relatively insensitive to cGMP-induced growth suppression, but reconstituting siRNA-resistant Smad1 fully restored cGMP anti-proliferative effects (Fig. 2D; knockdown efficiencies of Smad1 and 5 siRNAs are shown in supplemental Fig. 2G). Consistent with these results, the BMPR1 inhibitor dorsomorphin (19) blocked basal Smad phosphorylation and reduced cGMP anti-proliferative effects (supplemental Fig. 2, H and I). Furthermore, growth suppression by cGMP required Id1/3 (supplemental Fig. 2J; knockdown efficiencies of Id1 and 3 siRNAs are shown in supplemental Fig. 2K), similar to the obligatory role of Id1 in BMP-mediated inhibition of SMC proliferation (20, 21). These data indicate that basal PKG activity is necessary for BMP-induced growth suppression and apoptosis in PASMCs and that cGMP/PKGI and BMP/Smad cooperate to suppress PASMC growth. Their cooperative effect may be partly explained by their convergence on Id1/2/3 (Fig. 1H).
FIGURE 2.

Cooperation between cGMP and BMP to regulate PASMC growth and survival. A, human PASMCs were treated with BMP2 and/or Rp-pCPT-PET-cGMPS (Rp) for 48 h. BrdU incorporation was assessed by immunofluorescence labeling; the percent decrease of BrdU-positive cells compared with untreated cells is shown (n = 5, **, p < 0.01; ***, p < 0.001, Friedman test). Error bars, S.E. B, the percentage of trypan blue-positive, dead cells was determined after 48 h (n = 4, **, p < 0.01, one-way ANOVA). C, rat PASMCs were transfected with siRNAs targeting GFP or PKGI and infected with virus encoding LacZ or siRNA-resistant PKGIα. After receiving BMP2 and/or 8-pCPT-cGMP (cGMP) for the last 24 h, cells were counted at 72 h, relative to untreated cells with GFP siRNA/LacZ virus (n = 5, **, p < 0.01; ***, p < 0.001, one-way ANOVA); the immunoblot shows PKGI expression. D, cells were transfected with siRNAs targeting GFP or Smad1/5 and infected with LacZ or siRNA-resistant human Smad1 adenovirus; some cells received cGMP for the last 24 h. Cell numbers are expressed as in C (*, p < 0.05; ***, p < 0.001, one-way ANOVA); the immunoblot shows Smad1 expression. E, cell cycle control genes were analyzed in human PASMCs transfected with control or PKGI siRNA and cultured in the absence or presence of BMP2 for 24 h. β-actin, Gapdh, and β2-microglobulin served as controls in a quantitative RT-PCR-based array. F, expression of Anapc2, Mre11A, Rad1, and Bcl2 mRNA is shown, with levels in control siRNA-transfected, untreated cells assigned a value of 1 (n = 4–5, *, p < 0.05; **, p < 0.01; ***, p < 0.001, one-way ANOVA). G, human PASMCs treated with 8-pCTP-cGMP for 24 h were analyzed as in F (n = 3–4, *, p < 0.05; **, p < 0.01, two-tailed Student's t test).
To search for additional genes mediating anti-proliferative effects of cGMP and BMP, we examined a panel of 72 cell cycle-relevant genes in primary human PASMCs cultured with and without BMP and/or PKGI siRNA (Fig. 2E; Fig. 3D shows PKGI knockdown). BMP decreased expression of 42% of the genes (including Cdk2 and Bcl2; significance cut-off 1.3-fold; supplemental Table 1). Of the BMP2-affected genes, 29% were sensitive to PKGI depletion. Quantitative RT-PCR analysis confirmed that four genes were co-regulated by BMP and cGMP (Fig. 2, F and G): (i) Anapc2 encodes a catalytic subunit of the anaphase-promoting complex (22); (ii) Mre11A is part of a DNA protection-repair complex (23); (iii) Rad1 is a component of a cell cycle checkpoint complex activated by DNA damage (24); and (iv) Bcl2 is an anti-apoptotic protein (16, 18). All four genes are essential for regulation of cell growth (16, 18, 22–24). Thus, BMP/cGMP co-regulate several genes involved in growth and survival of PASMCs.
FIGURE 3.

PKGI is a critical mediator of BMP-induced contractile gene expression. A, human PASMCs received BMP2 and/or Rp-pCPT-PET-cGMPS (Rp) for 24 h and were analyzed for α2-actin by immunofluorescence staining (green, with nuclei counterstained in blue). Scale bar, 5 μm. B, SMC precursors were stimulated and stained as in A, and the number of α2-actin-positive cells was determined (relative to cells receiving only BMP2; median ± S.E. (error bar), n = 6, *, p < 0.05, two-tailed Wilcoxon Rank test). C, human PASMCs were treated with BMP2 and/or Rp for 24 h and examined for Acta2, Tagln, and Cnn mRNA expression by quantitative RT-PCR (normalized to Gapdh and to unstimulated cells; n = 4, **, p < 0.01; ***, p < 0.001; one-way ANOVA). D, Acta2, Tagln, Cnn, and Prkg1 mRNA expression was measured by real-time RT-PCR in human PASMCs transfected with siRNAs targeting GFP or PKGI and treated with BMP2 for 24 h. mRNA levels were normalized to Gapdh and to control siRNA-treated, unstimulated cells (*, p < 0.05; **, p < 0.01; ***, p < 0.001, one-way ANOVA). E, after treatment with BMP2 and/or Rp for 48 h, equal numbers of PASMCs were embedded into collagen matrices; gel contraction was measured relative to untreated controls (n = 6, **, p < 0.01; ***, p < 0.001, one-way ANOVA).
PKGI Is Essential for BMP-induced SM-specific Contractile Gene Expression
In response to environmental stimuli, SMCs undergo reversible switching between a differentiated, contractile state with high SM-specific gene expression, and a de-differentiated, proliferative state characterized by low SM-specific gene expression (3, 4). De-differentiation and proliferation of PASMCs contribute to the progressive narrowing of small pulmonary arteries in PAH due to thickening of the muscle layer (1, 2). BMP induces SM-specific gene transcription in a Smad-dependent manner, thereby maintaining a differentiated PASMC phenotype (25). We hypothesized that cGMP/PKGI could be involved in BMP regulation of the PASMC phenotype, because PKGI sustains a differentiated state in aortic SMCs (3, 12). We found that pharmacological inhibition or siRNA depletion of PKGI in primary human PASMCs and SMC precursors blocked BMP-induced Acta2 (α2-actin or SM-α-actin), Tagln (transgelin or SM22), and Cnn (calponin) expression (Fig. 3, A and B, shows immunofluorescence staining of α2-actin, with a longer excitation time shown in supplemental Fig. 3A and Western blot analysis of α2-actin expression in supplemental Fig. 3B; Fig. 3, C and D, shows RT-PCR quantification of all three mRNAs, and PKGI knockdown efficiency).
Increased contractile gene expression in SMCs induces shrinkage of collagen lattices (13), and we found that inhibiting PKG activity reduced BMP-induced collagen gel contraction (Fig. 3E). Thus, cGMP and BMP signaling cooperate to enhance contractile gene expression, thereby maintaining PASMC differentiation.
PKGI Deficiency Correlates with Impaired Smad Phosphorylation and Smad-dependent Gene Expression in Vivo, Causing Vascular Remodeling
To explore cGMP/PKGI interaction with BMP/Smad signaling in vivo, we examined Smad phosphorylation in lungs from homo- or heterozygous Prkg1 (PKGI) knock-out mice. We found reduced Smad1/5/8 phosphorylation in lungs of 6-week-old homo- and heterozygous Prkg1 knock-out mice compared with wild type littermates (Fig. 4A). Because Prkg1−/− mice die within 8 weeks of birth from severe gastrointestinal dysfunction (14), we concentrated subsequently on adult (16-week-old) Prkg1+/− mice, which appear phenotypically normal. We found that basal Smad1/5/8 phosphorylation in lung tissue was down-regulated in 7 of 26 heterozygotes compared with matched wild type littermates (significance cut-off 1.3-fold; Fig. 4B; see supplemental Fig. 4A for a representative Western blot and supplemental Fig. 4B for immunohistochemical detection of phospho-Smad1/5/8 in representative lung sections). The heterozygotes with reduced Smad1/5/8 phosphorylation had significantly lower mean Prkg1 mRNA expression compared with heterozygotes with normal Smad1/5/8 phosphorylation (Fig. 4C).
FIGURE 4.
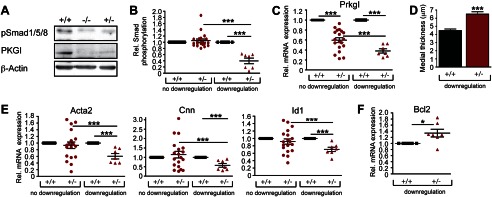
Homo- and heterozygous Prkg1 knock-out mice show impaired Smad activation and Smad-dependent gene expression, with remodeling of distal pulmonary arteries. A, lung homogenates of 6-week-old sex- and weight-matched Prkg1−/−, Prkg1+/−, and wild type littermates were analyzed by immunoblotting for Smad1/5/8 phosphorylation and PKGI expression. B, phospho-Smad1/5/8 was measured in 26 pairs of Prkg1+/− and wild type lungs (from 16-week-old sex- and weight-matched littermates). Two populations are plotted: one without phospho-Smad “down-regulation” (n = 19) and one with phospho-Smad levels <77% of the matched wild type (n = 7). ***, p < 0.001 by one-way ANOVA. Error bars, S.E. C, Prkg1 mRNA expression was measured by quantitative RT-PCR in the populations defined in B (***, p < 0.001, one-way ANOVA). D, histomorphometric analysis was performed of the medial thickness of small pulmonary arteries (<50 μm in diameter) using α2-actin immunohistochemical staining of lung sections to visualize vascular smooth muscle layers. At least 100 distal pulmonary arteries of matched Prkg1+/− and wild type littermates were measured using ImageJ (the Prkg1+/− population with phospho-Smad down-regulation was defined as in B, n = 4, ***, p < 0.0001, two-tailed Student's t test). E and F, Acta2, Cnn, Id1 (E), and Bcl2 (F) mRNA levels were quantified by RT-PCR in the populations defined as in B (*, p < 0.05; ***, p < 0.001, one-way ANOVA).
Homozygous PKGI knock-out mice develop PAH medial thickening (“muscularization”) of small pulmonary arteries, but Prkg1+/− heterozygotes were not examined (26). To determine whether impaired Smad signaling in Prkg1+/− mice correlated with vascular remodeling, we examined lung sections stained for α2-actin (supplemental Fig. 4B). We found a significant increase in the medial thickness of distal pulmonary arteries (<50 μm in diameter) in Prkg1+/− mice with down-regulation of phospho-Smad compared with sex-matched wild type littermates (Fig. 4D).
In mice with reduced Smad phosphorylation, Acta2, Cnn, and Id1 mRNA levels were significantly lower than in paired wild type littermates, whereas Bcl2 mRNA was increased, suggesting a less differentiated and apoptosis-resistant phenotype (Fig. 4, E and F). Expression of other genes important for NO/cGMP signaling, BMP/Smad signaling, or SM-specific gene transcription (including RhoA) did not differ between the two Prkg1+/− populations and wild type mice (supplemental Fig. 4, C–E). Similarly, expression of Spp1 (osteopontin) and Thbs1 (thrombospondin) mRNA was unaffected by low PKGI; Col1α1 (collagen 1α1) mRNA expression showed a trend toward higher levels in lungs of Prkg1+/− mice, but this did not reach statistical significance (supplemental Fig. 4F).
The correlation among Prkg1 gene expression, Smad phosphorylation, and Smad-dependent gene expression indicates that residual PKGI activity in some heterozygotes was insufficient to maintain normal Smad activity, and reduced Smad signaling was associated with vascular remodeling.
PKG Activation Restores Impaired Smad Phosphorylation in PAH Rats
Similar to findings in PAH patients, Smad1/5/8 phosphorylation is decreased in PASMCs from rats with hypoxia- or monocrotaline-induced PAH (8, 20, 21, 27). However, animals exposed to monocrotaline or hypoxia do not develop vascular lesions characteristic of human PAH (5, 28). In contrast, rodents kept under hypoxic conditions and treated with the VEGF receptor antagonist SU5416 better mirror the pathological vascular changes seen in PAH patients, and these changes are progressive even after cessation of hypoxia and SU5416 treatment (5, 29). We hypothesized that treating PAH rats with a cGMP-elevating agent may improve defective BMP/Smad signaling in this model. Rats were kept under hypoxic conditions and were treated with SU5416 for 21 days; then, animals were randomized to receive either vehicle or the soluble guanylate cyclase stimulator riociguat for 14 days (5). We showed previously that riociguat increases cGMP concentrations in lung tissue and reduces vascular remodeling, right ventricular systolic pressure, and right ventricular hypertrophy in this PAH model (5). We now demonstrate that riociguat significantly increased Smad1/5/8 phosphorylation in lungs of hypoxic rats treated with SU5416, without changing Smad1 and PKGI expression (Fig. 5, A and B, and supplemental Fig. 5A). Immunohistochemical staining of lung sections showed decreased phospho-Smad1/5 in PASMCs in the thickened media of PAH vascular lesions, and riociguat increased phospho-Smad1/5 while decreasing medial thickness (Fig. 5C; PASMCs were identified by α2-actin staining in adjacent sections). Immunostaining for phospho-Smad1/5 was also apparent in endothelial cells and bronchoalveolar epithelial cells, as observed previously (8, 30). However, co-immunofluorescence staining with phospho-Smad1/5 and α2-actin antibodies demonstrated that phospho-Smad1/5 was present in the nuclei of PASMCs (supplemental Fig. 5B). Thus, riociguat-induced activation of PKGI improved dysfunctional Smad signaling in vascular lesions of this rat PAH model.
FIGURE 5.

The cGMP-elevating agent riociguat restores defective Smad phosphorylation in vivo. A–C, rats were injected with SU5416 (SU) and exposed to 10% oxygen (Hx) for 21 days and then were randomized to receive either vehicle or riociguat (Rio) at 10 mg/kg per day for 14 days. Control rats were injected with saline and exposed to normoxic gas for 35 days. Smad1/5/8 phosphorylation in lung tissue was assessed by immunoblotting (A; B shows densitometry results normalized to β-actin; n = 6, *, p < 0.05, one-way ANOVA) or immunohistochemistry (C; α2-actin staining of adjacent sections identified SMCs). Scale bars, 10 μm). The insets in the photographs on the left are magnified on the right. D, model shows BMP/cGMP-mediated control of PASMC phenotype.
DISCUSSION
Using pharmacological and genetic approaches, we found that BMP/Smad and cGMP/PKGI signaling pathways cooperate in PASMCs to regulate expression of multiple genes involved in proliferation, apoptosis, and maintenance of a differentiated, contractile phenotype (Fig. 5D). Consistent with these results, we showed previously that PKGI overexpression in C2C12 cells enhances BMP-induced transcription of a BMP-responsive reporter gene, even in cells co-transfected with BMPR2 constructs containing loss-of-function mutations found in PAH patients (11).
The anti-proliferative and pro-apoptotic effects of BMP are attenuated in PASMCs from PAH patients (15, 16). Decreased BMPR2 expression and deficient Smad1/5/8 phosphorylation are observed in small pulmonary arteries of PAH patients and in several animal models of PAH, suggesting that impaired BMP signaling plays a central role in the pathophysiology of PAH (15, 27, 31). Microarray gene expression analysis comparing BMP-treated PASMCs derived from normal subjects and PAH patients also supports a proliferative, apoptosis-resistant phenotype in PAH (32). Id proteins are essential mediators of BMP-induced growth inhibition in PASMCs, and reduced Id1 expression occurs in vascular lesions from PAH patients with Bmpr2 mutations (20, 21). We found that the anti-proliferative effect of BMP was prevented when PKG was inhibited; inversely, the anti-proliferative effect of cGMP in PASMCs required Smad1/5 and Id1/3. Thus, both BMP and cGMP converge to induce Id proteins to inhibit PASMC proliferation. Furthermore, we identified several pro-proliferative and anti-apoptotic genes that were down-regulated by BMP in a PKGI-dependent fashion, namely Anapc2, Mre11A, Rad1, and Bcl2. Studies in different cell types have demonstrated that each of these proteins is essential for sustaining proliferation (16, 18, 22–24). Others have shown that BMP suppresses Bcl2 expression in PASMCs (16, 18), and Bcl2 mRNA is up-regulated in lung tissue from patients with heritable or idiopathic PAH, likely contributing to the apoptosis resistance of vascular cells (33).
Heterozygous BMPR2-deficient mice display either normal hemodynamics or mild PAH at base line but show enhanced susceptibility to inflammation-induced PAH (9, 34). Mice expressing an inducible dominant negative form of BMPR2 in smooth muscle develop PAH when the transgene is activated and demonstrate vascular lesions with medial hypertrophy and decreased SM-specific gene expression (35, 36). We found that BMP stimulated SM-specific gene expression in PASMCs 2–3-fold, consistent with results reported by Lagna et al. (25). We and others demonstrated previously that PKGI activity is required for maintenance of SM-specific gene expression in aortic SM cells (3, 12, 37). We now show that pharmacological inhibition or siRNA depletion of PKGI prevented BMP-induced SM-specific gene expression in PASMCs. The reduction of SM-specific gene expression in untreated, serum-starved PASMCs transfected with PKGI siRNA was less pronounced than the reduction observed in aortic SMCs grown in serum-containing medium (12).
Heterozygous PKGI knock-out mice with low expression of the wild type Prkg1 allele and impaired Smad phosphorylation expressed less Id1, Acta2, and Cnn mRNA compared with wild type mice or Prkg1+/− mice with higher expression of the wild type allele. These data are consistent with the finding that PKGI is essential for BMP-induced Id, Acta2, and Cnn mRNA expression in primary PASMCs. However, we noted a discrepancy between α2-actin staining in the thickened media of peripheral pulmonary arteries and the decrease in α2-actin mRNA measured in whole lung extracts of Prkg1+/− mice with impaired Smad signaling. It is possible that α2-actin mRNA is reduced primarily in larger pulmonary arteries and/or that α2-actin is regulated post-transcriptionally.
When PKGI is transfected into de-differentiated aortic SMCs lacking endogenous PKGI, Acta2 and Cnn mRNA increase more dramatically (6–9-fold) than suggested by the differences between wild type and Prkg1+/− mice, likely because higher amounts of PKGI are introduced (3). In the same model of de-differentiated aortic SMCs, transfection of constitutively active PKGI suppresses synthesis of osteopontin and thrombospondin proteins, but these changes were associated with minor reductions in mRNA levels (38). We did not observe significant changes in Spp1 and Thbs1 mRNA in Prkg1+/− lungs with impaired Smad phosphorylation compared with wild type lungs.
Homozygous Prkg1 knock-out mice exhibit PAH and impaired pulmonary vascular perfusion at an early age due to vasoconstriction and vascular remodeling (26). Vasoconstriction is partly caused by increased RhoA/Rho kinase activity, but the mechanism(s) of vascular remodeling were not examined (26). PKGI inactivation from nitration due to overactive NO synthase in Cav-1 (caveolin-1) knock-out mice also leads to PAH (39), emphasizing a key role of PKG in this disease. We found an increase in medial thickness of distal pulmonary arteries in Prkg1+/− mice with impaired Smad phosphorylation and low expression of the wild type Prkg1 allele. Because medial thickening (muscularization) of small pulmonary arteries is a hallmark of PAH and a linear relationship between the proportion of occluding vessels and right ventricular pressure has been demonstrated (5), these data suggest that low Prkg1 expression may be sufficient for the development of PAH.
We found that 27% of the Prkg1+/− mice had defective Smad phosphorylation and that these mice had lower mean Prkg1 mRNA expression compared with heterozygotes with normal Smad signaling. Expression of the wild type Prkg1 allele may be modified by epigenetic mechanisms; phenotypic heterogeneity has been observed previously in inbred mouse models with heterozygous targeted disruption of other genes (40, 41). Similarly, in families with a heterozygous Bmpr2 mutation leading to an unstable transcript, unaffected mutation carriers express significantly more wild type Bmpr2 mRNA than affected PAH patients (42). This heterogeneity, together with other genetic and environmental modifiers, likely explains incomplete penetrance of the disease. Because some patients with heritable or idiopathic PAH express less Prkg1 mRNA in lung than normal subjects (33), we propose that PKGI expression below a critical threshold contributes to reduced BMP/Smad signaling, leading to abnormal PASMC gene expression, proliferation, and survival.
Multiple studies have demonstrated that cGMP-elevating agents, including NO, phosphodiesterase inhibitors, and guanylate cyclase stimulators, can induce pulmonary vasodilation and reverse vascular remodeling and right heart hypertrophy in rodent models of PAH (43–46). We used a rat model of PAH induced by hypoxia and SU5416 that closely mimics the vascular changes seen in human PAH patients and found that riociguat not only improved pulmonary hemodynamics and vascular remodeling (5), but also increased Smad phosphorylation. Riociguat, the first guanylate stimulator to enter clinical development, has yielded promising results in phase II and III clinical trials in PAH patients (47, 48).
In conclusion, we found that cooperation between the cGMP/PKGI and BMP/Smad pathways is essential for maintaining a low proliferative, differentiated PASMC phenotype (Fig. 5D) and that a critical threshold of basal PKGI activity is required for normal Smad activation and Smad-dependent gene expression. PKGI activity in PASMCs may modify disease penetrance in Bmpr2 mutation carriers. This work provides a molecular basis for reversal of vascular remodeling by PKG activation in PAH animal models and at least partly explains the beneficial effects of cGMP-elevating agents in PAH patients (1, 47). Combining PKG activators and BMP agonists may lead to novel, personalized medical strategies for PAH patients.
Acknowledgments
We thank Drs. Hema Rangaswami and Darren E. Casteel for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-AR051300 (to R. B. P.) and NS047101 (to the University of California San Diego Neuroscience Microscopy Facility). This work was also supported by a Deutsche Forschungsgemeinschaft fellowship grant (to R. S.) and a BONFOR grant from the University of Bonn (to A. K.). R. S. is an inventor on the patent “A method for restoring BMP-receptor signalling in a cell” (PCT/EP2009/001408, Germany). R. T. S. has received honoraria from Actelion, Pfizer, Bayer-Schering, Novartis and Solvay and research grant support from Bayer-Schering.

This article contains supplemental Material, additional references, Figs. 1–5, and Table 1.
- PAH
- pulmonary arterial hypertension
- BMP
- bone morphogenetic protein
- BMPR1
- BMP receptor type 1
- BMPR2
- BMP receptor type 2
- PASMC
- pulmonary artery smooth muscle cell
- 8-pCPT-cGMP
- 8-(4-chlorophenylthio)-guanosine-3′,5′-cyclic monophosphate
- PKG
- cGMP-dependent protein kinase
- Rp-pCPT-PET-cGMPS
- 8-(4-chlo-rophenylthio)-β-phenyl-1,N2-ethenoguanosine-3′,5′-cyclic monophosphorothioate
- SM
- smooth muscle
- SMC
- smooth muscle cell
- TRITC
- tetramethylrhodamine isothiocyanate.
REFERENCES
- 1. Schermuly R. T., Ghofrani H. A., Wilkins M. R., Grimminger F. (2011) Mechanisms of disease: pulmonary arterial hypertension. Nat. Rev. Cardiol. 8, 443–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rabinovitch M. (2008) Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Invest. 118, 2372–2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boerth N. J., Dey N. B., Cornwell T. L., Lincoln T. M. (1997) Cyclic GMP-dependent protein kinase regulates vascular smooth muscle cell phenotype. J. Vasc. Res. 34, 245–259 [DOI] [PubMed] [Google Scholar]
- 4. Owens G. K., Kumar M. S., Wamhoff B. R. (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801 [DOI] [PubMed] [Google Scholar]
- 5. Lang M., Kojonazarov B., Tian X., Kalymbetov A., Weissmann N., Grimminger F., Kretschmer A., Stasch J. P., Seeger W., Ghofrani H. A., Schermuly R. T. (2012) The soluble guanylate cyclase stimulator riociguat ameliorates pulmonary hypertension induced by hypoxia and SU5416 in rats. PLoS One 7, e43433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Francis S. H., Busch J. L., Corbin J. D., Sibley D. (2010) cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 62, 525–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sieber C., Kopf J., Hiepen C., Knaus P. (2009) Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev. 20, 343–355 [DOI] [PubMed] [Google Scholar]
- 8. Yang X., Long L., Southwood M., Rudarakanchana N., Upton P. D., Jeffery T. K., Atkinson C., Chen H., Trembath R. C., Morrell N. W. (2005) Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ. Res. 96, 1053–1063 [DOI] [PubMed] [Google Scholar]
- 9. Song Y., Jones J. E., Beppu H., Keaney J. F., Jr., Loscalzo J., Zhang Y. Y. (2005) Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation 112, 553–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hansmann G., de Jesus Perez V. A., Alastalo T. P., Alvira C. M., Guignabert C., Bekker J. M., Schellong S., Urashima T., Wang L., Morrell N. W., Rabinovitch M. (2008) An antiproliferative BMP-2/PPARγ/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J. Clin. Invest. 118, 1846–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schwappacher R., Weiske J., Heining E., Ezerski V., Marom B., Henis Y. I., Huber O., Knaus P. (2009) Novel cross-talk to BMP signalling: cGMP-dependent kinase I modulates BMP receptor and Smad activity. EMBO J. 28, 1537–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang T., Zhuang S., Casteel D. E., Looney D. J., Boss G. R., Pilz R. B. (2007) A cysteine-rich LIM-only protein mediates regulation of smooth muscle-specific gene expression by cGMP-dependent protein kinase. J. Biol. Chem. 282, 33367–33380 [DOI] [PubMed] [Google Scholar]
- 13. Neuman N. A., Ma S., Schnitzler G. R., Zhu Y., Lagna G., Hata A. (2009) The four-and-a-half LIM domain protein 2 regulates vascular smooth muscle phenotype and vascular tone. J. Biol. Chem. 284, 13202–13212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pfeifer A., Klatt P., Massberg S., Ny L., Sausbier M., Hirneiss C., Wang G. X., Korth M., Aszódi A., Andersson K. E., Krombach F., Mayerhofer A., Ruth P., Fässler R., Hofmann F. (1998) Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 17, 3045–3051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morrell N. W., Yang X., Upton P. D., Jourdan K. B., Morgan N., Sheares K. K., Trembath R. C. (2001) Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-β1 and bone morphogenetic proteins. Circulation 104, 790–795 [DOI] [PubMed] [Google Scholar]
- 16. Zhang S., Fantozzi I., Tigno D. D., Yi E. S., Platoshyn O., Thistlethwaite P. A., Kriett J. M., Yung G., Rubin L. J., Yuan J. X. (2003) Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 285, L740–754 [DOI] [PubMed] [Google Scholar]
- 17. Chiche J. D., Schlutsmeyer S. M., Bloch D. B., de la Monte S. M., Roberts J. D., Jr., Filippov G., Janssens S. P., Rosenzweig A., Bloch K. D. (1998) Adenovirus-mediated gene transfer of cGMP-dependent protein kinase increases the sensitivity of cultured vascular smooth muscle cells to the anti-proliferative and pro-apoptotic effects of nitric oxide/cGMP. J. Biol. Chem. 273, 34263–34271 [DOI] [PubMed] [Google Scholar]
- 18. Lagna G., Nguyen P. H., Ni W., Hata A. (2006) BMP-dependent activation of caspase-9 and caspase-8 mediates apoptosis in pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 291, L1059–1067 [DOI] [PubMed] [Google Scholar]
- 19. Yu P. B., Hong C. C., Sachidanandan C., Babitt J. L., Deng D. Y., Hoyng S. A., Lin H. Y., Bloch K. D., Peterson R. T. (2008) Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 4, 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang J., Li X., Al-Lamki R. S., Southwood M., Zhao J., Lever A. M., Grimminger F., Schermuly R. T., Morrell N. W. (2010) Smad-dependent and Smad-independent induction of id1 by prostacyclin analogues inhibits proliferation of pulmonary artery smooth muscle cells in vitro and in vivo. Circ. Res. 107, 252–262 [DOI] [PubMed] [Google Scholar]
- 21. Yang J., Davies R. J., Southwood M., Long L., Yang X., Sobolewski A., Upton P. D., Trembath R. C., Morrell N. W. (2008) Mutations in bone morphogenetic protein type II receptor cause dysregulation of Id gene expression in pulmonary artery smooth muscle cells: implications for familial pulmonary arterial hypertension. Circ. Res. 102, 1212–1221 [DOI] [PubMed] [Google Scholar]
- 22. Barford D. (2011) Structure, function and mechanism of the anaphase promoting complex (APC/C). Q. Rev. Biophys. 44, 153–190 [DOI] [PubMed] [Google Scholar]
- 23. Buis J., Wu Y., Deng Y., Leddon J., Westfield G., Eckersdorff M., Sekiguchi J. M., Chang S., Ferguson D. O. (2008) Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell 135, 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bao S., Lu T., Wang X., Zheng H., Wang L. E., Wei Q., Hittelman W. N., Li L. (2004) Disruption of the Rad9/Rad1/Hus1 (9-1-1) complex leads to checkpoint signaling and replication defects. Oncogene 23, 5586–5593 [DOI] [PubMed] [Google Scholar]
- 25. Lagna G., Ku M. M., Nguyen P. H., Neuman N. A., Davis B. N., Hata A. (2007) Control of phenotypic plasticity of smooth muscle cells by bone morphogenetic protein signaling through the myocardin-related transcription factors. J. Biol. Chem. 282, 37244–37255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao Y. D., Cai L., Mirza M. K., Huang X., Geenen D. L., Hofmann F., Yuan J. X., Zhao Y. Y. (2012) Protein kinase G-I deficiency induces pulmonary hypertension through RhoA/Rho kinase activation. Am. J. Pathol. 180, 2268–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morty R. E., Nejman B., Kwapiszewska G., Hecker M., Zakrzewicz A., Kouri F. M., Peters D. M., Dumitrascu R., Seeger W., Knaus P., Schermuly R. T., Eickelberg O. (2007) Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arterioscler. Thromb. Vasc. Biol. 27, 1072–1078 [DOI] [PubMed] [Google Scholar]
- 28. Bauer N. R., Moore T. M., McMurtry I. F. (2007) Rodent models of PAH: are we there yet? Am. J. Physiol. Lung Cell Mol. Physiol. 293, L580–582 [DOI] [PubMed] [Google Scholar]
- 29. Abe K., Toba M., Alzoubi A., Ito M., Fagan K. A., Cool C. D., Voelkel N. F., McMurtry I. F., Oka M. (2010) Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121, 2747–2754 [DOI] [PubMed] [Google Scholar]
- 30. Frank D. B., Lowery J., Anderson L., Brink M., Reese J., de Caestecker M. (2008) Increased susceptibility to hypoxic pulmonary hypertension in Bmpr2 mutant mice is associated with endothelial dysfunction in the pulmonary vasculature. Am. J. Physiol. Lung Cell Mol. Physiol. 294, L98–109 [DOI] [PubMed] [Google Scholar]
- 31. Atkinson C., Stewart S., Upton P. D., Machado R., Thomson J. R., Trembath R. C., Morrell N. W. (2002) Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 105, 1672–1678 [DOI] [PubMed] [Google Scholar]
- 32. Fantozzi I., Huang W., Zhang J., Zhang S., Platoshyn O., Remillard C. V., Thistlethwaite P. A., Yuan J. X. (2005) Divergent effects of BMP-2 on gene expression in pulmonary artery smooth muscle cells from normal subjects and patients with idiopathic pulmonary arterial hypertension. Exp. Lung Res. 31, 783–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Geraci M. W., Moore M., Gesell T., Yeager M. E., Alger L., Golpon H., Gao B., Loyd J. E., Tuder R. M., Voelkel N. F. (2001) Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ. Res. 88, 555–562 [DOI] [PubMed] [Google Scholar]
- 34. Beppu H., Ichinose F., Kawai N., Jones R. C., Yu P. B., Zapol W. M., Miyazono K., Li E., Bloch K. D. (2004) BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am. J. Physiol. Lung Cell Mol. Physiol. 287, L1241–1247 [DOI] [PubMed] [Google Scholar]
- 35. Tada Y., Majka S., Carr M., Harral J., Crona D., Kuriyama T., West J. (2007) Molecular effects of loss of BMPR2 signaling in smooth muscle in a transgenic mouse model of PAH. Am. J. Physiol. Lung Cell Mol. Physiol. 292, L1556–1563 [DOI] [PubMed] [Google Scholar]
- 36. West J., Fagan K., Steudel W., Fouty B., Lane K., Harral J., Hoedt-Miller M., Tada Y., Ozimek J., Tuder R., Rodman D. M. (2004) Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ. Res. 94, 1109–1114 [DOI] [PubMed] [Google Scholar]
- 37. Lincoln T. M., Wu X., Sellak H., Dey N., Choi C. S. (2006) Regulation of vascular smooth muscle cell phenotype by cyclic GMP and cyclic GMP-dependent protein kinase. Front. Biosci. 11, 356–367 [DOI] [PubMed] [Google Scholar]
- 38. Dey N. B., Boerth N. J., Murphy-Ullrich J. E., Chang P. L., Prince C. W., Lincoln T. M. (1998) Cyclic GMP-dependent protein kinase inhibits osteopontin and thrombospondin production in rat aortic smooth muscle cells. Circ. Res. 82, 139–146 [DOI] [PubMed] [Google Scholar]
- 39. Zhao Y. Y., Zhao Y. D., Mirza M. K., Huang J. H., Potula H. H., Vogel S. M., Brovkovych V., Yuan J. X., Wharton J., Malik A. B. (2009) Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J. Clin. Invest. 119, 2009–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leoni A. L., Gavillet B., Rougier J. S., Marionneau C., Probst V., Le Scouarnec S., Schott J. J., Demolombe S., Bruneval P., Huang C. L., Colledge W. H., Grace A. A., Le Marec H., Wilde A. A., Mohler P. J., Escande D., Abriel H., Charpentier F. (2010) Variable Nav1.5 protein expression from the wild-type allele correlates with the penetrance of cardiac conduction disease in the Scn5a+/− mouse model. PLoS One 5, e9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Whitelaw N. C., Chong S., Morgan D. K., Nestor C., Bruxner T. J., Ashe A., Lambley E., Meehan R., Whitelaw E. (2010) Reduced levels of two modifiers of epigenetic gene silencing, Dnmt3a and Trim28, cause increased phenotypic noise. Genome Biol. 11, R111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hamid R., Cogan J. D., Hedges L. K., Austin E., Phillips J. A., 3rd, Newman J. H., Loyd J. E. (2009) Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele. Hum. Mutat. 30, 649–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Baliga R. S., Zhao L., Madhani M., Lopez-Torondel B., Visintin C., Selwood D., Wilkins M. R., MacAllister R. J., Hobbs A. J. (2008) Synergy between natriuretic peptides and phosphodiesterase 5 inhibitors ameliorates pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 178, 861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dumitrascu R., Weissmann N., Ghofrani H. A., Dony E., Beuerlein K., Schmidt H., Stasch J. P., Gnoth M. J., Seeger W., Grimminger F., Schermuly R. T. (2006) Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation 113, 286–295 [DOI] [PubMed] [Google Scholar]
- 45. Weissmann N., Hackemack S., Dahal B. K., Pullamsetti S. S., Savai R., Mittal M., Fuchs B., Medebach T., Dumitrascu R., Eickels M., Ghofrani H. A., Seeger W., Grimminger F., Schermuly R. T. (2009) The soluble guanylate cyclase activator HMR1766 reverses hypoxia-induced experimental pulmonary hypertension in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L658–665 [DOI] [PubMed] [Google Scholar]
- 46. Zuckerbraun B. S., Shiva S., Ifedigbo E., Mathier M. A., Mollen K. P., Rao J., Bauer P. M., Choi J. J., Curtis E., Choi A. M., Gladwin M. T. (2010) Nitrite potently inhibits hypoxic and inflammatory pulmonary arterial hypertension and smooth muscle proliferation via xanthine oxidoreductase-dependent nitric oxide generation. Circulation 121, 98–109 [DOI] [PubMed] [Google Scholar]
- 47. Chen C. N., Watson G., Zhao L. (2013) Cyclic guanosine monophosphate signalling pathway in pulmonary arterial hypertension. Vascul. Pharmacol. 58, 211–218 [DOI] [PubMed] [Google Scholar]
- 48. Stasch J. P., Pacher P., Evgenov O. V. (2011) Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 123, 2263–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]