

Background: The active site of rhomboid protease is embedded in the membrane and closed.
Results: Cross-linking transmembrane helices S5 and S2 does not significantly affect the protease activity in reconstituted membrane vesicles.
Conclusion: Contrary to the lateral gating model, large movement of the S5 helix is not required for substrate access to the active site of the protease.
Significance: Clarifying the movement of S5 helix during catalysis improves our mechanistic understanding of rhomboid intramembrane protease.
Keywords: Enzyme Mechanisms, Intramembrane Proteolysis, Protein Conformation, Rhomboid Protease, X-ray Crystallography
Abstract
Rhomboids represent an evolutionarily ancient protease family. Unlike most other proteases, they are polytopic membrane proteins and specialize in cleaving transmembrane protein substrates. The polar active site of rhomboid protease is embedded in the membrane and normally closed. For the bacterial rhomboid GlpG, it has been proposed that one of the transmembrane helices (S5) of the protease can rotate to open a lateral gate, enabling substrate to enter the protease from inside the membrane. Here, we studied the conformational change in GlpG by solving the cocrystal structure of the protease with a mechanism-based inhibitor. We also examined the lateral gating model by cross-linking S5 to a neighboring helix (S2). The crystal structure shows that inhibitor binding displaces a capping loop (L5) from the active site but causes only minor shifts in the transmembrane helices. Cross-linking S5 and S2, which not only restricts the lateral movement of S5 but also prevents substrate from passing between the two helices, does not hinder the ability of the protease to cleave a membrane protein substrate in detergent solution and in reconstituted membrane vesicles. Taken together, these data suggest that a large lateral movement of the S5 helix is not required for substrate access to the active site of rhomboid protease.
Introduction
The evolutionarily ancient rhomboid protein family represents a unique class of membrane-bound proteases (1, 2). The proteolytic function of rhomboids was first recognized and extensively studied in Drosophila, where the proteases play a critical role in epidermal growth factor receptor signaling by controlling the release of membrane-bound growth factors (3). In other species, rhomboids have been shown to be involved in bacterial quorum sensing (Providencia stuartii) (4), parasite invasion of host cells (e.g. Plasmodium falciparum) (5–8), and mitochondrial functions (9–11). Besides the active proteases, the protein family also includes a group of proteolytically inactive pseudoenzymes called iRhoms (2), which have been shown to have regulatory functions (12–14). The versatility of rhomboid proteins has generated great interest in their structure and biochemical mechanism.
Rhomboid proteases are polytopic membrane proteins and specialize in processing transmembrane (TM)2 protein substrates. The crystal structure of the bacterial rhomboid GlpG reveals that the Ser-His catalytic dyad of the protease is positioned in a polar cavity surrounded by TM helices (15–18). This structural feature is consistent with the function of the protein as an intramembrane-cleaving protease (I-CLiP) but raises the question of how a TM substrate, whose diffusion is restricted to the membrane plane, gains access to the active site of the protease. At least two models have been proposed. According to one hypothesis, a surface loop (L5), which caps the active site from the extracellular side of the membrane (Fig. 1A, yellow), can be lifted to expose the catalytic dyad to aqueous solution (19). The substrate cleavage site, often located near the end of the TM helix, partitions initially into solution and enters the active site from above the membrane plane (20, 21). According to another hypothesis, one of the TM helices (S5) of the protease can rotate significantly to the side (Fig. 1B). This movement opens a gate within the membrane and allows substrate to enter laterally (16, 22). Here, we examined the S5 gating model and critically evaluated published data that appear to support it. Our experimental results suggest that a large lateral movement of the S5 helix is not required for substrate access to the active site of the rhomboid protease.
FIGURE 1.
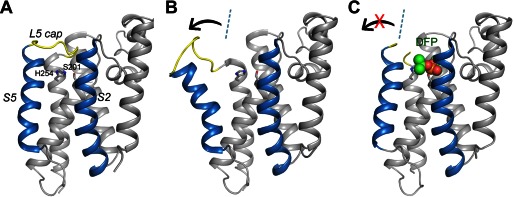
Lateral movement of TM helix S5 in the protease-inhibitor complex is small. A, the apoprotease adopts a closed conformation (Protein Data Bank code 2IC8) (15). The S2 and S5 helices are highlighted in blue. The L5 cap is highlighted in yellow. The catalytic residues are represented by stick models. B, in the open conformation (Protein Data Bank code 2NRF) (16), the C-terminal end of the S5 helix rotate way from the main body of the protease (arrow). The dashed line indicates the orientation of S5 in the closed conformation. C, the cocrystal structure of GlpG with DFP. The inhibitor is represented by a space-filling model. The L5 cap is disordered in the cocrystal structure.
EXPERIMENTAL PROCEDURES
Reagents
The detergents used in membrane protein purification and crystallization were purchased from Affymetrix, Inc. (Santa Clara, CA). Diisopropyl fluorophosphate (DFP) was purchased from EMD Chemicals, Inc. 1,2-Dimyristoylphosphatidylcholine (DMPC) was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). 1,2-Ethanediyl bismethanethiosulfonate (M2M) was purchased from Toronto Research Chemicals (Toronto, Canada). 4-Acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) was purchased from Invitrogen.
Protein Purification
The core catalytic domain of Escherichia coli GlpG and the fusion substrate maltose-binding protein-Gurken-thioredoxin were prepared as described previously (23). All mutants were generated by QuikChange. GlpG mutants were similarly purified as the wild-type protein.
Cocrystallization and Structure Determination
The GlpG core domain was prepared as described previously (15). The purified protein was concentrated to 5 mg/ml and dialyzed against 0.5% n-nonyl-β-d-glucopyranoside (NG) in 10 mm Tris (pH 7.4) for 7 days. After dialysis, freshly prepared DFP (50 mm in Me2SO) was added to the protein solution (5:1 inhibitor/protease molar ratio) to completely inactivate the protease (24). After incubation at room temperature for 30 min, the reaction mixture was used in crystallization screens in which 0.4 μl of protein solution (the DFP adduct) was mixed with 0.4 μl of well solution in a sitting drop vapor diffusion format. Tiny crystals started to appear after 1 week at room temperature over a well solution of 3 m NaCl and 0.1 m Bis-Tris propane (pH 7.0) and continued to grow for another 3 weeks to full size. A single crystal (∼50 μm in size) was harvested and stepwise transferred to a cryoprotection solution containing 3 m NaCl, 10 mm Tris (pH 7.4), 0.5% NG, and 20% glycerol. The crystal was flash-frozen in liquid nitrogen. X-ray diffraction data were collected from beamline X29 at the National Synchrotron Light Source and processed by HKL2000 (see Table 1) (25). The crystal structure was solved by molecular replacement using the known GlpG-DFP structure as the probe (Protein Data Bank code 3TXT) (24). Difference Fourier map, calculated without ligand and water molecules, confirmed the presence of the inhibitor in the active site and showed that the side chain of His-254 points toward the catalytic serine, instead of away from it as in the search probe. Model building and refinement were performed using Coot and PHENIX (26, 27).
TABLE 1.
Crystallographic statistics
GlpG and DFP cocrystallized in space group R32. r.m.s.d., root mean square deviation.
| GlpG-DFP | |
|---|---|
| Data collection | |
| Cell dimensions (Å) | a = b = 110.1, c = 124.0 |
| Wavelength (Å) | 1.10 |
| Resolution (Å)a | 40.0–2.9 (3.0–2.9) |
| Redundancy | 9.1 (8.5) |
| Completeness (%)a | 99.7 (98.0) |
| 〈I/σ〉a | 13.1 (3.4) |
| Rmergea,b | 0.064 (0.652) |
| Refinement | |
| Resolution (Å) | 40.0–2.9 |
| Total reflections/test set | 6580/311 |
| Rwork/Rfreec | 0.232/0.269 |
| No. of atoms | |
| Protein | 1354 |
| DFP | 10 |
| Water | 30 |
| B-factors | |
| Protein | 82 |
| DFP | 76 |
| Water | 75 |
| r.m.s.d. | |
| Bond lengths (Å) | 0.009 |
| Bond angle | 1.158° |
a The highest resolution shell is shown in parentheses.
b Rmerge = Σ|Ii − 〈I〉|/ΣIi.
c Rwork = Σ|Fo − Fc|/ΣFo. Rfree is the cross-validation R-factor for the test set of reflections (5% of the total) omitted in model refinement.
Chemical Cross-linking
M2M was dissolved in Me2SO to prepare a 20 mm stock solution. To achieve cross-linking, freshly prepared cysteine mutants (0.2 mg/ml) were mixed with M2M (50 μm) at room temperature for 30 min in assay buffer (50 mm Tris (pH 8.0), 0.1 m NaCl, and 0.5% NG). To break the cross-linking, the protein was incubated with 50 mm DTT at 37 °C for 2 h.
AMS Gel Shift Assay
50-μl cross-linked or uncross-linked protein samples (0.1 mg/ml) were precipitated by TCA (10 min on ice) and collected by centrifugation. The precipitants were washed three times with ice-cold acetone, air-dried, and resuspended in 20 μl of denaturation buffer (0.1 m Tris (pH 8) and 8 m urea). Complete unfolding was achieved by three rounds of 5-min heating at 95 °C. AMS was dissolved in denaturation buffer (30 mm final concentration). 20 μl of AMS solution was added to the 20-μl protein resuspension and incubated for 1 h at room temperature in darkness before gel electrophoresis.
Proteoliposome Preparation
The DMPC powder (40 mg) was first dissolved in 2 ml of chloroform. The organic solvent was then removed in a rotary evaporator. The dried lipid, evenly deposited on the wall of the flask, was resuspended in 10 ml of water. The hydrated lipid suspension was subject to three freeze-thaw cycles by alternately placing the sample in liquid nitrogen and a warm water bath. The suspension was extruded 20 times through a 0.4-μm polycarbonate membrane filter to generate a stock solution of medium-sized unilamellar vesicles (4 mg/ml). To prepare proteoliposomes, n-decyl-β-d-maltoside was added to the liposome solution with a detergent/lipid weight ratio of 1.5:1 to completely dissolve the vesicles. Protein solution was then added to generate a final mixture containing 1 mg/ml lipid and 0.1 mg/ml protein. The mixture was incubated for 1 h at room temperature and dialyzed against 1 liter of 50 mm Tris (pH 8.0) and 0.1 m NaCl for 3 days at 4 °C. The dialysis buffer was changed once after 24 h. The resulting proteoliposome preparation was extruded again through the 0.4-μm filter to generate medium-sized unilamellar vesicles. The membrane vesicles were collected by ultracentrifugation at 60,000 rpm for 1 h at 4 °C in an Optima L-90K ultracentrifuge (Beckman Coulter) and a type 75 Ti rotor.
Activity Assay and Quantification
The reaction in detergent was initiated by mixing 1 μg of protease with 2 μg of substrate (1.6:1 protease/substrate molar ratio) in 15 μl of assay buffer at 37 °C (in Fig. 5B (lower panels), 0.1 μg of protease was used). The reaction was stopped by the addition of SDS-PAGE loading buffer. To study the reaction in lipid bilayers, 10 μl of substrate-containing proteoliposomes (0.1 mg/ml protein concentration) was mixed with 5 μl of protease-containing proteoliposomes (0.1 mg/ml) in the presence of 0.02% Triton-X 100 at 37 °C. The reaction was stopped by the addition of SDS-PAGE loading buffer. In Fig. 4C (right panel), to fully reduce the cross-linked F153C/W236C mutant, the proteoliposome sample was incubated with 50 mm freshly prepared DTT at 37 °C for 12 h. The intensities of the protein bands were quantified by ImageJ (28). Each gel contained a reference lane in which the same amount of substrate was loaded, and all other lanes were calibrated against this lane. The N-terminal cleavage fragment was used to monitor the progression of the reaction. All intensity measurements (N-terminal fragment) were made within the linear range of the working curve.
FIGURE 5.

Effects of S5-S2 interface mutations on activity. A, detailed view of side chain contacts at the interface. B, comparison of the activities of wild-type and mutant proteases (assayed in detergent). In the lower panels, less protease (10%) was used to prevent substrate depletion (the reaction was ∼10 times slower under this condition). C, the relative activity (wild-type protease = 1) is based on the initial reaction rates calculated from the gels shown in B.
FIGURE 4.

Proteolysis in reconstituted membrane vesicles. A, schematic diagram of the experiment. The TM domain of the fusion substrate (Gurken) is highlighted in yellow. The red arrow indicates the cleavage site (31). MBP, maltose-binding protein; Trx, thioredoxin. B, reconstitution of GlpG and maltose-binding protein-Gurken-thioredoxin into DMPC vesicles. ↑, supernatant after ultracentrifugation (the soluble fraction); ↓, membrane pellet (the insoluble fraction). C, left panel, fusion of membrane vesicles (induced by 0.02% Triton X-100) initiates the reaction. CC*, cross-linked F153C/W236C double mutant (note that cross-linked protease migrated faster than wild-type GlpG). Right panel, comparison of the cleavage reactions catalyzed by different amounts of reduced protease (CC) and protease not treated with DTT (CC*; cross-linked). F, full-length substrate; N, N-terminal cleavage fragment; C, C-terminal fragment; E, enzyme; E*, cross-linked F153C/W236C. D, left panels, the proteolytic reaction was more efficient in lipid bilayers. Right panel, time course of the reaction. The experiment was performed in triplicate. E, the small amount of detergent (0.02% Triton X-100) used to induce vesicle fusion was not sufficient to solubilize either the protease or the substrate.
RESULTS
Cocrystallization of GlpG with a Class-specific Inhibitor
The crystal structures of three rhomboid protease-inhibitor complexes, those between E. coli GlpG and 7-amino-4-chloro-3-methoxyisocoumarin (29), DFP (24), and benzyloxycarbonyl-AlaP(O-iPr)F (23), have been described in the literature. In these structures, the L5 loop is displaced from the active site of the protease, but TM helix S5 shows little movement. The published complexes were all generated by “soaking,” a technique in which the inhibitor is diffused into a preformed crystal of the apoprotease to bind and react with it. S5 does not have much room to move laterally in the crystal because it is adjacent to the TM helices of a neighboring molecule. Is it possible that conformational changes in the solution or in membrane bilayers involve a larger movement of the helix? To help address this question, we set out to cocrystallize GlpG with DFP, as cocrystallization would allow us to capture the structure of the protease after its conformational change had completed in solution. After mixing the GlpG-DFP complex with the crystallization solution, small protein crystals started to appear after 1 week (growth to full size would take another 3 weeks). We can rule out the possibility that these crystals were initially formed by apo-GlpG but modified later, as in a soaking experiment, by the excess DFP present in the solution based on the following observations. (i) GlpG was completely inactivated by DFP within minutes of their mixing;3 (ii) the covalent complex between GlpG and DFP is stable (24); and (iii) the unbound inhibitor was expected to completely hydrolyze within the first day of the crystallization experiment (the half-life of DFP in water is ∼1 h), long before the crystals formed.
A 2.9 Å resolution diffraction data set was collected from a single GlpG-DFP cocrystal (Table 1), and difference Fourier analysis confirmed the presence of the inhibitor in the active site of the protease (Fig. 2A). The cocrystal structure is similar, but not identical, to the structure of the GlpG-DFP complex generated by soaking (the Cα atoms have a root mean square deviation of 0.504 Å) (24). The side chain of the catalytic histidine (His-254) in the cocrystal structure does not rotate away from the catalytic serine (Ser-201) as in the soaking structure (Fig. 2B). Although it is no longer hydrogen-bonded to Ser-201, the histidine side chain points in the general direction of the serine, which is more compatible with its hypothesized role later during catalysis as a proton donor to the leaving amine group (30). In the cocrystal structure, TM helix S5 does not tilt laterally away from the rest of the protein (Fig. 1C). Therefore, none of the solved protease-inhibitor complex structures, obtained either through soaking or by cocrystallization, supports the model in which S5 tilts and functions as a lateral substrate gate (Fig. 1B).
FIGURE 2.
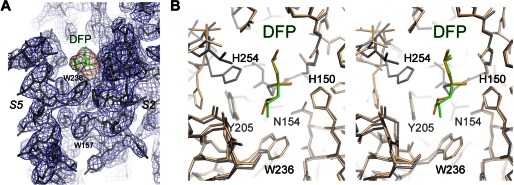
GlpG-DFP cocrystal structure. A, the final 2Fo − Fc electron density map contoured at 1.2σ levels (blue). The omit difference map, calculated without the inhibitor, is contoured at 3.0σ levels (orange). B, comparison of the DFP complexes generated by cocrystallization (gray and green) and by soaking (wheat and orange; Protein Data Bank code 3TXT) (24). The most pronounced difference is at His-254. The superposition is shown as a stereo pair.
Cross-linking S5 and S2 Does Not Abrogate GlpG Proteolytic Activity
Because the crystal structure captures only a snapshot of the conformational changes in the protein, is it possible that S5 opens only transiently to enable the inhibitor to diffuse into the active site but closes after it is bound? Because the inhibitors differ from peptide substrate in chemical structure (e.g. lacking the TM domain), there is also the concern that they may induce a different type of conformational change in the protease. To examine whether S5 undergoes any major conformational change during the binding and hydrolysis of real protein substrate, we studied the effect of cross-linking S5 to a neighboring helix (S2) on the activity of the protease against a fusion protein containing the TM region of Gurken, a natural substrate of Drosophila rhomboids (31). This approach has been attempted in an earlier study, but the result was not conclusive (22). In the published study, S5 was cross-linked to S2 through disulfide bonds between pairs of cysteines introduced to their interface by mutagenesis (Y160C/L229C, W157C/F232C, and F153C/W236C). A careful study of the crystal structure shows, however, that none of the pairs is close enough to form a disulfide bond (Fig. 3A): the distance between the Cα atoms of the disulfide-bonded cysteines is usually between 4.5 and 7.5 Å (32). To force a disulfide bond between the engineered cysteines in GlpG, the two helices will have to move closer by at least 1 Å. The inward movement of the helices may distort the active site and thus produce a false positive result (see “Discussion”).
FIGURE 3.
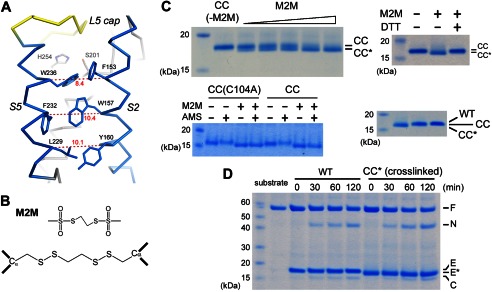
Cross-linking TM helices S2 and S5. A, distances (Å) between the Cα atoms are indicated for the 153/236, 157/232 and 160/229 residue pairs at the interface between S2 and S5. In this view, the Ser-His catalytic dyad is behind Phe-153 and Trp-236. B, the chemical structures of M2M and its reaction product with a pair of cysteines. C, cross-linked GlpG migrated faster on SDS-polyacrylamide gel. The M2M concentrations were 5, 10, 20, 30, and 70 μm. CC, the F153C/W236C double mutant; CC*, the cross-linked double mutant; CC(C104A), the C104A/F153C/W236C triple mutant; WT, wild-type. In the lower right panel, the wild-type protease also migrated slower than the cross-linked mutant. D, the cross-linked double mutant efficiently cleaved the maltose-binding protein-Gurken-thioredoxin fusion protein substrate in detergent solution. F, full-length substrate; N, N-terminal cleavage fragment; C, C-terminal fragment; E, enzyme; E*, cross-linked F153C/W236C.
Instead of using a disulfide bond, we used the methanesulfonate-based cross-linker M2M to tether the two helices (Fig. 3B). The spacer arm of M2M is 5.2 Å long and contains two flexible methylene groups (33, 34). The helices were cross-linked between residues 153 and 236 (in the F153C/W236C double mutant) because this pair is the closest to the active site, and its cross-linking offers the most stringent test of the S5 gating hypothesis (cross-linking at a lower position may not be sufficient to prevent the top of S5 from tilting away from S2). After reacting with M2M, the double mutant migrated slightly faster on SDS-polyacrylamide gel, probably because the unfolded protein was no longer a linear chain but contained an 84-amino acid loop (Fig. 3C). The addition of the reducing agent DTT caused the band to shift back to its original position. Wild-type GlpG also migrated slower than the cross-linked double cysteine mutant (Fig. 3C, lower right panel). The difference in electrophoretic mobility provided a convenient way to monitor the completeness of cross-linking. Besides Cys-153 and Cys-236, the double mutant contained a naturally occurring cysteine at position 104 (distal to Cys-153 and Cys-236). To demonstrate that cross-linking occurred between Cys-153 and Cys-236, we generated the triple mutant C104A/F153C/W236C. Reaction with M2M caused the gel mobility of the triple mutant to shift in an identical fashion to that of the double mutant (Fig. 3C). To further prove that Cys-153 and Cys-236 quantitatively reacted with the cross-linker, we performed AMS alkylation gel shift assay on both mutants. In the absence of M2M, alkylation of Cys-153 and Cys-236 in the triple mutant by AMS caused the protein band to shift slightly higher. After M2M treatment, AMS had no effect on the gel mobility of the triple mutant, confirming that both cysteines had completely reacted with M2M. For the double mutant, M2M-cross-linked protein could still be up-shifted by AMS because it contained the free Cys-104. Cys-104 did not react with M2M because it was not solvent-accessible in the folded protein. After denaturation in 8 m urea and SDS, Cys-104 became exposed and could react with AMS. Like wild-type GlpG, the cross-linked double mutant could efficiently cleave the TM protein substrate in detergent solution (Fig. 3D).
Cleavage of TM Substrate in Reconstituted Membrane Vesicles
A caveat of conducting the proteolytic reaction in detergent solution is that substrate may enter the protease differently than it normally does inside the membrane (the orientation of the solubilized protease and substrate is no longer restricted by the membrane plane). This is a serious concern especially because GlpG is also known to cleave soluble peptides (whose entry into the active site may or may not be the same) (35–37). To address this concern, we developed an assay to study the reaction in reconstituted membrane vesicles using purified protease and substrate (Fig. 4A). After reconstitution into unilamellar DMPC vesicles through dialysis, the protease and substrate could be collected separately by ultracentrifugation (the procedure did not cause protein denaturation and precipitation because both proteins could be solubilized completely from the proteoliposome pellets by 1% Triton X-100) (Fig. 4B). Simply mixing protease- and substrate-containing vesicles was not sufficient to initiate the reaction(Fig. 4C), demonstrating that the substrate cannot be cleaved in trans by the protease. The addition of 0.02% Triton X-100 to the mixture, which caused vesicle fusion (38), enabled the protease to cleave the substrate (the substrate could be completely processed upon longer incubation) (Fig. 4C). N-terminal sequencing of the cleavage products confirmed that the chimeric substrate was cleaved at the same Ala–His bond in detergent solution and in the lipid bilayer (31). We quantified and compared the initial rates of proteolysis and found that, under our assay conditions, the reaction in DMPC vesicles was about twice as fast as that in the detergent NG (Fig. 4D). The faster reaction rate in DMPC was probably because the diffusion of the proteins was restricted to a two-dimensional membrane plane. It is also possible that lipids subtly affected the structure of the protease or its conformational change. The small amount of detergent (0.02% Triton X-100) used to induce membrane fusion was not sufficient to dissolve the vesicles or to extract the proteins from the membrane (Fig. 4E). On the basis of these observations, we concluded that the proteolytic reaction must have taken place within the membrane bilayer.
Fully cross-linked F153C/W236C was reconstituted into DMPC vesicles under identical conditions. The reconstitution was conducted in an oxidizing environment and should not have altered the state of cross-linking. After membrane fusion, the cross-linked protease was also able to cleave the substrate (Fig. 4C). To rule out the possibility that a tiny amount of contaminating uncross-linked protease was responsible for the observed cleavage, we treated the protease-containing proteoliposomes with DTT, which removed the cross-linker. We then titrated down the reduced protease (50–500 ng of enzyme) (Fig. 4C, right panel, lanes 2–5) and compared the amount of proteolytic product (N-terminal fragment) with that generated by untreated cross-linked protease (500 ng of enzyme) (lane 6; note the difference in mobility between the protease bands). This comparison showed that, to generate the amount of product observed in lane 6, the untreated sample should contain at least 50–100 ng of uncross-linked enzyme (assuming that cross-linked protease was inactive). It was clear from the stained gel that, in lane 6, such an amount of reduced protease did not exist. Therefore, the observed proteolytic activity must be due to the cross-linked protease.
On the basis of the results presented above, we conclude that a large lateral movement of S5 is not required for substrate entry into the active site of the rhomboid protease. Furthermore, because the cross-linker prevented the passing of the substrate TM domain between S2 and S5 as an intact helix, our results suggest that the substrate can bend over residues 153 and 236 (the top part of the substrate TM helix needs to unwind), thus avoiding the proposed lateral gate, to reach into the active site to become cleaved (the catalytic dyad is roughly at the same level as Phe-153 and Trp-236) (Fig. 3A). In the native crystal structure, S2 and S5 do not interact directly with each other above Phe-153 and Trp-236, and their gap is blocked by the L5 cap.
Mutations at the S2-S5 Interface
An earlier study (22) had identified two double mutants (F153A/W236A and W157A/F232A) at the interface between TM helices S2 and S5 that significantly enhanced GlpG enzymatic activity. It was hypothesized that the mutations promoted gate opening by weakening the interaction between the two helices (Fig. 3A). We tested this hypothesis by studying the activities of both the single and double mutants. The assay we used also offered several experimental advantages. (i) The cleavage of Gurken by GlpG occurs at a single site and has simple enzyme kinetics (31). The earlier study used the sequence of Spitz as substrate, which is cleaved at multiple sites. (ii) The reaction is quantified by measuring the intensity of Coomassie Blue-stained product band. This is more reliable than Western blotting. (iii) We used the initial reaction rate to compare activities; the kinetic data were all collected when <10% of the substrate was cleaved (Fig. 4D). At later time points when more substrate was consumed, product formation was no longer linear with time (the reaction slowed down), and the difference between wild-type and mutant proteases became smaller.
We first examined the Trp-157 and Phe-232 pair. The crystal structure shows that the only contact between the two residues is through the indole ring of the tryptophan and the CH2 group (Cβ) of the phenylalanine (Fig. 5A). Consistent with this observation, we found that the W157A single mutation, which removed the contact, altered the protease activity, increasing it by 2-fold, whereas the F232A mutation, which was expected to have a minimum structural impact (the contacting Cβ group is retained in the mutant), did not affect the activity (Fig. 5, B and C). Combining the two single mutations did not produce any further activity enhancement. The 2-fold increase in the double mutant is in general agreement with the published study (22), which documented a 7-fold activity enhancement.
We studied the Phe-153 and Trp-236 pair next. Here, once again, the contact is mediated through Cβ of the phenylalanine and the indole ring of the tryptophan (Fig. 5A). Our assay showed that W236A was four times more active than the wild-type protease, but F153A, unlike the F232A mutation studied above, was even more active (20-fold increase; the assay had to be conducted with less protease to prevent substrate depletion) (Fig. 5B). Combining F153A with W236A, unlike the situation with the previous pair, again enhanced the activity further (40-fold). The enhancement caused by F153A alone and the additivity of the effects of the two mutations cannot be explained by a weakened interaction between the two helices. Of all the mutants studied here, F153A/W236A produced the largest activity enhancement, which is in agreement with the published study (22). Nevertheless, our results suggest that this enhancement is due mostly to a substitution (F153A) that does not significantly alter the packing of the S5 helix.
In summary, our data show that single mutations at the packing interface between S2 and S5 can generate complex functional effects depending on the contribution of the affected residue to the three-dimensional structure. We confirmed that the GlpG activity is sensitive to perturbations at the interface (W157A and W236A). However, does this necessarily mean that the enzyme mechanism involves a separation of the two TM helices (22)? The sensitivity can be caused by other mechanisms. For example, the cocrystal structure shows that inhibitor binding caused S5 to tilt slightly toward S4 and S6 (not away from them as predicted by the S5 gating model) (Fig. 6, A and B). Because this movement changes how S5 is packed against S2 (see “Discussion”), it is conceivable that mutations that alter the packing of the two helices may influence the overall conformational change in GlpG and thus affect its enzymatic activity.
FIGURE 6.
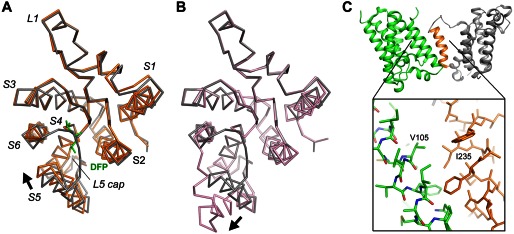
Comparison of the movement of S5. A, the GlpG-DFP cocrystal structure (orange) is superimposed onto the apo structure (gray) based on the Cα atoms of the central S4 helix (residues 201–216). The inhibitor is shown as a stick model (green). The arrow indicates the direction of the movement of S5. This image corresponds to a view from the extracellular side onto the membrane plane. B, a similar comparison between the open conformation (pink) and the closed conformation (gray). C, the heavily tilted S5 helix (orange) of one GlpG molecule (gray) interacts extensively with a neighboring molecule (green) in the crystal lattice. Favorable hydrophobic interactions, e.g. between Ile-235 and Val-105, may have caused the top of S5 to separate from the rest of the protein.
DISCUSSION
The S5 lateral gating model originated from a crystallographic observation (16): in one of the crystal forms of apo-GlpG, the protease appears to have adopted an “open conformation” in which the S5 helix is rotated 35° (Fig. 1B). This open conformation is unusual because at least 10 other apo-GlpG structures have been deposited in the Protein Data Bank, and none shows a similar rotation of the S5 helix. Even in the same crystal where the tilted S5 helix is observed, a second GlpG molecule in the asymmetric unit adopts a closed conformation identical to the others. The open conformation is unusual also because it would widely expose the polar active site of the protease to the hydrophobic membrane environment. The observation that the tilted S5 helix interacts extensively with a neighboring molecule in the crystal raises the possibility that the open conformation may result from crystal packing: the TM helices that S5 packs against are not positioned in front of the active site of the protease and thus cannot mimic the substrate (Fig. 6C).
The results of the disulfide cross-linking experiment, designed to test the relevance of the open conformation, were also inconclusive (22). Oxidization of W157C/F232C and Y160C/L229C by copper phenanthroline caused a strong decrease in activity, but for F153C/W236C (the most critical pair close to the active site) (Fig. 3A), the treatment had no effect. It was argued that maybe residues 153 and 236 are too far apart to be cross-linked, but this explanation is contradictory to the fact that the distance between Phe-153 and Trp-236 (8.4 Å) is the shortest among the three pairs (the Cα-Cα distances in residues 157/232 and 160/229 are 10.4 and 10.1 Å, respectively). It was also surprising that, after copper phenanthroline treatment, both W157C/F232C and Y160C/L229C retained some residual activity. If substrate has to pass between the two helices to access the active site, one might expect that keeping the gate closed would completely eliminate activity (because data were not presented to show the extent of disulfide bond formation, it remains possible that residual activity was due to incomplete cross-linking). It should be pointed out that, regardless of the role of S5 in the enzyme mechanism, disulfide cross-linking between residues 157 and 232 or between residues 160 and 229 will invariably have a large structural impact. To enable disulfide bond formation, TM helices S2 and S5 have to move closer by at least 3 Å, and there is some evidence suggesting that perturbation of protein structure in this general region (opposite to the active site) can adversely affect the protease activity (39).
The data presented in this study strongly suggest that a large conformational change in S5 is not required for substrate access to the active site of GlpG. If S5 does not function as a lateral gate, can it play another role in the mechanism of the protease? Comparison of known crystal structures indicates that the helix possesses a high degree of flexibility, and our cross-linking experiment did not rule out the possibility that the conformational change in the protease may involve a smaller movement of the S5 helix. The cocrystal structure generated in this study shows that, upon inhibitor binding, the top of S5 tilted slightly inward (the amplitude of the rotation is much smaller than that found in the open conformation) and became more tightly packed against S4 and S6 (instead of breaking off contact with them). This was accompanied by a series of side chain rotations within the interior of the protein (Fig. 7A). The phenol ring of Tyr-205 rotated ∼90° about the Cβ–Cγ bond to avoid clash with Ile-237 (from the S5 helix); the rotation of Tyr-205 caused the side chain of Trp-236 to flip outwards; and the side chain of Trp-157, which is in contact with the moving helix, also rotated slightly. To investigate if substrate binding also causes S5 to tilt inward, we introduced a bulky amino acid into the packing interface between S5 and S4 (A233F and I237F) to try to hinder the rotation of the helix toward S4. Both A233F and I237F showed reduced activities (Fig. 7, B and C). We also examined the effect of replacing Ile-237 with a smaller amino acid (I237A), which may facilitate the inward movement of S5 by reducing the resistance from Tyr-205. Our assay showed that I237A was twice as active as the wild-type protease (Fig. 7, B and C). These results suggest that substrate and inhibitor may induce a similar type of movement in the S5 helix.
FIGURE 7.

Movement of the S5 helix in the inhibitor complex. A, the cocrystal structure (orange) is superimposed onto the apo structure (gray). The Cα traces and a subset of affected side chains are shown. The inhibitor carbons are shown in green. B and C, mutations at the S5-S4 interface affect the protease activity (assayed in detergent). F, full-length substrate; N, N-terminal cleavage fragment; C, C-terminal fragment; E, enzyme.
The significance of the S5 inward tilt and the repacking of nearby side chains in the enzyme mechanism is unclear at this time. However, the possibility that S5 may move subtly during catalysis provides an explanation for those mutations that alter the packing of the helix. It is important to note that activating mutations have been found not only at the interface between S5 and S2 (W157A and W236A) but also in other places (L234P and I237A). W157A and W236A affect side chains directly involved in the conformational change (Fig. 7A); L234P creates a kink in the S5 helix (22); I237A, which alters S5 packing against S4, produces a similar level of activity change as the tryptophan mutants. Can the mutations affect other aspects of enzyme mechanism? To address this question, we compared our results with those from two previous studies that used Spitz as substrate (22, 40). If the mutations affected only the conformational change in GlpG, one might predict that the degree of activity enhancement should follow a similar pattern (the exact -fold increase may still be different). However, this does not appear to be the case. When Gurken was used as substrate, F153A/W236A showed a greater effect on activity (40-fold increase versus 10-fold increase with Spitz), whereas W157A/F232A had a smaller effect (2-fold versus 7-fold). The discrepancy suggests that the mutations may have altered the structure of the protease in such a way that the binding of one substrate is more affected than the other. This complicates the interpretation of the effects of the mutations because we cannot yet predict how they influence substrate binding.
The finding that F153A has a strong activating effect is unexpected (according to our assay, F153A is more active than those mutations that affect S5 packing). It seems unlikely that Phe-153 can be directly involved in the conformational change in GlpG because its side chain points toward the lipid and does not interact strongly with other parts of the protease. On the basis of its location in the three-dimensional structure (Fig. 3A), we can only speculate that the mutation somehow affected substrate binding. However, this does not appear to be a general mechanism because the same mutation in a highly homologous rhomboid (Haemophilus influenzae GlpG F68A) does not activate the protease (39). The crystal structure of F153A or F153A/W236A may be helpful in explaining why these mutants have such extraordinary enzymatic activities.
Acknowledgments
We thank Drs. Gary Rudnick and Barbara Ehlich for suggestions on membrane fusion and Dr. Satinder Singh for sharing the membrane extruder. The F153C/W236C double mutant construct was made by Dr. Sangwon Lee.
This work was supported, in whole or in part, by National Institutes of Health Grant GM082839 (to Y. H.).
The atomic coordinates and structure factors (code 4H1D) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Y. Xue and Y. Ha, unpublished data.
- TM
- transmembrane
- DFP
- diisopropyl fluorophosphate
- DMPC
- 1,2-dimyristoylphosphatidylcholine
- M2M
- 1,2-ethanediyl bismethanethiosulfonate
- AMS
- 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid
- NG
- n-nonyl-β-d-glucopyranoside
- Bis-Tris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane.
REFERENCES
- 1. Koonin E. V., Makarova K. S., Rogozin I. B., Davidovic L., Letellier M. C., Pellegrini L. (2003) The rhomboids: a nearly ubiquitous family of intramembrane serine proteases that probably evolved by multiple ancient horizontal gene transfers. Genome Biol. 4, R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lemberg M. K., Freeman M. (2007) Functional and evolutionary implications of enhanced genomic analysis of rhomboid intramembrane proteases. Genome Res. 17, 1634–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Urban S., Lee J. R., Freeman M. (2001) Drosophila Rhomboid-1 defines a family of putative intramembrane serine proteases. Cell 107, 173–182 [DOI] [PubMed] [Google Scholar]
- 4. Stevenson L. G., Strisovsky K., Clemmer K. M., Bhatt S., Freeman M., Rather P. N. (2007) Rhomboid protease AarA mediates quorum-sensing in Providencia stuartii by activating TatA of the twin-arginine translocase. Proc. Natl. Acad. Sci. U.S.A. 104, 1003–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Donnell R. A., Hackett F., Howell S. A., Treeck M., Struck N., Krnajski Z., Withers-Martinez C., Gilberger T. W., Blackman M. J. (2006) Intramembrane proteolysis mediates shedding of a key adhesin during erythrocyte invasion by the malaria parasite. J. Cell Biol. 174, 1023–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baker R. P., Wijetilaka R., Urban S. (2006) Two Plasmodium rhomboid proteases preferentially cleave different adhesins implicated in all invasive stages of malaria. PLoS Pathog. 2, e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buguliskis J. S., Brossier F., Shuman J., Sibley L. D. (2010) Rhomboid 4 (ROM4) affects the processing of surface adhesins and facilitates host cell invasion by Toxoplasma gondii. PLoS Pathog. 6, e1000858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Santos J. M., Ferguson D. J., Blackman M. J., Soldati-Favre D. (2011) Intramembrane cleavage of AMA1 triggers Toxoplasma to switch from an invasive to a replicative mode. Science 331, 473–477 [DOI] [PubMed] [Google Scholar]
- 9. McQuibban G. A., Saurya S., Freeman M. (2003) Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature 423, 537–541 [DOI] [PubMed] [Google Scholar]
- 10. Cipolat S., Rudka T., Hartmann D., Costa V., Serneels L., Craessaerts K., Metzger K., Frezza C., Annaert W., D'Adamio L., Derks C., Dejaegere T., Pellegrini L., D'Hooge R., Scorrano L., De Strooper B. (2006) Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 126, 163–175 [DOI] [PubMed] [Google Scholar]
- 11. Chao J. R., Parganas E., Boyd K., Hong C. Y., Opferman J. T., Ihle J. N. (2008) Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature 452, 98–102 [DOI] [PubMed] [Google Scholar]
- 12. Zettl M., Adrain C., Strisovsky K., Lastun V., Freeman M. (2011) Rhomboid family pseudoproteases use the ER quality control machinery to regulate intercellular signaling. Cell 145, 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adrain C., Zettl M., Christova Y., Taylor N., Freeman M. (2012) Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 335, 225–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McIlwain D. R., Lang P. A., Maretzky T., Hamada K., Ohishi K., Maney S. K., Berger T., Murthy A., Duncan G., Xu H. C., Lang K. S., Häussinger D., Wakeham A., Itie-Youten A., Khokha R., Ohashi P. S., Blobel C. P., Mak T. W. (2012) iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 335, 229–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y., Zhang Y., Ha Y. (2006) Crystal structure of a rhomboid family intramembrane protease. Nature 444, 179–180 [DOI] [PubMed] [Google Scholar]
- 16. Wu Z., Yan N., Feng L., Oberstein A., Yan H., Baker R. P., Gu L., Jeffrey P. D., Urban S., Shi Y. (2006) Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat. Struct. Mol. Biol. 13, 1084–1091 [DOI] [PubMed] [Google Scholar]
- 17. Ben-Shem A., Fass D., Bibi E. (2007) Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc. Natl. Acad. Sci. U.S.A. 104, 462–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lemieux M. J., Fischer S. J., Cherney M. M., Bateman K. S., James M. N. (2007) The crystal structure of the rhomboid peptidase from Haemophilus influenzae provides insight into intramembrane proteolysis. Proc. Natl. Acad. Sci. U.S.A. 104, 750–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Y., Ha Y. (2007) Open-cap conformation of intramembrane protease GlpG. Proc. Natl. Acad. Sci. U.S.A. 104, 2098–2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Y., Maegawa S., Akiyama Y., Ha Y. (2007) The role of L1 loop in the mechanism of rhomboid intramembrane protease GlpG. J. Mol. Biol. 374, 1104–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ha Y. (2009) Structure and mechanism of intramembrane protease. Semin. Cell Dev. Biol. 20, 240–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baker R. P., Young K., Feng L., Shi Y., Urban S. (2007) Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate. Proc. Natl. Acad. Sci. U.S.A. 104, 8257–8262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xue Y., Chowdhury S., Liu X., Akiyama Y., Ellman J., Ha Y. (2012) Conformational change in rhomboid protease GlpG induced by inhibitor binding to its S′ subsites. Biochemistry 51, 3723–3731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xue Y., Ha Y. (2012) Catalytic mechanism of rhomboid protease GlpG probed by 3,4-dichloroisocoumarin and diisopropyl fluorophosphonate. J. Biol. Chem. 287, 3099–3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 26. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Winn M. D., Murshudov G. N., Papiz M. Z. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 [DOI] [PubMed] [Google Scholar]
- 28. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vinothkumar K. R., Strisovsky K., Andreeva A., Christova Y., Verhelst S., Freeman M. (2010) The structural basis for catalysis and substrate specificity of a rhomboid protease. EMBO J. 29, 3797–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fersht A. (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, W. H. Freeman Publishers, New York [Google Scholar]
- 31. Strisovsky K., Sharpe H. J., Freeman M. (2009) Sequence-specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol. Cell 36, 1048–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thornton J. M. (1981) Disulphide bridges in globular proteins. J. Mol. Biol. 151, 261–287 [DOI] [PubMed] [Google Scholar]
- 33. Loo T. W., Clarke D. M. (2001) Determining the dimensions of the drug-binding domain of human P-glycoprotein using thiol cross-linking compounds as molecular rulers. J. Biol. Chem. 276, 36877–36880 [DOI] [PubMed] [Google Scholar]
- 34. Green N. S., Reisler E., Houk K. N. (2001) Quantitative evaluation of the lengths of homobifunctional protein cross-linking reagents used as molecular rulers. Protein Sci. 10, 1293–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maegawa S., Ito K., Akiyama Y. (2005) Proteolytic action of GlpG, a rhomboid protease in the Escherichia coli cytoplasmic membrane. Biochemistry 44, 13543–13552 [DOI] [PubMed] [Google Scholar]
- 36. Maegawa S., Koide K., Ito K., Akiyama Y. (2007) The intramembrane active site of GlpG, an E. coli rhomboid protease, is accessible to water and hydrolyses an extramembrane peptide bond of substrates. Mol. Microbiol. 64, 435–447 [DOI] [PubMed] [Google Scholar]
- 37. Adrain C., Strisovsky K., Zettl M., Hu L., Lemberg M. K., Freeman M. (2011) Mammalian EGF receptor activation by the rhomboid protease RHBDL2. EMBO Rep. 12, 421–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saez R., Goñi F. M., Alonso A. (1985) The effect of bilayer order and fluidity on detergent-induced liposome fusion. FEBS Letters 179, 311–315 [DOI] [PubMed] [Google Scholar]
- 39. Brooks C. L., Lazareno-Saez C., Lamoureux J. S., Mak M. W., Lemieux M. J. (2011) Insights into substrate gating in H. influenzae rhomboid. J. Mol. Biol. 407, 687–697 [DOI] [PubMed] [Google Scholar]
- 40. Baker R. P., Urban S. (2012) Architectural and thermodynamic principles underlying intramembrane protease function. Nat. Chem. Biol. 8, 759–768 [DOI] [PMC free article] [PubMed] [Google Scholar]