

Background: Biological consequences of novel signature mutations in the p53 gene of tumors from patients exposed to Aristolochia medicines are not yet known.
Results: N131Y, a prominent Aristolochia-linked mutation, confers cancer-associated behavior to cells.
Conclusion: Rare mutations in cancer genes can have profound consequences to protein function.
Significance: N131Y is a candidate biomarker of Aristolochia-related disease and target for pharmacological rescue of p53 function.
Keywords: Cancer Biology, Cellular Senescence, Molecular Cell Biology, Mouse Genetics, Mutagenesis Mechanisms, Tumor Suppressor Gene, Biomarkers, Site-specific Recombination, Suppressor Mutations
Abstract
Exposure to herbal remedies containing the carcinogen aristolochic acid (AA) has been widespread in some regions of the world. Rare A→T TP53 mutations were recently discovered in AA-associated urothelial cancers. The near absence of these mutations among all other sequenced human tumors suggests that they could be biologically silent. There are no cell banks with established lines derived from human tumors with which to explore the influence of the novel mutants on p53 function and cellular behavior. To investigate their impact, we generated isogenic mutant clones by integrase-mediated cassette exchange at the p53 locus of platform (null) murine embryonic fibroblasts and kidney epithelial cells. Common tumor mutants (R248W, R273C) were compared with the AA-associated mutants N131Y, R249W, and Q104L. Assays of cell proliferation, migration, growth in soft agar, apoptosis, senescence, and gene expression revealed contrasting outcomes on cellular behavior following introduction of N131Y or Q104L. The N131Y mutant demonstrated a phenotype akin to common tumor mutants, whereas Q104L clone behavior resembled that of cells with wild-type p53. Wild-type p53 responses were restored in double-mutant cells harboring N131Y and N239Y, a second-site rescue mutation, suggesting that pharmaceutical reactivation of p53 function in tumors expressing N131Y could have therapeutic benefit. N131Y is likely to contribute directly to tumor phenotype and is a promising candidate biomarker of AA exposure and disease. Rare mutations thus do not necessarily point to sites where amino acid exchanges are phenotypically neutral. Encounter with mutagenic insults targeting cryptic sites can reveal specific signature hotspots.
Introduction
It is well appreciated that common TP53 mutations in human cancers confer neoplastic properties to the mutant cells (1–3). As expected for driver mutations in cancer, TP53 missense mutations that are common have been shown to obliterate wild-type p53 tumor suppressor functions. In addition, some mutant proteins, which usually accumulate to abnormally high levels, even acquire new oncogenic activities that contribute to the cancer phenotype (4). In a landmark comprehensive study of all possible amino acid substitutions in p53, an excellent correlation was shown between the frequency with which a TP53 mutation has been observed in tumors and the extent to which the mutation compromises p53 wild-type function (5). In contrast, various amino acid substitutions in p53 that have never or rarely been found in cancers are considered more likely to allow retention of normal p53 function (6). At sites where mutations in tumors are infrequent, amino acid substitutions may have no serious deleterious consequences for the cancer-suppressing activities of the p53 protein. Sequence changes at these locations thus could behave as passenger mutations that are not specifically selected for during carcinogenesis (7, 8). In specific circumstances, however, certain mutations may have been observed infrequently simply because endogenous mutagenic processes or external exposures encountered thus far do not target the base change and sequence context in question.
In a recent study of aristolochic acid (AA)3-associated urothelial cancers in Taiwanese patients, an uncommon TP53 missense mutation, N131Y, was discovered in multiple tumors (9). This is a remarkable finding, considering that none of the >20,000 p53 mutations found in human cancers during the initial two decades of research were N131Y mutations (International Agency for Research on Cancer database, version R12). Eight tumors with the N131Y mutation have now been found in urothelial cancer patients (9, 10). A key factor these patients have in common is exposure to AA, either from medicinal herbal preparations containing extracts of Aristolochia sp., as is the case for the Taiwanese patients, or from consuming grains from fields where Aristolochia also flourishes. The codon 131 AAC to TAC (N131Y) mutation remains an exceedingly rare mutation in other cohorts and cancers (11, 12) (International Agency for Research on Cancer database, version R15; supplemental Table 1). The characteristic feature of the AA-associated tumor mutation spectrum is the high proportion (59%) of A→T substitutions. A→T transversion is an uncommon spontaneous sequence change in mammalian cells, and most known human carcinogens preferentially induce other types of base changes. In addition to N131Y, several other unusual A→T mutations emerged from analysis of AA-associated tumors, such as an A→T transversion in exon 4, Q104L, which has been observed only once among >25,000 non-AA associated cancers with TP53 mutations (supplemental Table 1). There are no human cell lines in repositories or archives derived from tumors that harbor N131Y or Q104L, so it is not surprising that there is little experimental data on their impact, if any, on cellular behavior.
Our objective here was to determine whether N131Y and Q104L are simply passenger mutations or are indeed driver mutations, and if the latter, to describe their detrimental effects. To achieve this goal, we first generated a panel of p53 mutant clones from primary mouse embryonic fibroblasts (MEFs) using a procedure that allows precise importation of human TP53 sequences into the endogenous murine p53 locus (see Fig. 1A). Integrase-mediated cassette exchange (IMCE) in p53 platform (p53 null) MEFs allows for isolation of distinct mutant clones with a common genetic background (13). We then examined the rare AA-associated TP53 mutations for their impact on the properties of the different clones. The functional consequences of harboring the N131Y mutation were confirmed in integrase-targeted baby platform mouse kidney (BMK) cells as well as the p53 null human lung carcinoma cell line, H1299. Importantly, we were able to recover several wild-type p53 functions by introducing a “global” secondary suppressor mutation, N239Y, onto the N131Y mutant. This switch was sufficient to blunt the growth promoting effects of the mutant. An AA signature mutation that is otherwise rare yet displays biochemical and phenotypic hallmarks typical of universally common TP53 tumor mutants would be a biomarker candidate of choice and would represent a potential target for specific therapies directed against dysfunctional p53.
FIGURE 1.
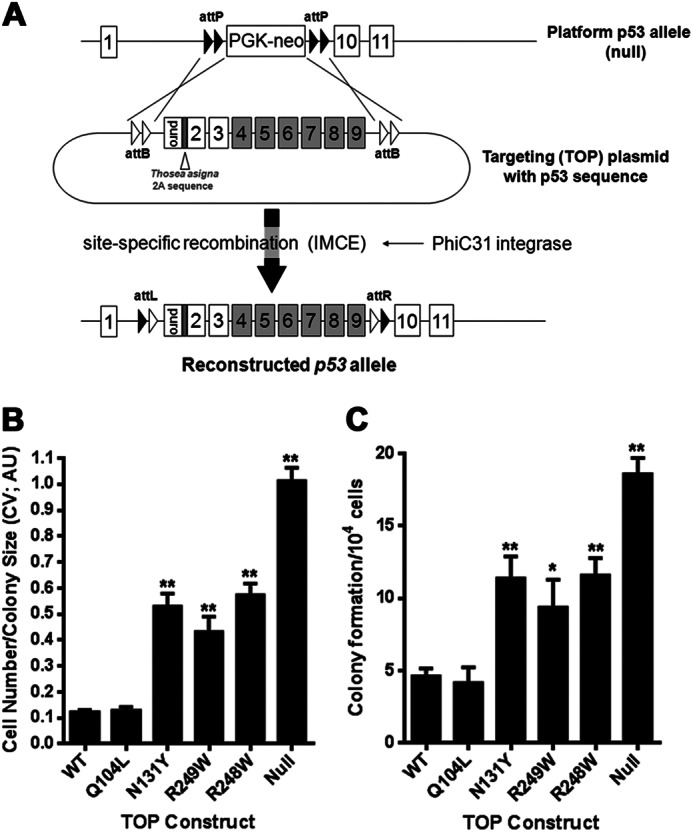
Establishing AA-induced p53 mutants in platform MEFs by IMCE. A, strategy for generating sets of p53 mutants in a common genetic background with the p53 platform/IMCE procedure. B and C, platform MEFs transfected with TOP plasmids containing the indicated p53 mutations together with PhiC31 integrase plasmid were selected for 3 days with puromycin before growth for a further 3 days in the absence of antibiotic selection. The ability of various TOP constructs containing the indicated p53 mutations to produce colonies after 7 days in antibiotic selection was determined and expressed as colonies formed per 1 × 104 cells seeded for transfection. Colonies were fixed and stained with crystal violet (CV) before extraction with 10% acetic acid. Absorbance was read at 590 nm (arbitrary units; AU) and taken as a measure of both colony number and size.
EXPERIMENTAL PROCEDURES
Plasmids, Transfection, and Cell Culture
Plasmids for use in IMCE have been described previously (14). Briefly, plasmids designed for IMCE at the Trp53 locus of the p53 (null) platform mouse are referred to as TOP plasmids for “targeting on platform” (see Fig. 1A). AA mutations were generated in TOP plasmids, pcDNA3-p53 and pWt1-p53 (pX42) by site-directed mutagenesis (QuikChange II XL site-directed mutagenesis kit; Stratagene, Leicester UK) and confirmed by DNA sequencing. Plasmids were transfected into platform MEFs, BMKs, and H1299 cells using either Lipofectamine 2000 (Invitrogen) or Nanofectin (PAA Laboratories; GE Healthcare) and selected as described previously (13) or with G418 (1 mg/ml). H1299 cells were grown in RPMI supplemented with 10% FCS. IMCE-derived cells are referred to either as clones, when single clones are used, or as pooled when the whole resistant population is used in a biological assay. MEF IMCE clones with dysfunctional p53 mutations can grow indefinitely as cell lines. Transient knockdown of p53 was performed using siRNA transfected with RNAiMAX (Invitrogen). Primary platform MEFs used for introduction of TOP constructs were prepared from 13.5-day-old embryos of plf/plf mice and grown as described previously (14) (European Mouse Mutant Archive; B6;129P2 (p53)TM182/DKFZ)). BMKs were isolated from 5-day-old mice by dispase II/collagenase I treatment and grown in DMEM/F12 supplemented with 5% FCS. In some experiments, BMKs were co-transfected with either c-Myc or Ad12E1A constructs (15).
Quantitative Reverse Transcription PCR (qRT-PCR)
Total RNA was extracted with the RNeasy Plus mini kit (Qiagen, Crawley, UK) according to the manufacturer's instructions. First-strand cDNA was prepared from total RNA (0.5–1 μg) using the GoScript Reverse Transcription system (Promega, Southhampton, UK). Real time quantitative reverse transcription PCR was performed using Power SYBR Green Master Mix (Applied Biosystems, Warrington, UK) with primer sets shown in supplemental Table 2. Gene expression determination was performed on an ABI 7900HT Fast real time PCR system (Applied Biosystems) and analyzed using the ΔΔCT method against GAPDH.
Cell Migration and Proliferation Assays
Cell migration toward a 10% FCS gradient was determined using modified Transwell filter assay (Falcon, BD Biosciences). Filters were fixed after 18 h and stained with crystal violet, and migrated cells were counted across four independent fields/filter. To analyze cell growth over a 4-day period, 4000–6000 cells per well were seeded in triplicate into 24-well plates. In 24-h intervals, cells were fixed and stained in a 0.2% (w/v) crystal violet/10% methanol solution for 30 min before washing. Dried samples were extracted with 10% acetic acid, and absorbance was measured at 590 nm. Cell proliferation was also determined using a BrdU ELISA assay performed in 96-well plates according to the manufacturer's instructions (Roche Applied Science) (13).
Colony Formation Assays
Following transfection of TOP constructs and PhiC31 integrase into platform MEFs, 5 × 104 cells were seeded in triplicate into 24-well plates. Puromycin selection was maintained for 5 days, after which cells were fixed in formaldehyde and stained with 0.2% crystal violet solution. Colonies were either counted or stain-extracted and measured at 590 nm. For analysis of anchorage-independent cell growth, 1 × 104 cells were mixed with 1.4% (w/v) agarose to form a 0.35% agarose final solution containing 10% FCS supplemented with basic fibroblast growth factor and insulin. This cell solution was overlaid onto a 0.7% (w/v) agarose/DMEM solution preformed in triplicate in six-well plates. Wells were overlaid with normal growth medium, which was replaced every 5–7 days. After 14–21 days, colonies were scored, counted, and size-determined using NIH ImageJ analysis software.
Analysis of Apoptosis Induction and Determination of Senescence
Cells subjected to overnight camptothecin treatment (10 μm) were trypsinized and labeled on ice for 30 min with annexin V-FITC solution (Molecular Probes). Cells were counterstained with propidium iodide prior to analysis by flow cytometry using a BD-LSRFortessa (BD Biosciences). The percentage of apoptotic cells was determined using DiVa software (version 6, BD Biosciences). For senescence determination, untreated or treated cells were fixed in 3% (w/v) paraformaldehyde and stained for β-galactosidase expression using a β-galactosidase staining kit according to the manufacturer's instructions (Sigma-Aldrich). Random fields from each condition were recorded using digital light microscopy, and percentage of blue cells was determined.
Antibodies, Western Blot, and Immunoprecipitation
Anti-p53 antibodies FL393 and pab1801, anti-PUMAα, anti-p21/WAF1, anti-MDM2, anti-E-cadherin, anti-N-cadherin, anti-transferrin receptor, anti-cyclin A, and anti-pRb were from Santa Cruz Biotechnology. Anti-phosphorylated serine 15, serine 37, and acetylated lysine 379 p53, anti-γH2A.x, anti-cyclin D1, anti-cdk1, anti-cdk6, anti-phosphorylated serine 807/811 pRb, and all HRP-conjugated secondary antibodies were from Cell Signaling Technology (New England Biolabs). Anti-p19arf antibody was from Abcam. Anti-α-tubulin, anti-β-catenin, and anti-actin were from Sigma-Aldrich. Alexa Fluor-conjugated secondary antibodies (donkey anti-rabbit Alexa Fluor 488 and donkey anti-mouse Alexa Fluor 594) were from BD Biosciences. Cells were lysed using 2% SDS lysis buffer (2% SDS, 50 mm Tris, 150 mm NaCl, 2 mm EDTA, 2 mm EGTA, pH 7.4), quantified using BCA reagent (Pierce) and 25–35 μg of total protein separated by 6–18% gradient SDS-PAGE followed by Western blotting with indicated antibodies.
Immunofluorescence Microscopy
Cells grown on glass coverslips were treated with camptothecin (1 μm; 18 h) prior to fixation in 10% formalin and staining with anti-p53 (FL393 or pab1801) and goat anti-p21 antibodies followed by Alexa Fluor-conjugated secondary antibodies. Nuclei were visualized with DAPI, and cells were analyzed on a Zeiss MetaFluor LSM510 confocal microscope.
Statistical Analysis
Data are presented as mean ± S.E. Statistical significance was evaluated using the Student's t test with SPSS software (IBM) or by analysis of variance with GraphPad Prism (GraphPad, Inc., San Diego, CA) and denoted as p < 0.05 (*) or p < 0.01 (**).
RESULTS
The AA-associated p53 Mutants, N131Y and Q104L, Have Contrasting Outcomes on Cell Behavior
To begin this study, we used IMCE to create a panel of MEF clones containing specific AA-associated TP53 missense mutations that encode the mutant p53 proteins Q104L, N131Y, R249W, and R273C (Fig. 1A). A p53 null clone containing the puromycin selection marker, but no p53 sequences from exons 4 to 9, and positive control lines expressing either the common tumor mutant, R248W, or WT p53 were also generated. The immortalized Hupki (human p53 knock-in) cell line COMN72 with wild-type human TP53 sequences and primary wild-type Trp53 MEFs served as additional controls (16). We have shown previously with the IMCE/MEF platform procedure that cassettes with common tumor p53 mutants produce greater numbers of colonies, and the cells proliferate more quickly than cells derived from cassettes encoding functional p53. Clonogenic potential in the IMCE protocol is thus an initial readout for loss of wild-type p53 function. As shown in Fig. 1, B and C, N131Y, R249W, and R248W cassettes demonstrate similar robust colony-forming ability. They produced more than twice the number of colonies (Fig. 1B) containing over five times the cells (Fig. 1C) compared with either a wild-type p53 construct or the Q104L construct. The p53 null TOP construct is even more effective than the mutants in producing colonies, possibly attributable to higher transfection/integration efficiency from the reduced cassette size.
The cell lines were examined by Western blotting for p53 protein expression and activation of p53 response genes following DNA damage (Fig. 2A). As anticipated, all mutant p53 proteins were expressed. Upon treatment with doxorubicin, the p53 variants underwent different levels of phosphorylation and acetylation, with N131Y and R248W exhibiting reduced serine 15 phosphorylation and Lys-379 acetylation (Fig. 2A). The Q104L mutant was the only mutant that showed substantial induction of wild-type p53 targets p21/WAF1 and BBC3/PUMA at levels similar to wild-type p53 (Fig. 2A). All IMCE-derived p53 mutant lines displayed high p19/ARF expression compared with either the (WT) COMN72 cell line or primary WT MEFs. Furthermore, integration and expansion occurred in the absence of any dramatic alteration in either pRb or cyclin D1 expression. Substantial phosphorylation of H2A.x (γH2A.x) was evident, indicating DNA damage following doxorubicin treatment (Fig. 2A).
FIGURE 2.

Immunoblot and immunocytofluorescence analysis of IMCE clones. A, untreated or treated (doxorubicin, 0.5 μm, 24 h) IMCE-derived MEF clones were lysed and analyzed by immunoblotting with the indicated antibodies together with wild-type Hupki MEFs (COMN72) and primary wild-type MEFs (WT). B, cell clones derived from IMCE as in A were analyzed for p53 and p21/WAF1 expression by confocal immunofluorescence microscopy before and after treatment with camptothecin (1 μm, 18 h). Scale bar represents 20 μm. C, expression of p53 response genes, p21/Waf1, Mdm2, cyclin G1/Ccng1, and Bbc3/Puma were analyzed by qRT-PCR following doxorubicin treatment (0.5 μm, 20 h). Fold changes were expressed as a ratio relative to untreated (control) p53 null cells. Target genes were standardized against GAPDH. Results are compiled from three independently generated cell lines for each mutant.
The mutant clones were examined by confocal microscopy for nuclear accumulation of p53 and p21/WAF1 induction following DNA damage with camptothecin (Fig. 2B). In agreement with immunoblots shown in Fig. 2A, WT and Q104L displayed marked nuclear p53 and p21/WAF1 accumulation following DNA damage. In contrast, the clones expressing common tumor mutants or the N131Y mutant showed substantial nuclear p53 levels but little or no p21/WAF1 expression under control conditions, although slight enhancement of p53 expression was evident after treatment. To assess the influence of the various mutants on p53 transcriptional transactivation, qRT-PCR was performed to examine induction of p21/Waf1, Mdm2, Cyclin G1/Ccng1, and Bbc3/Puma following DNA damage. Expression changes were compared against responses in untreated p53 null cells and expressed as ratios relative to GAPDH. Only cell lines expressing Q104L demonstrated significantly elevated p21/Waf1, Cyclin G1/Ccng1, and Mdm2 mRNA amounts compared with p53 null cells, with levels approaching wild-type expressing cells (Fig. 2C). However, very little induction of Bbc3/Puma was detected for any of the mutants.
Introduction of N131Y but Not Q104L Promotes Cell Proliferation and Migration
Cell proliferation was examined over a 4-day growth period. Cells derived from TOP constructs (Q104L, N131Y, R248W, R273C, R248W, WT, and null) were stained, and levels were expressed as a ratio change relative to day 1 levels. The N131Y mutant displayed the highest level of growth, becoming particularly evident as the cells neared confluence at day 4. This was significantly greater than what we observed with p53 null cells (Fig. 3A). The Q104L clones displayed growth similar to wild-type expressing cells, and this rate of growth was significantly lower than when p53 was absent (Fig. 3A). Proliferation analysis by BrdU incorporation also revealed highest incorporation in N131Y cells (Fig. 3B). Cell migration toward serum-containing media was determined using a Transwell assay, and the number of migrated cells was expressed as a ratio relative to the p53 null cells. N131Y-expressing cells migrated 80% more effectively than p53 null cells (Fig. 3C). The R249W, R273C, and R248W cells showed between 40–50% enhancement in cell migration relative to p53 null cells. There was no significant influence of either WT p53 or Q104L on cell migration (Fig. 3C). Taken together, these findings indicate that the N131Y mutation has a gain-of-function effect on cells in enhancing proliferation and migration when compared against cells with no p53 expression.
FIGURE 3.
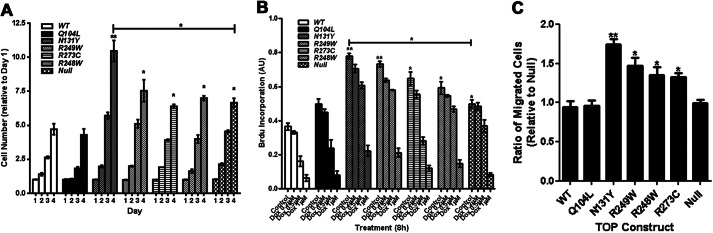
Introduction of N131Y mutation into p53 null MEFs confers a gain-of-function phenotype. A, cell growth was measured over a 4-day period and expressed as a ratio of day 1 levels. B, proliferation was measured by BrdU ELISA following treatment with doxorubicin for 8 h. BrdU was added for 2 h before fixation and detection. Results are expressed as arbitrary units (AU). C, cell migration toward serum was assessed using a Transwell-based assay over an 18-h period. Results are expressed as a ratio relative to p53 null cell migration. All results are compiled from at least three independent experiments.
N131Y Gain-of-function Activity Is Reduced by Introduction of the Global Second-site Suppressor Mutation, N239Y
The introduction of secondary intragenic mutations suppressing the effects of primary tumor mutations has the potential to restore wild-type p53 activity and would allow investigation into the effects of recovering p53 wild-type activity in mutant cells (17). We introduced the global suppressor mutation N239Y into the N131Y construct to create cells expressing the double mutant N131Y/N239Y. Introduction of this construct into platform MEFs significantly reduced both colony size (Fig. 4A) and number (Fig. 4B), compared with N131Y alone, although colony formation remained higher than for either WT or N239Y cells (Fig. 4, A and B). Although total p53 protein expression or accumulation following DNA damage with doxorubicin was unchanged between the mutants (Fig. 4C), significant restoration of serine 15 (serine 18 in the mouse) and serine 37 phosphorylation occurred in the double mutant (Fig. 4C and supplemental Fig. 1). Curiously, acetylation of lysine 379 (Lys-379) was almost completely absent in the N131Y mutant (Fig. 4C and supplemental Fig. 1). Note that exposure times were longer than those used for presenting a comparison of all mutants and WT p53 in Fig. 2A). Surprisingly, the addition of N239Y was able to significantly enhance acetylation at this site (Fig. 4C and supplemental Fig. 1). However, attenuated acetylation at lysine 379 does not appear to be a general attribute of p53 mutant cells because cells with some common tumor mutants (i.e. R249W and R273C) responded to DNA damage by acetylating this residue to levels equivalent to WT cells (Fig. 2A). Regardless, the alterations in p53 post-translational modifications evident between the single and double mutants were accompanied by elevated protein levels of the p53 target gene products, p21/WAF1, MDM2, and BBC3/PUMA (Fig. 4C and supplemental Fig. 1). The increased post-translational site modifications exhibited by the double mutant may contribute to the restoration of wild-type activity.
FIGURE 4.

Introduction of a second-site suppressor mutation to N131Y increases wild-type p53 activity. Platform MEFs transfected with indicated TOP plasmids were selected in puromycin-containing media for 7 days before assessment of colony size (A) and colony formation (B) by crystal violet (CV) staining and expressed as arbitrary units (AU) per 104 cells transfected. C, control or doxorubicin-treated (0.5 μm, 18 h) p53 Null, N131Y and N131Y/N239Y cells were analyzed for changes in p53 signaling by immunoblotting with antibodies against indicated proteins. D, expression of p21/Waf1), cyclin G1/Ccng1), Mdm2, and Bbc3/Puma mRNA was determined before and after doxorubicin (Dox; 0.5 μm, 20 h) treatment by qRT-PCR. Changes were expressed as log ratio relative to untreated null cells with target genes standardized against GAPDH. Results are compiled from three independently generated cell lines for each mutant.
To further assess restoration of p53 transcriptional activity to N131Y in the presence of the N239Y suppressor mutation, qRT-PCR was performed. Substantial increases in p21/Waf1, cyclin G1/Ccng1, and Mdm2 mRNA were observed in N131Y/N239Y cells compared with either N131Y alone or p53 null cells (Fig. 4D and supplemental Fig. 1). Expression levels generally approached half of those achieved upon re-expression of wild-type p53 regardless of DNA damage status (Fig. 4D and supplemental Fig. 1). However, only small changes in Bbc3/Puma mRNA expression were observed for the double mutant, with slight transcriptional trans-activation noted upon doxorubicin treatment (18 h, 0.5 μm). Thus, the presence of the N239Y mutation together with N131Y recovers several wild-type p53 activities in the mutant.
To evaluate the consequences to cell behavior of partially restoring wild-type p53 activity to N131Y, cellular proliferation was assessed by crystal violet staining over a 4-day period to compare p53 null, N131Y, and N131Y/N239Y cell growth (Fig. 5A). N131Y grew the most rapidly, whereas the double mutant grew at a rate even lower than observed for p53 null cells (Fig. 5A). Analysis of proliferation by monitoring BrdU uptake also showed a reduction in S phase cells upon introduction of N239Y to N131Y (data not shown). Next, we assessed whether the presence of N239Y in conjunction with N131Y could blunt the enhanced cell migration observed for N131Y in Transwell assays (see Fig. 3C). Indeed, the N131Y/N239Y double mutant displayed a 30% reduction in migration compared with N131Y alone (Fig. 5B). Finally, to compare the apoptotic response in the single and double mutants, we treated N131Y, N131Y/N239Y, and p53 null cells with camptothecin. Whereas the apoptotic fraction of N131Y cells doubled upon treatment to 12% of cells (a level slightly higher than exhibited by p53 null cells), >20% of cells expressing the double mutant showed induction of apoptosis as measured by annexin V binding (Fig. 5C).
FIGURE 5.

N131Y gain-of-function activity is reduced when the N239Y mutation is also present. A, growth of p53 null, N131Y, and N131Y/N239Y cells was determined over a 4-day period, and changes were expressed as a ratio relative to day 1. B, Transwell migration assays were performed toward a serum gradient over 18 h. Migrated cells are expressed as a ratio relative to N131Y. Representative images of stained filters are shown on the right. C, N131Y, N131Y/N239Y, and p53 null cells were treated for 20 h with camptothecin (1 μm) before staining with annexin V-FITC and propidium iodide and analysis by flow cytometry. Apoptotic fractions are expressed as a percentage of total cell number. D, null, N131Y, or N131Y/N239Y cells were treated for 48 h with either scrambled (Scr) or p53 (p53) siRNA before treatment with doxorubicin (dox) and analysis of p53, p21, pRb and γH2A.x, PCNA and α-tubulin expression by Western blotting. E, cell proliferation was measured by BrdU incorporation 48 h after siRNA treatment. (AU; arbitrary units). F, migration of siRNA-treated cells toward an FCS gradient was determined by Transwell filter assay. Migrated cells are expressed relative to scrambled siRNA-treated N131Y cells. G, cyclin A, cdk1, cdk6, p21, pRb, and p19arf levels were determined by Western blotting following siRNA treatment of null, N131Y, and N131Y/N239Y MEFs and compared against levels of actin and tubulin. Levels of cyclin A (H) and cdk1 (I) were quantified and expressed relative to null scrambled siRNA (H) controls (I). Results are an average of four independent experiments.
To confirm that the increased proliferative and migratory response was due to N131Y expression, we depleted p53 from IMCE-targeted MEFs using siRNA (control scrambled or p53-specific). Efficient knockdown of p53 was achieved following 48 h of treatment (Fig. 5D). There was altered functional output as demonstrated by knockdown of N131Y reducing cell proliferation by 15% (Fig. 5E) and cell migration by 22% (Fig. 5F). In contrast, loss of N131Y/N239Y increased both proliferation and migration, perhaps reflecting release from p21-mediated inhibition (Fig. 5, D–F). There was no significant effect on null cells upon siRNA treatment.
To explore the molecular mechanisms by which N131Y influences cell proliferation, we next examined expression of the cell cycle regulators, cyclin A and cdk1, by Western blotting (Fig. 5G). No significant difference in either cyclin A or cdk1 between null and N131Y cells under normal growth conditions was observed. However, following depletion of N131Y p53 by siRNA, both cyclin A and cdk1 levels were reduced by 30% (Fig. 5, H and I). No change in cdk6 expression was evident. Expression in Null cells were unaffected by siRNA treatment. These results suggest N131Y actively influences levels of specific cell cycle regulators, thereby promoting proliferation.
A critical gauge of p53 activity in primary MEFs is the induction of senescence upon prolonged culturing. Sustained treatment with a low dose of DNA damaging agent can enhance this senescent population. We examined whether early passage N131Y/N239Y cells showed elevated levels of senescence compared with N131Y and p53 null cells. The senescent population was also compared against cells expressing WT p53. As expected, the expression of N131Y/N239Y significantly increased the percentage of β-galactosidase-positive cells following doxorubicin treatment (72 h, 0.1 μm; Fig. 6A) compared with N131Y and p53 null cells, which failed to show significant changes in senescent population. Cells expressing wild-type p53 were the most effective in inducing senescence following DNA damage (Fig. 6A). The second-site mutation may thus contribute to a reduction in N131Y oncogenic potential by partially recovering p53 control of senescence.
FIGURE 6.

Effect of the N239Y second-site mutation on the impact of N131Y on cell senescence and growth in soft agar. A, percentage of senescent cells before and after doxorubicin (Dox; 0.1 μm, 3 days) treatment was determined as a percentage of β-galactosidase-positive cells relative to total cell number per field. Representative images are shown to the left. B, representative images of colonies produced following 21-day (21d) growth in soft agar. Introduced mutations are indicated. C, colony forming ability of various p53 mutants and wild-type Hupki MEF cells (E67) was determined by expressing colonies per field relative to total cell number per field. Results are presented as a ratio of p53 null cell colony formation (null). D, average size of colonies from C was determined using NIH ImageJ software and expressed as a ratio of the average single cell area. All experiments were performed in triplicate and are compiled from three independent experiments. *, p < 0.05, **, p < 0.01.
The ability of cells to survive and proliferate in soft agar (i.e. anchorage-independent growth) is a well characterized hallmark of an oncogenic phenotype. We assessed whether the presence of any of the mutations characteristic of AA exposure could evoke colony formation. Double mutant N131Y/N239Y cells were also examined. In these experiments, we included a control cell population consisting of genetically un-manipulated MEFs expressing the normal endogenous Trp53 gene (E67). Cells were examined 21 days after seeding. Examples of colony morphology are shown in Fig. 6B, which shows single cells or representative colonies from Null, N131Y, N131Y/N239Y, R249W, R248W, Q104L, and WT cells. Colonies per field were counted and colony-forming ability was expressed as a ratio relative to p53 null cells. N131Y expression resulted in a 2.7-fold increase in colony number (Fig. 6C). This was completely ablated by the co-presence of the N239Y mutation (Fig. 6C). Surprisingly, R249W was unable to stimulate further colony growth above that of the p53 null state. As expected, WT MEFs (E67) failed to produce any colonies (Fig. 6C). Average colony size was also determined and expressed relative to single cell averages (Fig. 6D). Both N131Y and R249W cells produced colonies larger than p53 null cells, whereas N131Y/N239Y, WT, and Q104L colonies were on average smaller than the null colonies. These findings show that the N131Y mutation allows for anchorage-independent growth in soft agar and that the effect is blocked when the N239Y intragenic mutation is also present.
To confirm the gain-of-function effect of N131Y p53 in additional non-fibroblastic cellular models, baby mouse kidney cells were isolated from platform mice and subjected to IMCE. Similar to MEFs, N131Y was highly expressed in the absence of DNA damage and was unable to induce p21 accumulation beyond that of p53 null cells. N131Y/N239Y was stabilized following DNA damage with doxorubicin and cells exhibited elevated p21 and pRb expression compared with null or N131Y cells (Fig. 7A). Immunofluorescence microscopy demonstrated nuclear p53 expression in BMKs coupled with peripheral accumulation of N-cadherin at cell-cell contacts reminiscent of epithelial cells (Fig. 7B and supplemental Fig. 2). Importantly, N131Y was able to increase BMK proliferation as measured by both short term BrdU incorporation (Fig. 7C) and analysis of cell growth during a 4-day period (Fig. 7D). Crucially, N131Y/N239Y cells had a significant growth disadvantage compared with either null or N131Y cells (Fig. 7, C and D). Similar to MEFs, N131Y BMK cells also displayed elevated cyclin A and cdk1 levels compared with the p53 null state (Fig. 7, E and F).
FIGURE 7.

N131Y promotes cell growth and migration of BMK cells. A, BMKs were isolated from 5-day-old mice and subjected to IMCE with null, N131Y, or N131Y/N239Y constructs. Protein expression was analyzed by Western blotting with indicated antibodies. B, confocal immunofluorescence microscopy was performed on BMKs with antibodies against p53 and N-cadherin. Cell proliferation was measured by both BrdU incorporation (C) or growth (D) over 4 days with crystal violet. E, expression of p53, cyclin A, phospho-cdk1, cdk1, E-cadherin, and damage DNA foci (γH2A.x) were determined by Western blotting. F, cyclin A levels were quantified from seven independent experiments.
Finally, we examined whether N131Y could influence the growth and migration of human cancer epithelial cells. The p53 null lung carcinoma line, H1299, was stably transfected with either N131Y or empty vector control (supplemental Fig. 2C). Cells were examined for p53 expression by Western blotting (supplemental Fig. 3A) and immunofluorescence microscopy (supplemental Fig. 3B). No change in p21 was evident; however, a small, reproducible increase in cyclin A/cdk1 could be detected. Strong nuclear expression of N131Y was achieved in the absence of DNA damage (supplemental Figs. 2C and 3B). Cells expressing N131Y displayed increased proliferation (∼20%) as determined by elevated BrdU incorporation (supplemental Fig. 3C). Cell migration was also significantly enhanced upon expression of N131Y (supplemental Fig. 3D). A 23% increase in cells migrating toward a serum gradient was noted (supplemental Fig. 3E). When taken together, these results suggest that the N131Y mutant retains its proliferative and migratory advantages when expressed in epithelial cells.
DISCUSSION
To discover the effects of N131Y and Q104L on p53 biology, we generated a panel of isogenic TP53 mutant cell clones by site-specific recombination at the Trp53 locus of primary MEFs isolated from p53 platform mice (13). The targeting plasmid includes human TP53 sequences from exons 4–9 (human p53 knock-in, Hupki) and restores a complete p53 gene under control of the endogenous Trp53 promoter in the recombinant clones. Previous work has demonstrated that cells and mice with p53 alleles containing wild-type human p53 sequences are similar to their genetically unmodified Trp53 counterparts, whereas Hupki alleles harboring human tumor mutations produce cancer-prone mice (Li-Fraumeni syndrome mouse models) and cells with compromised wild-type p53 activities (18–20).
In the present study, we found that IMCE-derived N131Y MEFs but not Q104L MEFs display dysfunctional properties typical of common TP53 human tumor mutations. Apoptotic and senescence responses to DNA damage and induction of key p53 target genes are compromised in the N131Y mutant, and it proliferates rapidly, even in soft agar. This latter property may be due to the combined loss of p21 expression and a novel ability of N131Y to promote or stabilize cyclin A/cdk1 expression. Furthermore, the second-site rescue mutation N239Y partially recovers wild-type activity to N131Y in both a MEF and BMK cell system. Suppressor TP53 mutations have been constructed and examined in yeast for their ability to restore the transcriptional profile typical of wild-type p53 to common tumor mutants (21, 22). Second-site mutation modeling studies are being pursued to assist in design of drugs that recover p53 function by mimicking the effects of suppressor amino acid substitutions on p53 conformation, DNA binding, and interaction with protein partners (23, 24).
We have previously reported the isolation of rare AA hot spot mutant MEFs following exposure of primary MEFs from the human p53 knock-in (Hupki) strain to the carcinogen and recovery of immortalized clones that harbor p53 mutants (25). Examination of human mutations in Hupki MEFs has proved to be instructive in elucidating p53 mutant behavior and the role of cancer agents in causing human tumors (26–28). In support of data shown here with the precisely mutated IMCE clones, comparisons among Hupki MEF immortalized cell lines isolated following AA exposure showed that the AA-N131Y cell line proliferated much more rapidly than the Q104L cells, accumulated more nuclear p53 under control conditions (a characteristic of well studied common tumor mutations in neoplastic cells), and was more refractory to induction of p53 target gene expression following DNA damage.4
Molecular modeling suggests that the N131Y mutation alters p53 global conformation, an effect opposed by N239Y (supplemental Fig. 4). The Asn-131 residue is located in the S2–S2′ β hairpin, and the side chain amide displays hydrogen bonding interactions with Ser-10 via the backbone carbonyl of Ser-269 and Ser-2 through the side chain OH in Thr-126 (supplemental Fig. 4A). The hydrogen-bonding network in the wild-type structure is expected to be lost in N131Y, and the tyrosine residue may lead to a conformational change and destabilization of the p53/DNA complex and/or a decrease in DNA binding. The second-site suppressor mutation, N239Y, is located in the L3 loop between β-strands Ser-8 and Ser-9 (supplemental Fig. 4B). In the native structure, the side chain amide sits in a hydrophobic cleft between the side chain of Leu-139 and the CHCH2 group, which links deoxyribose and the phosphate in DNA. In the N239Y structure, the phenyl ring is located in this cleft, which is expected to result in more favorable hydrophobic interactions, relative to wild type, as reported previously (29, 30). Thus, the addition of N239Y to N131Y may partially compensate for the destabilizing effect of the latter mutation and help restore DNA binding. The Q104L mutation, which confers a phenotype approaching that of cells with WT p53, is located in the N terminus at the protein-solvent interface, and no significant effects on conformation are predicted. Modeling of the two rare AA-associated mutants thus supports the experimental data, indicating that the N131Y mutant has a stronger detrimental impact on cellular homeostasis than the Q104L mutant.
The most plausible conclusion of our findings is that N131Y contributes directly to neoplastic development in urothelial tumorigenesis, and data from second-site suppressor experiments suggest that targeting this mutant in cancer cells could have therapeutic benefit. AA is now classified by the International Agency for Research on Cancer as a group 1A carcinogen (International Agency for Research on Cancer Monograph Series); nevertheless, it is feared that due to use of herbal preparations containing Aristolochia, exposure was, and perhaps remains, widespread, with estimates in the millions in Taiwan alone (9, 31). Given that the A→T mutation at codon 131 is tightly linked to AA exposure and appears to be otherwise so rare that it has the potential to serve as a specific biological marker of AA exposure and early disease, analogous to the R249S hot spot mutant in aflatoxin-exposed patients at high risk of hepatocellular carcinoma (32). The second reiterated AA-linked mutation we investigated that is likewise exceedingly rare in all other cancers, Q104L, appears to have small or negligible effects on cell proliferation, senescence, apoptosis, or cell migration. Conceivably, the A→T Q104L mutations in AA-associated urothelial cancers are passengers, attributable to preferred electrophilic attack by AA on adenine at this sequence context, and contributing to overall mutation load but not to phenotypic change. Our observations on the biological impact of N131Y support the choice of this mutant as a biomarker of AA exposure and early disease. On a more general note, this study argues that there are still cryptic hotspots in the TP53 human tumor mutation spectrum.
Acknowledgments
We are grateful for the technical assistance of A. Weninger, K. Muehlbauer, and N. Pechlivani.
This work was supported by Yorkshire Cancer Research Grant L351 (to M. H.), a White Rose University Consortium Grant (to A. F. O. and M. H.), and the Deutsches Krebsforschungszentrum (German Cancer Research Center).

This article contains supplemental Tables 1 and 2 and Figs. 1–4.
A. F. Odell, J. M. Askham, S. Ponnambalam, and M. Hollstein, manuscript in preparation.
- AA
- aristolochic acid
- IMCE
- integrase-mediated cassette exchange
- MEF
- murine embryonic fibroblast
- Hupki
- human p53 knock-in
- BMK
- baby platform mouse kidney
- TOP
- targeting on platform
- qRT-PCR
- quantitative RT-PCR.
REFERENCES
- 1. Oren M., Rotter V. (2010) Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2, a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vogelstein B., Lane D., Levine A. J. (2000) Surfing the p53 Network. Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- 3. Freed-Pastor W. A., Prives C. (2012) Mutant p53: one name, many proteins. Genes Dev. 26, 1268–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brosh R., Rotter V. (2009) When mutants gain new powers; news from the mutant p53 field. Nat. Rev. Cancer 9, 701–713 [DOI] [PubMed] [Google Scholar]
- 5. Kato S., Han S. Y., Liu W., Otsuka K., Shibata H., Kanamaru R., Ishioka C. (2003) Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. U.S.A. 100, 8424–8429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Petitjean A., Mathe E., Kato S., Ishioka C., Tavtigian S. V., Hainaut P., Olivier M. (2007) Impact of mutant p53 functional properties on TP53 mutation patterns and tumour phenotype: Lessons from recent developments in the IARC TP53 database. Human Mutation 28, 622–629 [DOI] [PubMed] [Google Scholar]
- 7. Greenman C., Stephens P., Smith R., Dalgliesh G. L., Hunter C., Bignell G., Davies H., Teague J., Butler A., Stevens C., Edkins S., O'Meara S., Vastrik I., Schmidt E. E., Avis T., Barthorpe S., Bhamra G., Buck G., Choudhury B., Clements J., Cole J., Dicks E., Forbes S., Gray K., Halliday K., Harrison R., Hills K., Hinton J., Jenkinson A., Jones D., Menzies A., Mironenko T., Perry J., Raine K., Richardson D., Shepherd R., Small A., Tofts C., Varian J., Webb T., West S., Widaa S., Yates A., Cahill D. P., Louis D. N., Goldstraw P., Nicholson A. G., Brasseur F., Looijenga L., Weber B. L., Chiew Y. E. (2007) Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hainaut P., Wiman K. (eds) (2006) 25 Years of p53 Research, Springer, Netherlands [Google Scholar]
- 9. Chen C. H., Dickman K. G., Moriya M., Zavadil J., Sidorenko V. S., Edwards K. L., Gnatenko D. V., Wu L., Turesky R. J., Wu X. R., Pu Y. S., Grollman A. P. (2012) Aristolochic acid-associated urothelial cancer in Taiwan. Proc. Natl. Acad. Sci. U.S.A. 109, 8241–8246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hollstein M., Moriya M., Grollman A. P., Olivier M. (2013) Analysis of TP53 mutation spectra reveals the fingerprint of the potent carcinogen, aristolochic acid. Mutat. Res. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olivier M., Hollstein M., Schmeiser H. H., Straif K., Wild C. P. (2012) Upper urinary tract urothelial cancers: where it's A:T. Nat. Rev. Cancer 12, 503–504 [DOI] [PubMed] [Google Scholar]
- 12. Schetter A. J., Harris C. C. (2012) Tumor suppressor p53 (TP53) at the crossroads of the exposome and the cancer genome. Proc. Natl. Acad. Sci. U.S.A. 109, 7955–7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wei Q. X., van der Hoeven F., Hollstein M., Odell A. (2012) Efficient introduction of specific p53 mutations into mouse embryonic fibroblasts and embryonic stem cells. Nat. Protoc. 7, 1145–1160 [DOI] [PubMed] [Google Scholar]
- 14. Wei Q. X., Odell A. F., van der Hoeven F., Hollstein M. (2011) Rapid derivation of genetically related mutants from embryonic cells harbouring a recombinase-specific Trp53 platform. Cell Cycle 10, 1261–1270 [DOI] [PubMed] [Google Scholar]
- 15. Mathew R., Degenhardt K., Haramaty L., Karp C. M., White E. (2008) Immortalized mouse epithelial cell models to study the role of apoptosis in cancer. Methods Enzymol. 446, 77–106 [DOI] [PubMed] [Google Scholar]
- 16. Whibley C., Odell A. F., Nedelko T., Balaburski G., Murphy M., Liu Z., Stevens L., Walker J. H., Routledge M., Hollstein M. (2010) Wild-type and Hupki (human p53 knock-in) murine embryonic fibroblatsts: p53/ARF pathway disruption in spontaneous escape from senescence. J. Biol. Chem. 285, 11326–11335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Joerger A. C., Fersht A. R. (2010) The tumour suppressor p53: from structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2, a000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luo J. L., Yang Q., Tong W. M., Hergenhahn M., Wang Z. Q., Hollstein M. (2001) Knock-in Hupki mice with a humanised p53 gene develop normally and show wild-type p53 responses to DNA damaging agents. Oncogene 20, 320–328 [DOI] [PubMed] [Google Scholar]
- 19. Song H., Hollstein M., Xu Y. (2007) P53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 9, 573–580 [DOI] [PubMed] [Google Scholar]
- 20. Hollstein M., Xu Y. (2013) Humanised mouse models: Targeting the murine p53 locus with human sequences. p53 in the Clinics (Hainaut P., Olivier M., Wiman K. G., eds), Vol. 2013, pp. 95–108, Springer, New York [Google Scholar]
- 21. Baroni T. E., Wang T., Qian H., Dearth L. R., Truong L. N., Zeng J., Denes A. E., Chen S. W., Brachmann R. K. (2004) A global suppressor motif for p53 cancer mutants. Proc. Natl. Acad. Sci. U.S.A. 101, 4930–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Otsuka K., Kato S., Kakudo Y., Mashiko S., Shibata H., Ishioka C. (2007) The screening of the second-site suppressor mutations of the common p53 mutants. Int. J. Cancer 121, 559–566 [DOI] [PubMed] [Google Scholar]
- 23. Danziger S. A., Baronio R., Ho L., Hall L., Salmon K., Hatfield G. W., Kaiser P., Lathrop R. H. (2009) Predicting positive p53 cancer rescue regions using Most Informative Positive (MIP) active learning. PLoS Comput. Biol. 5, e1000498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Merabet A., Houlleberghs H., Maclagan K., Akanho E., Bui T. T., Pagano B., Drake A. F., Fraternali F., Nikolova P. V. (2010) Mutants of the tumour suppressor p53 L1 loop as second-site suppressors for Restoring DNA binding to oncogenic p53 mutations: structural and biochemical insights. Biochem. J. 427, 225–236 [DOI] [PubMed] [Google Scholar]
- 25. Nedelko T., Arlt V. M., Phillips D. H., Hollstein M. (2009) TP53 mutation signature supports involvement of aristolochic acid in the aetiology of endemic nephropathy-associated tumours. Int. J. Cancer 124, 987–990 [DOI] [PubMed] [Google Scholar]
- 26. Liu Z., Hergenhahn M., Schmeiser H. H., Wogan G. N., Hong A., Hollstein M. (2004) Human tumour p53 mutations are selected for in mouse embryonic fibroblasts harbouring a humanized p53 gene. Proc. Natl. Acad. Sci. U.S.A. 101, 2963–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Besaratinia A., Pfeifer G. P. (2010) Applications of the human p53 knock-in (Hupki) mouse model for human carcinogen testing. FASEB J. 24, 2612–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Olivier M., Hollstein M., Hainaut P. (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2, a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nikolova P. V., Henckel J., Lane D. P., Fersht A. R. (1998) Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc. Natl. Acad. Sci. U.S.A. 95, 14675–14680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Joerger A. C., Ang H. C., Fersht A. R. (2006) Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. U.S.A. 103, 15056–15061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lai M. N., Wang S. M., Chen P. C., Chen Y. Y., Wang J. D. (2010) Population-based case-control study of Chinese herbal products containing aristolochic acid and urinary tract cancer risk. J Natl. Cancer Inst. 102, 179–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hussain S. P., Schwank J., Staib F., Wang X. W., Harris C. C. (2007) TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene 26, 2166–2176 [DOI] [PubMed] [Google Scholar]