

Background: DUF2233 domain is present in bacteria and human UCE, which is implicated in lysosomal storage disorders.
Results: Functional residues in DUF2233 and UCE identified in the structure of a bacterial DUF2233 domain were investigated.
Conclusion: A function for this domain in bacteria is proposed, and functional residues in human UCE were identified.
Significance: This is the first structure/function study of this protein domain.
Keywords: Crystal Structure, Functional Genomics, Genomics, Glycoprotein, Structural Biology, DUF2233, Structural Genomics, Uncovering Enzyme
Abstract
DUF2233, a domain of unknown function (DUF), is present in many bacterial and several viral proteins and was also identified in the mammalian transmembrane glycoprotein N-acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase (“uncovering enzyme” (UCE)). We report the crystal structure of BACOVA_00430, a 315-residue protein from the human gut bacterium Bacteroides ovatus that is the first structural representative of the DUF2233 protein family. A notable feature of this structure is the presence of a surface cavity that is populated by residues that are highly conserved across the entire family. The crystal structure was used to model the luminal portion of human UCE (hUCE), which is involved in targeting of lysosomal enzymes. Mutational analysis of several residues in a highly conserved surface cavity of hUCE revealed that they are essential for function. The bacterial enzyme (BACOVA_00430) has ∼1% of the catalytic activity of hUCE toward the substrate GlcNAc-P-mannose, the precursor of the Man-6-P lysosomal targeting signal. GlcNAc-1-P is a poor substrate for both enzymes. We conclude that, for at least a subset of proteins in this family, DUF2233 functions as a phosphodiester glycosidase.
Introduction
N-Acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase (uncovering enzyme (UCE)4; EC 3.1.4.45) converts N-acetylglucosamine-P-mannose diester to mannose-6-P monoester on newly synthesized lysosomal acid hydrolases, a key step in the targeting of these hydrolases to lysosomes (1). Disruption of the N-acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase (Nagpa) gene that encodes UCE leads to excessive cellular secretion of acid hydrolases (2), and mutations have recently been associated with persistent stuttering in humans (3). Despite the importance of UCE, very limited information is available concerning its structure.
UCE, a type 1 transmembrane glycoprotein localized in the trans-Golgi network (4, 5) is synthesized as a 515-residue preproenzyme and subsequently processed by furin, which removes the 49-residue propiece (6). Analysis of the domain architecture of mature UCE revealed that ∼50% of the luminal region of the protein (residues 130–325) is related to the domain of unknown function, DUF2233 (Pfam (7) protein family PF09992). This domain has been identified in proteins ranging in size from ∼300 to 2,000 residues, is present in 48 unique domain organizations and combinations, and has been identified in ∼1,200 bacterial proteins in addition to several viral and eukaryotic proteins.
We report the crystal structure of the first structural representative of the DUF2233 protein family, BACOVA_00430 from Bacteroides ovatus, at a resolution of 1.80 Å. Comparative sequence analysis of the bacterial members of this family covering a sequence identity range of 30–95% revealed several conserved residues located in a cleft on the surface of the BACOVA_00430 structure, indicating some involvement in its function. The BACOVA_00430 structure was used as a template for modeling the luminal region of human UCE (hUCE). Site-directed mutagenesis of hUCE based on this model confirmed the predicted functional importance of some of these conserved residues. Similar mutational analyses were performed on BACOVA_00430. These studies provide the first structure-function analysis of DUF2233 proteins.
EXPERIMENTAL PROCEDURES
Materials
UDP-6-[3H]GlcNAc (37Ci/mmol) was obtained from PerkinElmer Life Sciences. GalNAc, GlcNAc, GlcNAc-1-phosphate, ManNAc, UDP-GlcNAc, and p-nitrophenyl N-acetyl-α- and -β-d-glucosaminide and methyl α-d-mannopyranoside were purchased from Sigma. Monoclonal anti-HA antibody was obtained from Covance. Recombinant purified human UCE was generously provided by W. Canfield (Genzyme, Oklahoma City, OK). The HRP substrate used for the Western blot was Immobilon® and was obtained from Millipore (Billerica, MA).
Protein Production and Crystallization
Clones were generated using the polymerase incomplete primer extension (PIPE) cloning method (8). The gene encoding BACOVA_00430 was amplified by polymerase chain reaction (PCR) from B. ovatus ATCC 8483 genomic DNA using PfuTurbo DNA polymerase (Stratagene) and I-PIPE (insert) primers (forward primer, 5′-ctgtacttccagggcATGCCACAAACCGCCATAGGACGGC-3′; reverse primer, 5′-aattaagtcgcgttaCTTCTTTTCTATAATCAACATACTGTTG-3′ where the target sequence is in uppercase) that include sequences for the predicted 5′- and 3′-ends of the gene encoding the full-length protein. The expression vector, pSpeedET, which encodes an N-terminal tobacco etch virus protease-cleavable expression and purification tag (MGSDKIHHHHHHENLYFQ↓G), was PCR-amplified with V-PIPE (vector) primers (forward primer, 5′-taacgcgacttaattaactcgtttaaacggtctccagc-3′; reverse primer, 5′-gccctggaagtacaggttttcgtgatgatgatgatgatg-3′). V-PIPE and I-PIPE PCR products were mixed to anneal the amplified DNA fragments together. Escherichia coli GeneHogs (Invitrogen) competent cells were transformed with the I-PIPE/V-PIPE mixture and dispensed on selective LB agar plates. The cloning junctions were confirmed by DNA sequencing. Using the PIPE cloning method, the gene segment encoding residues Met1–Ala31 was deleted. Expression was performed in a selenomethionine-containing medium at 37 °C. Selenomethionine was incorporated via inhibition of methionine biosynthesis (9), which does not require a methionine auxotrophic strain. At the end of fermentation, lysozyme was added to the culture to a final concentration of 250 μg/ml, and the cells were harvested and frozen. After one freeze/thaw cycle, the cells were homogenized and sonicated in lysis buffer (50 mm HEPES, pH 8.0, 50 mm NaCl, 10 mm imidazole, and 1 mm tris(2-carboxyethyl)phosphine HCl (TCEP)). The lysate was clarified by centrifugation at 32,500 × g for 30 min. The soluble fraction was passed over nickel-chelating resin (GE Healthcare) pre-equilibrated with lysis buffer, the resin was washed with wash buffer (50 mm HEPES, pH 8.0, 300 mm NaCl, 40 mm imidazole, 10% (v/v) glycerol, and 1 mm TCEP), and the protein was eluted with elution buffer (20 mm HEPES, pH 8.0, 300 mm imidazole, 10% (v/v) glycerol, and 1 mm TCEP). The eluate was buffer-exchanged with tobacco etch virus buffer (20 mm HEPES, pH 8.0, 200 mm NaCl, 40 mm imidazole, and 1 mm TCEP) using a PD-10 column (GE Healthcare) and incubated with 1 mg of tobacco etch virus protease/15 mg of eluted protein for 2 h at ambient temperature followed by overnight incubation at 4 °C. The protease-treated eluate was passed over nickel-chelating resin (GE Healthcare) pre-equilibrated with HEPES crystallization buffer (20 mm HEPES, pH 8.0, 200 mm NaCl, 40 mm imidazole, and 1 mm TCEP), and the resin was washed with the same buffer. The flow-through and wash fractions were combined and concentrated to 21 mg/ml by centrifugal ultrafiltration (Millipore) for crystallization trials.
BACOVA_00430 was crystallized using the nanodroplet vapor diffusion method (10) with standard Joint Center for Structural Genomics crystallization protocols (11, 12). Sitting drops composed of 200 nl of protein solution mixed with 200 nl of crystallization solution in a sitting drop format were equilibrated against a 50-μl reservoir at 277 K for 15 days prior to harvest. The crystallization reagent consisted of 0.2 m Li2SO4, 30% PEG 4000, and 0.1 m Tris, pH 8.5. Ethylene glycol was added to a final concentration of 10% (v/v) as a cryoprotectant. Initial screening for diffraction and data collection was carried out using the Stanford automated mounting system (SAM) (13) at the Stanford Synchrotron Radiation Lightsource (Menlo Park, CA). Diffraction data were collected from a rod-shaped crystal of dimensions 200 × 20 × 20 μm and indexed in space group P61. The oligomeric state of BACOVA_00430 in solution was determined using size exclusion chromatography with a 1 × 30-cm2 Superdex 200 size exclusion column (GE Healthcare) coupled with miniDAWN (Wyatt Technology) static light scattering and Optilab differential refractive index detectors (Wyatt Technology). The mobile phase consisted of 20 mm Tris, pH 8.0, 150 mm NaCl, and 0.02% (w/v) sodium azide. The molecular weight was calculated using ASTRA 5.1.5 software (Wyatt Technology).
X-ray Data Collection, Structure Determination, and Refinement
Multiwavelength anomalous diffraction (MAD) data were collected at the Stanford Synchrotron Radiation Lightsource on beamline 9-2 at wavelengths corresponding to the high energy remote (λ1), inflection point (λ2), and peak (λ3) of a selenium MAD experiment using the Beamline User Integrated Control Environment (BLU-ICE) (14) data collection environment. The data sets were collected at 100 K using a MarMosaic 325 charge-coupled device detector (Rayonix). The MAD data were integrated and reduced using MOSFLM (15) and scaled with the program SCALA (16). The heavy atom substructure was determined with SHELXD (17). Phasing was performed with autoSHARP (18), SOLOMON (19) (implemented in autoSHARP) was used for density modification, and ARP/wARP (20) was used for automatic model building to 1.80 Å resolution. Model completion and crystallographic refinement were performed with the λ1 data set using Coot (21) and REFMAC5 (22). The refinement protocol included the experimental phase restraints in the form of Hendrickson-Lattman coefficients from autoSHARP and TLS refinement with one TLS group for the whole molecule. Data and refinement statistics are summarized in Table 1 (23–26).
TABLE 1.
Data collection and refinement statistics for BACOVA_00430 (Protein Data Bank code 3ohg)
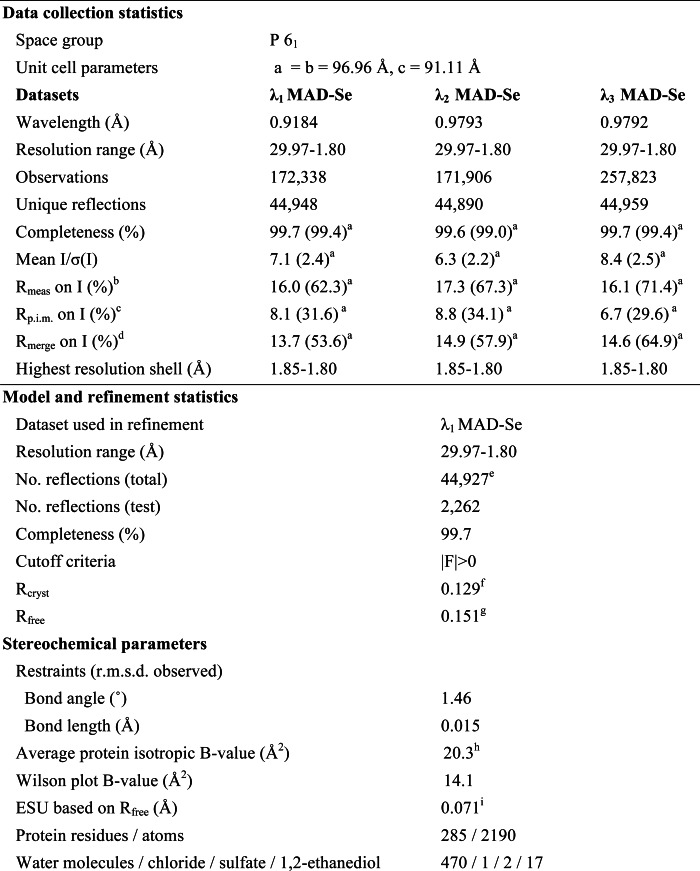
a Highest resolution shell.
b Rmeas (redundancy-independent Rmerge) = Σhkl(n/(n − 1))1/2 Σ|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl) (24), where n is the number of observations of a given reflection.
c Rp.i.m. (precision-indicating Rmerge) = Σhkl((1/(n − 1))1/2 Σi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl) (25, 26).
d Rmerge = ΣhklΣi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl).
e Typically, the number of unique reflections used in refinement is slightly less than the total number that were integrated and scaled. Reflections are excluded due to negative intensities and rounding errors in the resolution limits and cell parameters.
f Rcryst = Σ‖Fobs| − |Fcalc‖/Σ |Fobs|, where Fcalc and Fobs are the calculated and observed structure factor amplitudes, respectively.
g Rfree as for Rcryst, but for 5.0% of the total reflections chosen at random and omitted from refinement.
h This value represents the total B that includes overall TLS refinement and residual B components.
i ESU, estimated overall coordinate error (23).
Validation and Deposition
The quality of the crystal structure was analyzed using the Joint Center for Structural Genomics Quality Control server. This server verifies the stereochemical quality of the model using AutoDepInputTool (27), MolProbity (28), and WHATIF 5.0 (29); agreement between the atomic model and the data using SFcheck 4.0 (30) and RESOLVE (31); the protein sequence using ClustalW (32); atom occupancies using MOLEMAN2 (33); and the consistency of non-crystallographic symmetry pairs and evaluates difference in Rcryst/Rfree, expected Rfree/Rcryst, and maximum/minimum B-factors by parsing the refinement log file and Protein Data Bank header. Protein quaternary structure analysis was performed using the PISA server (34). Fig. 1B was adapted from an analysis using PDBsum (35), and Figs. 1A, 2, and 3A were prepared with PyMOL (36). Atomic coordinates and experimental structure factors were deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ under accession code 3ohg.
FIGURE 1.

Crystal structure of BACOVA_00430. A, stereo ribbon diagram of the DUF2233 protein BACOVA_00430 colored in yellow, orange, cyan, and green by domain from the N terminus to C terminus. The cysteine side chains and bound sulfate molecules from the crystallization reagent are shown as yellow sticks. B, diagram showing the secondary structure elements of BACOVA_00430 superimposed on its primary sequence adapted from PDBsum. α-Helices and 310-helices are sequentially labeled H1, H2, H3, etc.; β-strands are labeled β1, β2, β3, etc.; β- and γ-turns are labeled β and γ; and β-hairpins are indicated by red loops.
FIGURE 2.

Residue conservation analysis. A residue conservation analysis was performed using ConSurf (54–56) using the MAFFT (57) alignment program, the UniProt database (58) (UniProt release November 2010), and 31 unique sequences with 30–95% sequence identity range (search range) identified in one iteration of PSI-BLAST with an expectation value cutoff of 0.001. The protein with the highest sequence identity was F7M5I1_9BACE (from Bacteroides sp. 1_1_30) at 55% (over the full-length protein; expectation value = 1e−86), and the sequence identities of the other hits were ∼27–45% (including hits to shorter segments from other proteins, resulting in higher sequence identity value; expectation values ranged from 1e−24 to 9e−15). A, the most highly conserved residues are shown in stick representation. The view is rotated ∼90° anticlockwise relative to Fig. 1A. B, a putative functional site lined with the most highly conserved residues is visible on one surface of the protein (the views are rotated 180° along the vertical axis, and the left panel has approximately the same orientation as A). The surface is colored based on the conservation scale ranging from magenta (highest conservation) to cyan (most variable).
FIGURE 3.

Structural model of the human UCE. A, three-dimensional model of the hUCE color-coded and oriented similarly to the BACOVA_00430 x-ray structure in Fig. 1. Of the four disulfide bonds predicted from an earlier mass spectrometry study, the model contains one disulfide bond between Cys132-Cys323, but Cys307/Cys314 and Cys115/Cys148 are relatively close to each other (yellow sticks). The other two free Cys residues that could potentially form a disulfide, Cys51 and Cys221, are quite far apart. Cys221 is in a 13-residue insert (residues 217–229) in the model of hUCE compared with BACOVA_00430; the conservation of two residues in this loop and the loss of activity when mutated suggest that this loop might actually be closer to the putative active site (where it could form a disulfide and move functionally important residues toward the active site region) than what is modeled here. The solvent-exposed asparagine residues that are predicted to be glycosylated based on mass spectrometry analysis are shown as red sticks (Asn208, Asn214, and Asn296). The side chains of the conserved residues that represent the potential functional site are depicted as blue sticks. Residues Arg328 and His84, whose mutations (R328C and H84Q) have been associated with stuttering, are shown as magenta sticks. B, structure-based sequence alignment of BACOVA_00430 and hUCE based on superimposing the crystal structure and the model using DaliLite (44), which resulted in a Z-score of ∼31 and r.m.s.d of 1.7 Å over 252/285 Cα residues with a sequence identity of 17%. Residues in the strictly conserved A(I/L)NLDGGGS(T/S/A)T motif in the DUF2233 family are noted by a black bar. The figure was prepared using the BOXSHADE server with identical residues shown as white letters on a black background and similar residues shown as black letters on a gray background.
Cell Lines and Human UCE and BACOVA_00430 Constructs
HeLa cells were obtained from the ATCC. The cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 100 μg/ml penicillin, and 100 units/ml streptomycin. For mutational analysis, hUCE cDNA (4) was modified by addition of a C-terminal HA tag (YPYDVPDYA) and subcloned into the pcDNA3.1(−) expression vector (Invitrogen) using EcoRI and HindIII restriction sites. BACOVA_00430 cDNA was obtained from the Protein Structure Initiative Biology Materials Repository/DNASU (37) (Clone ID BoCD00384454). All mutations in the hUCE construct and the G288A mutation of BACOVA_00430 construct were introduced using QuikChange (Stratagene) site-directed mutagenesis protocols. For the H225Q and R227T double mutation BACOVA_00430 construct, the fragment of BACOVA between the EcoO109I site (264th base) and SpeI restriction site (612th base) was substituted with a fragment encoding Gln225 and Thr227 using the following primers (forward primer, 5′-GCA ACA GGA CCT GAA TCT AGT GCG CCT CTC CCG-3′; reverse primer, 5′-CAC ACT AGT CGG TTG GGT ATT CTG CAA ATC-3′).
BACOVA Protein Expression and Purification
Wild-type (WT) and mutant BACOVA constructs in DH5α were transformed into E. coli BL21 for the production of BACOVA protein. One milliliter of preculture was inoculated into 100 ml of LB containing 30 μg/ml kanamycin. At an A600 nm of around 1.0, 0.1% l-(+)-arabinose was added, and the induction proceeded overnight. The cells were centrifuged, and the pellet was resuspended in 2 ml of lysis buffer composed of 50 mm NaH2PO4, pH 8.0, 300 mm NaCl, 10 mm Imidazole, and 0.5% Triton X-100. After sonication and centrifugation at 29,000 × g for 10 min, the supernatant was incubated with 1 ml of nickel-nitrilotriacetic acid-agarose (Qiagen) at 4 °C for 1 h. The beads were washed three times with 1 ml of wash buffer containing 50 mm NaH2PO4, pH 8.0, 300 mm NaCl, and 20 mm imidazole and eluted with 0.7 ml of elution buffer composed of 50 mm NaH2PO4, pH 8.0, 300 mm NaCl, and 250 mm imidazole. The eluate was dialyzed against Milli-Q water. The yield was 4–7 mg of purified protein.
Enzyme Assays
UCE assays were performed as described previously (38) using 0.41 mm [3H]GlcNAc-P-α-Me-Man (2,510 cpm/nmol). Assays using 3 mm p-nitrophenyl N-acetyl-α- or -β-d-glucosaminide were carried out in 50 mm citrate buffer, pH 4.5 and 0.5% Triton X-100 or 50 mm Tris maleate, pH 7.0 and 0.5% Triton X-100. The reactions were terminated by the addition of 200 μl of 0.2 m Na2CO3, and the absorbance at 410 nm was measured. Inorganic phosphate release from GlcNAc-1-P was quantitated by the method of Lowry and Lopez as outlined by Leloir and Cardini (39).
Transfection, PNGase F Digestion, and Western Blot Analysis
HeLa cells cultured in 6-well plates at 37 °C for 20 h (95% confluent) were transfected with 3.1 μg of DNA using 7.7 μl of Lipofectamine 2000 (Invitrogen). At 24 h post-transfection, the cells were solubilized with a buffer containing 0.1 m Tris, pH 8.0, 150 mm NaCl, 1% Triton X-100, and protease inhibitor mixture (Complete®, Roche Applied Science). Ten micrograms of transfected HeLa cell lysates were treated with 1,000 units of PNGase F overnight at 37 °C. Treated and control samples were subjected to 12% SDS-PAGE and Western blot analysis using monoclonal anti-HA antibody.
RESULTS
Structure of BACOVA_00430
The cloning, expression, purification, and crystallization of BACOVA_00430 was carried out using standard Joint Center for Structural Genomics protocols as detailed under “Experimental Procedures.” The crystal structure of BACOVA_00430 was determined by MAD phasing to 1.80-Å resolution. Data collection, model, and refinement statistics are summarized in Table 1 (23–26). BACOVA_00430 is present as a monomer in the crystallographic asymmetric unit. Crystal packing analysis and analytical size exclusion chromatography support a monomer as the predominant oligomerization state in solution. The final model (Fig. 1) includes Gly0 (which remains after cleavage of the expression and purification tag), residues 32–315 of the full-length protein (the predicted lipoprotein signal peptide, residues 1–31, was excluded from the protein construct), one chloride ion from the purification buffer, two sulfate ions from the crystallization reagent, 17 1,2-ethanediol molecules from the cryoprotectant, and 470 water molecules. The Matthews' coefficient (40) is ∼4.0 Å3/Da with an estimated solvent content of ∼70%. The Ramachandran plot produced by MolProbity (28) shows that 96.8% of the residues are in favored regions with none in the disallowed regions.
BACOVA_00430 consist of four domains, each of which bears some resemblance to the cystatin fold (SCOP code 54402) (41), which consists of a curved antiparallel β-sheet wrapped around an α-helix (Fig. 1). The first domain, constituted by H1, β1, β2, and β3, resembles more closely the prototypical cystatin-like fold and is not included in the DUF2233 definition, which covers only domains 2–4 (residues 123–312) of BACOVA_00430. Interestingly, the C terminus of the bacterial protein reaches over from domain 4 and inserts its tail into domain 2 and probably serves to stabilize the DUF2233 portion of the protein. Of the four domains, only domains 3 and 4 (residues 131–194 and 195–268, respectively) can be superimposed onto each other (r.m.s.d. of 2.2 Å over 41 Cα atoms and 22% sequence identity), suggesting possible gene duplication in this portion of the protein.
Structural Comparisons
A search for other proteins of similar structure was carried out using FATCAT (42) (flexible alignment mode) against the SCOP database (43) and DALI (44). When queried using the entire BACOVA_00430 structure, FATCAT returned only two hits with significant p value scores (<0.05): human latexin (Protein Data Bank code 2bo9; p = 0.0286; Cα r.m.s.d., ∼3 Å; sequence identity, ∼3%) and a protein of unknown function, YpmB, from Bacillus subtilis (Protein Data Bank code 2gu3; p = 0.04; Cα r.m.s.d., ∼2 Å; sequence identity, ∼3%). However, in both cases, the coverage is restricted to domain 1 of BACOVA_00430 because latexin and YpmB are both α and β (α + β) proteins belonging to the cystatin-like fold (and cystatin/monellin superfamily). No significant hits were found by FATCAT when the search was restricted to the DUF2233 domain. Similar results were obtained with a DALI search for the full BACOVA_00430 structure; all structural similarities were again limited to the N-terminal domain, which most closely resembles the prototypical cystatin-like fold. Thus, BACOVA_00430 is the first structural representative of the novel DUF2233 domain architecture.
Sequence Analysis and Putative Functional Site
Analysis of sequence conservation in the structure (Fig. 2) of 31 unique DUF2233 family proteins (27–55% sequence identity; closest PSI-BLAST hits to BACOVA_00430) indicated the identity and location of residues that are likely functionally important. The most conserved residues are Asn130, His217, Arg219, Arg239, Asn268, Asp270, Gly271, Gly272, Gly273, Ser274, Arg303, and Val305. These residues are clustered on one side of the protein, and almost all have significant surface exposure. Of these, Asn268, Asp270, Gly271–273, and Ser274 are part of the highly conserved A(I/L)NLDGGGS(T/S/A)T motif present throughout the DUF2233 family and located in helix H7 and in the preceding loop between β17 and H7 near the center of the conserved site. Interestingly, a sulfate ion is bound near Gly272-Gly273 of the GGGS sequence and anchored by conserved residues Arg239 and Arg303 and may represent the binding site for the phosphate moiety of the substrate. Asn130, Asp270, and Ser274 are potential catalytic residues (see proposed function and nature of putative substrates under “Discussion”).
Modeling Human UCE
Mammalian UCEs are highly conserved type 1 transmembrane glycoproteins of ∼515 residues with ∼85% sequence identity. hUCE consists of a signal peptide (residues 1–25), a propeptide (residues 26–49), a luminal region (residues 50–448), a single TM region (residues 449–469), and a cytoplasmic tail (residues 470–515) (4). A sequence profile-based search for homologs of the entire luminal portion of hUCE (residues 50–448) using FFAS (45), which is very useful for finding remote homologs, identified a single significant hit to our BACOVA_00430 structure with a score of −39.3 (scores <−9.5 indicate less than 3% false positives) and 14% sequence identity between residues 50–298 of hUCE (comprising ∼62% of the luminal region) and residues 36–282 of BACOVA_00430. When only those residues of hUCE that are included in the DUF2233-like domain definition (luminal residues 130–325) are queried using FFAS, only one significant hit (score −58.7) is again found with ∼20% sequence identity to residues 123–311 of BACOVA_00430. These results indicated that we could confidently use BACOVA_00430 as a template for modeling the corresponding region of hUCE. A model for hUCE residues 50–335, which accounts for ∼70% of the Golgi luminal region, was built using I-TASSER (46) using explicit disulfide restraints between Cys51-Cys221, Cys115-Cys148, Cys132-Cys323, and Cys307-Cys314 (numbering is based on the hUCE sequence), corresponding to potential disulfide bonds identified in an earlier mass spectrometry study of a monomeric soluble construct of hUCE (47). The C-score of this final model (energy-minimized and optimized hydrogen-bonding contacts by I-TASSER) was −0.13 (C-scores usually range from −5 to 2, and a higher C-score indicates higher confidence), and the TM-score for estimated accuracy of the model (48) was 0.70 ± 0.12 (a TM-score higher than 0.5 indicates correct topology of the model) with an r.m.s.d. of 6.4 ± 3.9 Å compared with the template. I-TASSER using disulfide restraints produced the most complete model, although several other procedures were tested including Modeler, M4T, Swiss-Model, and HHPred (using the Protein Structure Initiative Protein Model Portal), which produced only partial models.
Despite attempts to use the disulfide restraints, the I-TASSER model does not contain all four expected disulfide bonds. The model (Fig. 3) contains a disulfide bond between Cys132-Cys323, and Cys307/Cys314 and Cys115/Cys148 are relatively close to each other (∼15 and 11 Å between Cα atoms, respectively). Arg328 and His84 when mutated to Cys and Gln, respectively, are associated with persistent stuttering, and their location is visualized in the model. The major consequence of these mutations is impaired folding in the endoplasmic reticulum (ER) followed by degradation by the ER-associated protein degradation system (49). Thus, based on our model, we speculate that the impaired folding induced by these mutations could be a result of destabilization of the β-sheet in which His84 resides as well as the potential to affect proper disulfide formation. In addition, three of the four N-linked glycosylation sites found by mass spectrometry in hUCE (Asn208, Asn214, Asn296, and Asn366) are solvent-exposed in our model. The conservation of disulfide and potential glycosylation sites indicates that the registry of the alignment used for modeling is likely correct. However, because of remote homology between the template and hUCE, it is expected that the accuracy may be low in some regions of the model. Nevertheless, the model provides the first three-dimensional view of the putative functional site of UCE and helped guide the mutational analysis.
Mutational Analysis of Human UCE
Next, we tested the consequences of mutating a number of the residues located in or near the surface cavity identified in the hUCE model that appears to be a putative active site. Most of these residues are conserved across the DUF2233 family (Asn137, Gln225, Thr227, Arg247, Asn284, Asp286, Gly287, Gly288, Gly289, Ser290, Thr320, and Val322) (Fig. 3). It was essential to take into account that hUCE is synthesized as an inactive preproenzyme that forms a tetramer in the ER and then traffics to the trans-Golgi network where the propiece is cleaved by furin to generate the active enzyme (6). Consequently, it was necessary to determine whether or not the mutant proteins could exit the ER and be processed by furin to ensure the mutation could be correlated with UCE activity.
HeLa cells transfected with plasmids encoding full-length WT or mutant hUCE containing a C-terminal HA tag were harvested, solubilized with Triton X-100, and analyzed to evaluate the effects of the mutations on cellular localization and activity of the proteins. First, aliquots were incubated with or without PNGase F to excise the N-linked glycans (high mannose and complex oligosaccharides) and then subjected to SDS-PAGE and Western blotting with anti-HA antibody. With two exceptions, N281A and V318A mutants, the untreated samples gave rise to two bands; the faster migrating band represents the ER form with high mannose glycans, and the slower migrating band represents the Golgi species with complex glycans (49) (Fig. 4). Following PNGase F treatment, the Golgi species migrated faster than the ER form, reflecting cleavage of the 24-residue propiece by furin in the trans-Golgi network in addition to the removal of the glycans. Evidence that the designation of these bands is correct is shown by the N281A and V318A mutants, which only exhibit the ER forms, with a single faster migrating band in the untreated samples and a single slower migrating band following PNGase F treatment. Exiting the ER seems to have been partially impaired for the G287A and G289A mutants, whereas all the other mutants trafficked to the Golgi and underwent furin cleavage similarly to the WT hUCE.
FIGURE 4.

Effect of mutations on UCE maturation. Ten micrograms of transfected HeLa cell lysates were treated with denaturing buffer containing 0.5% SDS. The samples were then incubated with 1,000 units of PNGase F (+) or without (−) overnight at 37 °C followed by SDS-PAGE and Western blot analysis. Each mutant was assayed on two occasions with the same result.
The remaining extract was used for hUCE activity measurements as summarized in Table 2. Among the residues in the most highly conserved patch, mutation of Asp286, Gly288, Gly289, and Ser290 to Ala resulted in the complete loss of hUCE activity with either no or only partial impairment of trafficking to the Golgi and furin cleavage. The G287A mutant exhibited about 16% of WT hUCE activity when taking into account its partial impairment in trafficking to the Golgi, whereas the N284A mutant had only 22% of WT activity.
TABLE 2.
Mutational analysis of UCE
| Mutation | Relative UCE activitya | Traffic to Golgib |
|---|---|---|
| WT | 100 | Yes |
| N137A | 11.1 ± 1.2 | Yes |
| N281A | ND | No |
| N284A | 22.0 ± 4.6 | Yes |
| D286A | 0.2 ± 0.4 | Yes |
| G287A | 9.8 ± 1.0 | ∼60% WT |
| G288A | 0.2 ± 0.4 | Yes |
| G289A | 0.4 ± 0.3 | ∼50% WT |
| S290A | 0.6 ± 0.7 | Yes |
| Q225H | 5.8 ± 0.3 | Yes |
| T227R | 0.1 ± 0.1 | Yes |
| R247A | 87.0 ± 1.4 | Yes |
| V318A | ND | No |
| T320A | 43.0 ± 7.8 | Yes |
| V322A | 67.0 ± 2.1 | Yes |
a The values represent units of enzyme activity divided by the intensity of the band of mature UCE determined by Western blot analysis with WT set to 100 and uniform sample loading. The values are the average of two to four determinations, each done in duplicate. ND, not detected, indicating failure of protein to traffic to the Golgi and undergo furin cleavage.
b Transport to the Golgi is equivalent to WT as determined by Western blot.
Among the other mutants, the N137A, R247A, T320A, and V322A mutants exhibited 11, 87, 43, and 67% of WT activity, respectively. The effect of the N281A and V318A mutations on hUCE activity could not be explored because these constructs were retained in the ER. Interestingly, when Gln225 and Thr227 were mutated to the residues at equivalent positions in BACOVA_00430 (His and Arg, respectively), the hUCE activity was greatly decreased to 5.8 and 0.1%, respectively, relative to WT.
Mutation of Cys51 to Met had only a small effect on hUCE trafficking and activity (65% of WT), indicating that the Cys51-Cys221 disulfide bond is not absolutely essential for folding or enzyme activity (Fig. 4 and Table 3). The double mutant C51M/C221L also folded adequately and trafficked to the Golgi where it was cleaved by furin, but it had only 9.7 ± 1.0% of WT activity. This result indicates that the C221L mutation leads to loss of enzymatic activity. By contrast, mutation of Cys115 and Cys132 greatly impaired folding of the enzyme as reflected by retention in the ER.
TABLE 3.
Effect of disulfide bond disruption on UCE activity
| Relative UCE activitya | Traffic to Golgib | |
|---|---|---|
| WT | 100 | Yes |
| C51M | 64.7 ± 5.2 | Yes |
| C51M/C221L | 9.7 ± 1.0 | Yes |
| C115S | 15.4 ± 1.6 | Trace |
| C115S/C148V | ND | No |
| C132V | ND | No |
| C132V/C323L | ND | No |
a The values represent units of enzyme activity divided by the density of the band of mature UCE determined by Western blot analysis with WT set to 100. The values are the average of three determinations, each done in duplicate. ND not detected, indicating failure of protein to traffic to the Golgi and undergo furin cleavage.
b “Yes” means transport to the Golgi is equivalent to WT as analyzed by Western blot.
BACOVA_00430 Exhibits Low Activity toward GlcNAc-P-Man
The BACOVA_00430 protein was able to cleave GlcNAc from GlcNAc-P-Man but did so at a much slower rate than hUCE. Kinetic studies showed that its Vmax toward this substrate was only 1% of the UCE value, whereas the apparent Km for GlcNAc-P-Man was 10.9 versus 0.64 mm for hUCE (Table 4). Both proteins had much lower activity toward GlcNAc-1-P, and neither exhibited any activity toward p-nitrophenyl N-acetyl-α- and -β-d-glucosaminide substrates. This result is consistent with our previous finding that hUCE has a strong preference for substrates with the underlying phosphate in a diester linkage (50). To establish that the activity of the BACOVA_00430 protein toward these substrates was not the consequence of a contaminant in the preparation, a G288A BACOVA_00430 mutant was prepared and found to be completely inactive in this assay. Because the hUCE Q225H and T227R mutants were inactive, we tested the possibility that these residues may be important for increased activity toward GlcNAc-P-Man. However, mutating His225 and Arg227 of BACOVA_00430 to the corresponding residues in hUCE (Gln and Thr, respectively) also resulted in a complete loss of enzyme activity.
TABLE 4.
Activity of UCE and BACOVA_00430 toward GlcNAc-P-mannose and GlcNAc-1-P
The kinetic analyses were carried out in 50 mm Tris maleate, pH 6.7, 0.5% Triton X-100 buffer containing various concentrations of substrates in a final volume of 30 μl. The reactions contained 3 μg of hUCE and 9 μg of BACOVA_00430 for the GlcNAc-P-mannose assays and 0.5 μg of UCE and 30 μg of BACOVA_00430 for the GlcNAc-1-P assays. The values for GlcNAc-P-Man are the average of two determinations, and the values for GlcNAc-1-P are the average of five determinations.
| Substrates |
Km apparent |
Vmax |
||
|---|---|---|---|---|
| hUCE | BACOVA_00430 | hUCE | BACOVA_00430 | |
| mm | μmol/h/mg | |||
| GlcNAc-P-Man | 0.64 | 10.9 | 1,400 | 14 |
| GlcNAc-1-P | 2.5 | 5.7 | 100 | 0.77 |
The effects of a number of sugars, sugar phosphates, nucleotide sugars, and inorganic Pi as inhibitors of the activity of BACOVA_00430 toward GlcNAc-P-Man were also investigated. All of the sugar phosphates as well as UDP-GlcNAc and inorganic Pi were effective inhibitors, whereas the other nucleotide sugars and the simple sugars were very weak inhibitors (Fig. 5). This pattern differs from that observed for hUCE where GlcNAc-1-P is a more potent inhibitor than the other sugar phosphates, GlcNAc is a relatively good inhibitor, and inorganic phosphate is a weaker inhibitor. This pattern of inhibition of hUCE agrees with that reported previously for a partially purified preparation of rat liver UCE (51).
FIGURE 5.

Effect of various compounds on BACOVA_00430 and hUCE catalytic activity. The assays contained 0.41 mm [3H]GlcNAc-P-Man and the indicated concentration of the various compounds. Results are expressed as the percentage of control reactions lacking the respective compounds. *, not tested. The values are the average of two to three determinations.
DISCUSSION
Elucidation of the crystal structure of BACOVA_00430 provides the first structural insights into proteins that contain DUF2233 domains. By focusing on the pattern and location of amino acids that are conserved within the DUF2233 family, we identified a surface cavity that is likely functionally important. Remarkably, among all known mammalian proteins, only one possesses the DUF2233 domain, namely UCE, a phosphodiester α-N-acetylglucosaminidase that catalyzes a critical step in the generation of the Man-6-P recognition signal on lysosomal acid hydrolases. The structure of BACOVA_00430 was used to model the catalytic domain of hUCE. This in turn provided a template for a structure-based mutational analysis and established that the highly conserved residues in the UCE surface cavity are essential for the catalytic function of the enzyme toward GlcNAc-P-Man.
The BACOVA_00430 protein exhibits only weak activity toward the GlcNAc-P-Man substrate. Kinetic analyses revealed that the apparent Km for this substrate is about 17 times higher than the corresponding value for hUCE, and the Vmax is only 1% of the value for hUCE. The finding that GlcNAc-1-P inhibited the enzymatic activity of BACOVA_00430 much more strongly than did GlcNAc points to a preference for the phosphorylated form of this aminosugar. This conclusion was confirmed by the fact that neither BACOVA_00430 nor hUCE has any detectable activity toward p-nitrophenyl N-acetyl-α- or -β-d-glucosaminide. However, both proteins exhibited poor activity toward GlcNAc-1-P, indicating that they function as phosphodiester glycosidases. Whereas BACOVA_00430 is inhibited equally by a variety of sugar phosphates, GlcNAc-1-P inhibits UCE much more than other sugar phosphates, consistent with having evolved to specifically recognize and act on GlcNAc-P-mannose. At this point, the physiological substrate(s) of BACOVA_00430 is unknown. In this regard, a number of bacterial cell wall components with sugar-P-sugar repeating structures could potentially be substrates for BACOVA_00430 and related bacterial proteins (52, 53), and the sulfate from the crystallization reagents that is near Gly272-Gly273 and Arg239/Arg303 may mimic the phosphate from the physiologically relevant substrate. Our results strongly hint at this possibility.
Acknowledgments
We thank the members of the Joint Center for Structural Genomics high throughput structural biology pipeline for contributions to this work. Genomic DNA from B. ovatus (ATCC number 8483) was extracted from cells (ATCC number 8483T) obtained from the American Type Culture Collection. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a directorate of the SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the United States Department of Energy Office of Science by Stanford University. The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the United States Department of Energy Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including Grant P41GM103393) and the National Center for Research Resources (Grant P41RR001209).
This work was supported, in whole or in part, by National Institutes of Health Grants R21DC011332 (to S. K.) and U54 GM094586 from the NIGMS Protein Structure Initiative (to the Joint Center for Structural Genomics). This work was carried out as part of the Protein Structure Initiative Biology Community Nominated Targets program.
The atomic coordinates and structure factors (code 3ohg) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- UCE
- uncovering enzyme
- hUCE
- human UCE
- DUF
- domain of unknown function
- BACOVA
- B. ovatus
- Man-6-P
- mannose 6-phosphate
- GlcNAc-1-P
- N-acetylglucosamine 1-phosphate
- MAD
- multiwavelength anomalous diffraction
- FATCAT
- flexible structure alignment by chaining aligned fragment pairs allowing twists
- SCOP
- Structural Classification of Proteins
- r.m.s.d.
- root mean square deviation
- TM
- transmembrane
- ER
- endoplasmic reticulum
- HeLa
- Henrietta Lacks
- PNGase F
- peptide:N-glycosidase F
- PIPE
- polymerase incomplete primer extension
- TLS
- translation/libration/screw
- ManNAc
- N-acetylmannosamine
- TCEP
- tris(2-carboxyethyl)phosphine HCl.
REFERENCES
- 1. Braulke T., Bonifacino J. S. (2009) Sorting of lysosomal proteins. Biochim. Biophys. Acta 1793, 605–614 [DOI] [PubMed] [Google Scholar]
- 2. Boonen M., Vogel P., Platt K. A., Dahms N., Kornfeld S. (2009) Mice lacking mannose 6-phosphate uncovering enzyme activity have a milder phenotype than mice deficient for N-acetylglucosamine-1-phosphotransferase activity. Mol. Biol. Cell 20, 4381–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kang C., Riazuddin S., Mundorff J., Krasnewich D., Friedman P., Mullikin J. C., Drayna D. (2010) Mutations in the lysosomal enzyme-targeting pathway and persistent stuttering. N. Engl. J. Med. 362, 677–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kornfeld R., Bao M., Brewer K., Noll C., Canfield W. (1999) Molecular cloning and functional expression of two splice forms of human N-acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase. J. Biol. Chem. 274, 32778–32785 [DOI] [PubMed] [Google Scholar]
- 5. Rohrer J., Kornfeld R. (2001) Lysosomal hydrolase mannose 6-phosphate uncovering enzyme resides in the trans-Golgi network. Mol. Biol. Cell 12, 1623–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Do H., Lee W. S., Ghosh P., Hollowell T., Canfield W., Kornfeld S. (2002) Human mannose 6-phosphate-uncovering enzyme is synthesized as a proenzyme that is activated by the endoprotease furin. J. Biol. Chem. 277, 29737–29744 [DOI] [PubMed] [Google Scholar]
- 7. Finn R. D., Tate J., Mistry J., Coggill P. C., Sammut S. J., Hotz H. R., Ceric G., Forslund K., Eddy S. R., Sonnhammer E. L., Bateman A. (2008) The Pfam protein families database. Nucleic Acids Res. 36, D281–D288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klock H. E., Koesema E. J., Knuth M. W., Lesley S. A. (2008) Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins. 71, 982–994 [DOI] [PubMed] [Google Scholar]
- 9. Van Duyne G. D., Standaert R. F., Karplus P. A., Schreiber S. L., Clardy J. (1993) Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229, 105–124 [DOI] [PubMed] [Google Scholar]
- 10. Santarsiero B. D., Yegian D. T., Lee C. C., Spraggon G., Gu J., Scheibe D., Uber D. C., Cornell E. W., Nordmeyer R. A., Kolbe W. F., Jin J., Jones A. L., Jaklevic J. M., Schultz P. G., Stevens R. C. (2002) An approach to rapid protein crystallization using nanodroplets. J. Appl. Crystallogr. 35, 278–281 [Google Scholar]
- 11. Elsliger M. A., Deacon A. M., Godzik A., Lesley S. A., Wooley J., Wüthrich K., Wilson I. A. (2010) The JCSG high-throughput structural biology pipeline. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 1137–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lesley S. A., Kuhn P., Godzik A., Deacon A. M., Mathews I., Kreusch A., Spraggon G., Klock H. E., McMullan D., Shin T., Vincent J., Robb A., Brinen L. S., Miller M. D., McPhillips T. M., Miller M. A., Scheibe D., Canaves J. M., Guda C., Jaroszewski L., Selby T. L., Elsliger M. A., Wooley J., Taylor S. S., Hodgson K. O., Wilson I. A., Schultz P. G., Stevens R. C. (2002) Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. Proc. Natl. Acad. Sci. U.S.A. 99, 11664–11669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cohen A. E., Ellis P. J., Miller M. D., Deacon A. M., Phizackerley R. P. (2002) An automated system to mount cryo-cooled protein crystals on a synchrotron beamline, using compact sample cassettes and a small-scale robot. J. Appl. Crystallogr. 35, 720–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McPhillips T. M., McPhillips S. E., Chiu H. J., Cohen A. E., Deacon A. M., Ellis P. J., Garman E., Gonzalez A., Sauter N. K., Phizackerley R. P., Soltis S. M., Kuhn P. (2002) Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 9, 401–406 [DOI] [PubMed] [Google Scholar]
- 15. Leslie A. G. W., Powell H. R. (2007) Processing diffraction data with mosflm. In Evolving Methods for Macromolecular Crystallography (Read R. J., Sussman J. L., eds) NATO Science Series, Volume 245, pp. 41–51, Springer, Dordrecht, The Netherlands [Google Scholar]
- 16. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 17. Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A 64, 112–122 [DOI] [PubMed] [Google Scholar]
- 18. Vonrhein C., Blanc E., Roversi P., Bricogne G. (2007) Automated structure solution with autoSHARP. Methods Mol. Biol. 364, 215–230 [DOI] [PubMed] [Google Scholar]
- 19. Abrahams J. P., Leslie A. G. (1996) Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D Biol. Crystallogr. 52, 30–42 [DOI] [PubMed] [Google Scholar]
- 20. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 22. Winn M. D., Murshudov G. N., Papiz M. Z. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 [DOI] [PubMed] [Google Scholar]
- 23. Cruickshank D. W. (1999) Remarks about protein structure precision. Acta Crystallogr. D Biol. Crystallogr. 55, 583–601 [DOI] [PubMed] [Google Scholar]
- 24. Diederichs K., Karplus P. A. (1997) Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 4, 269–275 [DOI] [PubMed] [Google Scholar]
- 25. Weiss M. S., Hilgenfeld R. (1997) On the use of the merging R factor as a quality indicator for x-ray data. J. Appl. Crystallogr. 30, 203–205 [Google Scholar]
- 26. Weiss M. S., Metzner H. J., Hilgenfeld R. (1998) Two non-proline cis peptide bonds may be important for factor XIII function. FEBS Lett. 423, 291–296 [DOI] [PubMed] [Google Scholar]
- 27. Yang H., Guranovic V., Dutta S., Feng Z., Berman H. M., Westbrook J. D. (2004) Automated and accurate deposition of structures solved by x-ray diffraction to the Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 60, 1833–1839 [DOI] [PubMed] [Google Scholar]
- 28. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vriend G. (1990) WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 8, 52–56, 29 [DOI] [PubMed] [Google Scholar]
- 30. Vaguine A. A., Richelle J., Wodak S. J. (1999) SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. D Biol. Crystallogr. 55, 191–205 [DOI] [PubMed] [Google Scholar]
- 31. Terwilliger T. C. (2000) Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thompson J. D., Higgins D. G., Gibson T. J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kleywegt G. J. (2000) Validation of protein crystal structures. Acta Crystallogr. D Biol. Crystallogr. 56, 249–265 [DOI] [PubMed] [Google Scholar]
- 34. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 35. Laskowski R. A., Chistyakov V. V., Thornton J. M. (2005) PDBsum more: new summaries and analyses of the known 3D structures of proteins and nucleic acids. Nucleic Acids Res. 33, D266–D268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. DeLano W. L. (2008) The PyMOL Molecular Graphics System, Schrödinger, LLC, New York [Google Scholar]
- 37. Cormier C. Y., Park J. G., Fiacco M., Steel J., Hunter P., Kramer J., Singla R., LaBaer J. (2011) PSI:Biology-materials repository: a biologist's resource for protein expression plasmids. J. Struct. Funct. Genomics. 12, 55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mullis K. G., Ketcham C. M. (1992) The synthesis of substrates and two assays for the detection of N-acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase (uncovering enzyme). Anal. Biochem. 205, 200–207 [DOI] [PubMed] [Google Scholar]
- 39. Leloir L. F., Cardini C. E. (1957) Characterization of phosphorus compounds by acid lability. Methods Enzymol. 3, 840–850 [Google Scholar]
- 40. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 41. Murzin A. G., Brenner S. E., Hubbard T., Chothia C. (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247, 536–540 [DOI] [PubMed] [Google Scholar]
- 42. Ye Y., Godzik A. (2003) Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics 19, Suppl. 2, ii246–ii255 [DOI] [PubMed] [Google Scholar]
- 43. Andreeva A., Howorth D., Chandonia J. M., Brenner S. E., Hubbard T. J., Chothia C., Murzin A. G. (2008) Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 36, D419–D425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Holm L., Kääriäinen S., Rosenström P., Schenkel A. (2008) Searching protein structure databases with DaliLite v.3. Bioinformatics 24, 2780–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jaroszewski L., Rychlewski L., Li Z., Li W., Godzik A. (2005) FFAS03: a server for profile-profile sequence alignments. Nucleic Acids Res. 33, W284–W288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wei Y., Yen T. Y., Cai J., Trent J. O., Pierce W. M., Young W. W., Jr. (2005) Structural features of the lysosomal hydrolase mannose 6-phosphate uncovering enzyme. Glycoconj. J. 22, 13–19 [DOI] [PubMed] [Google Scholar]
- 48. Zhang Y., Skolnick J. (2004) Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710 [DOI] [PubMed] [Google Scholar]
- 49. Lee W. S., Kang C., Drayna D., Kornfeld S. (2011) Analysis of mannose 6-phosphate uncovering enzyme mutations associated with persistent stuttering. J. Biol. Chem. 286, 39786–39793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Varki A., Sherman W., Kornfeld S. (1983) Demonstration of the enzymatic mechanisms of α-N-acetyl-d-glucosamine-1-phosphodiester N-acetylglucosaminidase (formerly called α-N-acetylglucosaminylphosphodiesterase) and lysosomal α-N-acetylglucosaminidase. Arch. Biochem. Biophys. 222, 145–149 [DOI] [PubMed] [Google Scholar]
- 51. Varki A., Kornfeld S. (1981) Purification and characterization of rat liver α-N-acetylglucosaminyl phosphodiesterase. J. Biol. Chem. 256, 9937–9943 [PubMed] [Google Scholar]
- 52. Swartley J. S., Liu L. J., Miller Y. K., Martin L. E., Edupuganti S., Stephens D. S. (1998) Characterization of the gene cassette required for biosynthesis of the (α1→6)-linked N-acetyl-d-mannosamine-1-phosphate capsule of serogroup A Neisseria meningitidis. J. Bacteriol. 180, 1533–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tzeng Y. L., Noble C., Stephens D. S. (2003) Genetic basis for biosynthesis of the (α1→4)-linked N-acetyl-d-glucosamine 1-phosphate capsule of Neisseria meningitidis serogroup X. Infect. Immun. 71, 6712–6720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, (suppl.) W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., Ben-Tal N. (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33, W299–W302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Glaser F., Pupko T., Paz I., Bell R. E., Bechor-Shental D., Martz E., Ben-Tal N. (2003) ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19, 163–164 [DOI] [PubMed] [Google Scholar]
- 57. Katoh K., Misawa K., Kuma K., Miyata T. (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. UniProt Consortium (2010) The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 38, D142–D148 [DOI] [PMC free article] [PubMed] [Google Scholar]