

Background: The type IIIe CohE-XDoc interaction connects cellulosomal components to the cell wall.
Results: The dockerin structure in the CohE-XDoc complex exhibits an atypical calcium-binding loop disrupted by a 13-residue insert.
Conclusion: The dockerin inserts evolved to serve as novel structural buttresses that support the stalklike X-module conformation.
Significance: The type IIIe CohE-XDoc complex underscores dockerin divergence and provides insight into the determinants for cohesin-dockerin specificity.
Keywords: Biofuel, Calcium-binding Proteins, Carbohydrate-binding Protein, Cellulase, X-ray Crystallography, Multienzyme Complex, Ruminococcus flavefaciens, Cellulose Degradation, Lignocellulosic Biomass, Modular Proteins
Abstract
The rumen bacterium Ruminococcus flavefaciens produces a highly organized multienzyme cellulosome complex that plays a key role in the degradation of plant cell wall polysaccharides, notably cellulose. The R. flavefaciens cellulosomal system is anchored to the bacterial cell wall through a relatively small ScaE scaffoldin subunit, which bears a single type IIIe cohesin responsible for the attachment of two major dockerin-containing scaffoldin proteins, ScaB and the cellulose-binding protein CttA. Although ScaB recruits the catalytic machinery onto the complex, CttA mediates attachment of the bacterial substrate via its two putative carbohydrate-binding modules. In an effort to understand the structural basis for assembly and cell surface attachment of the cellulosome in R. flavefaciens, we determined the crystal structure of the high affinity complex (Kd = 20.83 nm) between the cohesin module of ScaE (CohE) and its cognate X-dockerin (XDoc) modular dyad from CttA at 1.97-Å resolution. The structure reveals an atypical calcium-binding loop containing a 13-residue insert. The results further pinpoint two charged specificity-related residues on the surface of the cohesin module that are responsible for specific versus promiscuous cross-strain binding of the dockerin module. In addition, a combined functional role for the three enigmatic dockerin inserts was established whereby these extraneous segments serve as structural buttresses that reinforce the stalklike conformation of the X-module, thus segregating its tethered complement of cellulosomal components from the cell surface. The novel structure of the RfCohE-XDoc complex sheds light on divergent dockerin structure and function and provides insight into the specificity features of the type IIIe cohesin-dockerin interaction.
Introduction
The specific high affinity cohesin-dockerin (Coh-Doc)3 interaction is a fundamental component of cellulosome assembly. This interaction dictates the interconnection between the different cellulosomal subunits to form a functional and particularly efficient exocellular multienzyme cellulose-degrading complex. Extensive structural and biophysical characterization of type I and type II Coh-Doc interactions has revealed the determinants dictating cellulosome assembly in Clostridium thermocellum and Clostridium cellulolyticum (1–4). The growing demand for renewable sources of bioenergy (5) has prompted an increase in studies on the mechanism of cellulosome action (2, 6–9) and approaches for improvement of its overall performance (10, 11). Extensive genetic and genome sequencing investigations have promoted the study of cellulase systems of rumen bacteria, in particular Ruminococcus flavefaciens, because of its predominant fiber-degrading activity in the rumen, the intricate nature of its cellulosomal architecture, and its industrial potential (12–16).
The cellulosome system of R. flavefaciens is currently the only confirmed example of a cellulosome organization in gut bacteria. This rumen bacterium was discovered to bear one of the most elaborate complexes in the cellulosomal world; it is much more elaborate than those reported previously for cellulolytic Clostridium species (17, 18). Gene clustering analysis of several R. flavefaciens strains revealed that the cellulosome system is generally composed of four major scaffoldin subunits, ScaA, ScaB, ScaC, and ScaE, and of a cellulose-binding protein, CttA (19, 20). The cohesin, dockerin, and X-modules of these cellulosomal components were found to be divergent in sequence from previously known type I and type II cellulosomal modules, and the Coh-Doc interactions were therefore collectively designated type III on the respective phylogenetic trees (21–24).
The cellulosome complex of R. flavefaciens strain FD-1 is anchored to the bacterial cell wall via the interaction between the primary/adaptor scaffoldin ScaB and the anchoring scaffoldin ScaE (Fig. 1). ScaE possesses a C-terminal Gram-positive LPXTG-like motif, a site for proteolytic cleavage involved in covalent binding of the scaffoldin to the bacterial cell wall via a sortase-mediated attachment mechanism (25). Likewise, the CttA protein is also attached separately to the bacterium via ScaE. In both cases, the single cohesin module of ScaE (CohE) binds to a conserved X-dockerin (XDoc) modular dyad located at the C termini of both ScaB and CttA. Consecutive Coh-Doc interactions among ScaB, ScaA, and ScaC regulate the assembly of the different enzymatic units onto the complex. ScaE thus fulfills a key role in the assembly and function of the greater cellulosome system in R. flavefaciens, and its interactions with the dockerins are hereby classified as type IIIe.
FIGURE 1.

Schematic overview of the proposed modular interactions in the R. flavefaciens FD-1 cellulosome system. A, the dockerins of ScaC and the enzyme equivalent of Cel44A can both bind ScaA cohesins and ScaB cohesins 1–4. B, another enzyme, Ce3B, interacts with the cohesin of ScaC. C, the ScaA dockerin interacts with cohesins 5–9 of ScaB. D, the bimodular XDoc dyad of ScaB binds the cohesin of the anchoring scaffoldin ScaE. E, the cellulose-binding protein CttA bears CBMs and a C-terminal XDoc dyad that is also bound to CohE. The complex of RfCohE-XDoc is marked with a red circle.
Through its interactions with ScaB and CttA, ScaE facilitates the proximity between the bacterial enzymatic machinery and the insoluble substrate, thereby enhancing the potential for synergy among the different sets of catalytic enzymes. The XDoc modular dyads of ScaB and CttA share high sequence homology (47% identity over a 222-residue stretch) and comprise an X-module of unknown function and an unconventional dockerin sequence that includes three similarly conserved insertions unique to this type of protein; one of these insertions disrupts the putative second calcium (Ca2+)-binding site (24).
To date, only the single type IIIe CohE crystal structure from R. flavefaciens strain 17 has been reported (26, 27). A planar region at the 8-3-6-5 face of the molecule bordered by β-flap 8 has been proposed to play a role in type IIIe dockerin recognition and specificity. However, to fully determine the structural elements that dictate the type IIIe interaction, a crystal structure of the type IIIe Coh-Doc complex is essential.
Here we report the 1.97-Å resolution crystal structure of the complex between the CohE and the XDoc modular dyad of CttA from the cellulosome system of R. flavefaciens strain FD-1 (RfCohE-XDoc). The structure of the RfCohE-XDoc complex reveals that the dockerin exhibits an atypical Ca2+-binding site due to several sequence alterations and implantation of a 13-residue insert in the midst of the Ca2+-binding loop. The crystal structure of the complex provides molecular insight into the specific versus promiscuous binding properties of the CohEs from strains 17 and FD-1 toward their respective dockerins. A structural role for the dockerin inserts was established as providing structural “buttresses” for the elongated stalklike conformation of the X-module, which maintains the external modular elements of the parent protein at an adequate distance from the cell surface.
EXPERIMENTAL PROCEDURES
Cloning
A different set of cloning procedures was designed for each purpose, i.e. for the ELISA binding/affinity assay and for the crystallization protocol. For ELISA, the CohE modules were fused to the family 3a carbohydrate-binding module (CBM3a) from the C. thermocellum scaffoldin that exhibits strong binding to crystalline cellulose matrices, whereas complementary dockerins (together with the adjacent X-module) were fused to the high expression enzyme xylanase T6 of Geobacillus stearothermophilus (Xyn) (28). For crystallization purposes, the cohesins and XDoc derivatives were cloned and expressed on their own without an adjacent protein. A full list of the primers used in this study is provided in supplemental Table 1.
Cloning, Protein Expression, and Purification for Isothermal Titration Calorimetry/Differential Scanning Calorimetry Experiments
The gene fragments encoding the CohE module from the scaE scaffoldin gene of R. flavefaciens and the XDoc dyad from the CttA scaffoldin gene of R. flavefaciens strain FD-1 were produced as described previously (29) with minor modification where the resulting XDoc gene fusion encoded an N-terminal His6 tag to be independently purified. The resultant plasmids were separately transferred to Escherichia coli strain BL21(DE3). Induction was carried out at 37 °C for 4 h with 0.5 mm isopropyl 1-thio-β-d-galactoside.
Cells expressing either CohE or XDoc were harvested by centrifugation, and cell pellets were frozen at −20 °C. Frozen cell pellets were disrupted by sonication (4 × 30 s with 10-s intervals) on ice in 20 mm HEPES, pH 7.0, 200 mm NaCl (Buffer A). The suspension was clarified for 20 min at 48,000 × g using a Beckman JA25.5 rotor. The pellet was then resuspended in Buffer B (Buffer A supplemented with 8 m urea) and left to rock overnight to solubilize the inclusion bodies. Non-solubilized matter was removed by centrifugation, and the clarified supernatant was loaded onto a Ni2+-Sepharose column pre-equilibrated in Buffer B. The column was washed with Buffer B containing 40 mm imidazole and eluted with 500 mm imidazole. The denatured proteins were dialyzed into 20 mm Tris, pH 7.5, 50 mm NaCl, 5 mm CaCl2 (Buffer C). The refolded proteins were concentrated using an Amicon Centricon with a 3-kDa cutoff and loaded onto a Superdex 75 size exclusion column (GE Healthcare) equilibrated in Buffer C. Protein samples were prepared in 20 mm Tris, pH 7.5, 20 mm NaCl, 5 mm CaCl2 for subsequent use in differential scanning and isothermal titration calorimetry-based experiments.
Cloning, Protein Expression, and Purification for ELISA Experiments
The gene encoding for CohE from R. flavefaciens strains FD-1 and 17 was amplified from the genomic DNA of each bacterium and inserted into the BamHI and XhoI restriction sites of the pET28a-CBM3a vector described previously by Barak et al. (28). Cloning of the CBM-CohE double homology-swapped constructs was achieved with two rounds of mutations using standard restriction-free methods. The method included a “one-step” PCR procedure that amplifies the entire plasmid using phosphorylated primers that included the sequence of mutations (supplemental Table 1). Next, the original (methylated) template was digested with DpnI. The resultant plasmids were purified and ligated and then transformed to XL-1-Blue cells.
The genes encoding for the XDoc of CttA and ScaB from R. flavefaciens strains FD-1 and 17 were amplified from the genomic DNA of each bacterium. The resultant PCR products were separately inserted into the KpnI and BamHI restriction sites of the pET9D-Xyn vector (28). Expression and purification of the resultant Xyn-XDoc and CBM-CohE constructs were performed as described previously by Barak et al. (28) with the exception that CBM-CohE fusion proteins were purified using a nickel-nitrilotriacetic acid column similarly to the purification procedure of the Xyn-XDoc fusion proteins.
Crystallization and Structure Determination
Cloning, protein expression, co-purification, and crystallization of the trimodular RfCohE-XDoc complex as well as x-ray data collection and processing were described previously (29). The crystal structure of the type IIIe RfCohE-XDoc complex was determined by single wavelength anomalous diffraction using the anomalous data measured from a single selenomethionyl (SeMet) crystal. The SHELXC/D/E pipeline (30) was used for the determination of the selenium substructure followed by phasing, electron density modification, and initial tracing of the complex structure as a polyalanine string. The initial structure was rebuilt using ARP/wARP (31) and refined to a final model with PHENIX (32). Root mean square deviation (r.m.s.d.) values between the different cohesin modules were calculated using DaliLite (33). Consequently, the crystal structure of the native RfCohE-XDoc was refined starting with the final coordinates of the SeMet derivative.
Isothermal Titration and Differential Scanning Calorimetry
CohE and XDoc protein solutions were prepared in 20 mm Tris-HCl, pH 7.5, 20 mm NaCl, 5 mm CaCl2,filtered, and degassed. Isothermal titration calorimetry-based experiments were performed using a MicroCal VP-ITC calorimeter (GE Healthcare) at 30 °C. Two protein solutions containing 90 μm XDoc and 9.1 μm CohE were extensively degassed and loaded into the titration syringe and reaction cell, respectively. 30 injections of 10 μl were made with 15 min of equilibration between injections. Integration of the thermograms after correction for heats of dilution yielded a binding isotherm that fit best to a one-site binding model using Origin 7.0 software (MicroCal).
The heat capacity (Cp) measurements of 6.7 μm CohE, 8.7 μm XDoc, and 6.7 μm CohE in the presence of 6.7 μm XDoc were taken from 20 to 110 °C with a scan rate of 60 °C/h on a MicroCal VP-DSC differential scanning calorimeter (MicroCal). The final denaturation thermogram profiles were corrected using reference buffer scans, normalized for concentration, and fitted to a non-two-state denaturation model to determine corresponding thermodynamic parameters associated with the folding transition in Origin 7.0 software (MicroCal).
Affinity-based ELISA Protocols
Analysis of dockerin binding using immobilized cohesins was preformed similarly to the method described previously (28). ELISA data were analyzed using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA). Dockerin binding to the double-swapped cohesin modules was standardized according to the binding of the wild-type cohesin. Data were fitted to a non-linear regression curve-sigmoidal dose-response curve according to Equation 1.
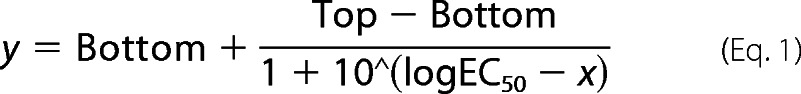 |
where Bottom is the minimal level of observed binding and Top is the maximum level of observed binding.
I-TASSER Modeling
The model of the XDoc dyad of CttA from R. flavefaciens strain 17 was predicted using the I-TASSER online server (34). The server used the coordinates of the XDoc dyad of CttA from the crystal structure of RfCohE-XDoc solved in this work. The best predicted model was evaluated by a C-score of −1.35 and TM-score of 0.55 ± 0.15 r.m.s.d.
Model Minimization
The model for the cognate XDoc17 was predicted using the I-TASSER server relying on the coordinates of the homologue XDocFD-1 from the complex. The predicted structure of R. flavefaciens strain 17 (CohE17) with XDoc17 was merged using the VMD software package (35). The psfgen module in VMD was used to generate a protein structure file. Subsequently, the structure was solvated in a water box (water model, TIP3P) with a solvent padding distance of at least 6 Å and ionized using sodium (Na+) and chloride (Cl−) counterions at a concentration of 0.15 mol/liter to achieve charge neutrality. The system was subjected to minimization for 50,000 steps using the conjugate gradient method. Five different minimization cycles were performed, and the average structure was used for further analysis. The NAMD program (36) and CHARMM27 force field (37) were used for molecular dynamics simulations.
Accession Codes
The structure factor and atomic coordinates of the native form and SeMet derivative of the type IIIe RfCohE-XDoc complex have been deposited in the Protein Data Bank under accession codes 4IU3 and 4IU2, respectively.
RESULTS
Biophysical Characterization of the RfCohE-XDoc Interaction
Thermodynamic analysis of the RfCohE-XDoc complex revealed a stoichiometric interaction between the two components with a dissociation constant (Kd) of 20.83 nm (supplemental Fig. 1A), which is comparable with that reported for the type I Coh-Doc interaction (2) but weaker than that reported for the type II interaction (3). Previous studies have shown that Coh-Doc interactions are highly thermostable (3). Indeed, analysis of the RfCohE-XDoc interaction by differential scanning calorimetry supported this finding whereby Tm values of 53.7 and 62.8 °C were obtained for the isolated CohE and XDoc constructs, respectively. Moreover, the RfCohE-XDoc complex exhibited an elevated Tm value of 86.8 °C (supplemental Fig. 1B), consistent with the elevated values for Coh-Doc interactions in general. Nonetheless, the particularly high Tm value associated with RfCohE-XDoc complex formation is somewhat surprising in view of the mesophilic properties of the bacterium.
Crystal Structure of the Type IIIe RfCohE-XDoc Complex
The crystal structure of the SeMet derivative of the type IIIe RfCohE-XDoc complex containing five selenium atoms was determined by single wavelength anomalous diffraction using the anomalous data measured on synchrotron. The structure of the native form of the RfCohE-XDoc complex was determined using the model of the SeMet derivative. The crystals of both the SeMet derivative and native RfCohE-XDoc were isomorphous and belonged to the tetragonal Bravais lattice of space group P43212, which contained eight symmetry operators producing eight molecules of the RfCohE-XDoc complex in the unit cell. The asymmetric unit of the unit cell comprised a single copy of the heterodimeric complex, which was oriented along the tetragonal axis with an inclination of about 18°. The final models were refined with PHENIX (32) to convergence. Except for differences between the SeMet and Met amino acid residues, the structures were similar with an r.m.s.d. of 0.273 Å. The model for the SeMet derivative was chosen as the final model. The type IIIe RfCohE-XDoc complex displays an elongated shape with overall dimensions of 32 × 40 × 113 Å and includes residues 30–230 from CohE and residues 565–803 from the XDoc dyad of the CttA scaffoldin from R. flavefaciens strain FD-1 (Fig. 2). Of note is the unusually protracted stalklike shape of the X-module. The RfCohE-XDoc complex was surrounded by six neighboring complexes in the unit cell, only three of which make intimate contacts with the X-module, thus stabilizing its position in the crystal lattice (supplemental Fig. 2). Crystal parameters and data collection statistics for the native and the SeMet models are summarized in Table 1.
FIGURE 2.

Structure of the type IIIe RfCohE-XDoc complex from R. flavefaciens FD-1. A, the type IIIe cohesin (in green) folds into an elongated nine-stranded β-sandwich in a classical jellyroll topology with a prominent helix between strands 8 and 9 enveloped by an extensive loop at the N terminus of the module. The dockerin (in red), which is composed of five α-helices, binds along the entire length of both its α1 and α3 helices to the 8-3-6-5 face of the cohesin module. The type IIIe dockerin contains two calcium-binding sites. Calcium ions bound to the dockerin and cohesin are shown as magenta spheres. The elongated stalklike type IIIe X-module (in blue) is composed of five β-strands. Hydrophobic (B), hydrogen-bonding (C), and electrostatic-interacting (D) residues at the interface of the type IIIe RfCohE-XDoc complex are shown. Residues involved in the Coh-Doc binding surface are labeled and shown as green (cohesin) and magenta (dockerin) sticks. Hydrogen bonds are shown as black dashed lines.
TABLE 1.
Data collection and refinement statistics for the native and selenomethionyl CohE-XDoc crystals
Data for the highest resolution shell are given in parentheses. ESRF, European Synchrotron Radiation Facility; r.m.s., root mean square.
| Data collection | ||
| Protein Data Bank accession code | 4IU3 | 4IU2 |
| X-ray source | ESRF, BM-14 | ESRF, BM-14 |
| Wavelength (Å) | 0.97833 (anomalous dispersion peak) | 0.97841 |
| Space group | P43212 | P43212 |
| Unit cell parameters (Å) | a = b = 78.53, c = 202.81 | a = b = 78.73, c = 203.24 |
| Resolution (Å) | 20-1.97 (1.99-1.97) | 48.8-2.00 (2.03-2.00) |
| Mosaicity (°) | 0.4-0.41 | 0.32-0.41 |
| Data processing | ||
| No. of measured reflections | 284,650 | 481,008 |
| No. of unique reflections | 46,047 | 44,177 |
| Completeness (%) | 98.5 (97.6) | 100 (100) |
| Redundancy | 6.3 (6.4) | 10.9 (11.0) |
| Rmergea | 0.107 (0.856) | 0.137 (0.948) |
| Mean I/σ(I) | 16.4 (2.0) | 17.1 (2.6) |
| Wilson B-factor (Å2) | 24.27 | 21.69 |
| Model refinement | ||
| Resolution range (Å) | 19.64–1.97 | 48.8–2.06 |
| No. atoms | 3,454 | 3,465 |
| No. water molecules | 580 | 568 |
| Overall average B-factor (Å2) | 29.9 | 28.6 |
| Average B-factor (protein) (Å2) | 24.9 | 26.6 |
| Average B-factor (solvent) (Å2) | 36.7 | 38.8 |
| Rwork | 0.170 | 0.153 |
| Rfree | 0.201 | 0.196 |
| Geometry | ||
| r.m.s. deviations, bond lengths (Å) | 0.007 | 0.009 |
| r.m.s. deviations, bond angles (°) | 1.023 | 0.945 |
| r.m.s. planar groups (Å) | 0.006 | 0.004 |
| Estimated coordinate error (Å) | 0.52 | 0.49 |
| Ramachandran favored (%) | 96.53 | 98.08 |
| Ramachandran allowed (%) | 2.98 | 1.68 |
| Ramachandran outliers (%) | 0.50 | 0.24 |
a Rmerge = ΣhklΣi|Ii(hkl) − I(hkl)|/ΣhklΣiIi(hkl) where Σhkl denotes the sum over all reflections and Σi is the sum over all equivalent and symmetry-related reflections.
CohE Structure in the Complex
The type IIIe CohE module in RfCohE-XDoc complex forms a nine-stranded β-sandwich in the classical jellyroll topology with an extensive hydrophobic core (Fig. 2, in green). The two faces of the β-sandwich comprise strands 8, 3, 6, and 5 and strands 9, 1, 2, 7, and 4, typical of cohesin modules (38). β-Strands 1–8 are aligned in an antiparallel arrangement, whereas β-strands 1 and 9 are aligned parallel to each other. The prominent α-helix between β-strands 8 and 9 and the two β-flaps that disrupt the normal course of strands 4 and 8 are also maintained in CohE structure. The structure of the CohE in the complex is essentially analogous to that of the isolated type IIIe CohE module from CohE17 with which it shares 45% sequence identity (27). Both CohEs also share the distinctive and extensive N-terminal loop formed by the first 24 amino acids that provides several hydrophobic contacts and hydrogen bonds with residues located on β-strands 3 and 8, β-flap 8, and the prominent α-helix. Additionally, CohE from the complex comprises an additional loop connecting the prominent α-helix with β-strand 9. This 17-residue loop contains a short 6-residue α-helix and a Ca2+-binding site where the Ca2+ ion is coordinated in a typical pentagonal bipyramid configuration. The remote location of this loop relative to the dockerin-binding site appears to preclude the possibility of a binding role for this loop, but it may act as a stabilizing element for the N-terminal loop and the 1-9-8 face of the molecule.
A structure similarity search using DALI (33) revealed that the CohE structure presented here and that from R. flavefaciens strain 17 (Protein Data Bank code 2ZF9) are indeed most similar to each other with an r.m.s.d. of 1.23 Å over 170 residues on Cα positions (Fig. 3). The main difference between the two CohE structures is the conformation of β-flap 8. In the complex structure of RfCohE-XDoc, β-flap 8 in CohE is diverted away from the occupied binding site by ∼10 Å with respect to the orientation of β-flap 8 from the uncomplexed CohE of strain 17.
FIGURE 3.

Structure-based alignment of type IIIe cohesin modules. Superposition of type III CohEFD-1 from the RfCohE-XDoc complex colored green with CohE17 (Protein Data Bank code 2ZF9) colored cyan is shown. A, front view of the alignment. B, 90° rotation about the x axis of A. β-Strands 8, 3, 6, and 5 are indicated by number. The position of the protruding β-flap 8 is noted.
CttA XDoc Dyad Structure in the Complex
The C-terminal R. flavefaciens FD-1 CttA XDoc modular dyad comprises an X-module of unknown function and a type IIIe dockerin module. The X-module in the RfCohE-XDoc (residues 1–119) exhibits a protracted five-β-strand topology aligned in antiparallel arrangement with two short α-helical regions connecting strands 1 and 2 and strands 3 and 4 (Fig. 2, in blue). The X-module lacks a true hydrophobic core but displays a small hydrophobic region concentrated at the bottom of the module adjacent to the dockerin module.
The structure of the CttA dockerin module (residues 120–237) is particularly elaborate compared with previously obtained dockerin structures. The type IIIe dockerin is composed of five α-helices in contrast to the three helices hitherto observed in the type I (1) and type II (3) dockerin folds (Fig. 4). Two α-helices (α1 and α3) are arranged in antiparallel orientation, forming a planar surface on one face of the type IIIe dockerin that interacts with CohE. These helices are reminiscent of the type I and type II dockerins and comprise portions of two repeating segments, each of which contains a bound Ca2+ ion in loops located at opposite ends of the dockerin module (Fig. 2, in red). However, the second repeated segment, which contains α3, appears to represent an unconventional variation of the F-hand motif composed of an atypical loop-helix motif that will be discussed further below.
FIGURE 4.

Comparison of type I, type II, and type IIIe dockerin modules. A, the WT type I dockerin from the type I Coh-DocWT complex (Protein Data Bank code 1OHZ). B, the S45A/T46A mutant type I dockerin module (Protein Data Bank code 2CCL). C, the type II dockerin from the type II Coh-Doc complex (Protein Data Bank code 2B59). D, the type IIIe dockerin from the type IIIe RfCohE-XDoc complex. The dockerins are aligned according to superposition of the their respective cognate cohesin. The α-helices of each module are numbered. The calcium ions in each module are shown as magenta spheres and numbered according to the first and second Ca2+-binding loops. The N and C termini of each module are labeled.
The orientation of the type IIIe dockerin relative to CohE in the complex is markedly different from those of the type I and type II dockerins. Previous studies on the type I Coh-Doc interaction, which assembles enzymes onto the main scaffoldin subunit of the C. thermocellum and C. cellulolyticum cellulosome, showed that the repetition of segments of the type I dockerin sequence bears functional consequence (2). In this context, a helix 3-disrupted dockerin will bind to its cohesin in 180° rotation relative to a helix 1-disrupted dockerin (Fig. 4, A and B, respectively). The type I dockerins thus exhibit a dual mode of binding with their corresponding cohesin modules. The dual binding mode hypothesis has been further supported experimentally in additional work by Currie et al. (39).
In contrast to the type I interaction, the complex of the type II Coh-Doc (3), which is responsible for the attachment of the cellulosome of C. thermocellum to the surface of the bacterium, showed that both dockerin helices interact along their entire length with the cohesin, forming an interface of multiple contacts. The lack of internal sequence symmetry of the type II dockerin interface residues suggests that it is unlikely that the interaction of type II Coh-Doc will exhibit a dual binding mechanism (Fig. 4C).
The dockerin in the type IIIe complex (Fig. 4D), however, appears in a ∼190°-rotated position relative to that of the type I complex where the N terminus of the type IIIe dockerin fits into a groove bordered by β-flap 8, β-strand 8b, and β-strand 3 (Fig. 2A). This orientation is thus more similar to that of the mutated (S45A/T46A) type I dockerin (Fig. 4B) from the C. thermocellum complex (Protein Data Bank code 2CCL) (2) and the native type I dockerin from the ternary complex (Protein Data Bank code 4FL4) (39). However, where helix 1 of the type I mutated dockerin dominates cohesin recognition in Protein Data Bank code 2CCL, the type IIIe dockerin interacts with its cognate CohE through both helices 1 and 3 where helix 3 and the loop connecting helices 3 and 4 dominate the interaction.
Type IIIe CohE-XDoc Complex Interface
The interface of the CohE module with the CttA XDoc dyad comprises portions of the N-terminal loop of CohE, its 8-3-6-5 “front” face, and the loop leading into the prominent α-helix located between β-strands 8 and 9 together with the planar surface of the CttA dockerin created by both helices (α1 and α3) of the traditional Ca2+-binding motif as well as the loop connecting helices α3 and α4 (Fig. 2A). Several hydrophobic residues contribute to complex formation (Fig. 2B). The interface also includes an extensive network of hydrogen-bonding contacts (Fig. 2C) and ionic interactions that also serve to stabilize the complex interface (Fig. 2D). A detailed list of the interacting residues is presented in Table 2.
TABLE 2.
Interacting residues in the RfCohE-XDoc complex and predicted interacting residues in the R. flavefaciens strain 17 CohE-XDoc putative complex interface
Type-IIIe conserved cohesin/dockerin residues are shown in bold. Backbone hydrogen-bonded interacting residues are shown in italics.
| Cohesin location | CohEFD1 residue | XDocFD1 residue | CohE17 residue | Predicted XDoc17 residue |
|---|---|---|---|---|
| Hydrophobic interactions | ||||
| Strand 5 | Leu-101 | Ala-203, Ala-133, Leu-207, Leu-137 | Leu-91 | Ala-199, Ala-130, Leu-203, Leu-134 |
| Strand 6 | Phe-112a | Tyr-210,a Val-215 | Phe-103a | Tyr-206,a Asn-211 |
| Strand 6 | Ala-114 | Tyr-210, Leu-207 | Ala-105 | Tyr-206 |
| Strand 8 | Leu-159 | Tyr-210 | Leu-150 | Tyr-206 |
| Hydrogen-bonding | ||||
| N′ loop | NH 1/2 Arg-15 | Oδ1/2 Asp-216 | Gln-8 | Ser-212 |
| Strand 3 | Oγ1 Thr-68 | Oγ1 Thr-134 | Ala-58 | Ser-131 |
| Strand 3 | Nϵ2 His-72 | O Ser-214 | His-62 | Met-210 |
| Strand 5 | Oϵ1 Glu-105, Oϵ2 Glu-105 | Oγ1 Thr-211, Nζ Lys-212 | Glu-95 | Ala-207, Lys-208 |
| Strand 5 | Oδ1 Asn-107 | N Asp-216 | Asp-97 | Ser-212 |
| Flap 8 | Nζ Lys-156 | O Ser-213 | Asn-147 | Arg-208 |
| Flap 8 | O Gly-157 | OH Tyr-210 | Glu-148 | Tyr-206 |
| Loop 8-helix 9 | Nδ2 Asn-163 | Oγ1 Thr-138 | Val-154 | Ala-135 |
| Loop 8-helix 9 | Oδ2 Asp-165 | NH 1 Arg-131 | Lys-156 | Val-128 |
| Ionic interactions | ||||
| N′ loop | Arg-15b | Asp-216b | Gln-8 | Ser-212 |
| Strand 5 | Glu-105b | Lys-212b | Glu-95 | Lys-208 |
| Flap 8 | Asp-153 | Lys-219 | Arg-144 | Asp-215 |
| Loop 8-helix 9 | Asp-165b | Arg-131b | Lys-156 | Val-128 |
a The residue is also involved with aromatic interaction.
b The residue is also involved with hydrogen-bonding interaction(s).
Role of Electrostatic Interactions in Cross-strain Specificity Versus Promiscuity
Although homologues of R. flavefaciens strains 17 and FD-1 were initially derived from different geographical locations and time frames (40, 41), they have been classified as the same species. Both strains share marked similarities in their properties. They produce elaborate cellulosomes of similar overall architecture and exhibit high sequence homology within the sca gene cluster (19, 20). Surprisingly, affinity-based assays showed that CohE from R. flavefaciens strain 17 recognizes dockerins of its own strain exclusively (supplemental Fig. 3A), whereas CohE from strain FD-1 also recognizes the dockerins of strain 17 but with lower affinity (supplemental Fig. 3B). Thus, CohE from strain 17 is faithful to its dockerin, whereas CohE from strain FD-1 is promiscuous. To account for the possible molecular consequences that might lead to the observed behavior of the two cohesin modules, we reassessed the available structural information.
Currently, only the structures of the uncomplexed CohE from strain 17 (Protein Data Bank code 2ZF9) and the complexed CohE from strain FD-1 have been solved. Although extensive attempts to solve the structures of the XDoc of CttA from strain 17 independently and in complex with its cognate cohesin were made, the crystals yielded poor and anisotropic diffraction in both cases. Therefore, we generated a model for the CttA-XDoc structure from strain 17 using the I-TASSER server (34) relying on the coordinates of its homologue from the solved complex of strain FD-1. Subsequently, the predicted model was superimposed on the known complex together with the known structure of its cognate CohE from strain 17.
The model allowed us to evaluate the interactions that are likely to be involved in the Coh-Doc interaction between the XDoc dyad and CohE from strain 17. Table 2 lists the interactions seen in the RfCohE-XDoc strain FD-1 complex structure presented here that were detected using the Protein Interaction Calculator (PIC) server (42) and the homologous residues in the putative RfCohE-XDoc complex from strain 17 that were predicted from the structure-based alignment. The data show an almost identical set of hydrophobic interactions on the interfaces of the two complexes and a similar set of hydrogen-bonding residues. However, the difference between these interfaces is evident in the character of the residues participating in ionic interactions located at β-flap 8 and the loop connecting β-strand 8 with the prominent α-helix (Fig. 5).
FIGURE 5.

Comparison of specificity residues in cognate and noncognate CohEFD-1 and CohE17-XDoc complexes. A, CohE-XDoc complex strain FD-1 (Protein Data Bank code 4IU2). B, predicted CohE17-XDoc17 complex. C, predicted CohEFD-1-XDoc17 complex. D, predicted CohE17-XDocFD-1 complex. Residues involved with electrostatic interactions are shown as sticks in the figure. See the text for details.
Examination of the cognate CohE-XDoc crystallographic complex from strain FD-1 reveals that CohEFD-1 Asp-153 and Asp-165 form ionic interactions with the XDocFD-1 Lys-219 and Arg-131, respectively (Fig. 5A). In our model of the cognate RfCohE-XDoc complex from strain 17 (Fig. 5B), residues Arg-144 and Lys-156 replace the equivalent dockerin-interacting residues on the putative CohE17 surface, whereas Asp-215 and Val-128 replace the equivalent cohesin-interacting residues on the predicted XDoc17 surface. In the noncognate CohEFD-1-XDoc17 (Fig. 5C), chemical frustration produced by the destabilizing specificity contact between the noncognate Asp-165 (CohEFD-1) and Val-128 (XDoc17) would appear to be tolerated because of the stabilizing interactions between the conserved hydrophobic residues as well as several possible hydrogen-bonding contacts on the binding interface. In addition, XDoc17 Asp-215 appears to be too remote (7.5 Å) from CohEFD-1 Asp-153 to form repulsive interactions because its side chain is predicted to rotate away from the interface. In our model of the transposed noncognate CohE17-XDocFD-1 complex (Fig. 5D), however, the repulsive interactions are more conspicuous whereby CohE17 Arg-144 and Lys-156 confront XDocFD-1 Lys-219 and Arg-131, respectively, and these opposing residues would likely contribute to a lack of recognition between these proteins. We conclude that the conserved hydrophobic core and multiplicity of the hydrogen-bonding network in addition to differences in charged residues help sculpt the respective binding interfaces and dictate specific versus promiscuous binding between the type IIIe components of the two strains.
To further support the premise that ionic interactions dictate binding and interstrain specificity, a double homology swapping approach was taken, i.e. mutual substitution of designated secondary structural elements between the CohE modules of both strains. The selected sequences are in the vicinity of the above described electrostatic residues: Arg-144 and Lys-156 from CohE17 and Asp-153 and Asp-165 from CohEFD-1. As expected, the resultant CohE17 chimera bearing the doubled-swapped mutations SRTTNE → WDPSKG (143–148) and NVKK → DNKD (153–156) (supplemental Fig. 3C) and the reverse chimera of CohEFD-1 bearing the doubled-swapped mutations WDPSKG → SRTTNE (152–157) and DNKD → NVKK (162–165) (supplemental Fig. 3D) both lost their ability to recognize their cognate dockerins, although their affinity toward the noncognate dockerins was not affected (data not shown). In this context, the swapped elements from both CohEFD-1 and CohE17 were located in β-flap 8 and the loop connecting β-strand 8b with the prominent α-helix. These results support the hypothesis that electrostatic interactions in these regions dictate the specificity of the type IIIe Coh-Doc interaction. Given the extent of hydrophobic and hydrogen bond contacts that characterize the interface, it is intriguing that mutation of only two short sequences would produce such a dramatic effect on the binding affinity. The replacements of electrostatic residues (Asp with Lys or Arg and vice versa) presumably caused strong local repulsive forces between the interacting cognate modules, resulting in the complete loss of binding.
The Atypical Second Calcium-binding Loop
The two Ca2+-binding loops of the type IIIe dockerin in the complex are markedly different. The first comprises a canonical 12-residue “F-hand” loop-helix Ca2+-binding dockerin motif that coordinates the Ca2+ ion in a pentagonal bipyramid configuration whereby positions 1 (Asp-121), 3 (Asp-123), and 5 (Asn-125) provide side-chain carboxylate oxygen ligands to the Ca2+ ion, whereas Asp-132 at position 12 serves as a bidentate ligand (Figs. 6 and 7). The main-chain carbonyl oxygen at position 7 (Ile-127) and a bridged water molecule at position 9 (via Asp-129) provide additional coordinating ligands in the loop.
FIGURE 6.

Sequence-based alignment of selected type I, type II, and type III dockerins. Sequences of ins1, ins2, and ins3 represent designated conserved inserts unique to the CttA and ScaB dockerins. The degree of conservation of each position within the repeated CttA and ScaB sequence is indicated as follows: vertical lines denote identity, colons indicate that the residues are conserved, and dots indicate that the residues are semiconserved as defined by the European Bioinformatics Institute server (ClustalW). Calcium-coordinating residues are highlighted in cyan, and suspected specificity residues are shown in bold and highlighted in yellow. Residues that interact with the X-module and CohE in the CohE-XDoc structure are colored blue and green, respectively. The location of α-helices is denoted along the sequence and numbered α-1 to α-5. The GenBank accession codes for C. thermocellum (Clotm) Xyn10B and CipA are P51584 and L08665, respectively. Rumfl, R. flavefaciens.
FIGURE 7.

Schematic representation of the first and second calcium-binding loops. The first Ca2+-binding loop is composed of a loop-helix with canonical Ca2+-coordinating residues Asp-121, Asp-123, Asn-125, and Asp-132 in positions 1, 3, 5, and 12 where the latter serves as a bidentate ligand. The backbone carbonyl oxygen at position 7 (Ile-127) and a bridged water molecule at position 9 (via Asp-129) provide another two coordinating ligands in the first loop. The second Ca2+-binding loop, however, exhibits an atypical helix-loop-helix Ca2+-binding motif. In addition to the traditional calcium-coordinating residues located at positions 1 (Asp-178), 7 (Leu-197), 9 (Asp-199), and 12 (Asp-202), the loop exhibits several alterations from the canonical motif: Lys-180 at position 3 that provides a backbone carbonyl oxygen ligand and a bridged water molecule at position 5 (via Asp-182 and Glu-59, the latter of which originates from the neighboring unit cell X-module molecule in the crystal; not shown). Moreover, the loop is disrupted in its midst by a 13-residue insert shown in orange. The dockerin molecule is represented in red ribbon, whereas the residues that coordinate the calcium ion are shown as yellow sticks. Calcium ions and water (W) molecules are shown as magenta and blue spheres, respectively.
The second Ca2+-binding motif comprises a complete “EF-hand” helix-loop-helix motif with an entering helix (α2) and exiting helix (α3) oriented perpendicular to each other wherein the Ca2+ ion is coordinated in a typical pentagonal bipyramid configuration (Fig. 7). However, it has several additional unconventional features. The Ca2+-binding loop is disrupted by a 13-residue insert, which displaces the traditional positions of the standard Ca2+-coordinating residues. A lysine residue (Lys-180) replaces the typical Asn/Asp Ca2+-coordinating group at position 3 and provides a backbone carbonyl oxygen ligand. Another atypical element is found in position 5 where a bridged water molecule (via Asp-182) provides another coordinating ligand in the loop. Typical coordinating residues that provide the side-chain carboxylate oxygens are found in positions 1 (Asp-178) and 12 (Asp-202) with the latter serving as a bidentate ligand. The main-chain carbonyl oxygen at positions 7 (Leu-197) and a bridged water molecule in position 9 (via Asp-199) provide coordinating ligands similar to that of the typical Ca2+-binding loop.
The Stabilizing Role of the Dockerin Inserts in the XDoc Intermodular Interface
The CttA XDoc is the first reported example of a dockerin that exhibits greater thermostability than its cognate cohesin partner (3, 6), presumably reflecting the presence of an extensive and intimate intermodular interaction with the X-module. This modular arrangement represents a common theme shared by XDoc dyads from the CttA and ScaB proteins of both R. flavefaciens strains 17 and FD-1. The dockerin sequences of the latter proteins and their inserts are highly conserved (24). The conserved insertions are unique to the type IIIe dockerins of CttA and ScaB and absent from other known dockerin modules. Sequence alignment with selected type I and type II dockerins shows that the first insert (ins1) is located at the end of the linker connecting the two Ca2+-binding loops and includes helix 2. As mentioned above, the second insert (ins2) is embedded in the midst of the second Ca2+-binding loop, whereas the third insert (ins3) is found at the C terminus of the proteins and comprises the C-terminal portion of helix 4 and helix 5 (Fig. 6).
The RfCohE-XDoc complex structure revealed that almost all of the dockerin residues that participate in the interface between the X- and dockerin modules are positioned in the insert regions and are conserved within the dockerin modules from CttA and ScaB of R. flavefaciens strains FD-1 and 17 (Fig. 6) (24). The XDoc interface residues essentially combine to form a common hydrophobic core involving residues Val-18, Phe-21, Phe-23, Ala-32, Phe-89, Tyr-115, and Leu-118 from the X-module and Leu-124, Val-128, Leu-160, Val-168, Tyr-169, Phe-172, Phe-175, Leu-176, and Phe-190 from the dockerin module. Leu-160 within insert 1 of the dockerin module is positioned in the linker connecting the first and second α-helices, whereas Val-168, Tyr-169, Phe-172, Phe-175, and Leu-176 in the same insert are located in the α-helix (α2) preceding the second Ca2+-binding loop. The common hydrophobic core is further stabilized by additional hydrogen-bonding and electrostatic contacts involving Phe-21, Thr-25, Glu26, Asp-30, and Val-85 of the X-module and Asp-121, Asn-126, Arg-189, and Asn-227 from the dockerin module. Asp-121, Leu-124, Asn-126, and Val-128 collectively represent an immediate continuum of the sequence of the X-module and are also involved in the first Ca2+-binding loop. Arg-189 and Phe-190 are positioned in the middle of insert 2, and Asn-227, which is the only non-conserved residue, is found in insert 3.
The inserts protrude from the CttA dockerin structure toward the X-module, providing structural support for interactions with the elongated X-module (Fig. 8). We postulate that these inserts have evolved in the CttA and ScaB dockerins to support and reinforce the intermodular XDoc interface, thus facilitating the extended conformation of the X-module.
FIGURE 8.

The elongated conformation of the X-module is buttressed by the three dockerin inserts. Surface (A and B) and schematic (C) representation of the RfCohE-XDoc complex showing the three inserts of the dockerin that support the extended stalklike conformation of the X-module (blue). Inserts 1, 2, and 3 are colored magenta, light orange, and cyan, respectively. The cohesin and dockerin modules are shown in green and red, respectively.
DISCUSSION
When the sequence of the ScaB scaffoldin was first reported in R. flavefaciens strain 17 (43), we initially failed to identify its dockerin sequence because of sequence aberrations in both of its Ca2+-binding motifs in this strain. In a later publication (21), the association of ScaB with the cell surface was confirmed, but the mechanism for cell surface attachment was still unclear. The presence of a cryptic N-terminal XDoc sequence for ScaB was discovered only when its interaction with CohE was established (23). Subsequent studies (24) demonstrated a second conserved XDoc sequence in CttA that also bound CohE. Bioinformatics analysis of these unconventional dockerin sequences on a background of standard dockerin sequences served to predict the Ca2+-binding residues, including those of the unconventional second Ca2+-binding loop, as well as the possible existence of three distinctive inserts in both CttA and ScaB. The functional significance of these putative sequence inserts remained elusive until the present work. The structure of the RfCohE-XDoc complex confirmed the previous prediction that the dockerin module contains a second atypical calcium-binding loop that is disrupted by a 13-residue insert. In addition, a combined functional role for the three enigmatic dockerin inserts was established whereby the extraneous segments serve as structural buttresses that support the extended conformation of the X-module via an extensive network of intermodular interactions.
Unlike the X-module in the type II Coh-XDoc interaction of C. thermocellum (3), the X-module in the type IIIe RfCohE-XDoc complex does not appear to contribute directly to the CohE-Doc binding surface in this structure. Rather, its elongated stalklike conformation appears to serve as an extended spacer, which separates the cellulose-binding modules at the N terminus of CttA and the bacterial cell wall. Indeed, a structural similarity search (33) indicated that the X-module does not share structural similarity with other known X-modules from cellulolytic bacteria, e.g. X-2 (44) or X-60 (3, 45). Instead, it exhibits significant similarity with the G5-1 module (p value = 0.04 with 49 equivalent positions and r.m.s.d. of 2.59 Å) of the multimodular protein StrH from the human pathogen Streptococcus pneumoniae (Protein Data Bank code 2LTJ) (46). StrH is a surface-attached exo-β-d-N-acetylglucosaminidase that cooperates with a sialidase (NanA) and a β-galactosidase (BgaA) to sequentially degrade the nonreducing terminal arms of complex N-linked glycans. In general, G5 modules are widespread among cell surface-binding proteins. The two G5 modules in the StrH structure adopt a linear and extended conformation, suggesting a structural role for these modules that would appear to position the two catalytic modules away from the cell surface for optimal processing of the glycans of soluble or host cell surface-presented glycoconjugates. The analogy of the G5 module supports a structural role of the stalklike X-module as a molecular spacer for the substrate binding event in R. flavefaciens. Likewise, the presence in ScaB of a similar X-module and set of three inserts in the dockerin module suggests a similar role whereby the ScaB cohesin modules and the remainder of the cellulosome assembly are separated physically from the cell surface by the reinforced X-module, thus facilitating the action of the cellulosomal enzymes on the plant cell polysaccharide substrates.
The specificity of the different types of Coh-Doc interactions is crucial for the correct assembly of the enzymatic cellulase machinery and its attachment to the bacterial cell wall. The crystal structure presented in this work enhances our understanding of the molecular architecture of the type IIIe Coh-Doc interfaces that exhibit specific high affinity binding. The results further pinpoint two charged specificity-related residues on the surface of CohE that are responsible for specific binding versus promiscuous cross-strain interaction. The ability to achieve high affinity binding toward a specific protein partner while precluding binding to other noncognate proteins is of fundamental importance in other biological processes as well, including cell regulation, the immune response, signal transduction, and others. One such example of interacting protein pairs from complementary families that exhibit specific high affinity binding is the colicin endonuclease/immunity pair (47). Colicin DNase binding by immunity proteins is characterized by a multiplicity of hydrophobic as well as hydrogen-bonding interactions where electrostatic frustration between noncognate pairs dictates specificity (48), similar to the phenomenon described herein for the Coh-Doc interaction.
It is of interest to consider whether or not the cross-strain fidelity-versus-promiscuity phenomenon has further reaching biological consequences. Fidelity would indicate that the bacterium would refuse to incorporate foreign dockerin-containing enzymes produced by other rumen strains, whereas promiscuous behavior among the different strains would indicate a degree of interstrain flexibility and cooperation. In this context, it is important to review the precise sources of the two strains and the prominence and significance of R. flavefaciens in the rumen environment. The digestive tract of herbivores, particularly ruminants, is one of the most prevalent cellulose-based ecosystems in nature. In this environment, a congested consortium of anaerobic carbohydrate-processing bacteria, fungi, and other microorganisms combines to rapidly deconstruct the tremendous quantities of plant biomass that are consumed by the animal. Of the true fiber-degrading species that comprise this diverse microbial community, R. flavefaciens plays a critically important role in the direct degradation of the otherwise recalcitrant cellulosic biomass in the rumen and hindgut of ruminants (12–16). Surprisingly, this bacterial species is present in the rumen in a multiplicity of functionally divergent subtypes that undergo dynamic fluctuations within a given animal (49). R. flavefaciens strains 17 and FD-1, however, were initially derived from different animals in different geographical locations and time frames (40, 41), indicating that their ScaE cohesin modules and XDoc derivatives would not have occupied the same locale. It thus seems that their sequence features that would account for the observed specific coupling and promiscuous behavior may be a function of simple evolutionary events. Nevertheless, there is evidence that the various subtypes may represent distinct lineages whose descendants are maintained in the rumen of subsequent generations (20). Indeed, such distinct lineages related to R. flavefaciens strains 17 and FD-1 have been identified in the rumens of contemporary animals, and the possibility would exist for similar cross-strain interactions with some strains exhibiting a propensity for sharing their components.
Acknowledgments
We thank the European Synchrotron Radiation Facility, Grenoble, France, for synchrotron beam time and the staff scientists of the BM-14 station for assistance. We appreciate the advice and assistance with the cloning and expression of Drs. Yoav Peleg and Tamar Unger of the Israel Structural Proteomics Center at the Weizmann Institute of Science and thank Dr. Ozgur Gul (Weizmann Institute) for model minimization experiments.
This work was supported in part by Israel Science Foundation (ISF) Grants 293/08 (to F. F.) and 966/09 (to E. A. B.); Natural Sciences and Engineering Research Council of Canada Grant RGPIN 398508 (to S. P. S.); Grant 24/11 from The Sidney E. Frank Foundation through the ISF (to R. L.); an F. Warren Hellman Grant for Alternative Energy Research in Israel in support of alternative energy research in Israel administered by the Israel Strategic Alternative Energy Foundation (to E. A. B.); the establishment of an Israeli Center of Research Excellence (I-CORE Center 152/11; to E. A. B.) managed by the Israel Science Foundation; the United States-Israel Binational Science Foundation, Jerusalem, Israel; the Weizmann Institute of Science Alternative Energy Research Initiative and the Helmsley Foundation; and a grant from the Israel Ministry of Science (to E. A. B. and R. L.).

This article contains supplemental Figs. 1–3 and Table 1.
The atomic coordinates and structure factors (codes 4IU3 and 4IU2) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Coh
- cohesin
- Doc
- dockerin
- CBM
- carbohydrate-binding module
- CBM3a
- family 3a carbohydrate-binding module
- CohE
- cohesin module of ScaE
- CttA
- cellulose-binding protein
- XDoc
- X-dockerin
- Xyn
- xylanase T6 of G. stearothermophilus
- Rf
- R. flavefaciens
- SeMet
- selenomethionyl
- r.m.s.d.
- root mean square deviation
- insert
- ins.
REFERENCES
- 1. Carvalho A. L., Dias F. M., Prates J. A., Nagy T., Gilbert H. J., Davies G. J., Ferreira L. M., Romão M. J., Fontes C. M. (2003) Cellulosome assembly revealed by the crystal structure of the cohesin-dockerin complex. Proc. Natl. Acad. Sci. U.S.A. 100, 13809–13814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carvalho A. L., Dias F. M., Nagy T., Prates J. A., Proctor M. R., Smith N., Bayer E. A., Davies G. J., Ferreira L. M., Romão M. J., Fontes C. M., Gilbert H. J. (2007) Evidence for a dual binding mode of dockerin modules to cohesins. Proc. Natl. Acad. Sci. U.S.A. 104, 3089–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adams J. J., Pal G., Jia Z., Smith S. P. (2006) Mechanism of bacterial cell-surface attachment revealed by the structure of cellulosomal type II cohesin-dockerin complex. Proc. Natl. Acad. Sci. U.S.A. 103, 305–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pinheiro B. A., Proctor M. R., Martinez-Fleites C., Prates J. A., Money V. A., Davies G. J., Bayer E. A., Fontesm C. M., Fierobe H. P., Gilbert H. J. (2008) The Clostridium cellulolyticum dockerin displays a dual binding mode for its cohesin partner. J. Biol. Chem. 283, 18422–18430 [DOI] [PubMed] [Google Scholar]
- 5. Himmel M. E., Bayer E. A. (2009) Lignocellulose conversion to biofuels: current challenges, global perspectives. Curr. Opin. Biotechnol. 20, 316–317 [DOI] [PubMed] [Google Scholar]
- 6. Adams J. J., Gregg K., Bayer E. A., Boraston A. B., Smith S. P. (2008) Structural basis of Clostridium perfringens toxin complex formation. Proc. Natl. Acad. Sci. U.S.A. 105, 12194–12199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nataf Y., Bahari L., Kahel-Raifer H., Borovok I., Lamed R., Bayer E. A., Sonenshein A. L., Shoham Y. (2010) Clostridium thermocellum cellulosomal genes are regulated by extracytoplasmic polysaccharides via alternative σ factors. Proc. Natl. Acad. Sci. U.S.A. 107, 18646–18651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ding S. Y., Liu Y. S., Zeng Y., Himmel M. E., Baker J. O., Bayer E. A. (2012) How does plant cell wall nanoscale architecture correlate with enzymatic digestibility? Science 338, 1055–1060 [DOI] [PubMed] [Google Scholar]
- 9. Stahl S. W., Nash M. A., Fried D. B., Slutzki M., Barak Y., Bayer E. A., Gaub H. E. (2012) Single-molecule dissection of the high-affinity cohesin-dockerin complex. Proc. Natl. Acad. Sci. U.S.A. 109, 20431–20436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gefen G., Anbar M., Morag E., Lamed R., Bayer E. A. (2012) Enhanced cellulose degradation by targeted integration of a cohesin-fused β-glucosidase into the Clostridium thermocellum cellulosome. Proc. Natl. Acad. Sci. U.S.A. 109, 10298–10303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moraïs S., Morag E., Barak Y., Goldman D., Hadar Y., Lamed R., Shoham Y., Wilson D. B., Bayer E. A. (2012) Deconstruction of lignocellulose into soluble sugars by native and designer cellulosomes. MBio 3, e00508–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Julliand V., de Vaux A., Millet L., Fonty G. (1999) Identification of Ruminococcus flavefaciens as the predominant cellulolytic bacterial species of the equine cecum. Appl. Environ. Microbiol. 65, 3738–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krause D. O., Dalrymple B. P., Smith W. J., Mackie R. I., McSweeney C. S. (1999) 16S rDNA sequencing of Ruminococcus albus and Ruminococcus flavefaciens: design of a signature probe and its application in adult sheep. Microbiology 145, 1797–1807 [DOI] [PubMed] [Google Scholar]
- 14. Nelson K. E., Zinder S. H., Hance I., Burr P., Odongo D., Wasawo D., Odenyo A., Bishop R. (2003) Phylogenetic analysis of the microbial populations in the wild herbivore gastrointestinal tract: insights into an unexplored niche. Environ. Microbiol. 5, 1212–1220 [DOI] [PubMed] [Google Scholar]
- 15. Flint H. J. (1997) The rumen microbial ecosystem—some recent developments. Trends Microbiol. 5, 483–488 [DOI] [PubMed] [Google Scholar]
- 16. Wedekind K. J., Mansfield H. R., Montgomery L. (1988) Enumeration and isolation of cellulolytic and hemicellulolytic bacteria from human feces. Appl. Environ. Microbiol. 54, 1530–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lamed R., Setter E., Bayer E. A. (1983) Characterization of a cellulose-binding, cellulase-containing complex in Clostridium thermocellum. J. Bacteriol. 156, 828–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bayer E. A., Kenig R., Lamed R. (1983) Adherence of Clostridium thermocellum to cellulose. J. Bacteriol. 156, 818–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jindou S., Borovok I., Rincon M. T., Flint H. J., Antonopoulos D. A., Berg M. E., White B. A., Bayer E. A., Lamed R. (2006) Conservation and divergence in cellulosome architecture between two strains of Ruminococcus flavefaciens. J. Bacteriol. 188, 7971–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jindou S., Brulc J. M., Levy-Assaraf M., Rincon M. T., Flint H. J., Berg M. E., Wilson M. K., White B. A., Bayer E. A., Lamed R., Borovok I. (2008) Cellulosome gene cluster analysis for gauging the diversity of the ruminal cellulolytic bacterium Ruminococcus flavefaciens. FEMS Microbiol. Lett. 285, 188–194 [DOI] [PubMed] [Google Scholar]
- 21. Rincon M. T., Ding S. Y., McCrae S. I., Martin J. C., Aurilia V., Lamed R., Shoham Y., Bayer E. A., Flint H. J. (2003) Novel organization and divergent dockerin specificities in the cellulosome system of Ruminococcus flavefaciens. J. Bacteriol. 185, 703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rincón M. T., Martin J. C., Aurilia V., McCrae S. I., Rucklidge G. J., Reid M. D., Bayer E. A., Lamed R., Flint H. J. (2004) ScaC, an adaptor protein carrying a novel cohesin that expands the dockerin-binding repertoire of the Ruminococcus flavefaciens 17 cellulosome. J. Bacteriol. 186, 2576–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rincon M. T., Cepeljnik T., Martin J. C., Lamed R., Barak Y., Bayer E. A., Flint H. J. (2005) Unconventional mode of attachment of the Ruminococcus flavefaciens cellulosome to the cell surface. J. Bacteriol. 187, 7569–7578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rincon M. T., Cepeljnik T., Martin J. C., Barak Y., Lamed R., Bayer E. A., Flint H. J. (2007) A novel cell surface-anchored cellulose-binding protein encoded by the sca gene cluster of Ruminococcus flavefaciens. J. Bacteriol. 189, 4774–4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Navarre W. W., Schneewind O. (1994) Proteolytic cleavage and cell wall anchoring at the LPXTG motif of surface proteins in gram-positive bacteria. Mol. Microbiol. 14, 115–121 [DOI] [PubMed] [Google Scholar]
- 26. Alber O., Noach I., Lamed R., Shimon L. J., Bayer E. A., Frolow F. (2008) Preliminary x-ray characterization of a novel type of anchoring cohesin from the cellulosome of Ruminococcus flavefaciens. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64, 77–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alber O., Noach I., Rincon M. T., Flint H. J., Shimon L. J., Lamed R., Frolow F., Bayer E. A. (2009) Cohesin diversity revealed by the crystal structure of the anchoring cohesin from Ruminococcus flavefaciens. Proteins 77, 699–709 [DOI] [PubMed] [Google Scholar]
- 28. Barak Y., Handelsman T., Nakar D., Mechaly A., Lamed R., Shoham Y., Bayer E. A. (2005) Matching fusion protein systems for affinity analysis of two interacting families of proteins: the cohesin-dockerin interaction. J. Mol. Recognit. 18, 491–501 [DOI] [PubMed] [Google Scholar]
- 29. Salama-Alber O., Gat Y., Lamed R., Shimon L. J., Bayer E. A., Frolow F. (2012) Crystallization and preliminary x-ray characterization of a type III cohesin-dockerin complex from the cellulosome system of Ruminococcus flavefaciens. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 1116–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sheldrick G. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holm L., Park J. (2000) DaliLite workbench for protein structure comparison. Bioinformatics 16, 566–567 [DOI] [PubMed] [Google Scholar]
- 34. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 36. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mackerell A. D., Jr., Feig M., Brooks C. L., 3rd. (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 [DOI] [PubMed] [Google Scholar]
- 38. Shimon L. J., Bayer E. A., Morag E., Lamed R., Yaron S., Shoham Y., Frolow F. (1997) A cohesin domain from Clostridium thermocellum: the crystal structure provides new insights into cellulosome assembly. Structure 5, 381–390 [DOI] [PubMed] [Google Scholar]
- 39. Currie M. A., Adams J. J., Faucher F., Bayer E. A., Jia Z., Smith S. P. (2012) Scaffoldin conformation and dynamics revealed by a ternary complex from the Clostridium thermocellum cellulosome. J. Biol. Chem. 287, 26953–26961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bryant M. P., Small N., Bouma C., Robinson I. M. (1958) Characteristics of ruminal anaerobic celluloytic cocci and Cillobacterium cellulosolvens n. sp. J. Bacteriol. 76, 529–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Flint H. J., McPherson C. A., Bisset J. (1989) Molecular cloning of genes from Ruminococcus flavefaciens encoding xylanase and β(1–3,1–4)glucanase activities. Appl. Environ. Microbiol. 55, 1230–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tina K. G., Bhadra R., Srinivasan N. (2007) PIC: Protein Interactions Calculator. Nucleic Acids Res. 35, W473–W476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ding S.-Y., Rincon M. T., Lamed R., Martin J. C., McCrae S. I., Aurilia V., Shoham Y., Bayer E. A., Flint H. J. (2001) Cellulosomal scaffoldin-like proteins from Ruminococcus flavefaciens. J. Bacteriol. 183, 1945–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mosbah A., Belaïch A., Bornet O., Belaïch J. P., Henrissat B., Darbon H. (2000) Solution structure of the module X2 1 of unknown function of the cellulosomal scaffolding protein CipC of Clostridium cellulolyticum. J. Mol. Biol. 304, 201–217 [DOI] [PubMed] [Google Scholar]
- 45. Adams J. J., Pal G., Yam K., Spencer H. L., Jia Z., Smith S. P. (2005) Purification and crystallization of a trimodular complex comprising the type II cohesin-dockerin interaction from the cellulosome of Clostridium thermocellum. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61, 46–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pluvinage B., Chitayat S., Ficko-Blean E., Abbott D. W., Kunjachen J. M., Grondin J., Spencer H. L., Smith S. P., Boraston A. B. (2013) Conformational analysis of StrH, the surface-attached exo-β-d-N-acetylglucosaminidase from Streptococcus pneumoniae. J. Mol. Biol. 425, 334–349 [DOI] [PubMed] [Google Scholar]
- 47. Cascales E., Buchanan S. K., Duché D., Kleanthous C., Lloubès R., Postle K., Riley M., Slatin S., Cavard D. (2007) Colicin biology. Microbiol. Mol. Biol. Rev. 71, 158–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wojdyla J. A., Fleishman S. J., Baker D., Kleanthous C. (2012) Structure of the ultra-high-affinity colicin E2 DNase-Im2 complex. J. Mol. Biol. 417, 79–94 [DOI] [PubMed] [Google Scholar]
- 49. Brulc J. M., Yeoman C. J., Wilson M. K., Berg Miller M. E., Jeraldo P., Jindou S., Goldenfeld N., Flint H. J., Lamed R., Borovok I., Vodovnik M., Nelson K. E., Bayer E. A., White B. A. (2011) Cellulosomics, a gene-centric approach to investigating the intraspecific diversity and adaptation of Ruminococcus flavefaciens within the rumen. PLoS One 6, e25329. [DOI] [PMC free article] [PubMed] [Google Scholar]