

Background: Snake venom protease batroxobin clots fibrinogen in a manner distinct from thrombin.
Results: Batroxobin binds fibrin(ogen) with higher affinity than thrombin and promotes greater clot expansion.
Conclusion: Batroxobin's distinctive interaction with fibrin(ogen) may contribute to its unique pattern of fibrinopeptide release.
Significance: Clinically, batroxobin is used as a defibrinogenating agent, but its capacity to promote clot expansion may promote microvascular thrombosis.
Keywords: Blood Coagulation Factors, Fibrin, Fibrinogen, Snake Venom, Thrombin, Batroxobin, Fibrinopeptides
Abstract
Batroxobin is a thrombin-like serine protease from the venom of Bothrops atrox moojeni that clots fibrinogen. In contrast to thrombin, which releases fibrinopeptide A and B from the NH2-terminal domains of the Aα- and Bβ-chains of fibrinogen, respectively, batroxobin only releases fibrinopeptide A. Because the mechanism responsible for these differences is unknown, we compared the interactions of batroxobin and thrombin with the predominant γA/γA isoform of fibrin(ogen) and the γA/γ′ variant with an extended γ-chain. Thrombin binds to the γ′-chain and forms a higher affinity interaction with γA/γ′-fibrin(ogen) than γA/γA-fibrin(ogen). In contrast, batroxobin binds both fibrin(ogen) isoforms with similar high affinity (Kd values of about 0.5 μm) even though it does not interact with the γ′-chain. The batroxobin-binding sites on fibrin(ogen) only partially overlap with those of thrombin because thrombin attenuates, but does not abrogate, the interaction of γA/γA-fibrinogen with batroxobin. Furthermore, although both thrombin and batroxobin bind to the central E-region of fibrinogen with a Kd value of 2–5 μm, the α(17–51) and Bβ(1–42) regions bind thrombin but not batroxobin. Once bound to fibrin, the capacity of batroxobin to promote fibrin accretion is 18-fold greater than that of thrombin, a finding that may explain the microvascular thrombosis that complicates envenomation by B. atrox moojeni. Therefore, batroxobin binds fibrin(ogen) in a manner distinct from thrombin, which may contribute to its higher affinity interaction, selective fibrinopeptide A release, and prothrombotic properties.
Introduction
Fibrinogen is important for optimal primary and secondary hemostasis because of its critical role in platelet aggregation and fibrin clot formation (1). Fibrinogen is a dimeric glycoprotein composed of two pairs of Aα-, Bβ-, and γ-chains connected by numerous disulfide bonds (2). The NH2 termini of the six chains form the central E-domain, which is connected by the coiled-coil regions to the peripheral D-domains formed by the COOH termini (2). There are two circulating isoforms of fibrinogen that differ with respect to their γ-chains. The predominant fibrinogen isoform is a homodimer that contains two γA-chains consisting of 411 residues and is designated γA/γA-fibrinogen. About 10–15% of circulating fibrinogen is heterodimeric, containing one γA-chain and one variant γ′-chain that possesses a 16-residue anionic extension at its COOH terminus. This minor fibrinogen population is designated γA/γ′-fibrinogen (3–5). Epidemiological studies suggest that elevated levels of circulating γA/γ′-fibrinogen are associated with an increased risk of cardiovascular disease (6, 7).
Thrombin is the protease that converts fibrinogen to insoluble fibrin by releasing fibrinopeptides (Fp)4 A and B from the NH2 termini of the Aα- and Bβ-chains, respectively (8). Fibrin polymerization occurs when the newly exposed NH2 termini on one fibrin monomer bind to pre-existing complementary sites on the D-domains of adjacent fibrin monomers (2, 8, 9). During this process, some thrombin remains bound to the fibrin clot (10). Thrombin possesses two anion-binding exosites that flank the active site and mediate substrate binding and catalysis (11, 12). Exosite I mediates thrombin binding to the Aα- and Bβ-chains in the central E-region of fibrin(ogen), whereas exosite 2 is responsible for thrombin's interaction with the unique COOH terminus of the γ′-chain of a second fibrin(ogen) molecule (13, 14). Consequently, thrombin binds γA/γA-fibrin solely via exosite 1 with a Kd value of 2–4 μm. In contrast, both exosites are engaged when thrombin binds to γA/γ′-fibrin(ogen), which results in an ∼20-fold higher affinity interaction than that with γA/γA-fibrin (Kd value of 80–200 nm) (15, 16). Because fibrin-bound thrombin retains activity and is protected from inhibition by antithrombin and heparin cofactor II, the fibrin clot serves as a reservoir of thrombin that promotes thrombus expansion by locally activating platelets and factor (f) XI, fVIII, and fV (10, 17). The higher affinity, bivalent interaction of thrombin with γA/γ′-fibrin affords thrombin more protection from fluid-phase inhibitors than the univalent interaction of thrombin with γA/γA-fibrin (18).
Batroxobin is a serine protease isolated from Bothrops atrox moojeni venom that clots fibrinogen (19). In contrast to thrombin, fibrinogen is the sole substrate for batroxobin, and batroxobin only releases FpA and is not inhibited by antithrombin or heparin cofactor II (20). Because of these properties, batroxobin is often used in clinical laboratories to determine whether prolonged thrombin clotting times are the result of heparin contamination or abnormal fibrinogen molecules (21). In addition, batroxobin has been used clinically for prevention and treatment of thrombosis because of its capacity to lower circulating fibrinogen levels (22–25).
Although it is well established that batroxobin induces defibrinogenation, the interaction of batroxobin with fibrinogen has not been studied nor is it known whether, like thrombin, batroxobin binds to fibrin and triggers thrombus expansion. The potential procoagulant activity of batroxobin is important because patients envenomated by B. atrox moojeni often experience thrombotic complications (26, 27). To address these questions, (a) we characterized the binding of batroxobin to γA/γA- and γA/γ′-fibrinogen to determine whether its fibrinogen-binding sites overlap with those of thrombin because both enzymes release FpA; (b) we assessed the binding of batroxobin to γA/γA- and γA/γ′-fibrin clots to determine whether the protease remains bound to the product following catalysis; and (c) we measured the procoagulant activity of fibrin-bound batroxobin to determine whether, like fibrin-bound thrombin, batroxobin promotes clot expansion.
EXPERIMENTAL PROCEDURES
Materials
Reagents
Human α-thrombin and plasminogen-free fibrinogen were from Enzyme Research Laboratories (South Bend, IN), and fXIII was from Haematologic Technologies Inc. (Essex Junction, VT). Fibrinogen was endered fXIII-free, and γA/γA- and γA/γ′-fibrinogens were separated by fractionation on DEAE-Sepharose (GE Healthcare) as described previously (18, 28). Batroxobin from the venom of B. atrox moojeni was from Pentapharm (Basel, Switzerland). A 20-amino acid analog of the COOH terminus of the γ′-chain of fibrinogen with a Cys at the NH2 terminus, termed γ′-peptide (VRPEHPAETEYDSLYPEDDL), and a γ′-peptide-directed IgG from sheep were prepared by Bachem Bioscience, Inc. (King of Prussia, PA). The two Tyr residues of the γ′-peptide were modified with phosphate groups to mimic the sulfated Tyr residues found in the native γ′-chain and to increase the affinity for thrombin (29). The γ′-peptide-directed IgG was subjected to affinity chromatography using immobilized γ′-peptide as described previously (30). The α(17–51)-peptide (GPRVVERHQSAAKDSDWPFASDEDWNYKAPSGCRM) with Cys-28, Cys-36, and Cys-45 changed to Ala and an NH2-terminal Cys residue was synthesized by LifeTein (Plainfield, NJ). Chromogenic substrates H-d-Phe-Pip-Arg-p-nitroaniline (S-2238) and pyro-Glu-Pro-Arg-nitroaniline (CS-21(66)) were from Chromogenix (Milano, Italy) and Aniaria (Neuville-sur-Oise, France), respectively. d-Phe-Pro-Arg-chloromethyl ketone (FPR) and d-Tyr-Pro-Arg-chloromethyl ketone (YPR) were from Calbiochem. Full-length hirudin was from Dade-Behring (Marburg, Germany). Citrated human plasma was prepared as described (31). Unless otherwise specified, other reagents were from Sigma.
Preparation of Fibrinogen Derivatives and Fragments
Fibrinogen derivatives and fragments were isolated from γA/γA-fibrinogen to eliminate the contribution of the γ′-chain. Des-Bβ(1–42) fibrinogen was prepared by incubating γA/γA-fibrinogen with protease III fraction from Crotalus atrox venom and purified using ethanol precipitation as described (16). Fragments D and E were generated by plasmin digestion and purified as reported previously (30). The NH2-terminal disulfide knot (NDSK) was obtained with cyanogen bromide treatment and purified as described (32).
Modification of Proteins and Peptides
Batroxobin and thrombin were radiolabeled by reaction with 125I-YPR, prepared using Na125I (McMaster University Nuclear Reactor, Hamilton, Ontario, Canada) and IODO-BEADs (Pierce) as described (18, 30). 125I-YPR-batroxobin and 125I-YPR-thrombin concentrations were 1–2 and 6–7 μm, respectively, as determined by absorbance at 280 nm, and had specific radioactivities of ∼500,000–700,000 cpm/μg. Unfractionated fibrinogen (0.5 mg in phosphate-buffered saline) was radiolabeled with 1 mCi of Na125I using 1 IODO-BEAD for 5 min at 23 °C. The sample was then subjected to gel filtration on a PD-10 column (GE Healthcare). The labeled fibrinogen was over 90% clottable and had a specific radioactivity of 925,000 cpm/μg. FPR-batroxobin, FPR-thrombin, and biotin conjugated γ′- and α(17–51)-peptides were prepared as described (33).
Methods
Characterization of Thrombin and Batroxobin
The integrity of thrombin and batroxobin was assessed by SDS-PAGE analysis on 4–15% polyacrylamide gradient gels (Bio-Rad) under nonreducing conditions. Thrombin and batroxobin migrated as single bands with expected apparent molecular weights of 37,000 and 32,000, respectively (Fig. 1). The identity of batroxobin was confirmed by amino acid sequence analysis. Briefly, batroxobin was resolved by SDS-PAGE, and protein bands were transferred to a PVDF membrane (Bio-Rad), and identified by Ponceau Red staining. The bands of interest were submitted for amino acid sequence analysis (Hospital for Sick Children Advanced Protein Technology Centre, Toronto, Ontario, Canada). In addition, SDS-PAGE analysis of batroxobin-treated fibrinogen revealed bands corresponding solely to α-, Bβ-, and γ-chains, indicating the absence of contaminating proteases in the batroxobin preparation (data not shown). The specificity of batroxobin was confirmed by comparing its capacity to cleave FpA and/or FpB from fibrinogen with that of thrombin. FpA and FpB release were quantified by HPLC using synthetic FpA (Bachem) and FpB (LifeTein) as internal standards (34), and the identities of released FpA and FpB were confirmed by mass spectrometry (Bioanalytical and Mass Spectrometry Laboratory, McMaster University). As expected, batroxobin only released FpA from fibrinogen, whereas thrombin released both FpA and FpB (data not shown); these findings are in agreement with previously published results (35).
FIGURE 1.
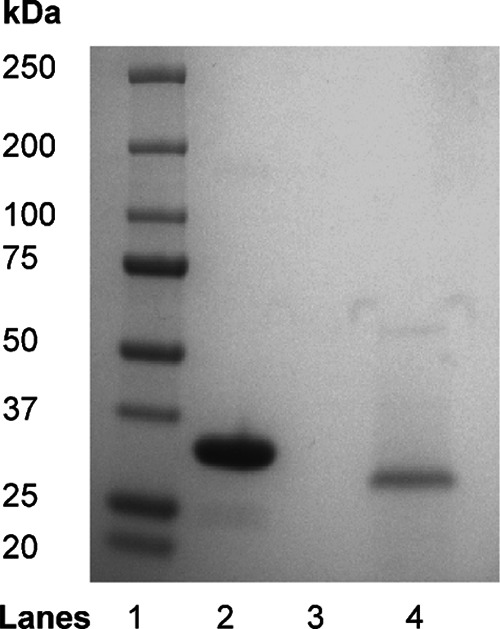
Integrity of thrombin and batroxobin as assessed by SDS-PAGE analysis. Thrombin (lane 2) and batroxobin (lane 4) were subjected to SDS-PAGE analysis on a 4–15% polyacrylamide gradient gel under nonreducing conditions. The molecular weights of the mobility markers are shown on the left (lane 1).
Surface Plasmon Resonance (SPR)
The interaction of FPR-batroxobin and FPR-thrombin with immobilized γA/γA- or γA/γ′-fibrin(ogen), biotinylated-γ′- and α(17–51)-peptides, NDSK, and fragments D or E was assessed by SPR on a BIAcore 1000 (GE Healthcare) as described (18, 30, 33), but with some modifications. Briefly, fibrinogen and fibrinogen fragments were covalently linked to separate flow cells of a carboxymethylated dextran CM5 biosensor chip at a flow rate of 5 μl/min using an amine coupling kit (GE Healthcare). Proteins were immobilized in 10 mm acetate buffer at varying pH values to maximize adsorption. γA/γA- and γA/γ′-fibrinogen and fragment D were immobilized at pH 5.5 to ∼6000–8000 response units (RU). Fragment E and NDSK were immobilized at pH 4.5 and 5.0 to ∼6000–8000 RU, respectively. For fibrin studies, immobilized γA/γA- and γA/γ′-fibrinogen were converted to fibrin by three successive 60-min injections of 100 nm α-thrombin, each followed by a wash with 0.5 m NaCl (30, 33). To prepare streptavidin-conjugated CM5 cells, 0.4 mg/ml streptavidin was immobilized at pH 4.5. Biotinylated γ′-peptide and α(17–51) were adsorbed onto separate streptavidin-containing cells to 200–600 RU, whereas 0.4 mg/ml ovalbumin was immobilized at pH 4.5 to ∼6000–8000 RU in control flow cells. The remaining reactive amine groups were neutralized with 1 m ethanolamine. All SPR procedures were done in 10 mm Hepes-NaOH, 150 mm NaCl at pH 7.4 (HBS) containing 0.005% Tween 20 and 2 mm CaCl2 (HBS-Tw-Ca). Flow cells were regenerated with 0.5 m NaCl between runs. To measure the affinity of proteases for immobilized fibrinogen, fibrin, and fibrinogen fragments, aliquots of FPR-batroxobin or FPR-thrombin (0–40 μm) were injected at a flow rate of 30 μl/min. In reciprocal experiments, FPR-batroxobin and FPR-thrombin were immobilized at pH 4.5 to ∼4000–6000 RU, and the binding of fibrinogen to the adsorbed proteins was assessed. The sensorgram tracings were obtained from the instrument software, and all experiments were performed at least twice.
To determine the extent to which the batroxobin and thrombin-binding sites on fibrinogen overlap, the binding of γA/γA-fibrinogen to immobilized FPR-batroxobin was measured as described previously in the absence or presence of FPR-thrombin up to 40 μm. As a positive control, the experiment was repeated in the presence of 80 μm hirudin; as a negative control, ovalbumin (0–40 μm) was used in place of FPR-thrombin. The amount of γA/γA-fibrinogen bound to adsorbed batroxobin in the presence of FPR-thrombin was normalized to that determined in the absence of competitor. All competition experiments were performed three times.
SPR Data Analysis
Kd values for one-site binding were determined by kinetic analysis of on- and off-rates of batroxobin or thrombin binding to immobilized ligands using Scrubber2 version 2.0a (Bio-Logic Software Co., Campbell, Australia) as described (33). To calculate two-site binding of thrombin to γA/γ′-fibrin(ogen), the amount of analyte bound at equilibrium (Req) was plotted against input protease concentrations and analyzed by nonlinear regression analysis of a two-site binding equation (Table Curve, Jandel Scientific, San Rafael, CA) as described (16).
Binding of Batroxobin and Thrombin to Fibrin Clots
In a series of microcentrifuge tubes, up to 30 μm γA/γA-, γA/γ′-, or des-Bβ(1–42) γA/γA-fibrinogen was clotted with 10 nm thrombin in the presence of 20–40 nm 125I-YPR-batroxobin or 125I-YPR-thrombin in 20 mm Tris-HCl, pH 7.4, 150 mm NaCl (TBS) containing 0.005% Tween 20 and 2 mm CaCl2 (TBS-Tw-Ca) (33). After 60 min of incubation at 23 °C, clots were compacted by centrifugation at 14,000 × g for 4 min. Fibrin-bound 125I-protease was quantified by subtracting the radioactivity in the clot supernatant from that obtained in controls prepared without fibrinogen. Plots of bound 125I-protease versus fibrin concentration were analyzed by nonlinear regression of a rectangular hyperbola to determine the composite Kd value. Experiments were performed twice in duplicate.
Effect of Competitors on the Binding of 125I-YPR-Batroxobin or 125I-YPR-Thrombin to Fibrin
Affinity-purified γ′-peptide-directed IgG (30) was used to assess the importance of the γ′-region in protease binding to fibrin clots. After preincubating 1 μm γA/γA- or γA/γ′-fibrinogen with γ′-peptide-directed IgG (0–8 μm) for 60 min at 23 °C, 40 nm 125I-YPR-thrombin or 125I-YPR-batroxobin was added, and clotting was initiated with 10 nm thrombin. The fraction of 125I-YPR-protease bound to the clot was then calculated as described above.
To determine whether batroxobin and thrombin have overlapping binding sites, clots were formed with 1 μm γA/γA- or γA/γ′-fibrinogen, 10 nm thrombin, 20–40 nm 125I-YPR-batroxobin or 125I-YPR-thrombin, and 0–25 μm FPR-thrombin in TBS-Tw-Ca. Fibrin-bound 125I-protease was quantified as described above. Experiments were performed twice in duplicate.
Batroxobin and Thrombin Diffusion from Preformed Fibrin Clots
The rate of 125I-protease diffusion from clots formed from γA/γA- or γA/γ′-fibrinogen was determined as described (30, 33). Briefly, 120-μl fibrin clots were formed around plastic loops (Bac-Loop, Thermo-Fisher Scientific, Waltham, MA) in microcentrifuge tubes by adding 100 nm thrombin to 9 μm γA/γA- or γA/γ′-fibrinogen, 30 nm fXIII, and 25 nm 125I-YPR-batroxobin or -thrombin. After incubation at 37 °C for 45 min, clots were removed and immersed in 5 ml of TBS-Ca or 2 m NaCl containing 5 mm EDTA. At intervals, the fraction of clot-bound 125I-YPR-protease was quantified, and time courses were fit to a two-phase exponential decay curve (Table Curve, Jandel scientific, San Rafael, CA). Experiments were repeated three times.
Fibrin Clot Accretion
The capacity of fibrin-bound batroxobin or thrombin to promote clot accretion was assessed by counting clots for radioactivity after incubation in plasma containing 125I-fibrinogen. Briefly, clots were formed around plastic loops with concentrations of batroxobin or thrombin that yielded comparable clot times. Thus, 8.3 μm γA/γA- or γA/γ′-fibrinogen in HBS-Tw-Ca containing 500 cpm 125I-fibrinogen (for clot standardization) and 30 nm fXIII was incubated with 76.5 units/ml batroxobin or 45 nm thrombin for 30 min at 37 °C. Clots were then incubated in citrated human plasma supplemented with ∼800,000 cpm/ml of 125I-fibrinogen for up to 3.3 h at 23 °C. At intervals, clots were removed from the plasma, washed with HBS-Tw, and counted for radioactivity. Experiments were performed three times, each in triplicate.
Statistical Analysis
Results are presented as the mean ± S.D. The extent of clot accretion over time induced by batroxobin and thrombin was compared by two-way repeated measures analysis of variance using the Greenhouse Geisser correction, whereas t tests were used for other analyses. In all cases, p < 0.05 was considered statistically significant.
RESULTS
Binding of Batroxobin and Thrombin to γA/γA- and γA/γ′-Fibrinogen
Although the interaction of thrombin with γA/γA- and γA/γ′-fibrinogen is well characterized, binding of batroxobin to fibrinogen has not been investigated. SPR was used to quantify binding, and Req values from the sensorgrams are plotted against input protease concentrations. As reported previously, the amount of FPR-thrombin bound to γA/γ′-fibrinogen is about 2-fold greater than that bound to γA/γA-fibrinogen (p < 0.05) (Fig. 2A). In contrast, similar amounts of FPR-batroxobin bind to both isoforms of fibrinogen (Fig. 2B). Sensorgrams showing real time binding to γA/γA-fibrinogen reveal rapid association and dissociation phases for both proteases. However, the off-rate of FPR-batroxobin is about 16-fold slower than that of FPR-thrombin (p < 0.005), which suggests that batroxobin binds γA/γA-fibrinogen with higher affinity than thrombin (Fig. 2, A and B, insets). Kinetic analyses of the on- and off-rates reveal that FPR-thrombin binds γA/γA-fibrinogen via a single site (Kd of 2.3 ± 0.3 μm), whereas thrombin binds γA/γ′-fibrinogen via high and low affinity sites (Kd values of 0.05 ± 0.02 and 1.5 ± 0.1 μm, respectively); these findings are in agreement with previous studies (16, 18). In contrast, FPR-batroxobin binds both γA/γA- and γA/γ′-fibrinogen via a single high affinity site (Kd values of 0.6 ± 0.1 and 0.5 ± 0.04 μm, respectively, p = 0.9). As illustrated in Table 1, the affinities of batroxobin for both isoforms of fibrinogen are 3–4-fold higher than that of thrombin for γA/γA-fibrinogen and resemble the high affinity interaction of thrombin with γA/γ′-fibrinogen. Similar results were obtained in reciprocal experiments, where the binding of fibrinogen to immobilized FPR-batroxobin or FPR-thrombin was assessed (data not shown). The γ′-chain, which distinguishes γA/γ′-fibrinogen from γA/γA-fibrinogen, affords thrombin a high affinity binding site. Because batroxobin binds γA/γA- and γA/γ′-fibrinogen with similar affinities, it is unlikely to bind to the γ′-chain. To verify this, SPR was used to compare the affinity of batroxobin for immobilized γ′-peptide with that of thrombin. Whereas thrombin binds γ′-peptide with a Kd value of 1.1 ± 0.1 μm, batroxobin does not bind (Table 1). These findings suggest that the γ′-region on γA/γ′-fibrinogen is important for thrombin but not batroxobin binding.
FIGURE 2.

SPR analysis of the interaction of FPR-batroxobin and FPR-thrombin with immobilized γA/γA- or γA/γ′-fibrinogen. γA/γA- (●) and γA/γ′-fibrinogen (▴) were adsorbed on individual flow cells to ∼6000–8000 RU. Increasing concentrations (0–15 μm) of FPR-thrombin (A) or FPR-batroxobin (B) were successively injected into flow cells for 2 min, followed by a 4-min wash to monitor dissociation. The amount of protease bound at equilibrium (Req) after background correction is plotted against the input FPR-protease concentration. The insets show representative sensorgrams for the interaction of FPR-thrombin (A) and FPR-batroxobin (B) with γA/γA-fibrinogen in concentrations up to 15 and 6.4 μm, respectively. Data points represent the mean ± S.D. of 2- 3 experiments, and the lines represent nonlinear regression analyses.
TABLE 1.
Dissociation constants for the binding of FPR-thrombin and FPR-batroxobin to fibrinogen, fibrin, and related fragments
The affinities of FPR-thrombin or FPR-batroxobin for immobilized fibrin(ogen), fibrinogen fragments, or peptides was determined using SPR. Kd values were determined by kinetic analysis of the binding sensorgrams. The interaction of thrombin with γA/γ′-fibrin(ogen) was analyzed using a two-site binding model. Each value represents the mean ± S.D. of two to three experiments. NB represents no binding.
| FPR-thrombin |
FPR-batroxobin Kd | ||
|---|---|---|---|
| Kd1 | Kd2 | ||
| μm | μm | μm | |
| γA/γA-Fibrinogen | 2.3 ± 0.3 | 0.6 ± 0.1 | |
| γA/γ′-Fibrinogen | 1.5 ± 0.1 | 0.05 ± 0.02 | 0.5 ± 0.04 |
| γA/γA-Fibrin | 2.0 ± 0.9 | 0.5 ± 0.9 | |
| γA/γ′-Fibrin | 2.1 ± 0.4 | 0.1 ± 0.06 | 0.5 ± 0.9 |
| Fragment D | NB | NB | |
| Fragment E | 3.2 ± 1.5 | 2.1 ± 0.5 | |
| NDSK | 3.0 ± 1.0 | 4.5 + 1.7 | |
| γ′-Peptide | 1.1 ± 0.1 | NB | |
| α(17–51)-Peptide | 2.2 ± 1.0 | >25 | |
Interaction of Batroxobin and Thrombin with γA/γA- and γA/γ′-Fibrin
Thrombin binds fibrinogen and fibrin with similar affinities. To determine whether the same is true for batroxobin, SPR was used to compare the affinity of batroxobin for immobilized γA/γA- and γA/γ′-fibrin with that of thrombin (30, 33). Like thrombin, batroxobin binds both isoforms of fibrin with affinities similar to those for fibrinogen (Table 1); these findings suggest that batroxobin binds fibrin as well as fibrinogen.
To confirm the SPR results, the binding of 125I-YPR-proteases to fibrin clots was assessed. 125I-YPR-thrombin binds γA/γA-fibrin clots with a Kd of 2.5 ± 0.3 μm, whereas it binds γA/γ′-fibrin clots with a composite Kd of 230 ± 70.0 nm (Fig. 3A), and 1.5-fold more thrombin binds γA/γ′-fibrin clots than γA/γA-fibrin clots; these findings are in agreement with previous work (33). In contrast, 125I-YPR-batroxobin binds γA/γA- and γA/γ′-fibrin clots with Kd values of 268 ± 18.5 and 409 ± 48.4 nm (p = 0.18), respectively (Fig. 3B), and similar amounts of batroxobin bind to clots formed from either isoform of fibrinogen. Consistent with their distinct modes of binding to γA/γ′-fibrin, the γ′-peptide-directed IgG has no effect on 125I-YPR-batroxobin binding to γA/γ′-fibrin clots, but it decreases the binding of 125I-YPR-thrombin to γA/γ′-fibrin clots by ∼70% at 8 μm (Fig. 4). In contrast, the γ′-peptide-directed IgG has only nonspecific effects on 125I-YPR-batroxobin and 125I-YPR-thrombin binding to γA/γA-fibrin clots (Fig. 4, inset). Thus, the findings with fibrin clots are similar to those with immobilized fibrin and demonstrate that batroxobin does not bind to the COOH terminus of the γ′-chain.
FIGURE 3.

Binding of 125I-YPR-batroxobin and 125I-YPR-thrombin to γA/γA- or γA/γ′-fibrin clots. 20 nm 125I-YPR-thrombin (A) or 40 nm 125I-YPR-batroxobin (B) was added to microcentrifuge tubes containing 0–8 μm γA/γA- (●) or γA/γ′-fibrinogen (▴), and 10 nm α-thrombin was used to initiate clotting. After incubation for 60 min, clots were pelleted by centrifugation, and free 125I-YPR-protease in the supernatant was used to calculate the bound fraction. Data are plotted as the percentage of 125I-YPR-protease bound to fibrin clots versus the fibrinogen concentration. Data points represent the mean ± S.D. of two experiments, each performed in duplicate, and the lines represent nonlinear regression analyses.
FIGURE 4.

Effect of γ′-peptide-directed IgG on 125I-YPR-batroxobin and 125I-YPR-thrombin binding to γA/γA- or γA/γ′-fibrin clots. The binding of 40 nm 125I-YPR-thrombin (●/▴) or 125I-YPR-batroxobin (○/Δ) to clots formed from 1 μm γA/γA- (circles) or γA/γ′-fibrinogen (triangles) was assessed in the absence or presence of γ′-peptide-directed IgG up to 8 μm. Clots were generated with thrombin and pelleted, and clot-bound 125I-YPR-protease was determined. The percentage of 125I-YPR-protease bound in the absence of antibody is plotted versus antibody concentration. The main figure illustrates the percentage of 125I-YPR-protease bound to γA/γ′-fibrin clots, and the inset shows the percentage of 125I-YPR-protease bound to γA/γA-fibrin clots. Symbols represent the mean ± S.D. of two experiments, each performed in duplicate, and the lines represent nonlinear regression analyses of the data.
Comparison of the Rates of Batroxobin and Thrombin Diffusion from γA/γA- and γA/γ′-Fibrin Clots
To further characterize batroxobin binding to fibrin, we compared the rates of dissociation of batroxobin and thrombin from preformed γA/γA- or γA/γ′-fibrin clots. Consistent with previously reported results, the rate of 125I-YPR-thrombin dissociation from γA/γ′-fibrin clots is 9-fold slower than that from γA/γA-fibrin clots (p < 0.005) (Fig. 5A); this is a difference that reflects the bivalent interaction of thrombin with γA/γ′-fibrin and its univalent interaction with γA/γA-fibrin (18). In contrast, the rates of 125I-YPR-batroxobin diffusion from γA/γA- and γA/γ′-fibrin clots are similar and comparable with the rate of 125I-YPR-thrombin diffusion from γA/γ′-fibrin clots (Fig. 5B). Thus, even though batroxobin does not interact with the γ′-chain, it binds both isoforms of fibrin with affinities similar to the high affinity bivalent interaction of thrombin with γA/γ′-fibrin. As a control, rates of diffusion were determined in the presence of 2 m NaCl and 5 mm EDTA. Under these conditions, the rates of diffusion of batroxobin from both γA/γA- and γA/γ′-fibrin clots are 3–4-fold slower than those of thrombin (p < 0.05) (Fig. 5). This finding raises the possibility that ionic interactions are less important for batroxobin binding to fibrin than they are for thrombin.
FIGURE 5.

Diffusion of 125I-YPR-batroxobin and 125I-YPR-thrombin from γA/γA- or γA/γ′-fibrin clots. Clots were formed around plastic loops by incubating 9 μm γA/γA- (●/○) or γA/γ′-fibrinogen (■/□) with 100 nm thrombin and 30 nm fXIII in the presence of 20 nm 125I-YPR-thrombin (A) or 40 nm 125I-YPR-batroxobin (B) and then immersed in solutions containing TBS-Ca (●/■) or 2 m NaCl and 5 mm EDTA (○/□). At the indicated time points, 0.5-ml aliquots of the bathing solutions were removed and counted for radioactivity to quantify bound 125I-YPR-protease. Data are plotted as the percentage of clot-bound 125I-YPR-protease versus time, and lines represent nonlinear regression analyses. Data points represent the mean ± S.D. of three separate determinations.
Thrombin Competition of Batroxobin Binding to Fibrin(ogen)
SPR was used to determine whether thrombin and batroxobin have overlapping binding sites. The extent to which increasing concentrations of FPR-thrombin attenuate γA/γA-fibrinogen (2.5 μm) binding to immobilized FPR-batroxobin was assessed. FPR-thrombin attenuates γA/γA-fibrinogen binding to adsorbed FPR-batroxobin in a concentration-dependent fashion with an IC50 of 2.5 ± 0.5 μm. At 40 μm, FPR-thrombin reduces fibrinogen binding by 39% (Fig. 6A). Because hirudin binds exosite 1 on thrombin (36), we hypothesized that hirudin would attenuate the capacity of FPR-thrombin to compete with adsorbed FPR-batroxobin for fibrinogen binding. As expected, hirudin reduces competition by FPR-thrombin by 1.3–1.7-fold. This is not a nonspecific protein effect because, even at 40 μm, ovalbumin has no effect on γA/γA-fibrinogen binding to immobilized FPR-batroxobin (data not shown). The finding that saturating concentrations of FPR-thrombin reduce γA/γA-fibrinogen binding to FPR-batroxobin by a maximum of 39% suggests that the batroxobin-binding sites on fibrinogen partially overlap with those of thrombin.
FIGURE 6.

Effect of FPR-thrombin on batroxobin binding to γA/γA-fibrinogen or γA/γA-fibrin clots. A, effect of FPR-thrombin on γA/γA-fibrinogen binding to immobilized FPR-batroxobin was determined using SPR. γA/γA-fibrinogen (2.5 μm) was incubated with FPR-thrombin concentrations up to 40 μm in the absence (●) or presence (■) of 80 μm hirudin, and the mixture was then injected into flow cells containing immobilized FPR-batroxobin. Binding in the presence of competitors was normalized relative to that determined in their absence. Data are plotted as the percentage of γA/γA-fibrinogen bound versus the FPR-thrombin concentration. B, binding of 125I-YPR-batroxobin (■) and 125I-YPR-thrombin (●) to γA/γA-fibrin clots was measured in the absence or presence of FPR-thrombin concentrations up to 25 μm. Clots were formed by incubating 2 or 0.06 μm γA/γA- fibrinogen with 20 nm 125I-YPR-thrombin or 40 nm 125I-YPR-batroxobin, respectively, and 10 nm thrombin. After 60 min, clots were counted for radioactivity to quantify bound 125I-YPR-protease. Data are plotted as the percentage of 125I-YPR-protease bound versus the FPR-thrombin concentration. Data points represent the mean ± S.D. of 3–4 experiments, and the lines represent nonlinear regression analyses.
We next examined the effect of increasing concentrations of FPR-thrombin on 125I-YPR-batroxobin or 125I-YPR-thrombin binding to γA/γA-fibrin clots. At 25 μm, FPR-thrombin reduces 125I-YPR-thrombin binding by ∼60%. In contrast, 25 μm FPR-thrombin reduces 125I-YPR-batroxobin binding by 40%; this value is consistent with the SPR results with fibrinogen (Fig. 6B). Collectively, these data suggest that the batroxobin-binding sites on fibrin(ogen) partially overlap with those of thrombin.
Localization of the Batroxobin-binding Sites on Fibrinogen
To begin to localize the batroxobin-binding sites on fibrinogen, the interaction of batroxobin with immobilized fibrinogen fragments was examined using SPR and compared with that of thrombin. The fibrinogen derivatives include fragment X, which lacks the αC-domains, fragment E, and NDSK, which consist of the NH2 termini of all six chains, and fragment D, which contains the COOH-terminal portions of the three chains. Fragments were generated from γA/γA-fibrinogen to avoid possible contribution of the γ′-chain to thrombin binding. FPR-batroxobin and FPR-thrombin bind to fragment E and NDSK (Kd values of 3–5 μm) but not to fragment D (Table 1). Thrombin binds fragment E and NDSK with affinities comparable with that of intact γA/γA-fibrinogen, suggesting that most of the sequences necessary for thrombin binding to fibrinogen are localized to the E-domain. Like thrombin, batroxobin binds fragment E and NDSK, suggesting that the protease primarily binds to the central region of fibrinogen. However, because batroxobin binds the central E-region with affinities 4–8-fold lower than that for intact γA/γA-fibrinogen, the batroxobin-binding sites may extend beyond the E-domain.
To further distinguish the batroxobin and thrombin-binding sites on fibrin(ogen), their interaction with α(17–51) was compared because this sequence contains a thrombin-binding site (37). Biotinylated α(17–51)-peptide was adsorbed onto a streptavidin-containing cell, and the binding of FPR-batroxobin and FPR-thrombin was assessed by SPR. Whereas FPR-thrombin binds α(17–51)-peptide (Kd value of 2.2 ± 1.0 μm), FPR-batroxobin displays minimal binding (Kd >25 μm), suggesting that the NH2 terminus of the α-chain is only important for thrombin binding (Table 1). To determine the role of the NH2 terminus of the β-chain, the binding of 125I-YPR-batroxobin and 125I-YPR-thrombin to clots generated from intact or des-Bβ(1–42) γA/γA-fibrinogen was examined. 125I-YPR-batroxobin binds intact and des-Bβ(1–42) γA/γA-fibrin with similar affinities (Kd values of 0.27 ± 0.02 and 0.24 ± 0.1 μm, respectively). In contrast, the affinity of thrombin for des-Bβ(1–42) γA/γA-fibrin is 2.6-fold lower than that for intact γA/γA-fibrin (Kd values of 6.5 ± 1.5 and 2.5 ± 0.3 μm, respectively; p = 0.07); this finding is in agreement with previous work (16). These results further support the contention that the batroxobin-binding sites on fibrin(ogen) are distinct from those of thrombin.
Comparison of the Capacities of Fibrin-bound Batroxobin and Thrombin to Promote Clot Accretion
To determine whether, like fibrin-bound thrombin, fibrin-bound batroxobin also promotes clot accretion, γA/γA- and γA/γ′-fibrinogen were clotted with batroxobin or thrombin, and the clots were counted for radioactivity after incubation in citrated plasma containing 125I-labeled fibrinogen. Both fibrin-bound batroxobin and thrombin trigger progressive fibrin accretion over time (Fig. 7). The extent of fibrin accretion onto γA/γA-fibrin clots prepared with thrombin is 1.5–2.0-fold greater than that onto γA/γ′-fibrin clots (Fig. 7, inset, p < 0.001). Therefore, thrombin bound to γA/γA-fibrin clots promotes more fibrin accretion than thrombin bound to γA/γ′-fibrin clots. In contrast, clot accretion induced by batroxobin is similar regardless of whether batroxobin is bound to γA/γA- or γA/γ′-fibrin clots (p = 0.18). It is notable that clot accretion induced by fibrin-bound batroxobin is ∼18-fold greater than that induced by fibrin-bound thrombin (p < 0.001); this indicates that the procoagulant activity of fibrin-bound batroxobin is greater than that of fibrin-bound thrombin, likely reflecting the fact that batroxobin is not inhibited by antithrombin or heparin cofactor II (20).
FIGURE 7.

Clot accretion induced by batroxobin or thrombin bound to γA/γA- or γA/γ′-fibrin clots. Clots were formed around plastic loops by incubating 8.3 μm γA/γA-fibrinogen (●/○) or γA/γ′-fibrinogen (■/□) with 76.5 units/ml of batroxobin (●/■) or 45 nm thrombin (○/□) in the presence of 2 mm CaCl2, 30 nm fXIII, and 500 cpm of 125I-fibrinogen for 30 min at 37 °C. After determining radioactivity, clots were incubated in 1 ml of citrated human plasma containing ∼800,000 cpm/ml of 125I-fibrinogen. At intervals, clots were washed twice with 1 ml of HBS and again counted for radioactivity so that fibrin accretion could be determined. The inset provides an expanded view of clot accretion induced by thrombin. Data points represent the mean ± S.D. of three experiments, each performed in triplicate, and the lines represent nonlinear regression analyses. (*, p < 0.001).
DISCUSSION
Although the thrombin-like activity of batroxobin is well known, the interaction of batroxobin with fibrinogen has not been studied. To address this, we compared the interaction of batroxobin with γA/γA- and γA/γ′-fibrin(ogen) with that of thrombin. Batroxobin forms a high affinity interaction with both isoforms of fibrin(ogen), which resembles the high affinity thrombin interaction with γA/γ′-fibrin(ogen). The batroxobin-binding sites on fibrin(ogen) only partially overlap with those of thrombin because thrombin incompletely displaces batroxobin from fibrinogen and fibrin clots. In keeping with the concept that the batroxobin-binding sites on fibrin(ogen) are distinct from those of thrombin, batroxobin binds NDSK and fragment E with lower affinity than intact γA/γA-fibrinogen, whereas the affinity of thrombin for these fragments is similar to that for fibrinogen, suggesting that the batroxobin-binding sites on fibrinogen extend beyond the E-domain. Furthermore, the α(17–51) and Bβ(1–42) regions in the E-domain are important for thrombin binding but not for batroxobin binding. In addition, fibrin-bound batroxobin retains catalytic activity and is a more potent stimulus for fibrin accretion than fibrin-bound thrombin.
Structural and functional studies have elucidated the structural determinants of thrombin's interaction with fibrinogen (12, 38–41). In the absence of a crystal structure for batroxobin, structural appraisal relies on sequence comparison and modeling. Based on primary sequence alignments, batroxobin exhibits 25 and 37% sequence identity to thrombin and trypsin, respectively. Notably, batroxobin lacks the 60- and γ-loops and the Na+-binding site, domains that endow thrombin with its specificity for a wide repertoire of macromolecular substrates (37, 41). The absence of these domains may explain why fibrinogen is the sole substrate of batroxobin (20). Without the 60- and γ-loops and the Na+-binding site, the interaction of batroxobin with fibrinogen is likely to depend on other residues in the vicinity of the active site of batroxobin (42, 43).
The exosites flanking the active site of thrombin also are critical determinants of its interaction with fibrin(ogen) and other substrates (11, 33, 41). Exosite 1 on thrombin contains hydrophobic and basic regions that mediate substrate and cofactor binding (39, 40). In particular, Tyr-76 of thrombin provides the anchor for a hydrophobic patch surrounded by basic residues that interact with a complementary region on fibrinogen (39, 40). Although only 50% of the corresponding residues are retained, a homologous fibrinogen recognition exosite in batroxobin has been proposed (43, 44). The observation that the interaction of batroxobin with γA/γA-fibrin clots is less readily disrupted by high salt than that of thrombin raises the possibility that hydrophobic interactions may be more important for batroxobin binding to the fibrinogen recognition exosite than they are for thrombin binding. Although both proteases have similar affinities for fibrinogen, batroxobin does not bind other thrombin exosite 1-mediated ligands, such as hirudin or HD1 aptamer (data not shown). These findings support the concept that exosite specificity contributes to the distinct functional properties of batroxobin and thrombin.
Although sequence alignments reveal conserved exosite regions consisting of hydrophobic and basic residues, snake venom proteases possess fewer basic residues in the region corresponding to exosite 1 (44–46). This is demonstrated by modeling the putative exosite residues in batroxobin onto the crystal structure of Trimeresurus stejnejeri venom plasminogen activator (TSV-PA), which has 64% sequence identity to batroxobin (42). The spatial organization of these residues reveals a putative exosite that resembles exosite 1 on thrombin and may contribute to the interaction of batroxobin with fibrin(ogen) (Fig. 8A) (46, 47). Although the existence of a second exosite on batroxobin cannot be excluded, there is little surface homology with thrombin in the exosite 2 region, and a thrombin variant with mutations in exosite 2 displays normal fibrin binding (16).
FIGURE 8.

Models of binding sites on batroxobin and fibrinogen. A, crystal structure of TSV-PA (Protein Data Bank accession number 1BQY) in complex with Glu-Gly-Arg chloromethyl ketone (green) is shown in space-filling format using PyMOL. The image is rotated clockwise ∼90° from standard view with the active site Ser-195 (magenta) facing left. Hydrophobic (yellow) and basic (blue) residues in batroxobin that constitute a putative exosite are mapped onto the TSV-PA structure. Residues are identified using chymotrypsin numbering system. B, crystal structure of the NH2 termini of fibrinogen α- and β-chains forming the thrombin-binding site (Protein Data Bank accession number 2A45) is shown in space-filling format. Both the γ-chains and the reciprocal α- and β-chains are omitted for clarity. The α- and β-chain surfaces are white and gray, respectively. Thrombin binding residues α-chain Phe-35 and Asp-38 and β-chain Ala-68, Asp-69, and Asp-71 are shown within a solid black oval with hydrophobic residues in yellow and acidic residues in red. Residues comprising the putative batroxobin-binding site include α-chain Asp-30, Asp-32, Trp-33, Pro-34, and Phe-35 (hatched black oval). The site where the unmapped α1–26 segment exits the α-chain at Αla-27 indicates the vicinity of FpA.
In addition to the structural differences between the proteases, binding site differences on fibrinogen may also affect specificity. The importance of the β-chain to binding is demonstrated by the observation that FpA release from fibrinogen Naples I, a variant with a point mutation at residue 68 in the Bβ-chain, is normal with batroxobin but impaired with thrombin (48). The crystal structure of thrombin bound to fragment E provides further insight into the differences between the batroxobin and thrombin-binding sites on fibrinogen (40). Thrombin exosite 1 interacts with a patch populated by residues from both the α- and β-chains, including αPhe-35, αAsp-38, βAla-68, βAsp-69, and βAsp-71. Likewise, the α-chain segment DSDWPF (α(30–35)), which lies proximal to where FpA resides, contains a similar combination of acidic and hydrophobic residues that may support batroxobin binding (Fig. 8B). However, our observation that the affinity of thrombin, but not batroxobin, for α(17–51) is comparable with its affinity for intact fibrinogen suggests that a tertiary structure consisting of multiple fibrinogen chains also may be necessary to optimally position batroxobin for FpA cleavage. These results suggest that the proteases have related, but unique, modes of interaction with fibrinogen.
The distinct batroxobin-binding sites in the E-domain of fibrinogen may explain why batroxobin only induces FpA release. Current thinking is that fibrin generation and fibrin monomer assembly in solution occur in two distinct steps. In the first step, thrombin releases FpA, thereby generating des-AA-fibrin monomers that associate to form protofibrils (2). This is followed by a second step whereby FpB is released, resulting in lateral growth of the fibrin protofibrils; this is a process dependent on des-AA-fibrin polymerization (2, 49). Thus, the generation of des-AA-fibrin facilitates FpB release either by rendering the BβArg-14–Gly-15 bond more susceptible to thrombin hydrolysis or by positioning FpB in close proximity to the active site of thrombin (8, 9, 50). A conformational change in the vicinity of the BβArg-14–Gly-15 bond cannot be the sole explanation for FpB release because, like thrombin, batroxobin also generates des-AA-fibrin polymers, yet it does not induce FpB release (20, 35). Whereas thrombin cleaves the BβArg-14–Gly-15 bond on Bβ(1–42)-peptide, thereby releasing FpB, batroxobin does not, which suggests that unlike thrombin, batroxobin does not bind to the Bβ(1–42) region (data not shown). In support of this concept, the affinity of batroxobin for intact and des-Bβ(1–42) γA/γA-fibrinogen is the same, whereas the affinity of thrombin for γA/γA-fibrinogen is reduced by 2.6-fold when the Bβ(1–42) sequence is removed. Therefore, these data suggest that thrombin interaction with the Bβ(1–42) region is an important determinant of its capacity to release FpB. Because batroxobin fails to bind to this region, it does not induce FpB release.
Batroxobin is a potent defibrinogenating enzyme, which is a property that has been exploited in studies of batroxobin for prevention or treatment of thrombosis. Batroxobin binds fibrin(ogen) with high affinity, and fibrin-bound batroxobin is more potent than fibrin-bound thrombin at triggering fibrin accretion, likely because batroxobin is not inhibited by antithrombin and heparin cofactor II (20). Therefore, the procoagulant activity of fibrin-bound batroxobin may contribute to the microvascular thrombosis observed in patients envenomated with B. atrox moojeni (26, 27). The defibrinogenating effects of batroxobin and other venom-derived thrombin-like serine proteases underlie their therapeutic potential. Our results suggest that treatment with these agents may be complicated by microvascular thrombosis, which may explain the disappointing results of studies that explored the use of fibrinogen-depleting snake venom enzymes for treatment of acute ischemic stroke (51).
In summary, we have characterized the interaction of batroxobin with fibrin(ogen) and have shown how it differs from that of thrombin. These differences likely reflect the fact that batroxobin is missing the surface loops and allosteric sites that modulate thrombin's interaction with fibrin(ogen) (37, 41). The structural features on batroxobin that mediate the high affinity interaction with fibrin(ogen) remain to be elucidated.
Acknowledgments
We thank Dr. Boris Zhorov (McMaster University) for helpful discussions about sequence alignments and modeling and Inga Kireeva (McMaster University) for fibrinopeptide sequencing by mass spectrometry.
This work was supported in part by Canadian Institutes of Health Research Grants MOP 3992, MOP 102735, and CTP 79846, Heart and Stroke Foundation of Ontario Grants T4792 and T4730, and the Ontario Research and Development Challenge Fund.
- Fp
- fibrinopeptide
- f
- factor
- FPR
- Phe-Pro-Arg-chloromethyl ketone
- HBS
- Hepes-buffered saline
- NDSK
- NH2-terminal disulfide knot
- Req
- amount of analyte bound at equilibrium
- RU
- response unit
- SPR
- surface plasmon resonance
- TSV-PA
- T. stejnejeri venom plasminogen activator
- Tw
- Tween
- YPR
- Tyr-Pro-Arg-chloromethyl ketone
- γ′-peptide
- synthetic analog of the COOH-terminal portion of fibrinogen γ′-chain.
REFERENCES
- 1. Wolberg A. S. (2012) Determinants of fibrin formation, structure, and function. Curr. Opin. Hematol. 19, 349–356 [DOI] [PubMed] [Google Scholar]
- 2. Mosesson M. W. (2005) Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 3, 1894–1904 [DOI] [PubMed] [Google Scholar]
- 3. Wolfenstein-Todel C., Mosesson M. W. (1980) Human plasma fibrinogen heterogeneity: evidence for an extended carboxyl-terminal sequence in a normal γ chain variant (γ′). Proc. Natl. Acad. Sci. U.S.A. 77, 5069–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chung D. W., Davie E. W. (1984) γ and γ′ chains of human fibrinogen are produced by alternative mRNA processing. Biochemistry 23, 4232–4236 [DOI] [PubMed] [Google Scholar]
- 5. Fornace A. J., Jr., Cummings D. E., Comeau C. M., Kant J. A., Crabtree G. R. (1984) Structure of the human γ-fibrinogen gene. Alternate mRNA splicing near the 3′ end of the gene produces γA and γB forms of γ-fibrinogen. J. Biol. Chem. 259, 12826–12830 [PubMed] [Google Scholar]
- 6. Lovely R. S., Kazmierczak S. C., Massaro J. M., D'Agostino R. B., Sr., O'Donnell C. J., Farrell D. H. (2010) γ′ fibrinogen: evaluation of a new assay for study of associations with cardiovascular disease. Clin. Chem. 56, 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van den Herik E. G., Cheung E. Y., de Lau L. M., den Hertog H. M., Leebeek F. W., Dippel D. W., Koudstaal P. J., de Maat M. P. (2011) γ′/total fibrinogen ratio is associated with short-term outcome in ischaemic stroke. Thromb. Haemost. 105, 430–434 [DOI] [PubMed] [Google Scholar]
- 8. Blombäck B., Hessel B., Hogg D., Therkildsen L. (1978) A two-step fibrinogen-fibrin transition in blood coagulation. Nature 275, 501–505 [DOI] [PubMed] [Google Scholar]
- 9. Mullin J. L., Gorkun O. V., Binnie C. G., Lord S. T. (2000) Recombinant fibrinogen studies reveal that thrombin specificity dictates order of fibrinopeptide release. J. Biol. Chem. 275, 25239–25246 [DOI] [PubMed] [Google Scholar]
- 10. Weitz J. I., Hudoba M., Massel D., Maraganore J., Hirsh J. (1990) Clot-bound thrombin is protected from inhibition by heparin-antithrombin III but is susceptible to inactivation by antithrombin III-independent inhibitors. J. Clin. Invest. 86, 385–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huntington J. A. (2005) Molecular recognition mechanisms of thrombin. J. Thromb. Haemost. 3, 1861–1872 [DOI] [PubMed] [Google Scholar]
- 12. Adams T. E., Huntington J. A. (2006) Thrombin-cofactor interactions: structural insights into regulatory mechanisms. Arterioscler. Thromb. Vasc. Biol. 26, 1738–1745 [DOI] [PubMed] [Google Scholar]
- 13. Fredenburgh J. C., Stafford A. R., Pospisil C. H., Weitz J. I. (2004) Modes and consequences of thrombin's interaction with fibrin. Biophys. Chem. 112, 277–284 [DOI] [PubMed] [Google Scholar]
- 14. Mosesson M. W. (2007) Update on antithrombin I (fibrin). Thromb. Haemost. 98, 105–108 [PubMed] [Google Scholar]
- 15. Meh D. A., Siebenlist K. R., Mosesson M. W. (1996) Identification and characterization of the thrombin-binding sites on fibrin. J. Biol. Chem. 271, 23121–23125 [DOI] [PubMed] [Google Scholar]
- 16. Pospisil C. H., Stafford A. R., Fredenburgh J. C., Weitz J. I. (2003) Evidence that both exosites on thrombin participate in its high affinity interaction with fibrin. J. Biol. Chem. 278, 21584–21591 [DOI] [PubMed] [Google Scholar]
- 17. Becker D. L., Fredenburgh J. C., Stafford A. R., Weitz J. I. (1999) Exosites 1 and 2 are essential for protection of fibrin-bound thrombin from heparin-catalyzed inhibition by antithrombin and heparin cofactor II. J. Biol. Chem. 274, 6226–6233 [DOI] [PubMed] [Google Scholar]
- 18. Fredenburgh J. C., Stafford A. R., Leslie B. A., Weitz J. I. (2008) Bivalent binding to γA/γ′-fibrin engages both exosites of thrombin and protects it from inhibition by the antithrombin-heparin complex. J. Biol. Chem. 283, 2470–2477 [DOI] [PubMed] [Google Scholar]
- 19. Da Graca Salomao M., Wuster W., Thorpe R. S., Touzet J.-M. (1997) DNA evolution of South American pitvipers of the genus Bothrops (Reptilia:Serpentes:Viperidae). Symp. Zool. Soc. Lond. 70, 89–98 [Google Scholar]
- 20. Aronson D. L. (1976) Comparison of the actions of thrombin and the thrombin-like venom enzymes ancrod and batroxobin. Thromb. Haemost. 36, 9–13 [PubMed] [Google Scholar]
- 21. Braud S., Bon C., Wisner A. (2000) Snake venom proteins acting on hemostasis. Biochimie 82, 851–859 [DOI] [PubMed] [Google Scholar]
- 22. Latallo Z. S. (1983) Retrospective study on complications and adverse effects of treatment with thrombin-like enzymes–a multicentre trial. Thromb. Haemost. 50, 604–609 [PubMed] [Google Scholar]
- 23. Stocker K. F. (1998) in Hemostasis and Animal Venoms (Pirkle H., Markland F., eds) pp. 525–540, Marcel Dekker, Inc., Los Angeles [Google Scholar]
- 24. Xu G., Liu X., Zhu W., Yin Q., Zhang R., Fan X. (2007) Feasibility of treating hyperfibrinogenemia with intermittently administered batroxobin in patients with ischemic stroke/transient ischemic attack for secondary prevention. Blood Coagul. Fibrinolysis 18, 193–197 [DOI] [PubMed] [Google Scholar]
- 25. Wang J., Zhu Y. Q., Liu F., Li M. H., Zhao J. G., Tan H. Q., Wang J. B., Cheng Y. S., Zhang P. L. (2010) Batroxobin for prevention of restenosis in diabetic patients after infrapopliteal arterial angioplasty: a small randomized pilot trial. Ann. Vasc. Surg. 24, 876–884 [DOI] [PubMed] [Google Scholar]
- 26. White J. (2005) Snake venoms and coagulopathy. Toxicon 45, 951–967 [DOI] [PubMed] [Google Scholar]
- 27. Isbister G. K. (2010) Snakebite doesn't cause disseminated intravascular coagulation: coagulopathy and thrombotic microangiopathy in snake envenoming. Semin. Thromb. Hemost. 36, 444–451 [DOI] [PubMed] [Google Scholar]
- 28. Schaefer A. V., Leslie B. A., Rischke J. A., Stafford A. R., Fredenburgh J. C., Weitz J. I. (2006) Incorporation of fragment X into fibrin clots renders them more susceptible to lysis by plasmin. Biochemistry 45, 4257–4265 [DOI] [PubMed] [Google Scholar]
- 29. Lovely L. P., Meyer W. R., Ekstrom R. D., Golden R. N. (2003) Effect of stress on pregnancy outcome among women undergoing assisted reproduction procedures. South. Med. J. 96, 548–551 [DOI] [PubMed] [Google Scholar]
- 30. Vu T. T., Stafford A. R., Leslie B. A., Kim P. Y., Fredenburgh J. C., Weitz J. I. (2011) Histidine-rich glycoprotein binds fibrin(ogen) with high affinity and competes with thrombin for binding to the γ′-chain. J. Biol. Chem. 286, 30314–30323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kretz C. A., Cuddy K. K., Stafford A. R., Fredenburgh J. C., Roberts R., Weitz J. I. (2010) HD1, a thrombin- and prothrombin-binding DNA aptamer, inhibits thrombin generation by attenuating prothrombin activation and thrombin feedback reactions. Thromb. Haemost. 103, 83–93 [DOI] [PubMed] [Google Scholar]
- 32. Olexa S. A., Budzynski A. Z. (1979) Binding phenomena of isolated unique plasmic degradation products of human cross-linked fibrin. J. Biol. Chem. 254, 4925–4932 [PubMed] [Google Scholar]
- 33. Petrera N. S., Stafford A. R., Leslie B. A., Kretz C. A., Fredenburgh J. C., Weitz J. I. (2009) Long range communication between exosites 1 and 2 modulates thrombin function. J. Biol. Chem. 284, 25620–25629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cooper A. V., Standeven K. F., Ariëns R. A. (2003) Fibrinogen γ-chain splice variant γ′ alters fibrin formation and structure. Blood 102, 535–540 [DOI] [PubMed] [Google Scholar]
- 35. Moen J. L., Gorkun O. V., Weisel J. W., Lord S. T. (2003) Recombinant BβArg14His fibrinogen implies participation of N terminus of Bβ chain in desA fibrin polymerization. Blood 102, 2466–2471 [DOI] [PubMed] [Google Scholar]
- 36. Stone S. R., Hofsteenge J. (1986) Kinetics of the inhibition of thrombin by hirudin. Biochemistry 25, 4622–4628 [DOI] [PubMed] [Google Scholar]
- 37. Lord S. T., Rooney M. M., Hopfner K. P., Di Cera E. (1995) Binding of fibrinogen Aα1–50-β-galactosidase fusion protein to thrombin stabilizes the slow form. J. Biol. Chem. 270, 24790–24793 [DOI] [PubMed] [Google Scholar]
- 38. Bode W., Mayr I., Baumann U., Huber R., Stone S. R., Hofsteenge J. (1989) The refined 1.9 Å crystal structure of human α-thrombin: interaction with d-Phe-Pro-Arg chloromethyl ketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 8, 3467–3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rose T., LeMosy E. K., Cantwell A. M., Banerjee-Roy D., Skeath J. B., Di Cera E. (2003) Three-dimensional models of proteases involved in patterning of the Drosophila embryo. Crucial role of predicted cation-binding sites. J. Biol. Chem. 278, 11320–11330 [DOI] [PubMed] [Google Scholar]
- 40. Pechik I., Madrazo J., Mosesson M. W., Hernandez I., Gilliland G. L., Medved L. (2004) Crystal structure of the complex between thrombin and the central “E”-region of fibrin. Proc. Natl. Acad. Sci. U.S.A. 101, 2718–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Di Cera E., Cantwell A. M. (2001) Determinants of thrombin specificity. Ann. N.Y. Acad. Sci. 936, 133–146 [DOI] [PubMed] [Google Scholar]
- 42. Parry M. A., Jacob U., Huber R., Wisner A., Bon C., Bode W. (1998) The crystal structure of the novel snake venom plasminogen activator TSV-PA: a prototype structure for snake venom serine proteinases. Structure 6, 1195–1206 [DOI] [PubMed] [Google Scholar]
- 43. Castro H. C., Zingali R. B., Albuquerque M. G., Pujol-Luz M., Rodrigues C. R. (2004) Snake venom thrombin-like enzymes: from reptilase to now. Cell. Mol. Life Sci. 61, 843–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Henschen-Edman A. H., Theodor I., Edwards B. F., Pirkle H. (1999) Crotalase, a fibrinogen-clotting snake venom enzyme: primary structure and evidence for a fibrinogen recognition exosite different from thrombin. Thromb. Haemost. 81, 81–86 [PubMed] [Google Scholar]
- 45. Serrano S. M., Maroun R. C. (2005) Snake venom serine proteinases: sequence homology vs. substrate specificity, a paradox to be solved. Toxicon 45, 1115–1132 [DOI] [PubMed] [Google Scholar]
- 46. Maroun R. C., Serrano S. M. (2004) Identification of the substrate-binding exosites of two snake venom serine proteinases: molecular basis for the partition of two essential functions of thrombin. J. Mol. Recognit. 17, 51–61 [DOI] [PubMed] [Google Scholar]
- 47. Zhang Y., Wisner A., Maroun R. C., Choumet V., Xiong Y., Bon C. (1997) Trimeresurus stejnegeri snake venom plasminogen activator. Site-directed mutagenesis and molecular modeling. J. Biol. Chem. 272, 20531–20537 [DOI] [PubMed] [Google Scholar]
- 48. Koopman J., Haverkate F., Lord S. T., Grimbergen J., Mannucci P. M. (1992) Molecular basis of fibrinogen Naples associated with defective thrombin binding and thrombophilia. Homozygous substitution of Bβ68Ala–Thr. J. Clin. Invest. 90, 238–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weisel J. W., Veklich Y., Gorkun O. (1993) The sequence of cleavage of fibrinopeptides from fibrinogen is important for protofibril formation and enhancement of lateral aggregation in fibrin clots. J. Mol. Biol. 232, 285–297 [DOI] [PubMed] [Google Scholar]
- 50. Riedel T., Suttnar J., Brynda E., Houska M., Medved L., Dyr J. E. (2011) Fibrinopeptides A and B release in the process of surface fibrin formation. Blood 117, 1700–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu M., Marder V. J., Levy D. E., Wang S-J., Yang F., Paganini-Hill A., Fisher M. (2011) Ancrod and fibrin formation: perspectives on mechanisms of action. Stroke 42, 3277–3280 [DOI] [PMC free article] [PubMed] [Google Scholar]