

Background: Traumatic brain injury (TBI) contributes to the development tauopathy-related dementia.
Results: Rapid formation of oligomeric and phosphorylated Tau proteins in a rodent model for TBI.
Conclusion: TBI triggers the formation of Tau oligomers, which may represent a link between TBI and sporadic tauopathies.
Significance: The results suggest that targeting Tau oligomers may be useful for the prevention of dementia following TBI.
Keywords: Neurodegeneration, Protein Aggregation, Protein Assembly, Protein Misfolding, Tau, Tau Aggregation, Tau Oligomers, Tauopathies, Traumatic Brain Injury
Abstract
Traumatic brain injury (TBI) is a serious problem that affects millions of people in the United States alone. Multiple concussions or even a single moderate to severe TBI can also predispose individuals to develop a pathologically distinct form of tauopathy-related dementia at an early age. No effective treatments are currently available for TBI or TBI-related dementia; moreover, only recently has insight been gained regarding the mechanisms behind their connection. Here, we used antibodies to detect oligomeric and phosphorylated Tau proteins in a non-transgenic rodent model of parasagittal fluid percussion injury. Oligomeric and phosphorylated Tau proteins were detected 4 and 24 h and 2 weeks post-TBI in injured, but not sham control rats. These findings suggest that diagnostic tools and therapeutics that target only toxic forms of Tau may provide earlier detection and safe, more effective treatments for tauopathies associated with repetitive neurotrauma.
Introduction
Traumatic brain injury (TBI)3 is often thought of as an acute event resulting from a motor vehicle accident, blast injury, fall, or gunshot wound to the head. In reality, however, the effects from even a mild TBI can be long lasting and negatively impact quality of life. Demonstration of neuronal and glial changes months after TBI (1, 2), in combination with evidence of seizures (3, 4), endocrine disorders (5, 6), and circadian rhythm disruption (7), have led to a paradigm shift in the field, and now TBI is acknowledged as a chronic disease, rather than a single event (8, 9). Repetitive neurotrauma, believed to contribute to the development of chronic traumatic encephalopathy in professional athletes, victims of domestic abuse, and combat veterans, can lead to the development of a progressive form of dementia reminiscent of early onset Alzheimer disease (AD) (10) (and reviewed in Ref. 11).
Neurofibrillary tangles (NFT) may play a role in the pathophysiology of chronic traumatic encephalopathy, but little is known about the mechanisms underlying their formation. Hyperphosphorylation contributes to NFT formation, and until recently, it has generally been accepted that NFT trigger processes that lead to neuronal cell death. However, NFT accumulation alone may be insufficient to cause cell death, as neuronal loss and cognitive deficits precede NFT formation (12–15). Tau oligomers precede the formation of NFT and contribute to learning and memory deficits and neuronal cell death (16–19).
In support of this idea, we demonstrated that oligomeric Tau causes neurotoxicity in vivo (20) and that increased oligomeric Tau species are present in postmortem brain samples from AD patients as compared with healthy controls (21). Moreover, we recently injected Tau oligomers (isolated from AD brains) into wild-type mice; these oligomers disrupt memory and propagate abnormal Tau conformation of endogenous Tau after prolonged incubation (22). Previous studies of Tau in brain, serum, or cerebrospinal fluid following TBI have only examined total native Tau species (23, 24).
Previously, with our antibody (T22) that specifically recognizes oligomeric Tau (21, 22, 25), we detected Tau oligomers in both the PBS and sarkosyl soluble fractions and showed data supporting that Tau oligomers as both intracellular and extracellular deposits (21). Recent reports have examined the involvement of extracellular Tau in the spreading of Tau pathology (26–28) from cell to cell. In the non-transgenic tauopathy model, extracellular Tau spreads by multiple mechanisms (29), including the release of Tau from healthy neurons upon stimulation conditions that simulate normal neuronal activity (30). We believe this release and spreading effect of Tau may contribute to the development of tauopathy following TBI. Here, we used antibodies against oligomeric and hyperphosphorylated Tau in a non-transgenic rodent model of parasagittal fluid percussion injury.
We detected oligomeric and phosphorylated Tau proteins as early as 4 h post-TBI. In our model of fluid percussion TBI, we have detected neuronal cell death as early as 4 h after TBI and do not normally see more than one or two injured neurons in the hippocampus of sham-injured animals (31, 32). Tau oligomers may well be a valuable diagnostic biomarker and therapeutic target for TBI. Strategies designed to prevent Tau aggregation and eliminate these oligomeric toxic forms of Tau specifically, while leaving the functional Tau protein intact and available for microtubule formation (19, 33–35), could lead to the development of more effective treatments for diseases involving Tau protein dysfunction.
EXPERIMENTAL PROCEDURES
Animals
This research was conducted in a facility approved by the American Association for the Accreditation of Laboratory Animal Care, and all experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the University of Texas Medical Branch. Male Sprague-Dawley (Charles Rivers, Wilmington, MA) rats (400–500 grams) were anesthetized (4% isoflurane), incubated, mechanically ventilated with 1.5% isoflurane in O2:air (20:80) using a volume ventilator (NEMI Scientific, New England Medical Instruments, Medway, MA), and prepared for moderate or sham parasagittal fluid percussion injury as described previously (36, 37). Rectal and temporalis muscle temperatures were monitored using telethermometers (Physitemp Instruments, Clifton, NJ), and temperatures were maintained within a range of 37.5 ± 0.5 °C using an overhead lamp and a thermostatically controlled water blanket (Gaymar, Orchard Park, NY). Rats were placed in a stereotaxic apparatus, a midline incision of the skin was performed, and the skull was exposed. With the use of a Michele trephine, a craniotomy was performed 1 mm lateral (right) to the sagittal suture, midway between the lambda and bregma. The bone chip was removed, leaving the dura intact. A modified 20-gauge needle hub was secured in place over the exposed dura with cyanoacrylic adhesive and cemented into place with hygienic dental acrylic.
Parasagittal Fluid Percussion Injury
TBI was administered by means of an fluid percussion injury device (38) consisting of a fluid-filled Plexiglas cylinder 60-cm-long and 4.5 cm in diameter, one end of which was connected to a hollow metal cylinder housing a pressure transducer (Statham PA856-100, Data Instruments, Acton, MA), with the other end closed by a Plexiglas piston mounted on O rings. The transducer housing was connected to the rat by a plenum tube at the craniotomy site. Each TBI was induced by dropping a 4.8-kg steel pendulum that struck the piston. The height of the pendulum determined the intensity of the injury. The fluid pressure pulse was recorded on an oscilloscope triggered photoelectrically by the descent of the pendulum. Four or 24 h after TBI or sham injury (n = 6 at each time point), rats were reanesthetized and transcardially perfused with chilled saline to remove blood, and then brains were collected and immediately frozen.
ELISA
For ELISA, plates were coated with 10 μl of the brain extract using 0.1 m sodium bicarbonate, pH 9.6, as coating buffer, followed by overnight incubation at 4 °C, washing three times with Tris-buffered saline with low Tween (TBST; 0.01%), then blocked for 3 h at room temperature with 10% nonfat dry milk in TBST. The plates were then washed with TBST. Antibodies used include anti-Tau oligomer antibody T22 (1:500), Tau1 (clone PC1C6/MAB3420 1:1000; Millipore, Billerica, MA), GAPDH (1:1000; Abcam, Cambridge, MA) AT8, and AT180 (1:1000; Thermo Scientific, Waltham, MA). All antibodies were diluted in 5% nonfat milk in TBST and were added and allowed to react for 1 h at room temperature. The plates were then washed three times with TBST and 100 μl of horseradish peroxidase-conjugated anti-rabbit IgG (1:3000; GE Healthcare) was added, followed by incubation for 1 h at room temperature. Finally, plates were washed three times with TBST and developed with 3,3′,5,5′,-tetramethylbenzidine (TMB-1 component substrate) from DAKO (Carpinteria, CA). The reaction was stopped with 100 μl of 1 m HCl, and samples were read at 450 nm using a POLARstar OMEGA plate reader (BMG Labtechnologies, Melbourne, Australia).
Western Blot Analysis
Tissue samples were homogenized in 5% ice-cold protease inhibitor. 16 μl of each of the brain extracts were mixed with 4 μl of 4× sample buffer (without boiling) and were run on 4–12% Bis-Tris SDS-PAGE gels and subsequently transferred onto nitrocellulose membranes. After blocking overnight at 4 °C with 10% nonfat milk, membranes were probed for 1 h at room temperature with T22 antibody (1:200), Tau-1 (1:1000; Millipore, Billerica, MA), GAPDH antibody (1:1000; Abcam, Cambridge, MA), AT8, or AT180 antibodies (1:1000; Thermo Scientific) diluted in 5% nonfat dried milk. For detection, horseradish peroxidase-conjugated anti-rabbit IgG and anti-mouse IgG (1:3000; GE Healthcare) and ECL plus (GE Healthcare) were used.
Western Blot Densitometry
Analysis was performed using Labworks software (version 4.5, UVP, Inc., Upland, CA). For protein quantification, the densitometry of each band in the Western blot was normalized with GAPDH. All densitometry results represent the mean and S.D. (n = 6).
Isolation and Characterization of Tau Oligomers Derived Tau Species
Immunoprecipitation experiments were performed as described previously (21, 39). Briefly, tosyl-activated magnetic Dynabeads (Dynal Biotech, Lafayette Hill, PA) were coated with 20 μg of T22 antibody (1.0 mg/ml) diluted in 0.1 m borate, pH 9.5, overnight at 37 °C. Beads were washed (0.2 m Tris, 0.1% bovine serum albumin, pH 8.5) and then incubated with either the 24-h TBI or sham brain homogenate with rotation at room temperature for 1 h. Beads were then washed three times with PBS and eluted using 0.1 m glycine, pH 2.8. The pH of each eluted fraction was adjusted using 1 m Tris, pH 8.0. Fractions were pooled and concentrated 5× using Amicon Ultra Centrifugal Filter units with 30-kDa cut-off (Millipore) (21). Characterization of Tau oligomers isolated by immunoprecipitation was performed by various methods as described previously (21, 40). Size-exclusion chromatography analysis was performed using an LC-6AD Shimadsu HPLC system fitted with a Superdex 200-10/300 GL column (GE Healthcare). 6 μl of isolated sample was adjusted to 50 μl of total volume with filtered 1× PBS for injecting into the column. The running buffer (1× PBS) was used with a flow rate of 0.8 ml/min. The approximate molecular weight of the peaks was estimated using gel filtration standard (Bio-Rad) was used for calibrations (21, 40). Various methods were as previously described (40). The morphology of Tau oligomers isolated by immunoprecipitation was assessed as described previously by atomic force microscopy (AFM) using a non-contact tapping method (ScanAsyst-air) with a Multimode 8 AFM machine (Veeco, CA) (21, 40).
Immunofluorescence
Frozen sections were fixed with chilled acetone for 10 min at room temperature. After blocking in normal goat serum for 1 h, sections were incubated overnight with T22 (1:300). The next day, the sections were washed in PBS three times for 10 min each and then incubated with goat anti-rabbit IgG Alexa Fluor 568 (1:350; Invitrogen) for 1 h. The sections were then washed three times for 10 min each in PBS before incubation overnight with mouse Tau-1 (1:200; Millipore). The next day, the sections were washed in PBS three times for 10 min each prior to incubation with goat anti-mouse IgG Alexa Fluor 488 (1:350; Invitrogen) for 1 h. The sections were then washed three times for 10 min with PBS. Sections were incubated with DAPI (1:3000 in PBS) for 5 min, washed, and mounted in Fluoromount-G mounting medium (Southern Biotech, Birmingham, AL). The sections were examined using a Bio-Rad Radiance 2100 confocal system mounted on a Nikon Eclipse E800 microscope equipped with a CoolSnap-FX monochrome CCD camera (Photometrics, Tucson, AZ) using standard Nikon FITC, Texas Red, and DAPI filters set for Alexa Fluor 488 and 568, and DAPI, respectively.
Peroxidase Immunohistochemistry
Peroxidase immunohistochemistry was performed on frozen sections. In brief, sections (5 μm) were fixed with chilled acetone for 10 min at room temperature and blocked for 1 h with 5% horse serum in PBS. The following antibodies were used for immunostaining: T22 (1:300) and mouse AT8 and AT180 (1:100; Thermo Scientific). Primary antibodies were incubated overnight at 4 °C and detected with biotinylated goat anti-mouse IgG (Vectastin ABC kit; Vector Laboratories, Burlingame, CA) or biotinylated goat anti-rabbit IgG (Vectastin ABC kit; Vector Laboratories) and visualized using a 3,3′-diaminobenzidine peroxidase substrate kit (Vector Laboratories) according to the manufacturer's instructions. Sections were counterstained with hematoxylin (Vector Laboratories) for nuclear staining. Brightfield images were acquired using a Nikon Eclipse 800 microscope equipped with a Nikon DXM1200 color CCD camera (Nikon Instruments, Inc., Melville, NY).
Statistical Analyses
Data were analyzed by one-way analysis of variance followed by Bonferroni's correction for multiple comparisons using GraphPad Prism software (version 5) and OriginPro software (version 8.0). Data in the text and figures are represented as mean ± S.D.
RESULTS
Tau abnormalities in TBI have been investigated extensively, but whether Tau oligomers form in non-transgenic animals in response to TBI is unknown. We investigated the presence of Tau oligomers in the rat brains 4 and 24 h post-injury. First, using a novel anti-Tau oligomer antibody, T22 (21), we observed that Tau oligomers were significantly increased (p < 0.001 TBI versus sham) 4 and 24 h after moderate parasagittal fluid percussion injury injury (Fig. 1A). We were able to detect large amounts of total Tau protein using Tau-1 in both the sham-operated and TBI samples at both time points by ELISA (Fig. 1B), indicative of Tau oligomerization after TBI rather than overproduction of Tau.
FIGURE 1.
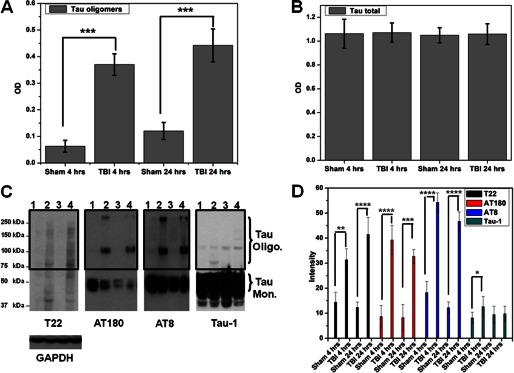
Tau oligomer accumulation after TBI. A, ELISA analysis of TBI and sham brains (PBS extracts) using T22 shows significant increase in Tau oligomers. B, ELISA analysis using Tau-1 antibody shows no differences in total Tau between sham and TBI (PBS extracts). C, representative Western blots of fractions from TBI and sham samples: lane 1, sham after 4 h; lane 2, TBI after 4 h; lane 3, sham after 24 h; lane 4, TBI after 24 h. Shown is a Western blot using T22; high molecular weight bands corresponding to Tau oligomers are detected in TBI brains. Western blots using AT180 show the formation of phosphorylated Tau dimer and large aggregates in TBI samples. Phosphorylated Tau monomers were decreased in sham as compared with TBI; Western blot using AT8 showed results similar to those for AT180, confirming the significant increase in SDS-stable phosphorylated Tau oligomers after TBI. Western blot using Tau-1 showed that non-phosphorylated monomeric Tau levels are unchanged in TBI compared with sham brains. D, quantifications of the oligomeric Tau species (boxed area) as detected by Western blots (n = 6). ****, p < 0.0001; ***, p < 0.0005; **, p < 0.001; *, p < 0.05.
To determine the conformation and aggregation state of Tau proteins following TBI, we performed Western blots using a variety of Tau antibodies (Fig. 1C). Using Western blotting, we detected a pattern of Tau protein following TBI that was similar to that obtained using ELISA, confirming that Tau oligomers were significantly increased after TBI at both 4 and 24 h after injury. There were no significant differences in the quantities of total Tau (Tau-1) detected in any of the groups tested (Fig. 1D). Because hyperphosphorylation of Tau plays a major role in NFT formation (41), we used two different phosphorylated Tau antibodies (Fig. 1C). Western blots using AT8 (42), which detects Tau phosphoylation at the Ser202 and Thr205 sites showed similar results to AT180 (43), which detects Tau phosphorylation at the Thr231 site, confirming increased SDS-stable phosphorylated Tau oligomers after TBI (Fig. 1D). All blots were normalized to GAPDH protein.
To further characterize Tau oligomers formed after TBI, we isolated Tau oligomers from rat brains 24 h post-TBI by immunoprecipitation (21) and analyzed them using size-exclusion chromatography (Fig. 2A) and atomic force microscopy (Fig. 2B). Both of the FPLC chromatogram images show that these oligomers are heterogeneous, with two major populations, with one at ∼100–150 kDa and with a second peak at ∼300 kDa. AFM images confirm the heterogeneous nature of the isolated oligomer, but despite this, no fibrillar material was detected in the isolated oligomers. Moreover, no oligomers were found in the immunoprecipitation samples from sham rat brains (Fig. 2C).
FIGURE 2.

Isolation and characterization of Tau oligomers from TBI brains. Tau oligomers were isolated by immunoprecipitation (IP) using anti-Tau oligomer antibody T22 from rat brains extracted 24 h after TBI. A, size exclusion chromatogram of Tau oligomers showing two main peaks, one at 75–150 kDa, which may include Tau dimer/trimers, and a second peak at 300 kDa. B, AFM image of the TBI brain-derived Tau oligomers. C, AFM image of the material isolated from sham brains.
We were able to localize Tau oligomers (T22) and total Tau (pan Tau antibody Tau-1) in the hippocampus and the cortex of TBI (24-h survival) and sham rats using immunofluorescence (Fig. 3). It is clear that the oligomer seeds detected by T22 are formed from Tau aggregations and represent a small fraction of the total Tau, consistent with the results obtained by Western blot (Fig. 1C).
FIGURE 3.

Tau oligomer accumulation in hippocampus and cortex in TBI. A–C, representative immunofluorescent images of hippocampus 24 h post-TBI showing Tau-1 (A, green), T22 (B, red), and merged signals (C, also including DAPI (blue)), confirming the presence of Tau oligomers in situ. D–F, representative immunofluorescent images of sham (24 h) hippocampal sections. G–I, representative immunofluorescent photomicrographs of cortex sections post TBI (24 h) showing Tau 1 (G, green) and T22 (H, red arrows indicate representative positive pyramidal neurons), and merged images (I, also including DAPI (blue)). J–L, representative immunofluorescent images of sham (24 h) cortical sections. Scale bar, 25 μm.
Phosphorylated Tau (AT180) was detected 24 h after TBI in the CA3 region (Fig. 4A) and dentate gyrus (Fig. 4B), as well as cortex (Fig. 4C). The presence of oligomeric Tau after TBI was further confirmed by T22 staining in the CA3 (Fig. 4D) and dentate gyrus (Fig. 4E) of the hippocampus and in the cortex (Fig. 4F).
FIGURE 4.

Detection of Tau oligomers within 24 h of TBI. A–F, phosphorylated Tau (AT180) and Tau oligomers (T22) were detected in TBI brains (24-h survival) using peroxidase immunohistochemistry in the CA3 (A and D), dentate gyrus (B and E), and cortex (C and F). Scale bar, 10 μm.
Finally, we investigated the presence of Tau oligomers at a longer time point after TBI (Fig. 5): oligomeric Tau (T22) and phosphorylated Tau (AT8) are present 2 weeks after TBI in both the cortex (Fig. 5, A–D) and hippocampus (Fig. 5, E–P). Sham brains were used as a control and displayed significantly less AT8 staining. Oligomeric Tau was present in the brain regions that were ipsilateral and contralateral to the injury site after TBI (24 h and 2-week survival) but not in the sham control brains. Our model of TBI induces cellular damage to the injured (ipsilateral) side of the brain and oftentimes causes nonlethal cellular changes to the uninjured (contralateral) hemisphere (44). These finding were confirmed by ELISA using T22 and AT8 (Fig. 5Q). The persistence of Tau oligomers and phosphorylated Tau 2 weeks after TBI suggests these entities may play a key role in the development of tauopathies following TBI. We have included a schematic of the proposed mechanism linking toxic oligomeric Tau release after neuronal cell injury and death, which may lead to dysfunction and death of the surrounding neurons (Fig. 6).
FIGURE 5.

Detection of Tau oligomers 2 weeks after TBI. Tau oligomers (T22) and phosphorylated Tau (AT8) were detected in TBI brains (2-week (2 wk) survival) using peroxidase immunohistochemistry in the cortex (contralateral (Contra) and ipsilateral (Ipsi) to the injury site) and in the dentate gyrus (DG) and CA1/2 (CA2) regions of the hippocampus (HC). Control sham-injured brains were stained with AT8. Scale bars, 200 μm (A–H) and 50 μm (I–P). Q, Tau oligomers and phosphorylated Tau were quantified by direct ELISA (PBS extracts) using T22 and AT8, respectively. Samples measured in triplicates (n = 2); ***, p < 0.0005.
FIGURE 6.

Schematic showing a proposed mechanism regarding the means by which neuronal injuries and cell death resulting from TBI may lead to rapid formation of toxic Tau oligomers, which may initiate the spread of Tau pathology.
DISCUSSION
This work was designed to explore the role of Tau oligomers in trauma-induced neurodegeneration. Very little is known about the mechanisms involved in the accumulation of toxic Tau proteins after one or multiple TBIs. Oligomeric Tau is now considered one of the major players in the development of tauopathy, possibly acting by a prion-like seeding mechanism (21, 26, 28) that allows for extracellular Tau to spread to neighboring cells (29, 45). Our data, demonstrating the rapid formation of neurotoxic oligomeric and phosphorylated Tau aggregates in a non-transgenic rodent model of parasagittal fluid percussion TBI, represent a novel contribution to our understanding of the early events that lead to Tau aggregation after TBI.
Neuronal Tau plays a key role in microtubule formation, axonal and dendritic transport, and growth and cytoskeletal stability (46). Post-translational modifications of Tau protein can alter its function and may result in microtubule destabilization and cellular dysfunction (47, 48). Hyperphosphorylated and oligomeric Tau proteins have been shown to be toxic to neuronal cells (49) and accumulate following TBI and in AD (21, 50). Hyperphosphorylated Tau increased between 1 and 7 days after cortical impact TBI in triple transgenic AD mice (51), and 3 weeks after repetitive mild TBI in the human Tau (hTau) tauopathy mouse model (52). AT8 was found in the cortex of Sprague-Dawley rats 6 months after fluid percussion injury (53). Even a single blast exposure resulted in Tau hyperphosphorylation and subsequent chronic traumatic encephalopathy development in veterans and in mice (54). Tau oligomerization is known as a key mechanism in the development of NFT, which consist of hyperphosphorylated Tau proteins and play important pathological roles in neurodegenerative tauopathies (18, 19, 21, 55–57). Here, we show that both hyperphosphorylated Tau and oligomeric Tau accumulate acutely after TBI.
Tau, which increases within hours after clinical TBI (58), may be a useful diagnostic marker for neuronal injury. Amyloid-β and Tau proteins were detected in nearly one-third of human brain samples measured years after a single TBI (59). Tau may be of use as a diagnostic tool given that increased levels of extracellular Tau predict a poor outcome following TBI, as confirmed by lower scores on the Glasgow Outcome Scale 60 months post-TBI (60).
Recently, two groups of investigators (61, 62) reported regional spread of Tau pathology. The proposed trans-synaptic mechanism of Tau spreading along anatomically connected networks, between connected and vulnerable neurons (61), and the seeded propagation of endogenous Tau in vivo downstream in interconnected synaptic circuits are intriguing processes that may occur after TBI. Tau aggregation and propagation in animal models can be induced using exogenous brain extracts containing Tau aggregates (26, 63), and our recent results demonstrate that Tau oligomers, not NFT, are responsible for the initiation and spread of Tau pathology in a manner reminiscent of sporadic tauopathies (22); thus, it is possible that elevated levels of extracellular Tau following TBI (60) accelerate the formation of oligomeric seeds, leading to the spread of Tau pathology in TBI via a prion-like mechanism (22, 27, 29). Fig. 6 shows a schematic of the proposed mechanism whereby neuronal injury and death resulting from TBI leads to rapid formation of toxic Tau oligomers, which may initiate the spread of Tau pathology in the brain (28, 29).
The characterization of these Tau species might lead to powerful new insights regarding the mechanisms underlying Tau aggregation, its acute and long term consequences following TBI, and the development of diagnostic tools and therapeutic interventions. Discovery of effective therapeutic interventions to reduce or abolish posttraumatic dementia would be of significant benefit to civilian patients with TBI as well as military veterans returning with blast-related brain injuries.
Acknowledgments
We thank Debbie Boone and Dr. Helen L. Hellmich for expertise with the traumatic brain injury samples. We are grateful to Professor Alan D. T. Barrett for suggestions and support.
This work was supported by the Cullen Family Trust for Health Care, the Mitchell Center for Neurodegenerative Diseases, and the Moody Center for Traumatic Brain & Spinal Cord Injury Research/Mission Connect. R. K. has patent applications on the compositions and methods related to Tau oligomers and antibodies.
- TBI
- traumatic brain injury
- AD
- Alzheimer disease
- NFT
- neurofibrillary tangles
- TBST
- Tris-buffered saline with low Tween
- AFM
- atomic force microscopy.
REFERENCES
- 1. Colicos M. A., Dixon C. E., Dash P. K. (1996) Delayed, selective neuronal death following experimental cortical impact injury in rats: possible role in memory deficits. Brain Res. 739, 111–119 [DOI] [PubMed] [Google Scholar]
- 2. Smith D. H., Chen X. H., Pierce J. E., Wolf J. A., Trojanowski J. Q., Graham D. I., McIntosh T. K. (1997) Progressive atrophy and neuron death for one year following brain trauma in the rat. J. Neurotrauma 14, 715–727 [DOI] [PubMed] [Google Scholar]
- 3. McKinney R. A., Debanne D., Gähwiler B. H., Thompson S. M. (1997) Lesion-induced axonal sprouting and hyperexcitability in the hippocampus in vitro: implications for the genesis of posttraumatic epilepsy. Nat. Med. 3, 990–996 [DOI] [PubMed] [Google Scholar]
- 4. Pitkänen A., McIntosh T. K. (2006) Animal models of post-traumatic epilepsy. J. Neurotrauma 23, 241–261 [DOI] [PubMed] [Google Scholar]
- 5. Behan L. A., Phillips J., Thompson C. J., Agha A. (2008) Neuroendocrine disorders after traumatic brain injury. J. Neurol. Neurosurg. Psychiatry 79, 753–759 [DOI] [PubMed] [Google Scholar]
- 6. Taylor A. N., Rahman S. U., Tio D. L., Sanders M. J., Bando J. K., Truong A. H., Prolo P. (2006) Lasting neuroendocrine-immune effects of traumatic brain injury in rats. J. Neurotrauma 23, 1802–1813 [DOI] [PubMed] [Google Scholar]
- 7. Orff H. J., Ayalon L., Drummond S. P. (2009) Traumatic brain injury and sleep disturbance: a review of current research. J. Head Trauma Rehabil. 24, 155–165 [DOI] [PubMed] [Google Scholar]
- 8. Masel B. E., DeWitt D. S. (2010) Traumatic brain injury: a disease process, not an event. J. Neurotrauma 27, 1529–1540 [DOI] [PubMed] [Google Scholar]
- 9. Johnson V. E., Stewart W., Smith D. H. (2010) Traumatic brain injury and amyloid-β pathology: a link to Alzheimer's disease? Nat. Rev. Neurosci. 11, 361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fleminger S., Oliver D. L., Lovestone S., Rabe-Hesketh S., Giora A. (2003) Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 74, 857–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McKee A. C., Cantu R. C., Nowinski C. J., Hedley-Whyte E. T., Gavett B. E., Budson A. E., Santini V. E., Lee H. S., Kubilus C. A., Stern R. A. (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 68, 709–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kril J. J., Patel S., Harding A. J., Halliday G. M. (2002) Neuron loss from the hippocampus of Alzheimer's disease exceeds extracellular neurofibrillary tangle formation. Acta Neuropathol. 103, 370–376 [DOI] [PubMed] [Google Scholar]
- 13. Haroutunian V., Davies P., Vianna C., Buxbaum J. D., Purohit D. P. (2007) Tau protein abnormalities associated with the progression of alzheimer disease type dementia. Neurobiol. Aging 28, 1–7 [DOI] [PubMed] [Google Scholar]
- 14. Morsch R., Simon W., Coleman P. D. (1999) Neurons may live for decades with neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 58, 188–197 [DOI] [PubMed] [Google Scholar]
- 15. Gómez-Isla T., Hollister R., West H., Mui S., Growdon J. H., Petersen R. C., Parisi J. E., Hyman B. T. (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann. Neurol. 41, 17–24 [DOI] [PubMed] [Google Scholar]
- 16. Spires T. L., Orne J. D., SantaCruz K., Pitstick R., Carlson G. A., Ashe K. H., Hyman B. T. (2006) Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am. J. Pathol. 168, 1598–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berger Z., Roder H., Hanna A., Carlson A., Rangachari V., Yue M., Wszolek Z., Ashe K., Knight J., Dickson D., Andorfer C., Rosenberry T. L., Lewis J., Hutton M., Janus C. (2007) Accumulation of pathological Tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 27, 3650–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cowan C. M., Quraishe S., Mudher A. (2012) What is the pathological significance of tau oligomers? Biochem. Soc. Trans. 40, 693–697 [DOI] [PubMed] [Google Scholar]
- 19. Lasagna-Reeves C. A., Castillo-Carranza D. L., Jackson G. R., Kayed R. (2011) Tau oligomers as potential targets for immunotherapy for Alzheimer's disease and tauopathies. Curr. Alzheimer Res. 8, 659–665 [DOI] [PubMed] [Google Scholar]
- 20. Lasagna-Reeves C. A., Castillo-Carranza D. L., Sengupta U., Clos A. L., Jackson G. R., Kayed R. (2011b) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lasagna-Reeves C. A., Castillo-Carranza D. L., Sengupta U., Sarmiento J., Troncoso J., Jackson G. R., Kayed R. (2012) Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. FASEB J. 26, 1946–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lasagna-Reeves C. A., Castillo-Carranza D. L., Sengupta U., Guerrero-Munoz M. J., Kiritoshi T., Neugebauer V., Jackson G. R., Kayed R. (2012) Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gabbita S. P., Scheff S. W., Menard R. M., Roberts K., Fugaccia I., Zemlan F. P. (2005) Cleaved-tau: a biomarker of neuronal damage after traumatic brain injury. J. Neurotrauma 22, 83–94 [DOI] [PubMed] [Google Scholar]
- 24. Liliang P. C., Liang C. L., Lu K., Wang K. W., Weng H. C., Hsieh C. H., Tsai Y. D., Chen H. J. (2010) Relationship between injury severity and serum tau protein levels in traumatic brain injured rats. Resuscitation 81, 1205–1208 [DOI] [PubMed] [Google Scholar]
- 25. Wu J. W., Herman M., Liu L., Simoes S., Acker C. M., Figueroa H., Steinberg J. I., Margittai M., Kayed R., Zurzolo C., Di Paolo G., Duff K. E. (2013) Small Misfolded Tau Species Are Internalized via Bulk Endocytosis and Anterogradely and Retrogradely Transported in Neurons. J. Biol. Chem. 288, 1856–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clavaguera F., Bolmont T., Crowther R. A., Abramowski D., Frank S., Probst A., Fraser G., Stalder A. K., Beibel M., Staufenbiel M., Jucker M., Goedert M., Tolnay M. (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hall G. F., Patuto B. A. (2012) Is tau ready for admission to the prion club? Prion 6, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frost B., Diamond M. I. (2010) Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 11, 155–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Le M. N., Kim W., Lee S., McKee A. C., Hall G. F. (2012) Multiple mechanisms of extracellular tau spreading in a non-transgenic tauopathy model. Am. J. Neurodegener. Dis. 1, 316–333 [PMC free article] [PubMed] [Google Scholar]
- 30. Pooler A. M., Phillips E. C., Lau D. H., Noble W., Hanger D. P. (2013) Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hellmich H. L., Capra B., Eidson K., Garcia J., Kennedy D., Uchida T., Parsley M., Cowart J., DeWitt D. S., Prough D. S. (2005) Dose-dependent neuronal injury after traumatic brain injury. Brain Res. 1044, 144–154 [DOI] [PubMed] [Google Scholar]
- 32. Hellmich H. L., Eidson K. A., Capra B. A., Garcia J. M., Boone D. R., Hawkins B. E., Uchida T., Dewitt D. S., Prough D. S. (2007) Injured Fluoro-Jade-positive hippocampal neurons contain high levels of zinc after traumatic brain injury. Brain Res. 1127, 119–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schneider A., Mandelkow E. (2008) Tau-based treatment strategies in neurodegenerative diseases. Neurotherapeutics 5, 443–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Castillo-Carranza D. L., Lasagna-Reeves C. A., Kayed R. (2013) Tau aggregates as immunotherapeutic targets. Front. Biosci. (Schol Ed) 5, 426–438 [DOI] [PubMed] [Google Scholar]
- 35. Ubhi K., Masliah E. (2011) Recent advances in the development of immunotherapies for tauopathies. Exp. Neurol. 230, 157–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hawkins B. E., Frederickson C. J., Dewitt D. S., Prough D. S. (2012) Fluorophilia: fluorophore-containing compounds adhere non-specifically to injured neurons. Brain Res. 1432, 28–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li Y., Hawkins B. E., DeWitt D. S., Prough D. S., Maret W. (2010) The relationship between transient zinc ion fluctuations and redox signaling in the pathways of secondary cellular injury: relevance to traumatic brain injury. Brain Res. 1330, 131–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dixon C. E., Lyeth B. G., Povlishock J. T., Findling R. L., Hamm R. J., Marmarou A., Young H. F., Hayes R. L. (1987) A fluid percussion model of experimental brain injury in the rat. J. Neurosurg. 67, 110–119 [DOI] [PubMed] [Google Scholar]
- 39. Lasagna-Reeves C. A., Glabe C. G., Kayed R. (2011) Amyloid-β annular protofibrils evade fibrillar fate in Alzheimer disease brain. J. Biol. Chem. 286, 22122–22130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lasagna-Reeves C. A., Castillo-Carranza D. L., Guerrero-Muoz M. J., Jackson G. R., Kayed R. (2010) Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49, 10039–10041 [DOI] [PubMed] [Google Scholar]
- 41. Augustinack J. C., Schneider A., Mandelkow E. M., Hyman B. T. (2002) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 103, 26–35 [DOI] [PubMed] [Google Scholar]
- 42. Biernat J., Mandelkow E. M., Schröter C., Lichtenberg-Kraag B., Steiner B., Berling B., Meyer H., Mercken M., Vandermeeren A., Goedert M. (1992) The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 11, 1593–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goedert M., Jakes R., Crowther R. A., Cohen P., Vanmechelen E., Vandermeeren M., Cras P. (1994) Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer's disease: identification of phosphorylation sites in tau protein. Biochem. J. 301, 871–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rojo D. R., Prough D. S., Falduto M. T., Boone D. R., Micci M. A., Kahrig K. M., Crookshanks J. M., Jimenez A., Uchida T., Cowart J. C., Hawkins B. E., Avila M., DeWitt D. S., Hellmich H. L. (2011) Influence of stochastic gene expression on the cell survival rheostat after traumatic brain injury. PLoS One 6, e23111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tran H. T., Sanchez L., Brody D. L. (2012) Inhibition of JNK by a peptide inhibitor reduces traumatic brain injury-induced tauopathy in transgenic mice. J. Neuropathol. Exp. Neurol. 71, 116–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Johnson G. V., Stoothoff W. H. (2004) Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 117, 5721–5729 [DOI] [PubMed] [Google Scholar]
- 47. Janke C., Bulinski J. C. (2011) Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat. Rev. Mol. Cell Biol. 12, 773–786 [DOI] [PubMed] [Google Scholar]
- 48. Wang J. Z., Xia Y. Y., Grundke-Iqbal I., Iqbal K. (2013) Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J. Alzheimers Dis. 33, S123–39 [DOI] [PubMed] [Google Scholar]
- 49. Castillo-Carranza D. L., Lasagna-Reeves C., Guerrero-Munoz M. J., Estes D. M., Barrett A., Dineley K., Jackson G. R., R. K. (2010) Modulation of tau oligomers by passive vaccination. Soc. Neurosci. Abstract 347.11 [Google Scholar]
- 50. Maeda S., Sahara N., Saito Y., Murayama S., Ikai A., Takashima A. (2006) Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci. Res. 54, 197–201 [DOI] [PubMed] [Google Scholar]
- 51. Tran H. T., LaFerla F. M., Holtzman D. M., Brody D. L. (2011) Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-β accumulation and independently accelerates the development of tau abnormalities. J. Neurosci. 31, 9513–9525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ojo J. O., Mouzon B., Greenberg M. B., Bachmeier C., Mullan M., Crawford F. (2013) Repetitive mild traumatic brain injury augments tau pathology and glial activation in aged hTau mice. J. Neuropathol. Exp. Neurol. 72, 137–151 [DOI] [PubMed] [Google Scholar]
- 53. Hoshino S., Tamaoka A., Takahashi M., Kobayashi S., Furukawa T., Oaki Y., Mori O., Matsuno S., Shoji S., Inomata M., Teramoto A. (1998) Emergence of immunoreactivities for phosphorylated tau and amyloid-β protein in chronic stage of fluid percussion injury in rat brain. Neuroreport 9, 1879–1883 [DOI] [PubMed] [Google Scholar]
- 54. Goldstein L. E., Fisher A. M., Tagge C. A., Zhang X. L., Velisek L., Sullivan J. A., Upreti C., Kracht J. M., Ericsson M., Wojnarowicz M. W., Goletiani C. J., Maglakelidze G. M., Casey N., Moncaster J. A., Minaeva O., Moir R. D., Nowinski C. J., Stern R. A., Cantu R. C., Geiling J., Blusztajn J. K., Wolozin B. L., Ikezu T., Stein T. D., Budson A. E., Kowall N. W., Chargin D., Sharon A., Saman S., Hall G. F., Moss W. C., Cleveland R. O., Tanzi R. E., Stanton P. K., McKee A. C. (2012) Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 4, 134ra160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Henkins K. M., Sokolow S., Miller C. A., Vinters H. V., Poon W. W., Cornwell L. B., Saing T., Gylys K. H. (2012) Extensive p-Tau Pathology and SDS-Stable p-Tau Oligomers in Alzheimer's Cortical Synapses. Brain Pathol. 22, 826–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Patterson K. R., Remmers C., Fu Y., Brooker S., Kanaan N. M., Vana L., Ward S., Reyes J. F., Philibert K., Glucksman M. J., Binder L. I. (2011) Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 286, 23063–23076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tai H. C., Serrano-Pozo A., Hashimoto T., Frosch M. P., Spires-Jones T. L., Hyman B. T. (2012) The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 181, 1426–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Uryu K., Chen X. H., Martinez D., Browne K. D., Johnson V. E., Graham D. I., Lee V. M., Trojanowski J. Q., Smith D. H. (2007) Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 208, 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Johnson V. E., Stewart W., Smith D. H. (2012) Widespread tau and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol. 22, 142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Magnoni S., Esparza T. J., Conte V., Carbonara M., Carrabba G., Holtzman D. M., Zipfel G. J., Stocchetti N., Brody D. L. (2012) Tau elevations in the brain extracellular space correlate with reduced amyloid-β levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain 135, 1268–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu L., Drouet V., Wu J. W., Witter M. P., Small S. A., Clelland C., Duff K. (2012) Trans-synaptic spread of tau pathology in vivo. PLoS One 7, e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. de Calignon A., Polydoro M., Suárez-Calvet M., William C., Adamowicz D. H., Kopeikina K. J., Pitstick R., Sahara N., Ashe K. H., Carlson G. A., Spires-Jones T. L., Hyman B. T. (2012) Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Iba M., Guo J. L., McBride J. D., Zhang B., Trojanowski J. Q., Lee V. M. (2013) Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J. Neurosci. 33, 1024–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]