

Abstract
Cellular iron homeostasis is maintained by iron regulatory proteins 1 and 2 (IRP1 and IRP2). IRPs bind to iron-responsive elements (IREs) located in the untranslated regions of mRNAs encoding protein involved in iron uptake, storage, utilization and export. Over the past decade, significant progress has been made in understanding how IRPs are regulated by iron-dependent and iron-independent mechanisms and the pathological consequences of IRP2 deficiency in mice. The identification of novel IREs involved in diverse cellular pathways has revealed that the IRP–IRE network extends to processes other than iron homeostasis. A mechanistic understanding of IRP regulation will likely yield important insights into the basis of disorders of iron metabolism. This article is part of a Special Issue entitled: Cell Biology of Metals.
Keywords: Iron, IRP, Iron-responsive element, RNA-binding protein, Iron-sulfur protein, Post-transcriptional regulation
1. Introduction
Iron is required by most organisms as it serves as a prosthetic group for proteins involved in central cellular processes, including respiration, DNA synthesis and oxygen transport. In excess, cellular iron catalyzes the generation of free radicals that damage protein, DNA and lipids, whereas cellular iron deficiency impairs cellular proliferation. In humans, with the inherited diseases hemochromatosis, excess cellular iron can result in cirrhosis, cardiomyopathy and diabetes mellitus (reviewed in [1]). Excess iron content in the brain is associated with several inherited neurodegenerative diseases, including neurodegeneration with brain iron accumulation (NBIA) and Friedreich’s ataxia (FA), as well as common neurodegenerative disorders such as Parkinson’s and Alzheimer’s diseases (reviewed in [1–3]). On the other hand, iron deficiency affects billions of people worldwide, and results in cognitive defects in children and anemia in adults. Cellular iron content must therefore be maintained within a narrow range to avoid the adverse consequences of iron deficiency or excess. Maintenance of cellular iron homeostasis is accomplished by the coordinated regulation of iron uptake, storage and export by iron-regulatory proteins 1 and 2 (IRP1 and IRP2, also known as ACO1 and IREB2). This review focuses on advances in IRP regulation of iron metabolism focusing on the past 6 years. We direct readers to several excellent reviews on IRPs [4–7] and to chapters in this volume on systematic iron homeostasis, iron–sulfur (Fe–S) cluster biogenesis, iron trafficking, and SKP1–CUL1–FBXL5 structure and function.
2. Overview of systemic and cellular iron metabolism
2.1. Systemic iron metabolism
Approximately, 1–3 mg of iron is absorbed by humans each day, in order to replace iron losses in the urine, sweat and from desquamated enterocytes. As mammals lack a regulated physiological mechanism for iron excretion, intestinal iron absorption is a highly regulated process. Dietary non-heme ferric ironis absorbed by enterocytes by divalent metal transporter 1 (SLC11A2, also known as DMT1 and NRAMP2) after reduction by membrane bound ferrireductases (such as duodenal cyto-chrome b (CYBRD1, also known as DCYTB)) (Fig. 1). Heme iron is also taken up by enterocytes by an undefined mechanism. Cellular iron is translocated through the enterocyte and is exported into the circulation by the basolateral exporter ferroportin (SLC40A1). Iron export is dependent on the oxidation of iron by the membrane bound multicopper oxidase hephaestin enabling it to bind to plasma transferrin (Tf). Diferric Tf (Tf-Fe(III) is the major form of iron for most cells with erythroid precursors being the major user as iron is required for heme synthesis. Tf-Fe(III) binds to cell surface transferrin receptor 1 (TFRC, also known as TfR1) followed by internalization of the Tf-Fe(III)–TfR1 complex by clathrin-mediated endocytosis. On acidification of the endosome, ferric iron is released from Tf and reduced by STEAP3, and exported into the cytosol by DMT1. Cytosolic iron is used for the formation of iron-containing proteins and by the mitochondria for biosynthesis of Fe–S clusters and heme. When body iron stores and erythropoiesis are adequate, iron export from enterocytes is reduced by hepcidin-mediated internalization and degradation of ferroportin, and instead iron is stored in ferritin. Iron in ferritin is lost after 3 days by desquamation of intestinal cells.
Fig. 1.
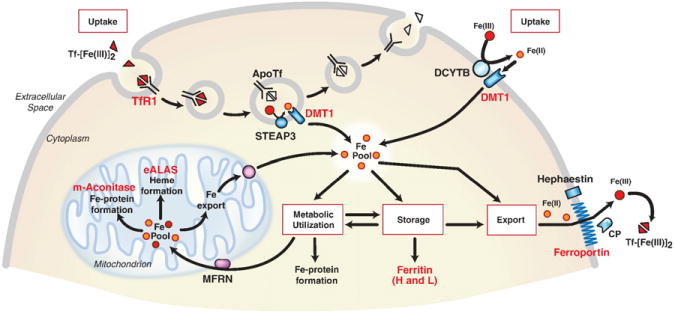
Control of mammalian cellular iron homeostasis by the IRE–IRP regulatory network. A generic mammalian cell depicting roles of proteins encoded by IRE-containing mRNAs (red lettering). Transferrin bound to two iron atoms (Tf-[Fe(III)2) binds to TfR1 on the cell surface where the Tf-[Fe(III)2–TfR1 complex is endocytosed. Acidification of the endosome causes the release of Fe(III) (red balls) from Tf where it is reduced to Fe(II) (orange balls) by the STEAP3 oxidoreductase before export by DMT1 (divalent metal transporter 1). ApoTf/TfR1 complex is returned to the cell surface where it dissociates and initiates another round of iron uptake. Tf-bound iron is taken up by most cells, but it is especially important in erythroid precursors where it is the primary source of iron for heme synthesis. DMT1 is also localized on the apical membrane of duodenal enterocytes where it transports Fe(II) after reduction by membrane reductases, such as DCYTB. Iron taken up by either TfR1 or DMT1 enters a cytosolic free labile iron pool thought to consist of Fe(II) bound to small molecular weight molecules. IRPs sense iron in this pool and regulate the translation of 5′ IRE-containing mRNAs (H-ferritin and L-ferritin, eALAS (erythroid aminolevulinate synthase), mitochondrial (m)-aconitase and ferroportin) or the stability of 3′ IRE-containing mRNAs (TfR1 and DMT1). eALAS serves as the rate-limiting enzyme in heme synthesis in erythroid precursors. Mitochondrial aconitase is an enzyme in the TCA cycle that requires a [4Fe–4S] cluster for activity. Iron that is not utilized or stored in ferritin is exported by ferroportin. Export of iron from cells is coupled to the oxidation of iron by membrane-bound hephaestin or serum multicopper oxidase ceruloplasmin (CP).
Modified from Wallander et al. [4].
The small amount of iron (1–3 mg) absorbed by humans each day represents only a fraction of the total body iron, and most of the circulating iron comes from the recycling of heme from senescent erythrocytes by reticuloendothelial macrophages. The iron released from heme is exported into the circulation by ferroportin where it binds to apoTf for delivery to bone marrow for hemoglobin synthesis. Elevated hepatic iron stores and inflammation increase hepcidin production, which mediates ferroportin degradation in intestinal cells and in reticuloendothelial macrophages, leading to reduced plasma iron levels.
2.2. IRPs are the principal cellular iron regulators in vertebrates
Maintaining cellular iron content requires precise mechanisms for regulating its uptake, storage and export. IRP1 and IRP2 are the principal regulators of cellular iron homeostasis in vertebrates. IRPs are cytosolic proteins that bind to iron-responsive elements (IREs) in the 5′ or 3′ untranslated regions of mRNAs encoding proteins involved in iron uptake (TfR1, DMT1), sequestration (H-ferritin subunit (FTH1) and L-ferritin subunit (FTL)) and export (ferroportin) (Fig. 2). When cells are iron deficient, IRPs bind to 5′ IREs in ferritin and ferroportin mRNAs with high affinity to repress translation, and to 3′ IREs in TfR1 mRNA to block its degradation. When iron is in excess, IRPs do not bind to IREs, increasing synthesis of ferritin and ferroportin, while promoting the degradation of TfR1 mRNA. The coordinated regulation of iron uptake, storage and export by the IRPs ensures that cells acquire adequate iron for their needs without reaching toxic levels.
Fig. 2.
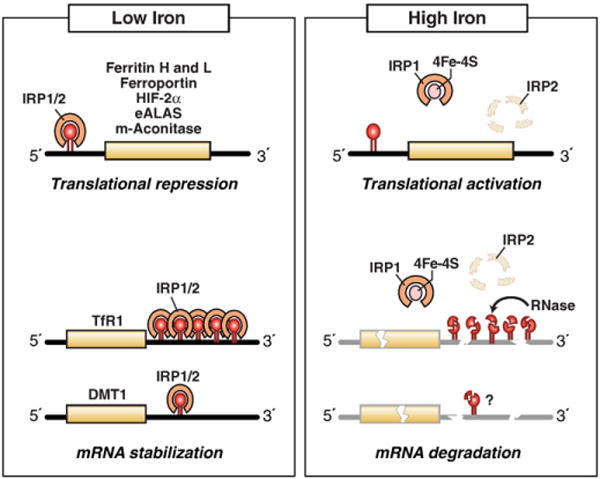
IRPs regulate translation and stability of IRE-containing mRNAs. IRPs bind to IREs located in either the 5′ or 3′ untranslated regions of specific mRNAs. When iron is limited, IRPs bind with high affinity to 5′ IRE mRNAs and repress translation, and to the five 3′ IREs in TfR1 mRNA and to the single IRE in DMT1 mRNA and stabilize these mRNAs. When iron is abundant, IRPs do not bind IREs, resulting in the translation of 5′ IRE-containing mRNAs and degradation of TfR1 mRNA. Iron mediates the conversion of the IRP1 RNA binding form into the [4Fe–4S] cluster c-aconitase form and the ubiquitination and targeted proteasomal degradation IRP2 by FBXL5 E3 ligase. IRE-containing mRNAs indicated are those that have been shown to be functional in vivo.
Modified from Wallander et al. [4].
IRPs also regulate other mRNAs that contain 5′ IREs, including mitochondrial aconitase (ACO2, also known as m-acon, tricarboxylic acid cycle (TCA)), erythroid aminolevulinate synthase (ALAS2, also known as eALAS, heme biosynthesis), hypoxia-inducible transcription factor-2α (EPAS1 also known as HIF-2α, hypoxia adaptation) and amyloid beta precursor protein (APP, Alzheimer’s disease), and 3′ IREs, including DMT1 (metal transport), CDC14A (mitotic phosphatase), CDC42-binding protein kinase α (also known as MRCKα, cytoskeletal dynamics) and hydroxyacid oxidase 1 (HAO1, peroxisomal enzyme) [8–17]. More recently, a transcriptome-wide approach was utilized to identify 35 novel putative IRE-containing mRNAs, some of which have exclusivity for IRP1 or IRP2 [18]. While these mRNAs each contain an IRE, the roles of many of these IREs in vivo remain to be determined. The presence of IREs in a variety of mRNAs indicates that IRP regulation extends to processes other than iron homeostasis.
3. Recent advances in IRP1 regulation
Although IRP1 and IRP2 share approximately 64% identity, bind RNA and are regulated by iron, they differ in several ways. First, IRP1 is a bifunctional protein serving either as a high affinity IRE binding protein in its apoprotein form or the cytosolic isoform of the iron– sulfur (Fe–S) enzyme aconitase (c-acon) (Fig. 3). Formation and loss of the [4Fe–4S] cluster regulates RNA binding and provides unique links between IRP1 and cellular Fe–S cluster biogenesis. Second, IRP1 has distinctive roles in the adaptive response to oxygen availability as well as in sensing reactive oxygen species (ROS) or reactive nitrogen species (RNS). Third, IRP1 is controlled by multiple iron-dependent mechanisms, including targeted protein degradation to limit accumulation of the RNA binding form of IRP1 [19]. Fourth, phosphorylation of IRP1 at serine 138 dictates the mechanism of its regulation by iron. Unlike IRP1, IRP2 lacks a [4Fe–4S] cluster and aconitase activity, and functions as an RNA binding protein. IRP2 is primarily regulated by iron-mediated degradation. Thus, IRP1 responds to iron-dependent and iron-independent signals that control its function through multiple mechanisms in order to maintain IRE-binding activity within a range that permits the optimal maintenance of cellular iron homeostasis.
Fig. 3.
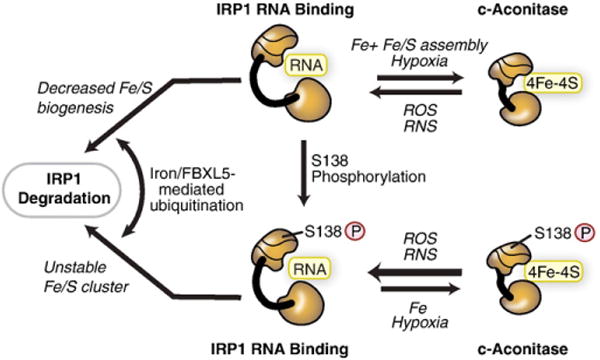
Iron-dependent and iron-independent mechanisms for regulating IRP1. IRP1 can be regulated through mechanisms dependent or independent of the [4Fe–4S] cluster. In the cluster-dependent pathway, IRP1 has dual roles either as a high affinity IRE-binding protein when devoid of the [4Fe–4S] cluster or as a c-aconitase when the [4Fe–4S] cluster is assembled. The c-aconitase form does not bind RNA. The [4Fe–4S] cluster is accessible to low molecular cluster perturbants, including reactive oxygen species (ROS), such as O2•−, H2O2 or reactive nitrogen species (RNS), such as NO• or ONOO−. Hypoxia stabilizes the c-aconitase form by reducing the level of oxygen or ROS. IRP1 can be phosphorylated at S138. IRP1 phosphomimetic mutants indicate that the Fe–S cluster can be assembled and the protein exhibits aconitase activity; however, the Fe–S cluster is more sensitive to disruption by oxygen and hydrogen peroxide. Iron stimulates the FBXL5-mediated degradation of IRP1 S138 phosphomimetic mutants and non-phosphorylated IRP1 when Fe–S cluster biogenesis is impaired.
Modified from Wallander et al. [4].
3.1. Regulation of IRP1 RNA binding activity by the 4Fe–4S cluster
The key role of the [4Fe–4S] cluster in IRP1 in controlling RNA binding activity provides a means for linking cellular pathways involved in formation of these cofactors to cellular iron status. In recent years, many studies have focused on unraveling the mechanism and pathways of Fe–S biogenesis in model organisms and their dysfunction in human disease [20–23] [see also Lill et al. in this volume]. In brief, the evidence indicates that the major pathway for biogenesis of Fe–S clusters and their insertion into cytosolic proteins, such as IRP1, first involves targeting of iron to the mitochondria where it is imported through the action of mitoferrin [24] [see also Paw et al. in this volume]. A transient Fe–S cluster is formed on the scaffold protein ISCU through the action of iron, the iron binding protein frataxin and sulfide derived from cysteine via the cysteine desulfurase NFS1 and its partner ISD11 [25–30]. The Fe–S cluster on ISCU, or in some cases other scaffold proteins [31], is donated to apoprotein targets with the assistance of protein chaperones (e.g. HSCB in humans) and electron donors (e.g. FDX, ferredoxin) [21]. Activities of the mitochondrial Fe–S cluster biogenesis machinery along with the export machinery components (e.g. ABCB7) are required for activation of the cytosolic Fe–S cluster assembly (CIA) pathway. Genetic approaches using IRP1 expressed in yeast led to discovery of the first CIA factor, Cfd1 [32]. Additional elegant studies primarily in yeast led to identification of Nbp35 that together with Cfd1 forms the initial Fe–S scaffold for cytosolic cluster formation with the assistance of DRE2 and TAH18 [30,32–36]. Transfer of the labile cluster from Cfd1/Nbp35 to some apoprotein targets requires the Nar1/Cia1 heteromer while for others Nbp35 suffices (reviewed in [22]). Interestingly, recent studies have begun to identify “specificity factors” that target specific apoFe–S proteins (e.g. complex 1) [37,38]. Whether or not separate pathways for Fe–S cluster biogenesis exist for sensory and regulatory roles of Fe–S proteins in mammalian cells, such as IRP1, remains to be determined.
Primarily through the use of RNAi-based knockdown approaches, the activity of the Fe–S biogenesis pathways have been shown to influence cellular iron sensing and distribution in mammalian cells. Disruption of components of the mitochondrial Fe–S biogenesis pathway, including the cysteine desulfurase NFS1 and its partner ISD11 [25,39], the Fe–S scaffolds ISCU [40] and ISCA [41], the iron binding protein frataxin [27,42], glutaredoxin 5 [43], protein chaperone HSC20 [44,45] and electron donors ferredoxins 1 and 2 [39,46], impairs the activity of both mitochondrial, cytosolic, and presumably nuclear Fe–S proteins in mammalian cells. The ensuing reduction in c-acon activity is reflected in IRP1 activation that is associated with an increase in TfR1 and reduction in ferritin expression. That IRP2 is stabilized suggests a general cytosolic iron deficiency [39,43,44,47,48]. While the increase in TfR1 and repression of ferritin induced by IRP activation may reflect a compensatory mechanism to combat cytosolic iron deficiency, failure to prevent further inappropriate iron accumulation in the face of excess iron reflects an inability to downregulate IRP activity that could be cytotoxic [40]. There is also evidence of cellular iron maldistribution with abnormal cellular iron deposits and mitochondrial overload coupled with cytosolic iron deficiency when mitochondrial Fe–S biogenesis is impaired [40,43,47]. Similar changes have been observed in yeast [49–53]. While these studies confirm the concept that cytosolic Fe–S cluster biogenesis is dependent on active mitochondrial cluster formation in mammalian cells, they also demonstrate that the impact of impaired mitochondrial Fe–S biogenesis may in part involve indirect effects on IRPs or IRP targets. Future studies should address questions concerning what pathways of Fe–S cluster biogenesis are sensed by IRP and whether trafficking of iron through mitochondria is an obligate process through which IRP senses iron.
The impact of impaired cytosolic Fe–S cluster biogenesis on IRP1 has also been examined [54–58]. Reduction of NUBP1 and NAR1 specifically impairs cytosolic Fe–S proteins, including c-acon and others [56–58]. Concomitant with this is the activation of IRP1 along with an increase in TfR1 and a reduction in ferritin expression. Interestingly, IRP2 is not affected suggesting that the response of IRP2 observed when mitochondrial Fe–S biogenesis is impaired is a secondary response to the maldistribution of cellular iron. Thus, as is the case when the cytosolic Fe–S cluster biogenesis system is inhibited in yeast, inhibition of the cognate proteins in mammalian cells has a selective impact on cytosolic Fe–S proteins. The finding that IRP2 is stabilized in ABCB7-deficient mammalian cells may reflect a feedback effect on mitochondrial Fe–S cluster formation or iron metabolism [55]. Deletion of the yeast homolog of ABCB7, Atm1p, also leads to an abnormal distribution of iron [51]. Taken together, these studies suggest that unlike IRP1,IRP2 does not sense the flux of iron through the cytosolic Fe–S biogenesis pathway.
Investigations of the etiology of FA provide the most clearly understood example linking a human disease of Fe–S cluster biogenesis and alterations in IRP function (reviewed in [59–61]). FA is the most common inherited ataxia in humans. It is primarily caused by formation of triplet expansion repeats in the frataxin gene leading to reduced expression of this small acidic mitochondrial protein although missense mutations of frataxin are also a cause. Frataxin binds to a complex containing the Fe–S scaffold protein ISCU and the cysteine desulfurase NFS1 and its partner ISD11, and in this process is believed to function as an iron chaperone [27–29,62]. One study reported that a small portion of frataxin also functions in the cytosol where it interacts with IRP1 and facilitates conversion to c-acon [42]. At the cellular level, FA is characterized by a loss of activities of mitochondrial, cytosolic, and presumably nuclear Fe–S proteins. Similar to what has been observed with the knockdown of other participants in mitochondrial Fe–S cluster biogenesis pathway, FA is associated with mitochondrial iron overload and signs of cytosolic iron deficiency [27,63,64]. Both IRPs are activated, and this is associated with a reduction in ferritin and increase in TfR1 expression. Furthermore, expression of the iron exporter ferroportin is reduced further contributing to the abnormal iron distribution, and presumably to oxidative stress observed in FA [64]. Interestingly, frataxin deficiency is associated with a reduction of other components of the mitochondrial Fe–S cluster machinery, including ISCU and NFS1, along with defects in heme synthetic enzymes [64]. The fact that frataxin expression is itself reduced in iron deficiency suggests that the induction of cytosolic iron deficiency due to impaired mitochondrial Fe–S cluster biogenesis could be amplified by a downstream impact of reduced frataxin expression [27]. An interesting area for further investigation concerns the extent to which enhanced accumulation of IRE RNA binding activity, especially that provided by the large latent pool of IRP1 RNA binding activity in the form of c-acon, coupled with impaired ability to down-regulate IRP due to inhibition of the iron-dependent SKP1–CUL1–FBXL5 (S-phase kinase-associated protein 1-Cullin 1-F-box-leucine rich repeat protein 5) E3 ubiquitin ligase further exacerbates disease etiology. This issue likely has relevance to numerous diseases of Fe–S cluster biogenesis (reviewed in [46,59]).
3.2. Fe–S cluster disassembly in c-acon facilitates sensing reactive oxygen and nitrogen species
The discovery that IRP1 RNA binding activity could be generated from c-acon raised a key issue of how, and under what conditions, could the Fe–S cluster be removed. Iron deficiency per se was not believed to be sufficient to promote loss of the cluster on its own. However, aconitases have long been known to sense ROS and RNS because their [4Fe–4S] cluster is solvent accessible and these reactive species can initiate cluster loss (reviewed in [65]). In fact, for one isoform of aconitase in Escherichia coli, the ease with which it was inactivated by superoxide anion appears to function as a feedback mechanism to modulate an overactive electron transport chain [66]. This only requires conversion to the [3Fe–4S] cluster species that, in the case of c-acon, is not sufficient to generate RNA binding activity. A further important fact is that aconitases differ in their sensitivity to ROS and RNS, where c-acon is more resistant [67–69]. Thus, although the Fe–S switch mechanism was accepted as a bonafide means for iron regulation of IRP1, the mechanistic basis through which it occurred was not clear.
Insight into this problem came from early studies showing that tumor cell aconitase was a target of NO generated by activated macrophages (reviewed in [70]). On this basis, a large number of studies were initiated demonstrating that NO and its reactive product ONOO- can facilitate conversion of c-acon to IRP1 (reviewed in [71]). NO also influenced IRP2, but the mechanism for this regulation and the impact on iron metabolism was unclear [72,73]. Recent studies using mice lacking IRP1 or IRP2 have assessed the role of each IRP in the NO-dependent regulation of macrophage iron metabolism. These studies demonstrated that IRP1 was solely responsible for alterations in macrophage iron metabolism in response to NO [74]. Furthermore, NO-dependent activation of IRP1 could ameliorate abnormal iron storage in Irp2−/− bone marrow macrophages ex vivo by repressing ferritin expression. Similarly, activation of IRP1 by the pharmacological nitroxide Tempol prevented the neurodegenerative symptoms in Irp2−/− mice [75]. Tempol is best known as a free radical scavenger; however, in Irp2−/− mice Tempol appeared to promote cluster loss from c-acon through the direct action of RNS.
While the ability of NO to activate IRP1 may be necessary for macrophage activation or action, the physiological role of ROS is less clear. Loss of the Fe–S cluster from aconitases can be initiated by superoxide leading to the generation of the [3Fe–4S] non-RNA binding form and H2O2 generates IRP1 from c-acon although the mechanism through which this occurs is not clear (reviewed in [76,77]). Increased conversion of c-acon to IRP1 by H2O2 may enhance iron uptake while impairing storage and export, leading to iron overload. However a recent report suggests a mechanism to partially evade IRP action under these conditions [78]. Perhaps the physiological role of ROS involves the constant conversion of a small fraction of c-acon into IRP1. Depending on the cellular iron status, IRP1 could then be converted back to c-acon or remain in the RNA binding form to regulate IRE-containing mRNAs. In other words, physiological generation of ROS may be necessary for iron sensing by IRP1.
An additional, but not necessarily alternative view is that the sensitivity of c-acon to ROS and RNS can be regulated allowing for the existence of pools of protein with a stable versus unstable Fe–S cluster. Previous studies demonstrated that two protein kinase C phosphorylation sites exist in IRP1, S138 and S711. S138 phosphomimetic mutants of c-acon are much more oxygen labile compared to the wildtype protein and are also at least 10-fold more sensitive to NO in terms of cluster disassembly [79,80]. Furthermore, these phosphomutants undergo spontaneous disassembly of the Fe–S cluster even in the absence of a perturbant [80]. Another study showed that IRP1 activation by H2O2 requires a signaling pathway possibly involving phosphorylation [81]. Taken together, these studies show that rapid recruitment of IRE-binding activity can be achieved by cluster perturbants or phosphorylation induced alterations in Fe–S cluster stability.
3.3. Iron regulation of IRP1 protein degradation
Early studies on the mechanism of IRP1 regulation by iron suggested that protein degradation was the primary mechanism for inactivation of RNA binding [82]. With the demonstration that IRP1 is the cytosolic isoform of aconitase, and that insertion and loss of the [4Fe–4S] cluster regulated RNA binding, the “Fe–S switch” mechanism was widely accepted as the dominant mechanism for controlling IRP1 RNA binding activity [83]. This view was further advanced by the observation that total levels of IRP1/c-acon protein did not change as a function of iron status. In many cases, however, the c-acon pool is in vast excess relative to IRP1 [84,85]. Thus, the much larger and more stable c-acon pool would mask iron-dependent changes in abundance of IRP1. Liver provides a case in point as c-acon exceeds IRP1 by more than 100-fold. Severe dietary iron deficiency results in an increase of IRP1 RNA binding activity of no more than 4-fold, indicating that a small portion of the c-acon pool was recruited [84]. Hence, iron-mediated degradation of IRP1 would not be routinely observed using immunoblots. Some evidence indicated that IRP1 could be regulated through protein degradation during iron overload or through phosphorylation [86,87]. The role of protein degradation in controlling IRP1 was examined anew using models where the Fe–S switch was disrupted either by mutation of cluster-ligating cysteines or through genetic manipulation of Fe–S cluster assembly or disassembly [54,88]. In this manner, regulation of IRP1 can be examined without c-acon. Under these conditions, IRP1 stability was clearly iron-regulated, and led to the view that iron-mediated IRP1 degradation was a compensatory mechanism to prevent excess RNA binding activity when the Fe–S cluster switch mechanism is not active, which can be lethal [19,54]. The relevance of this mechanism to IRP1 function was further substantiated with the finding that the FBXL5 E3 ligase that targets IRP2 for degradation also acts on IRP1 [89–91 ].
Given these findings, it is of interest that studies where Fe–S cluster biogenesis is impaired, IRP1 protein levels dropped significantly [25,41,44,47,48,54–58,92]. Under these conditions, IRP1 protein can be stabilized by iron chelation, suggesting that the loss of IRP1 is not merely due to instability of an apoprotein but instead is part of a second iron dependent mechanism to control accumulation of IRP1 RNA binding activity [54]. Impaired mitochondrial Fe–S biogenesis leads to cytosolic iron deficiency, presumably reducing FBXL5 activity as evidenced by increased IRP2 protein levels [23,27,39,44,47,48,55]. The mechanism by which IRP1 is degraded while IRP2 is stabilized under these conditions remains undetermined. Relative to normal cells, a reduction in FBXL5 activity in response to impaired mitochondrial Fe–S biogenesis could lead to increased IRP2 accumulation but still be sufficient to degrade excess IRP1 arising from an inability to accumulate c-acon. FBXL5 independent mechanisms may also account for this paradox, but further studies are required to determine FBXL5 function in response to impaired mitochondrial Fe–S cluster biogenesis. Regardless of the mechanism, the extentto which IRP1 and IRP2 RNA binding activity rises to pathological levels when Fe–S cluster formation is impaired is likely to be influenced by the abundance of c-acon and the extent to which FBXL5 is inactivated. These factors will likely be strong predictors of the extent to which IRP1 and IRP2 RNA binding activity rise to unacceptable levels in pathological states involving impaired Fe–S cluster formation or accelerated cluster disassembly.
3.4. S138 phosphorylation and the mechanism of IRP1 regulation by iron
The Fe–S switch and protein degradation mechanisms for controlling IRP1 function could each have unique uses in modulating iron metabolism in order to meet cellular needs. Hence, issues such as sensitivity to Fe–S biogenesis activity or the level of ROS or RNS may meet the distinctive iron requirements of specific cell types or physiological scenarios. A mechanism that could serve these needs involves S138 phosphorylation of IRP1 by protein kinase C because it leads to accumulation of the RNA binding form [93]. The finding that S138 phosphomimetic mutants of IRP1 could be converted to a form of c-acon with a highly unstable Fe–S cluster suggested an altered set-point for iron regulation [79]. Surprisingly, the S138E phosphomutant was still subjected to iron regulation but as a consequence of iron-stimulated protein degradation reminiscent of IRP2 regulation [87]. In order to determine if enhanced oxidative damage of IRP1 as a consequence of increased Fe–S cluster cycling in IRP1S138E promoted its degradation, the cluster-ligating cysteine residues were mutated to serine, blocking cluster insertion in S138E and wildtype IRP1 (IRP1S138E/3C > 3S and IRP13C > 3S). While this failed to block iron-dependent degradation of IRP1S138E, the more surprising result was that the protein level of IRP13C > 3S and another cluster mutant were iron-regulated [54,88]. These observations were confirmed by the subsequent demonstration that IRP1 is a substrate for the FBXL5 E3 ligase [89,90]. Recent studies indicate that IRP1S138E can be regulated either by protein degradation or Fe–S cluster insertion depending on the level of ROS or RNS [80]. These studies suggest that S138 phosphorylation of IRP1 is a mechanism to rapidly increase IRP1 RNA binding activity and to facilitate formation of a pool of c-acon that is sensitive to cluster perturbants and cellular iron levels. Coupled with the iron-mediated degradation of IRP1 by FBXL5, inducible phosphorylation broadens the physiological scenarios of IRP1 action.
3.5. IRPs and oxygen sensing
The discovery of a functional IRE in the 5′ UTR of the mRNA encoding HIF-2α suggested new roles for IRPs in the adaptive response to hypoxia and iron deficiency [10]. A recent study reported that HIF-2α mRNA is translationally repressed in liver from iron-deficient rats, and is associated with increased IRP1 and IRP2 activity [94]. Other studies have provided evidence that the HIF-2α IRE is preferentially targeted by IRP1 [11,95]. HIF-2α is primarily regulated by protein stabilization in response to hypoxia and iron deficiency (reviewed in [96,97]). Under these conditions, HIF-2α stimulates eryth-ropoiesis through activation of erythropoietin (EPO) and intestinal iron absorption through the activation of DMT1 and ferroportin [98–101], providing a mechanism to coordinate iron uptake with red cell production. IRP-dependent translational regulation of HIF-2α mRNA has been proposed as a mechanism to reduce EPO production during hypoxia resulting from impaired erythropoiesis in iron deficient anemia, thereby ensuring that microcytic hypochromic red blood cells are not produced [10]. Given the preferential regulation of HIF-2α mRNA by IRP1, this suggests that under hypoxic iron sufficient conditions IRP1 would exist in its c-acon form, and unable to bind RNA, ensuring that HIF-2α mRNA is translated and EPO is produced. The significance of the preferential regulation of the HIF-2α IRE by IRP1 requires further investigation.
3.6. Hierarchical regulation of IRE-containing mRNA
Vertebrate mRNAs containing consensus IREs encode proteins of widely varying function, from those with a direct role in iron metabolism to proteins involved in the regulation of citrate metabolism (m-acon), cell proliferation, cytoskeletal dynamics (MRCKα), cell cycle (CDC14A), and in the adaptive responses to hypoxia (HIF-2α). The broad functional spectrum of proteins encoded by IRE-containing mRNAs suggests that they are not identically regulated by IRPs. A comparison of the binding affinity of 5′ IRE-containing mRNAs to IRP1 provides a basis for determining the extent to which IRPs may act hierarchically in mammals.
One example of differential regulation of IRP target mRNAs is expression of ferritin and eALAS during erythroid differentiation. Erythroid precursors have a high iron requirement given the large quantity of heme and hemoglobin they produce. Thus, inerythroid precursors producing hemoglobin, iron would be diverted to mitochondria for heme formation and away from storage in ferritin. With regard to IRPs, the evidence indicates that eALAS mRNA is more weakly regulated than ferritin mRNA [9]. In primary murine erythroid progenitors upon differentiation, ferritin mRNA translation is tightly repressed with less than 5% of the mRNA polysome bound, whereas translation of eALAS mRNA is more efficiently translated with 40% of this mRNA associated with polysomes [102]. This 8-fold relative difference in translatability of these mRNAs occurred under conditions when IRPs are activated and TfR1 mRNA levels are enhanced. The higher affinity of the ferritin IRE for IRPs may in part explain its selective repression under these conditions [103]. It remains to be determined if the polysome bound eALAS mRNA present during normal erythroid differentiation has escaped IRP action due toits low-affinity IRE or because variants of the eALAS mRNA exist that lack the IRE. Regarding this point it is of interest that at least two studies indicate that both IRPs are critical for red cell heme formation. First, in Irp2−/− mice, eALAS biosynthesis is increased in erythroid bone marrow precursor cells, leading to inappropriate synthesis of protoporphyrin IX and anemia [104]. These mice also display reduced TfR1 and increased ferritin expression, indicating erythroid precursors are iron deficient. Although Irp1−/− mice do not develop microcytic anemia, the anemia is more severe in Irp2−/−Irp1+/− mice, indicating that IRP1 has a role in erythroid function [105]. Second, in zebrafish and humans, defects in glutaredoxin 5 lead to impairment of mitochondrial Fe–S biogenesis and cytosolic iron depletion that activates IRP1, and reduces eALAS expression [19,43]. Taken together, these studies suggest that differential targeting of 5′ IRE-containing mRNAs by IRPs is essential for optimal coordination of heme formation and iron storage during erythropoiesis.
Another example of differential regulation of IRP target mRNAs is the expression of ferritin and m-acon [84,106]. Dietary iron deficiency leads to a reduction of liver ferritin to undetectable levels. The decline of m-acon protein levels under these conditions is significant, but reflects only about a 50% decrease. Thus, in response to the same signal, proteins encoded by 5′ IRE-containing mRNAs, are selectively regulated. These differences in expression can be attributed to the selective regulation of these proteins at the translational level, as observed in cultured cells and in cell-free protein synthesis systems programmed with IRP1 or IRP2 [84,106,107] and the finding that IRP1 binds less well to the m-acon versus L-ferritin IRE [108,109]. Consistent with these findings, genetic ablation of both IRP1 and IRP2 in mice leads to a substantial enhancement not only of ferritin, but also ferroportin expression in intestine, while there was a smaller fold increase in m-acon expression [110]. These studies support the view that m-acon mRNA is less efficiently repressed in tissues compared with other 5′ IRE-containing mRNAs.
A third example of differential regulation of 5′ IRE-containing mRNAs in mammals comes from a comparison of HIF-2α and L-ferritin mRNA translation in liver [94]. In iron-replete rat liver, about 3-fold more HIF-2α mRNA was found associated with polysomes compared with L-ferritin mRNA. Iron deficiency leads to increased IRP RNA binding activity and a further translational repression of both mRNAs, yet HIF-2α mRNA remained more highly polysome associated. Finally, the comparison of succinate dehydrogenase iron protein subunit and ferritin expression in Drosophila further indicates that IRP hierarchically regulate mRNA translation in vivo [111]. Thus, differential translational control of 5′ IRE-containing mRNA by IRPs is essential for adaptive changes in iron metabolism and in other metabolic processes.
What dictates the differential translational control of 5′ IRE-containing mRNAs? To determine how mRNAs containing functional 5′ IREs are differentially regulated, the affinity of IRP1 for six 5′ IRE-containing mRNAs was determined [108]. These IREs bound to IRP1 in a hierarchical manner that related to the function of the encoded protein, such that ferritin IREs had the highest affinity while m-acon had the lowest. IRP1 bound to 5′ IREs over a 9-fold range: L-ferritin, H-ferritin, ferroportin, HIF-2α, eALAS and m-acon (Fig. 4A). Similar results were reported for IRP1 binding affinities for ferritin and m-acon IRE using an alternative binding assay [109]. Given the high affinity of IRP1 for binding the IRE (pM affinity), a valid concern is whether the 9-fold difference in affinity is sufficient to alter regulation. To address this, the affinity of IRP1 with a L-ferritin mutant IRE observed in a case of hereditary hyperferritinemia–cataract syndrome (HHCS) was examined. The affinity of the mutant L-ferritin IRE differed from the wildtype by only 1.9 fold yet this was sufficient to produce L-ferritin dysregulation in vivo[108,112]. A more striking evaluation of this is seen when the affinities of a large number of HHCS IRE mutants were compared to the rise in serum ferritin due to mRNA derepression [113] (Fig. 4B). The plot of serum ferritin as a function of Krel (relative binding affinity) revealed that the steepest slope occurred over the first 10-fold loss of affinity. Similar results were obtained for IRP2 [113]. This is essentially the same range of affinity differences observed for the binding of natural 5′ IREs to IRP1 (Fig. 4A). These findings indicate that small changes in IRP1 or IRP2 binding affinity lead to physiological and pathologically significant changes in the synthesis of proteins encoded by 5′ IRE-containing mRNAs.
Fig. 4.
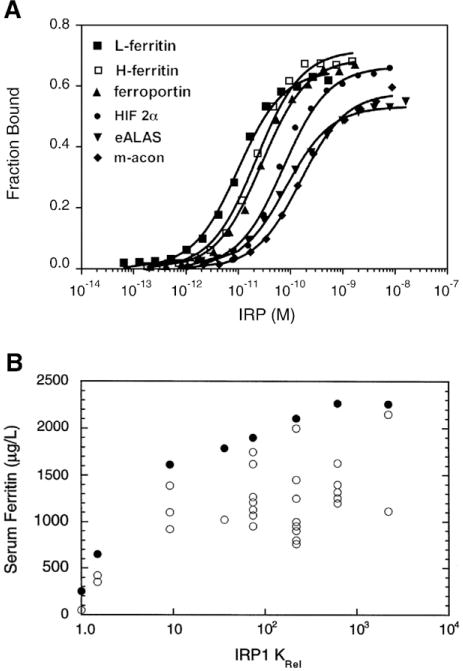
Altered affinity of IRP1 for mutant and natural IREs. A) The affinity of interaction of IRP1 for six vertebrate 5′ IREs as determined by electrophoretic mobility shift assays (EMSA) [108]. The KD for these IREs varied over a 9-fold range. B) Mutations in the human L-ferritin IRE were identified in seven patients with hereditary hyperferritinemia–cataract syndrome. The affinity of interaction of IRP1 with mutant human L-ferritin IREs was determined by EMSA and related to serum ferritin values reported for each patient on different occasions (open circles) [113]. Maximal serum ferritin for each patient (closed circles). Results on the x-axis are expressed as Krel which is KD,mutant/KD,wildtype and higher values for Krel indicate lower binding affinity of the mutant IRE with IRP1. Note the steeper slope of the relationship between serum ferritin and Krel over the range 1 to 10 for Krel.
A similar observation was made for IRP2 [113].
How are these changes in affinity of interaction between IRP1 and IRE established? Binding affinity is dependent on the number and strength of bonds between receptor and ligand, and biologically significant differences in affinity are usually assumed to involve a loss or gain of multiple bonds. In the case of IRP1, however, it can be inferred that the 9-fold difference in binding affinity would arise due to small differences in binding energy equivalent to a single H-bond [108]. Consequently, Goforth, et al. proposed that the difference in bond strength and/or in an induced fit mechanism, but not bond number, may give rise to the regulatory binding hierarchy [108]. Indeed, the recent report of the crystal structure of IRP1 bound to the TfR1 B IRE indicated that overall the number of bonds in this complex was essentially the same as observed in the complex of IRP1 and the H-ferritin IRE [114]. Thus, subtle differences in binding of IREs to IRP or in the pathway of binding are likely to be an important contributor to the hierarchical regulation of IRE-containing mRNA. A recent study reported that the ferritin IRE, but not the m-acon IRE directly binds ferrous iron, leading to a weakened interaction of IRP1 [109], suggesting an additional mechanism that contributes to the hierarchical regulation of IRE-containing mRNAs. There are also mechanisms to evade IRE action, such as mRNA isoforms that lack the IRE or those that contain internal ribosome entry sites [78,115].
3.7. What constitutes an IRE?
A key factor in the discovery of the IRE was the strong sequence identity of a short ~28–30 nt sequence in the 5′ UTR of both H- and L-ferritin mRNAs. Importantly, the sequence identity between these IREs (~90%) was substantially greater than that of the coding region (~50%) amongst vertebrate H- and L-ferritin mRNAs [116]. Of further significance were the accompanying “functional” discoveries of IRPs and that the IRE conferred iron regulation of translation when transferred to the 5′ UTR of a heterologous mRNA [117–119]. Shortly thereafter, multiple IREs were identified in the 3′ UTR of TfR1 mRNA where they served to control RNA stability yet could modulate translation if transferred to a 5′ UTR [120]. The conserved secondary structure of IREs established the concept that the canonical IRE was an RNA stem loop with a terminal hexanucleotide CAGUG(N) loop and an unpaired C (C8) in the stem, 5′ of the loop [121] (Fig. 5). Structural and biochemical studies indicated that formation of the AGU pseudotriloop, as a consequence of base pairing of the first and fifth nucleotides of the CAGUGN sequence, occurs in solution and is critical for the binding of IRP1 [122–127]. Molecular and in silico studies identified IREs with the same canonical loop sequence and overall secondary structure in multiple metazoan mRNAs, and demonstrated key roles of the terminal loop and unpaired C stem in IRP binding [108,128,129].
Fig. 5.
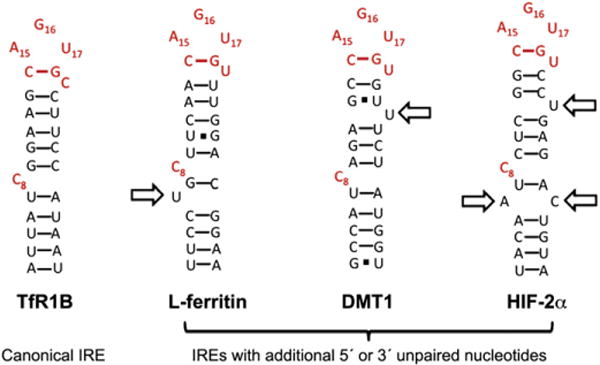
Comparison of the proposed secondary structure of IREs. The key structural elements required for recognition of the IRE by IRP1 include the unpaired C (C8) in the IRE stem and the A16G17U18 nucleotides of the pseudotriloop. The canonical IRE secondary structure is represented by one of the five TfR1 IREs (TfR1 B), where the RNA helix is interrupted by only the unpaired C8. The L-ferritin IRE secondary structure showing the conserved unpaired U at position 6 (arrow). This additional unpaired nucleotide has been observed in the NMR of the ferritin IRE and the crystal structure of IRP1 bound to the ferritin IRE [124,130,133]. Proposed secondary structures of DMT1 and HIF-2α IREs. Note the additional unpaired nucleotides (arrows).
IRE residue numbering is according to Walden et al. [130].
The high resolution crystal structures of IRP1 bound to the ferritin IRE and the TfR1 B IRE provide key insights into the nature of this unusually high affinity RNA:protein interaction [114,130] (reviewed in [128]) (Fig. 6). IRP1 binds to the IRE through two sets of interactions separated over about 30 Å. IRP1 makes the majority of its bonds with the IRE in a sequence specific manner and involves canonical features of known functional IREs. Thus, the exposed nucleotides, including AGU of the terminal pseudotriloop and the unpaired C8 residue in the IRE stem, make 15 of 22 contacts with IRP1. The additional seven bonds are largely with sites in the phosphodiester backbone of the IRE stem. The two-site binding mode of IRP1, which bears strong similarities with the recognition of tRNA by tRNA synthetases, allows for enhanced specificity of RNA recognition, and likely contributes to the unusual high affinity of interaction [130]. When bound to IRP1, the ferritin IRE exhibits significant additional bending and twisting of the RNA helix as a consequence of the larger inter-helical junction around C8 and due to the presence of the unpaired U6 in ferritin IRE compared with the TfR1 B IRE. This causes a significant difference in the degree to which the lower stem of the IRE approaches the so-called stem-binding domain (domain 4) of IRP1 [114] (Fig. 6). Deviations in structure of the lower stem of IREs have been proposed to make key contributions to regulation [131].
Fig. 6.
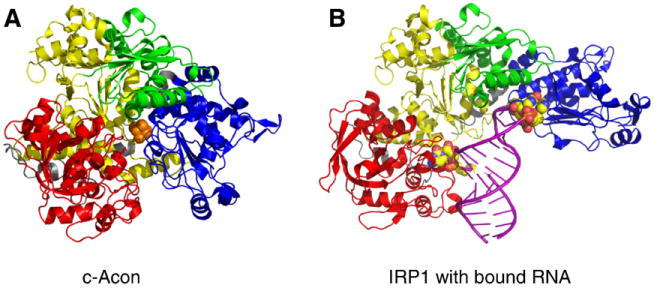
Crystal structure of c-aconitase and the IRP1:IRE complex. The crystal structures of c-aconitase [132] and the IRP1:IRE complex [121,130] are shown. A) Cytosolic aconitase structure showing domain 1 (yellow), domain 2 (green), domain 3 (blue) and domain 4 (red) with the [4Fe–4S] cluster in the center (orange balls). B) IRP1:IRE complex structure shown with domains 1 to 4 as in (A). The ferritin IRE helix (purple) is shown with the two major contact sites of C8 (left) and the A16G17U18 bases of the pseudotriloop shown (right) as enlarged balls.
Significant large scale structural changes occur in c-acon, particularly in domains 3 and 4, in order to accommodate the IRE [130] (Fig. 6). In comparing this structure to that of c-acon [132], Walden et al. provide unique insight into how structural plasticity of one polypeptide can support two widely different functions [130]. They proposed that residues within IRP1/c-acon that influence the ability to interconvert between these activities may dictate the ability of why some aconitases bind RNA but others do not. Importantly, this study establishes a structural framework from which to evaluate how the different canonical IREs as well as proposed IRE-like elements may make use of subtle differences in bonding or require major differences, possibly including additional binding partners, in order to allow for their unique regulation by IRPs.
The first indication of structural differences amongst the known IREs that suggested altered ability to regulate mRNA fate came from studies of the ferritin IRE [122]. Initial computer predictions had suggested that the highly conserved unpaired C8 formed a single nucleotide bulge and gave rise to the canonical view of IRE secondary structure (Fig. 5). However, secondary structure mapping with small chemical probes confirmed other in silico predictions of a more complex stem structure compared to the canonical IREs [103,122,124]. Vertebrate ferritin IREs contain an unpaired U two base pairs 5′ of C8 (Fig. 5) as confirmed in both NMR and X-ray crystallography [124,130,133]. This contrasts with all other known functional IREs and even with ferritin IREs in lower organisms that appear to only have the single C8 bulge [134]. Interestingly, deletion of U6 from the ferritin IRE substantially diminishes the affinity of interaction with IRPs, particularly IRP2 [103,108,109,133,135]. Chemical probing studies suggested that this “double bulge” feature of vertebrate ferritin IREs is dynamic and may interconvert with a larger UGC/C internal loop perhaps facilitating direct binding of ferrous iron [103,109]. These additional features likely facilitate structural alterations that may underlie the stronger regulatory role of the IRE in ferritin relative to other mRNAs. Indeed, the recent structural studies comparing a single C-bulge IRE from TfR1 with the H-ferritin IRE bound to IRP1 revealed a substantial difference in the bend-angle and helical twist of the IRE stem and the degree to which the lower stem approaches IRP1 [114]. Since these two IREs bind IRP1 with similar affinities [114], it appears that significant plasticity of the RNA and protein allows for the accommodation of IREs with different structures. It will be of interest to determine if alternative structural alterations provide a mechanistic basis for the substantially weaker binding of the m-acon IRE, and to determine the impact of unpaired nucleotide(s) in the upper stem, such as observed for DMT1 and HIF-2α IREs (Fig. 5) [18].
Given the observations that U6 enhances the interaction of both IRPs with the ferritin IRE, one question is what is the role for bulge nucleotides in other IREs? While structures of IREs other than H-ferritin and TfR1 B have not been determined, secondary structure predictions suggest that several IREs possess additional unpaired nucleotides in the stem, especially on the 3′ side (Fig. 5). The HIF-2α and DMT1 each contain a single IRE with an additional 3′ unpaired nucleotide for which protein expression and RNA binding data are available (Fig. 5). In the case of both IREs, there is evidence suggesting that they are preferentially regulated by IRP1 [10,11,136]. Given the critical role of hypoxia in regulating intestinal absorption and erythropoiesis, the possibility that IRP1 may preferentially regulate these mRNAs is intriguing. A second issue of interest concerns whether unpaired nucleotides in different IREs program specific changes in regulation. Deletion of U6 lowers the affinity of IRP1 for the ferritin IRE and may have an even stronger effect on IRP2 binding [108,109,133]. In the case of HIF-2α and DMT1 IREs, the impact of the 3′ unpaired nucleotides may bias binding toward IRP1 not IRP2. In addition, the presence of this feature in the HIF-2α and DMT1 IREs may be critical for limiting the strength of IRP1 binding in order to fine tune regulation. Finally, a recent transcriptome-wide examination of mRNAs that interact with IRPs in vitro identified novel mRNAs containing canonical IREs, and demonstrated that IRPs may bind RNAs with differences in the location of unpaired nucleotides or in upper stem base pairing capacity [18]. Similarly, a recent in silico study has pointed to additional potential IREs in previously unidentified mRNAs [129]. Thus, the reach of the IRP-IRE network may extend into new areas of cellular function.
3.8. Roles for non-canonical IREs in IRP action
Test-tube RNA evolution experiments (SELEX) provided the first indication of the sequence and structure requirements for IRP recognition of IRE elements [125,126,137,138]. In this regard, one of the more intriguing results came from studies where the entire IRE sequence was randomized and IRP1 was used to select high affinity RNA ligands. Intriguingly, an IRE-like RNA was identified that possessed a larger terminal loop (CAGUGUCA) along with a somewhat more complex pattern of unpaired nucleotides in the C8 region of the stem [138]. On this basis, studies on amyloid precursor protein (APP) and α-hemoglobin stabilizing protein (AHSPα) mRNAs, and the recent transcriptome-wide study potentially identifying a wide array of new IRP targets are of clear interest [18,139,140]. The evidence strongly suggests that both APP and AHSPα mRNAs are targets of IRP action. However, the proposed IRE-like element in these mRNA differs substantially from the canonical IRE, raising questions regarding the structural basis of their binding to IRP1. It is of interest in this regard that for the novel RNAs identified by SELEX that bind tightly to IRP1, this IRE contains the CAGUGU consensus sequence in a larger eight nucleotide terminal loop. This IRE is also proposed to have an unpaired pyrimidine on the 5′ side of the stem with the correct spacing from the critical AGU binding site in the terminal loop. In contrast, the APP and AHSPα IRE-like elements do not have these structural elements. Thus, while the evidence is strong that IRP1 associates with APP and AHSPα mRNAs in cells, the question as to whether IRP1 binds these elements indirectly or whether they require the assistance of protein partners remains to be determined. The recent transcriptome-wide analysis [18] suggested that IRPs may bind many non-canonical IRE RNAs, suggesting the presence of an expanded IRP-dependent post-transcriptional regulon.
4. Recent advances in IRP2 regulation
The last decade has seen major advances in IRP2 regulation. The identity of FBXL5 as the E3 ligase responsible for the iron-mediated degradation of IRP2 was discovered. New insights into the role of IRP2 in cell proliferation have been defined, and mouse models of IRP2 deficiency have provided mechanistic insight into its role in regulating cellular iron homeostasis.
4.1. Iron-mediated regulation of IRP2 stability
IRP2 is primarily regulated by protein stability: iron depletion and hypoxia stabilize IRP2, whereas iron promotes IRP2 ubiquitination and proteasomal degradation [141–144]. One early study reported that a cysteine rich 73-amino acid domain was required for iron-mediated degradation of IRP2 [144]. Several models were proposed whereby iron or heme oxidized specific cysteine residue(s) within the 73-amino acid domain, triggering the ubiquitination and degradation of IRP2 [145,146]. A protein designated heme-oxidized IRP2 Ub ligase (HOIL-1) was identified and shown to bind to heme-oxidized residues within the 73-amino acid domain [147,148]. Subsequent studies from several groups showed that mutation of cysteine residues within the 73-amino acid domain or deletion of the entire 73-amino acid domain did not affect iron-mediated degradation of IRP2 [149–151]. Furthermore, HOIL-1 silencing or overexpression in cultured cells had no effect on the degradation of IRP2 by iron [152]. These studies prompted investigation into other mechanisms to account for IRP2 degradation by iron.
Recently, a novel E3 ligase complex, SKP1–CUL1–FBXL5, was identified as the E3 ligase responsible for iron-mediated IRP2 degradation [89,90] (Fig. 7). FBXL5 was independently identified by two groups using different approaches. Vashisht et al. [90] used multidimensional protein identification technology (MUDPIT) to identify proteins that interact with epitope-tagged FBXL5. Salahudeen et al. [89] used a small interfering RNA (siRNA) screen to identify proteins that altered IRP2 degradation. FBXL5 is a member of the F-box family of adaptor proteins that confer substrate specificity to SCF E3 Ub ligases (reviewed in [153,154]). FBXL5 is conserved in vertebrates, and contains an N-terminal hemerythin domain, a F-box domain that mediates its association with SKP1 and four leucine-rich repeats that likely function in IRP2 binding. Hemerythrins are oxygen carrier proteins in marine organisms that bind to oxygen through a diiron L center [155]. Hemerythrin-like domains have been identified in bacterial proteins where they are thought to function as oxygen sensors [156,157]. FBXL5 represents the first eukaryotic protein to contain this domain.
Fig. 7.
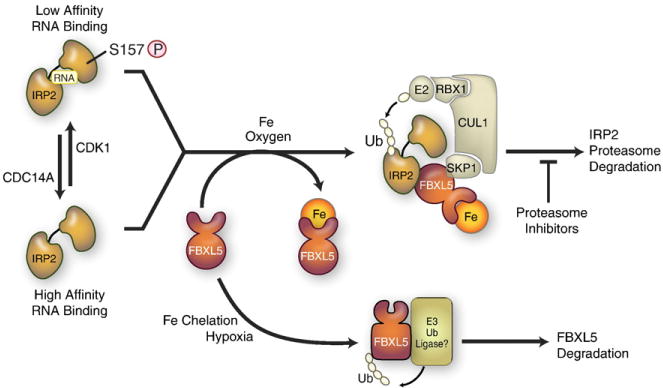
Mechanisms for iron-dependent and iron-independent IRP2 regulation. Iron-dependent degradation: when iron and oxygen are high, the FBXL5 hemerythrin-domain binds iron and leads to a conformational change that increases FBXL5 stability. FBXL5 binds to IRP2 and FBXL5-IRP2 associates with SKP1–CUL1 complex, catalyzing the ubiquitination and proteasomal degradation of IRP2. When iron or oxygen is limited, FBXL5 is destabilized and targeted for degradation by an unidentified E3 ligase. IRP2 iron-independent pathway: IRP2 is phosphorylated by cyclin-dependent kinase 1(CDK1/cyclinB1) during G2/M and dephosphorylated by CDC14A at the end of mitosis. S157 phosphorylation is independent of iron and is associated with reduced RNA binding activity. Non-phosphorylated and phosphorylated IRP2 are subjected to FBXL5 iron-mediated degradation.
Modified from Wallander et al. [4].
Several experiments showed that FBXL5 regulates IRP2 stability [89,90,158]. First, FBXL5 forms a SCF complex that interacts with IRP2 in vitro. The region of IRP2 that binds FBXL5 is not yet known, but the 73-amino acid domain is not involved. A study showed that sequences in the C-terminal region of IRP2 are necessary, but not sufficient for IRP2 degradation [159]. Because IRP1 is a FBXL5 substrate, it is likely that IRP1 and IRP2 share a similar degron. Second, FBXL5 silencing stabilized IRP2, whereas FBXL5overexpression promotedIRP2 degradation. Third, FBXL5 is stabilized in iron-replete cells, and destabilized by iron depletion and hypoxia. The hemerythrin domain is required for FBXL5 stability as mutations in specific histidine and glutamate residues within this domain reduced FBXL5 abundance and iron-dependent stability. Finally, reduced stability of FBXL5 during iron depletion or hypoxia provides an explanation for IRP2 accumulation during hypoxia [150,160,161]. Similarly, the enhanced degradation of IRP2 by antioxidants and stabilization of IRP2 by oxidants is also likely explained by changes in FBXL5 stability [88,161]. It should be noted that oxidative stress can also inactivate IRP2 RNA binding independent of changes in IRP2 protein levels [162–164], and in one study this was shown to be due to oxidation of cysteines that are predicted to lie in the RNA-binding cleft [164].
Structural analysis revealed that the FBXL5 hemerythrin domain adopts a hemerythrin-like alpha-helical bundle fold stabilized by a diiron center [158] [see also Bruick et al. in this volume]. Although many hemerythrins can reversibly bind oxygen at the diiron center, there was no evidence for oxygen binding to the FBXL5 hemerythrin domain. A current model for FBXL5 regulation of IRP2 proposes that in the presence of iron and oxygen, the hemerythrin domain binds iron, leading to the stabilization of a FBXL5 E3 ligase that catalyzes IRP2 ubiquitination and proteasomal degradation (Fig. 7). Iron depletion or hypoxia causes a conformational change in FBXL5 rendering it susceptible to proteasomal degradation by an unidentified E3 ligase. How oxygen contributes to FBXL5 iron binding remains to be determined.
Several other mechanisms have been proposed to regulate iron-mediated degradation of IRP2. One mechanism involves 2-oxoglutarate (2-OG) dependent dioxygenases, which are enzymes that catalyze the hydroxylation of protein substrates (reviewed in [165]). These enzymes require iron, oxygen and 2-OG for catalysis. A notable example of 2-OG dioxygenase activity is the proline hydroxylation of HIF-α subunits during normoxia by prolyl-hydroxylases, targeting HIF-α subunits for ubiquitination and proteasomal degradation by the von Hippel–Lindau E3 ligase (reviewed in [96]). Evidence for 2-OG dependent dioxygenases in IRP2 degradation comes from studies showing that the 2-OG dependent dioxygenase inhibitor, dimethyloxalylglycine, blocked IRP2 degradation by iron [150,151]. Heme has been shown to have a role in IRP2 iron-mediated degradation as the heme biosynthesis inhibitor, succinylacetone, partially stabilized IRP2 against iron degradation [73,149,166]. A calcium dependent non-proteasomal pathway has been reported to regulate the steady-state abundance of IRP2 during iron replete conditions [167]. This pathway was only operative in specific cell lines, and was proposed as a mechanism to limit IRP2 abundance during normal growth conditions. Lysosomal degradation of IRP2 has also been reported [168]. IRP2 was stabilized in cells treated with the lysosomal inhibitors ammonium chloride and chloroquine. IRP2 was found to be degraded by lysosomes only under normal growth conditions (for example, in the absence of added iron), while iron overload resulted in IRP2 degradation by the proteasome. A caveat is that lysosomal inhibitors increase lysosomal and endosomal pH, and can cause cellular iron deficiency by inhibiting iron release from transferrin [169]. In this scenario, FBXL5 would be destabilized, leading to increased IRP2 abundance. As the aforementioned studies relied on the use of pharmacological inhibitors to inhibit 2-OG-dependent dioxygenases, heme biosynthesis, proteasomal and lysosomal functions, experiments using alternative approaches are needed to corroborate these mechanisms.
4.2. IRP2 and cell proliferation
Iron is critical for growth of tumors (reviewed in [170]). As a result, many tumor cells satisfy their increased need for iron by increasing TfR1 expression as well as reducing ferritin and ferroportin expression [171–175]. Changes in iron uptake, sequestration and export provide mechanisms to increase cellular iron for proliferative processes. IRP2 has also been shown to have a role in cell proliferation. IRP2 overexpression in H1299 lung cancer cells stimulated their ability to form tumors when grown as xenografts in immunodeficient mice, whereas overexpression of IRP2 lacking the 73-amino acid domain suppressed tumor xenograft growth, suggesting a unique function for this domain [176]. By contrast, overexpression of IRP1 in H1299 lung cancer cells suppressed tumor xenograft growth in mice [177]. Surprisingly, both IRP1 and IRP2 expressing xenografts displayed elevated TfR1 levels, but IRE target mRNAs, such as ferritin and ferroportin, were not affected. c-MYC and phospho-ERK1/2 levels were elevated in IRP2-expressing tumors compared to tumors expressing IRP2 lacking the 73-amino acid domain. These studies raise the possibility that unidentified IRP-target mRNAs are involved in cell proliferation and/or that IRPs have roles in cell proliferation unrelated to their function as RNA binding proteins.
Another study showed that IRP2 is phosphorylated on Ser157 located within the 73-amino acid domain. IRP2 is phosphorylated by cyclin-dependent kinase 1 (Cdk1/cyclin B1) during G2/M and dephosphorylated by the phosphatase CDC14A during mitotic exit [178] (Fig. 7). S157 phosphorylation is not dependent on iron and is associated with reduced IRP2 RNA binding during G2/M as evidenced by increased ferritin synthesis. It is interesting to note that CDC14A mRNA harbors a 3′ IRE; however, regulation of CDC14A mRNA was not associated with Ser157 phosphorylation during the cell cycle. How IRP2 Ser157 contributes to cell cycle regulation and proliferation remains to be determined.
5. Pathological consequences of IRP and FBXL5 deficiencies in mice
Over the past decade, the generation of global and conditional mouse models of IRP2 and IRP2/IRP1 deficiency has demonstrated their importance in cellular iron metabolism. These studies have also underscored their critical role in mitochondrial iron metabolism.
5.1. Total body ablation of IRP1 and IRP2
Genetic inactivation of both Irp1 and Irp2 in mice leads to embryonic lethality at the blastocyst stage indicating a critical role for the IRP–IRE network during early development [110,179,180]. Irp1−/− mice lack an overt phenotype, despite mild ferritin dysregulation in kidneys and in brown fat [179]. The lack of an overt phenotype is surprising as c-acon is a highly conserved protein found in many diverse organisms and that c-aconitase is the principal form present in tissues of the body [160]. The function of c-acon is still not well understood, but apparently it is not required for normal physiology.
Two distinct strains of Irp2−/− mice have been generated. One strain was generated by the insertion of a PGK-neomycin gene into exon 3/4 of the Irp2 gene [181] and the other strain was generated by Cre–Lox technology [182]. Both Irp2−/− mouse strains develop mild microcytic anemia and altered body iron distribution [104,183]. Iron content is elevated in intestine and liver and is associated with increased ferritin expression and decreased TfR1 expression, and is reduced in splenic macrophages and is associated with decreased ferritin expression.
Microcytic anemia in Irp2−/− mice is associated with normal transferrin saturation and serum iron parameters, and is thought to be due to cellular iron deficiency in erythroid precursors as a consequence of reduced TfR1 levels and increased ferritin levels [104,183]. Because erythroid precursors are dependent on transferrin-bound iron for heme synthesis, increased iron sequestration in ferritin and reduced iron uptake would lead to a reduction in the cellular labile iron pool, impairing heme synthesis. Mice lacking IRP2 in enterocytes, hepato-cytes or macrophages do not develop microcytic anemia suggesting that microcytic anemia in Irp2−/− mice is an intrinsic defect of iron homeostasis in erythroid precursors [184]. Irp2−/− mice also develop erythropoietic protoporphyria characterized by increased protoporphryin IX in bone marrow, serum and liver [104]. Erythroid ALAS synthesis is upregulated in Irp2−/− erythroid precursors likely due to loss of IRP2 translational repression. The heme precursor protoporphyrin IX accumulates inerythroid precursors due to reduced iron availability required to complete heme synthesis.
The Irp2−/− strain generated by LaVaute et al. [181] develop a late onset neurodegenerative disorder characterized by abnormal gait, hind-limb weakness and tremors, and by reduced grooming and neuromuscular performance assayed by the hanging wire test. The cerebellum, substantia nigra, hippocampus and caudate putamen among other brains areas displayed elevated levels of ferric iron and ferritin that correlated with axonopathy and neuronal loss [105]. The onset and severity of neurodegeneration is exacerbated in Irp2−/−Irp1+/− mice, demonstrating a gene dosage effect with the IRPs [75,105]. Neu-rodegenerative symptoms in both Irp2−/− and Irp2−/−Irp1+/− mice were alleviated by Tempol, a membrane-permeable radical scavenger, where it reduced ferritin levels and lower iron content in cerebellar white matter tracts [75]. Tempol was shown to convert the c-acon form of IRP1 to the RNA binding form that stabilized TfR1 mRNA and repressed ferritin synthesis, and thus restored iron homeostasis. More recently, Irp2−/− mice were reported to have lower motor neuronal degeneration and spinal cord axonopathy that was more severe in Irp2−/−Irp1+/−[185]. Lumbar spinal sections in Irp2−/− mice showed the accumulation of myelin dense bodies, which are hallmarks of neurodegeneration, in the ventral and lateral white matter. Mitochondria in the lumbar spinal cord were swollen, and displayed disrupted and vacuolized cristae. Ferritin and TfR1 levels were increased and decreased, respectively, in motor neurons, and total iron content was reduced in the spinal cord, consistent with cellular iron deficiency in motor neurons of Irp2−/− mice. Respiratory complexes I and II activities were reduced in lumbar spine mitochondria in Irp2−/− mice. As complexes I and II contain Fe–S clusters and heme prosthetic groups essential for activity, this suggested that cellular iron deficiency in motor neurons might impair mitochondrial function, leading to neuronal degeneration. This hypothesis was tested by treating Irp2−/− mice with Tempol and by crossing Irp2−/− or Irp2−/−Irp1+/− mice into a Fth−/+ background [186]. Motor neuron survival improved using both approaches consistent with the notion that cellular iron deficiency has a role in motor neuronal degeneration in Irp2−/− mice.
In contrast to Irp2−/− mice generated by LaVaute et al. [181], Irp2−/− mice generated by Galy et al. [187] did not display an abnormal gait, hind-limb weakness and tremors or neuropathological changes at 13–14 months of age. Perls’ and TUNEL staining did not reveal signs of iron deposition or cellular degeneration in the brain, and electron microscopy showed no evidence of ultrastructural defects or mitochondriopathy. Irp2−/− mice also did not show impaired muscle strength when tested by the forepaw grip strength test (analogous to the hanging wire test used by LaVaute et al. [181]). In agreement with LaVaute et al. [181], the Irp2−/− strain generated by Galy et al. [187] displayed motor coordination and balance defects and reduced grooming activity when assayed by the rotarod and the modified-hole board tests. In addition, both strains displayed decreased TfR1 and increased ferritin expression in the brain [187]. Whether differences in severity of clinical and pathological signs of neurodegeneration between the two Irp2−/− strains are due to targeting strategies, genetic backgrounds or technical issues is not known.
5.2. Macrophage/monocyte ablation of IRPs
Splenic and bone marrow macrophages of Irp2−/− mice have reduced ferric iron stores with repressed levels of ferritin and ferroportin [104,183,184]. Interestingly, these mice have a slight increase in serum ferritin and normal hepcidin levels [104,183,184]. Ferritin secretion ensues when cellular ferritin synthesis occurs during iron depletion, and may explain reduced ferritin levels and splenic iron deposition seen in these animals [188]. A conditional deletion of IRP2 from macrophages/monocytes using LysozymeM-Cre did not recapitulate the splenic phenotype seen in Irp2−/− mice [184], suggesting that splenic iron mismanagement may be a result of IRP2 deficiency in another cell type or the combined effect of IRP2 deficiency in multiple tissues.
5.3. Intestinal ablation of IRPs
The function of IRPs in iron absorption was investigated by specific deletion of IRP2 alone or both IRP1 and IRP2 in intestinal epithelial cells [184]. Mice lacking IRP2 in the duodenum recapitulated the intestinal phenotype of duodenal iron loading and increased ferritin expression in Irp2−/− mice. Duodenal iron content and ferritin levels were not affected in mice lacking IRP2 in hepatocytes or in macrophages, suggesting that dysregulated iron homeostasis in IRP2 deficient duodenum is likely cell autonomous.
Mice lacking both IRP1 and IRP2 in intestinal epithelial cells were generated by breeding mice homozygous for floxed Aco1 and Ire2 alleles with mice expressing Cre-recombinase under control of the Villin promoter [110]. These mice displayed severe growth defects 7 days post-partum, and 80% of the animals died shortly after weaning likely due to dehydration, with the remaining 20% reaching adulthood due to incomplete IRP deletion. IRP deficient enterocytes from mice displayed less structured duodenal crypts and villi, and intestinal enterocytes exhibited mitochondriopathy along with increased apoptosis. Ferritin and ferroportin levels were profoundly increased, and TfR1 and DMT1-IRE isoform mRNAs and protein were decreased. The increase in ferroportin levels occurred without changes in mRNA levels and despite increased hepcidin levels. Increased hepcidin levels could not be accounted by increased hepatic iron loading, and was postulated to be due to decreased erythropoiesis or inflammation. Surprisingly, blood parameters revealed no signs of systemic iron defects given the profound changes in cellular iron homeostasis in mice with IRP deficient enterocytes. It is possible that increased iron export by ferroportin equalizes reduced iron uptake and increased iron sequestration in ferritin. This study demonstrated that dysregulation of iron uptake, storage and export in IRP deficient enterocytes is consistent with cellular iron starvation as a possible cause for mitochondriopathy and impaired enterocyte function.
The importance of ferritin in iron absorption was demonstrated in mice with a specific deletion of H-ferritin subunit in intestinal epithelial cells [189]. These mice showed increased body iron stores and transferrin saturation, and as expected, increased hepatic hepcidin mRNA levels. Further analysis showed reduced levels of DMT1, DYCTB and TfR1 mRNAs, while ferroportin and L-ferritin subunit increased. IRP2 levels were reduced, suggesting an increase in the cellular labile iron pool, which would lead to the destabilization of DMT1 and TfR1 mRNAs and increased synthesis of ferroportin and L-ferritin subunit. DMT1 and DCYTB mRNAs are transcriptionally activated by HIF-2α in iron-deficient mice [98,99]. Whether reduced DMT1 and DCYTB mRNAs in H-ferritin deficient enterocytes are in part due to reduced HIF-2α accumulation is not known.
5.4. Hepatic ablation of IRPs
Conditional deletion of IRP2 in hepatocytes recapitulated the hepatic phenotype in Irp2−/− mice where iron loading was associated with ferritin derepression. In these mice, expression of TfR1 and DMT1 was unchanged, and the mechanism for increased hepatic iron loading is not clear. Mice lacking IRP2 in enterocytes or macrophages showed no evidence of iron loading in hepatocytes, showing that dysregulated iron homeostasis in hepatocytes in IRP2 deficient mice is intrinsic to hepatocytes.
The deletion of both IRP1 and IRP2 in hepatocytes has provided insight into IRP function in vivo[190]. Hepatocyte IRP deficiency causes miceto die between8 and 12 days post-partum. Profound liver damage as characterized by internal bleeding, hepatic steatosis and increased apoptosis was apparent in these animals. Despite reduced hepcidin levels, there were no overt signs of systemic iron dysfunction. Analysis of IRP-target mRNAs showed that levels of ferritin and ferroportin were elevated, whereas levels of TfR1 and the DMT1-IRE mRNAs and protein are decreased. Zip14 (SLC39A14, iron transporter) and mitoferrin 2 (SLC25A28, mitochondrial iron transporter), which are not IRP regulated, were also downregulated. Total liver nonheme iron and mitochondrial nonheme iron were reduced 50% compared with control mice. The reduction in iron uptake coupled with increased iron sequestration in ferritin and increased iron export by ferroportin is consistent with severe cellular iron starvation in IRP deficient hepatocytes.
Mitochondrial dysfunction is a defining feature of many fatty liver diseases [191]. Consistent with this, mitochondriopathy is the major pathological consequence of hepatocyte IRP deficiency [190]. Mitochondria appear swollen, and contain hypodense matrices and abnormal cristae architecture, consistent with iron and copper deficiency in rodents [192]. Interestingly, the expression of the mitochondrial iron transporter mitoferrin 2 is reduced in IRP deficient hepatocytes and correlates with a reduction in mitochondrial nonheme iron. Mitochondria are responsible for the synthesis of Fe–S clusters and heme that are required by complexes I, II and III and by TCA cycle enzymes. IRP deficient hepatocytes display markedly reduced activities of complexes I, II and III and ferrochelatase, as well as the cytosolic Fe–S cluster containing protein xanthine dehydrogenase. It is likely that defects in the electron transport chain and TCA cycle are major contributing factors to the mitochondrial abnormalities seen in IRP deficient hepatocytes.
5.5. Mouse models of FBXL5 and FBXL5-IRP2 deficiencies
The importance of IRP2 in cellular iron metabolism was further highlighted by FBXL5 ablation in mice [91]. Total body ablation of FBXL5 in mice resulted in embryonic lethality at E8.5, and was associated with iron overload and oxidative stress in embryos. Embryonic lethality was prevented by simultaneous ablation of IRP2. Fbxl5−/−Irp2−/− mice developed normally and were physically indistinguishable from littermate controls. Mice with hepatocyte specific ablation of FBXL5 were viable, but developed hepatic steatosis and mitochondriopathy in hepatocytes. Increased IRP levels were associated with increased TfR1 levels and ferrous iron in hepatocytes. Unexpectedly, L-ferritin protein and mRNA levels increased in FBXL5 deficient hepatocytes compared with control mice despite increased IRP expression. The most likely explanation for increased ferritin expression is transcriptional activation of the L-ferritin gene induced by oxidative stress in hepatocytes [193]. Mice lacking hepatocyte FBXL5 displayed reduced hepcidin mRNA expression as a consequence of reduced BMP signaling. These mice also showed reduced survival when fed a high iron diet. Taken together, this study showed that embryonic lethality in Fbxl5−/− mice is primarily due to dysregulation of IRP2 degradation, which leads to increased cellular iron and oxidative stress, and show that FBXL5 regulation of IRP2 is critical for regulation of cellular iron homeostasis.
5.6. IRP-IRE network in human disease
To date there are no reports of mutations in IRP1 or IRP2 that cause human disease, but mutations in IREs or in coding sequences of IRE-encoded genes have been linked to human disorders. Mutations in L-ferritin subunit mRNA cause autosomal dominant hereditary hyperferritinemia-cataract syndrome (HHCS) that is characterized by juvenile bilateral cataracts and high levels of serum ferritin without iron overload. These mutations reduce IRP binding to the 5′ IRE and lead to abnormal L-ferritin subunit accumulation in tissues and serum [194–196]. A point mutation in the 5′ IRE of H-ferritin subunit that increases its affinity for IRPs causes autosomal dominant iron overload disorder [197]. Coding region mutations in L-ferritin subunit cause hereditary ferritinopathy or neuroferritinopathy characterized by abnormal ferritin inclusion aggregates and iron in the globus pallidus [198]. These mutations alter the amino acid sequence at the C-terminus of L-ferritin subunit, reducing its stability and ability to incorporate iron [199,200]. The neurodegeneration and iron overload phenotype was replicated in mice expressing a mutant L-ferritin subunit form [201 ]. A missense mutation in the L-ferritin subunit coding sequence was identified that leads to high levels of serum ferritin without iron overload [202]. Kannengiesser and colleagues [188] postulated that this mutation may modify a signal sequence that increases secretion of L-ferritin sub-units, and a subsequent study in cell culture shows that secretion of this mutant ferritin is increased as compared to wildtype ferritin [188]. Mutations have been reported in other IRE-encoded genes, such as eALAS (sideroblastic anemia) [203–205], ferroportin (hereditary hemochromatosis) [206,207] and DMT1 [208–210]. These studies show that abnormal expression of proteins encoded by IRE-containing mRNAs disrupts systematic and cellular iron metabolism and leads to iron overload and neurodegeneration.
A genome wide association study (GWAS) has associated various IRP2 polymorphisms with Alzheimer’s disease (AD) [211]. A potentially functional single nucleotide polymorphism (SNP) in the IRP2 promoter region was located within a consensus binding site for the AP-1 and SP1 transcription factors, and could possibly explain altered IRP2 expression seen in AD patients. This is of interest as a study showed IRP2 co-localization with pathological features of AD, including neurofibrillary tangles and senile plaques [212]. An additional link between IRP2 and neurodegenerative disease involves the regulation of amyloid precursor protein (APP). Studies have shown APP to be a functional ferroxidase that interacts with FPN in mediating iron export, and inhibition of ferroxidase activity leads to neuronal iron accumulation consistent with AD [213]. Similarly, neurons of Tau deficient mice retain iron and mislocalize APP, thus disrupting iron export by FPN [214]. More importantly, iron chelation with Clioquinol alleviated cognitive deficits seen in these Tau deficient mice. These observations highlight the importance of iron metabolism in maintaining proper neurological function, and provide therapeutic opportunities for treating neurodegenerative diseases such as Alzheimer’s and Parkinson’s. More recent GWAS have found strong associations between IRP2 and patients with chronic obstructive pulmonary disease (COPD) [215]. Three SNPs in IRP2 were found to correlate with reduced forced expiratory volume (FEV), a prominent feature of COPD [216]. It is known that IRP2 expression is altered in lung tissue from COPD patients, and iron deposition is increased in the lungs of smokers [215]. Further studies are needed to confirm a role for IRP2 in AD and COPD.
6. Outlook
Since their discovery over 20 years ago, IRPs still remain an active field of research. Iron-dependent and iron-independent mechanisms regulating IRP1 and IRP2 function have been identified and the physiological consequences of these mechanisms determined. The crystal structure of IRP1 bound to RNA has provided a basis to study the selectivity of IRE binding. Mouse models of IRP deficiency have shown the importance of IRPs in regulating cellular iron metabolism. The elusive E3 ligase mediating IRP2 degradation by iron has been identified, and the identification of novel IREs has broadened our vision of the IRP– IRE network.
At a mechanistic level, many questions regarding IRP function remain unanswered. One question regards the path ways of Fe–Scluster biogenesis that are directly sensed by IRPs and whether trafficking of iron through mitochondria is an obligate process through which IRPs sense iron. Additionally, the extent to which Fe–S cluster removal from c-acon versus de novo synthesis of IRP1 contribute to its role in homeostatic regulation of iron in animals needs attention. Another question is to determine how canonical and non-canonical IREs bind IRP1 to allow for their linkage to processes uniquely sensed by IRP1 (e.g. cytosolic Fe–S biogenesis). The discovery of novel IREs that preferentially bind to IRP1 or IRP2 is an opportunity to study links between iron homeostasis and other cellular or organismal processes. For example, what is the physiological significance of the preferential regulation of HIF-2α IRE by IRP1? The identification of FBXL5 E3 ligase raises the question whether other substrates are also targeted by FBXL5. The role of the IRP2 73-amino acid domain and Ser157 phosphorylation in cell proliferation requires further investigation. A better understanding of how iron contributes to neurodegeneration is needed with the goal of developing safer and effective therapies to treat these disorders.
Acknowledgments
This work was supported by grants from the NIH to EAL (GM45201 and DK068602), RSE (DK089212 and DK66600) and training grants to CPA (T32DK007115) and MS (T32DK007665). RSE also acknowledges support from the USDA Hatch Project WIS01324. We thank Josh Romney for critical reading of the review.
Footnotes
This article is part of a Special Issue entitled: Cell Biology of Metals.
References
- 1.Fleming RE, Ponka P. Iron overload in human disease. N Engl J Med. 2012;366:348–359. doi: 10.1056/NEJMra1004967. [DOI] [PubMed] [Google Scholar]
- 2.Madsen E, Gitlin JD. Copper and iron disorders of the brain. Annu Rev Neurosci. 2007;30:317–337. doi: 10.1146/annurev.neuro.30.051606.094232. [DOI] [PubMed] [Google Scholar]
- 3.Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia. 2007;55:1074–1084. doi: 10.1002/glia.20526. [DOI] [PubMed] [Google Scholar]
- 4.Wallander ML, Leibold EA, Eisenstein RS. Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim Biophys Acta. 2006;1763:668–689. doi: 10.1016/j.bbamcr.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 6.Muckenthaler MU, Galy B, Hentze MW. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr. 2008;28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521. [DOI] [PubMed] [Google Scholar]
- 7.Wang J, Pantopoulos K. Regulation of cellular iron metabolism. Biochem J. 2011;434:365–381. doi: 10.1042/BJ20101825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng L, Kennedy MC, Blondin Ga, Beinert H, Zalkin H. Binding of cytosolic aconitase to the iron responsive element of porcine mitochondrial aconitase mRNA, Arch. Biochem. Biophys. 1992;299:356–360. doi: 10.1016/0003-9861(92)90287-7. [DOI] [PubMed] [Google Scholar]
- 9.Cox TC, Bawden MJ, Martin A, May BK. Human erythroid 5-aminolevulinate synthase: promoter analysis and identification of an iron-responsive element in the mRNA. EMBO J. 1991;10:1891–1902. doi: 10.1002/j.1460-2075.1991.tb07715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanchez M, Galy B, Muckenthaler MU, Hentze MW. Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency. Nat Struct Mol Biol. 2007;14:420–426. doi: 10.1038/nsmb1222. [DOI] [PubMed] [Google Scholar]
- 11.Zimmer M, Ebert BL, Neil C, Brenner K, Papaioannou I, Melas A, Tolliday N, Lamb J, Pantopoulos K, Golub T, Iliopoulos O. Small-molecule inhibitors of HIF-2a translation link its 5′UTR iron-responsive element to oxygen sensing. Mol Cell. 2008;32:838–848. doi: 10.1016/j.molcel.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogers JT, Bush AI, Cho HH, Smith DH, Thomson AM, Friedlich AL, Lahiri DK, Leedman PJ, Huang X, Cahill CM. Iron and the translation of the amyloid precursor protein (APP) and ferritin mRNAs: riboregulation against neural oxidative damage in Alzheimer’s disease. Biochem Soc Trans. 2008;36:1282–1287. doi: 10.1042/BST0361282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gunshin H, Mackenzie B, Berger U, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez M, Galy B, Dandekar T, Bengert P, Vainshtein Y, Stolte J, Muckenthaler MU, Hentze MW. Iron regulation and the cell cycle: identification of an iron-responsive element in the 3′-untranslated region of human cell division cycle 14A mRNA by a refined microarray-based screening strategy. J Biol Chem. 2006;281:22865–22874. doi: 10.1074/jbc.M603876200. [DOI] [PubMed] [Google Scholar]
- 15.Cmejla R, Petrak J, Cmejlova J. A novel iron responsive element in the 3′UTR of human MRCKalpha. Biochem Biophys Res Commun. 2006;341:158–166. doi: 10.1016/j.bbrc.2005.12.155. [DOI] [PubMed] [Google Scholar]
- 16.Kohler SA, Menotti E, Kuhn LC. Molecular cloning of mouse glycolate oxidase. High evolutionary conservation and presence of an iron-responsive element-like sequence in the mRNA. J Biol Chem. 1999;274:2401–2407. doi: 10.1074/jbc.274.4.2401. [DOI] [PubMed] [Google Scholar]
- 17.Recalcati S, Tacchini L, Alberghini A, Conte D, Cairo G. Oxidative stress-mediated down-regulation of rat hydroxyacid oxidase 1, a liver-specific peroxisomal enzyme. Hepatology. 2003;38:1159–1166. doi: 10.1053/jhep.2003.50417. [DOI] [PubMed] [Google Scholar]
- 18.Sanchez M, Galy B, Schwanhaeusser B, Blake J, Bahr-Ivacevic T, Benes V, Selbach M, Muckenthaler MU, Hentze MW. Iron regulatory protein-1 and -2: transcriptome-wide definition of binding mRNAs and shaping of the cellular proteome by iron regulatory proteins. Blood. 2011;118:e168–e179. doi: 10.1182/blood-2011-04-343541. [DOI] [PubMed] [Google Scholar]
- 19.Wingert RA, Galloway JL, Barut B, Foott H, Fraenkel P, Axe JL, Weber GJ, Dooley K, Davidson AJ, Schmid B, Paw BH, Shaw GC, Kingsley P, Palis J, Schubert H, Chen O, Kaplan J, Zon LI. Deficiency of glutaredoxin 5 reveals Fe–S clusters are required for vertebrate haem synthesis. Nature. 2005;436:1035–1039. doi: 10.1038/nature03887. [DOI] [PubMed] [Google Scholar]
- 20.Johnson DC, Dean DR, Smith AD, Johnson MK. Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem. 2005;74:247–281. doi: 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- 21.Lill R. Function and biogenesis of iron-sulphur proteins. Nature. 2009;460:831–838. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- 22.Sharma AK, Pallesen LJ, Spang RJ, Walden WE. Cytosolic iron-sulfur cluster assembly (CA) system: factors, mechanism, and relevance to cellular iron regulation. J Biol Chem. 2010;285:26745–26751. doi: 10.1074/jbc.R110.122218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye H, Rouault TA. Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry. 2010;49:4945–4956. doi: 10.1021/bi1004798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schultz IJ, Chen C, Paw BH, Hamza I. Iron and porphyrin trafficking in heme biogenesis. J Biol Chem. 2010;285:26753–26759. doi: 10.1074/jbc.R110.119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fosset C, Chauveau MJ, Guillon B, Canal F, Drapier JC, Bouton C. RNA silencing of mitochondrial m-Nfs1 reduces Fe–S enzyme activity both in mitochondria and cytosol of mammalian cells. J Biol Chem. 2006;281:25398–25406. doi: 10.1074/jbc.M602979200. [DOI] [PubMed] [Google Scholar]
- 26.Gerber J, Muhlenhoff U, Lill R. An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 2003;4:906–911. doi: 10.1038/sj.embor.embor918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li K, Besse EK, Ha D, Kovtunovych G, Rouault TA. Iron-dependent regulation of frataxin expression: implications for treatment of Friedreich ataxia. Hum Mol Genet. 2008;17:2265–2273. doi: 10.1093/hmg/ddn127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmucker S, Martelli A, Colin F, Page A, Wattenhofer-Donze M, Reutenauer L, Puccio H. Mammalian frataxin: an essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLoS One. 2011;6:e16199. doi: 10.1371/journal.pone.0016199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai CL, Barondeau DP. Human frataxin is an allosteric switch that activates the Fe–S cluster biosynthetic complex. Biochemistry. 2010;49:9132–9139. doi: 10.1021/bi1013062. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Lyver ER, Nakamaru-Ogiso E, Yoon H, Amutha B, Lee DW, Bi E, Ohnishi T, Daldal F, Pain D, Dancis A. Dre2, a conserved eukaryotic Fe/S cluster protein, functions in cytosolic Fe/S protein biogenesis. Mol Cell Biol. 2008;28:5569–5582. doi: 10.1128/MCB.00642-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muhlenhoff U, Richter N, Pines O, Pierik AJ, Lill R. Specialized function of yeast Isa1 and Isa2 proteins in the maturation of mitochondrial [4Fe-4S] proteins. J Biol Chem. 2011;286:41205–41216. doi: 10.1074/jbc.M111.296152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roy A, Solodovnikova A, Nicholson T, Antholine W, Walden WE. A novel eukaryotic factor for cytosolic Fe–S assembly. EMBO J. 2003;22:2826–2835. doi: 10.1093/emboj/cdg455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balk J, Aguilar Netz DJ, Tepper K, Pierik AJ, Lill R. The essential WD40 protein Cia1 is involved in a late step of cytosolic and nuclear iron–sulfur protein assembly. Mol Cell Biol. 2005;25:10833–10841. doi: 10.1128/MCB.25.24.10833-10841.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balk J, Pierik AJ, Netz DJ, Muhlenhoff U, Lill R. The hydrogenase-like Nar1p is essential for maturation of cytosolic and nuclear iron–sulphur proteins. EMBO J. 2004;23:2105–2115. doi: 10.1038/sj.emboj.7600216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hausmann A, Aguilar Netz DJ, Balk J, Pierik AJ, Muhlenhoff U, Lill R. The eukaryotic P loop NTPase Nbp35: an essential component of the cytosolic and nuclear iron–sulfur protein assembly machinery. Proc Natl Acad Sci U S A. 2005;102:3266–3271. doi: 10.1073/pnas.0406447102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Netz DJ, Stumpfig M, Dore C, Muhlenhoff U, Pierik AJ, Lill R. Tah18 transfers electrons to Dre2 in cytosolic iron–sulfur protein biogenesis. Nat Chem Biol. 2010;6:758–765. doi: 10.1038/nchembio.432. [DOI] [PubMed] [Google Scholar]
- 37.Bych K, Kerscher S, Netz DJ, Pierik AJ, Zwicker K, Huynen MA, Lill R, Brandt U, Balk J. The iron–sulphur protein Ind1 is required for effective complex I assembly. EMBO J. 2008;27:1736–1746. doi: 10.1038/emboj.2008.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheftel AD, Stehling O, Pierik AJ, Netz DJ, Kerscher S, Elsasser HP, Wittig I, Balk J, Brandt U, Lill R. Human ind1, an iron–sulfur cluster assembly factor for respiratory complex I. Mol Cell Biol. 2009;29:6059–6073. doi: 10.1128/MCB.00817-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi Y, Ghosh M, Kovtunovych G, Crooks DR, Rouault TA. Both human ferredoxins 1 and 2 and ferredoxin reductase are important for iron–sulfur cluster biogenesis. Biochim Biophys Acta. 2011;1823:484–492. doi: 10.1016/j.bbamcr.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tong WH, Rouault TA. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron–sulfur cluster biogenesis and iron homeostasis. Cell Metab. 2006;3:199–210. doi: 10.1016/j.cmet.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 41.Song D, Tu Z, Lee FS. Human ISCA1 interacts with IOP1/NARFL and functions in both cytosolic and mitochondrial iron–sulfur protein biogenesis. J Biol Chem. 2009;284:35297–35307. doi: 10.1074/jbc.M109.040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Condo I, Malisan F, Guccini I, Serio D, Rufini A, Testi R. Molecular control of the cytosolic aconitase/IRP1 switch by extramitochondrial frataxin. Hum Mol Genet. 2010;19:1221–1229. doi: 10.1093/hmg/ddp592. [DOI] [PubMed] [Google Scholar]
- 43.Ye H, Jeong SY, Ghosh MC, Kovtunovych G, Silvestri L, Ortillo D, Uchida N, Tisdale J, Camaschella C, Rouault TA. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J Clin Invest. 2010;120:1749–1761. doi: 10.1172/JCI40372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shan Y, Cortopassi G. HSC20 interacts with frataxin and is involved in iron–sulfurcluster biogenesis and iron homeostasis. Hum Mol Genet. 2012;21:1457–1469. doi: 10.1093/hmg/ddr582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uhrigshardt H, Singh A, Kovtunovych G, Ghosh M, Rouault TA. Characterization of the human HSC20, an unusual DnaJ type III protein, involved in iron–sulfur cluster biogenesis. Hum Mol Genet. 2010;19:3816–3834. doi: 10.1093/hmg/ddq301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheftel A, Stehling O, Lill R. Iron–sulfur proteins in health and disease. Trends Endocrinol Metab. 2010;21:302–314. doi: 10.1016/j.tem.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 47.Shi Y, Ghosh MC, Tong WH, Rouault TA. Human ISD11 is essential for both iron–sulfur cluster assembly and maintenance of normal cellular iron homeostasis. Hum Mol Genet. 2009;18:3014–3025. doi: 10.1093/hmg/ddp239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheftel AD, Stehling O, Pierik AJ, Elsasser HP, Muhlenhoff U, Webert H, Hobler A, Hannemann F, Bernhardt R, Lill R. Humans possess two mito-chondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc Natl Acad Sci U S A. 2010;107:11775–11780. doi: 10.1073/pnas.1004250107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Babcock M, Silva Dd, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276:1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 50.Gerber J, Neumann K, Prohl C, Muhlenhoff U, Lill R. The yeast scaffold proteins Isu1p and Isu2p are required inside mitochondria for maturation of cytosolic Fe/S proteins. Mol Cell Biol. 2004;24:4848–4857. doi: 10.1128/MCB.24.11.4848-4857.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kispal G, Csere P, Prohl C, Lill R. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 1999;18:3981–3989. doi: 10.1093/emboj/18.14.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li J, Kogan M, Knight SA, Pain D, Dancis A. Yeast mitochondrial protein, Nfs1p, coordinately regulates iron–sulfur cluster proteins, cellular iron uptake, and iron distribution. J Biol Chem. 1999;274:33025–33034. doi: 10.1074/jbc.274.46.33025. [DOI] [PubMed] [Google Scholar]
- 53.Schilke B, Voisine C, Beinert H, Craig E. Evidence for a conserved system for iron metabolism in the mitochondria of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1999;96:10206–10211. doi: 10.1073/pnas.96.18.10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clarke SL, Vasanthakumar A, Anderson SA, Pondarre C, Koh CM, Deck KM, Pitula JS, Epstein CJ, Fleming MD, Eisenstein RS. Iron-responsive degradation of iron-regulatory protein 1 does not require the Fe–S cluster. EMBO J. 2006;25:544–553. doi: 10.1038/sj.emboj.7600954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pondarre C, Antiochos BB, Campagna DR, Clarke SL, Greer EL, Deck KM, McDonald A, Han AP, Medlock A, Kutok JL, Anderson SA, Eisenstein RS, Fleming MD. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron–sulfur cluster biogenesis. Hum Mol Genet. 2006;15:953–964. doi: 10.1093/hmg/ddl012. [DOI] [PubMed] [Google Scholar]
- 56.Song D, Lee FS. A role for IOP1 in mammalian cytosolic iron–sulfur protein biogenesis. J Biol Chem. 2008;283:9231–9238. doi: 10.1074/jbc.M708077200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Song D, Lee FS. Mouse knock-out of IOP1 protein reveals its essential role in mammalian cytosolic iron–sulfur protein biogenesis. J Biol Chem. 2011;286:15797–15805. doi: 10.1074/jbc.M110.201731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stehling O, Netz DJ, Niggemeyer B, Rosser R, Eisenstein RS, Puccio H, Pierik AJ, Lill R. Human Nbp35 is essential for both cytosolic iron–sulfur protein assembly and iron homeostasis. Mol Cell Biol. 2008;28:5517–5528. doi: 10.1128/MCB.00545-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rouault TA, Tong WH. Iron–sulfur cluster biogenesis and human disease. Trends Genet. 2008;24:398–407. doi: 10.1016/j.tig.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmucker S, Puccio H. Understanding the molecular mechanisms of Friedreich’s ataxia to develop therapeutic approaches. Hum Mol Genet. 2010;19:R103–R110. doi: 10.1093/hmg/ddq165. [DOI] [PubMed] [Google Scholar]
- 61.Stemmler TL, Lesuisse E, Pain D, Dancis A. Frataxin and mitochondrial FeS cluster biogenesis. J Biol Chem. 2010;285:26737–26743. doi: 10.1074/jbc.R110.118679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Lyver ER, Knight SA, Pain D, Lesuisse E, Dancis A. Mrs3p, Mrs4p, and frataxin provide iron for Fe–S cluster synthesis in mitochondria. J Biol Chem. 2006;281:22493–22502. doi: 10.1074/jbc.M604246200. [DOI] [PubMed] [Google Scholar]
- 63.Calmels N, Schmucker S, Wattenhofer-Donze M, Martelli A, Vaucamps N, Reutenauer L, Messaddeq N, Bouton C, Koenig M, Puccio H. The first cellular models based on frataxin missense mutations that reproduce spontaneously the defects associated with Friedreich ataxia. PLoS One. 2009;4:e6379. doi: 10.1371/journal.pone.0006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang ML, Becker EM, Whitnall M, Suryo Rahmanto Y, Ponka P, Richardson DR. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc Natl Acad Sci U S A. 2009;106:16381–16386. doi: 10.1073/pnas.0906784106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beinert H, Kennedy MC, Stout CD. Aconitase as iron–sulfur protein, enzyme, and iron regulatory protein. Chem Rev. 1996;96:2335–2373. doi: 10.1021/cr950040z. [DOI] [PubMed] [Google Scholar]
- 66.Gardner PR, Fridovich I. Superoxide sensitivity of the Escherichia coli aconitase. J Biol Chem. 1991;266:19328–19333. [PubMed] [Google Scholar]
- 67.Walden WE. From bacteria to mitochondria: aconitase yields surprises. Proc Natl Acad Sci U S A. 2002;99:4138–4140. doi: 10.1073/pnas.082108799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varghese S, Tang Y, Imlay JA. Contrasting sensitivities of Escherichia coli aconitases A and B to oxidation and iron depletion. J Bacteriol. 2003;185:221–230. doi: 10.1128/JB.185.1.221-230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brown NM, Kennedy MC, Antholine WE, Eisenstein RS, Walden WE. Detection of a [3Fe–4S] cluster intermediate of cytosolic aconitase in yeast expressing iron regulatory protein 1. Insights into the mechanism of Fe–S cluster cycling. J Biol Chem. 2002;277:7246–7254. doi: 10.1074/jbc.M110282200. [DOI] [PubMed] [Google Scholar]
- 70.Drapier JC, Hibbs JB. Murine cytotoxic activated macrophages inhibit aconitase in tumor cells. Inhibition involves the iron–sulfur prosthetic group and is reversible. J Clin Invest. 1986;78:790–797. doi: 10.1172/JCI112642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bouton C, Drapier JC. Iron regulatory proteins as NO signal transducers. Sci STKE. 2003;2003:e17. doi: 10.1126/stke.2003.182.pe17. [DOI] [PubMed] [Google Scholar]
- 72.Gray NK, Pantopoulos K, Dandekar T, Ackrell B, Hentze MW. Translational regulation of mammalian and Drosophila citric acid cycle enzymes via iron-responsive elements. Proc Natl Acad Sci U S A. 1996;93:4925–4930. doi: 10.1073/pnas.93.10.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang J, Fillebeen C, Chen G, Andriopoulos B, Pantopoulos K. Sodium nitroprusside promotes IRP2 degradation via an increase in intracellular iron and in the absence of S nitrosylation at C178. Mol Cell Biol. 2006;26:1948–1954. doi: 10.1128/MCB.26.5.1948-1954.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stys A, Galy B, Starzynski RR, Smuda E, Drapier JC, Lipinski P, Bouton C. Iron regulatory protein 1 outcompetes iron regulatory protein 2 in regulating cellular iron homeostasis in response to nitric oxide. J Biol Chem. 2011;286:22846–22854. doi: 10.1074/jbc.M111.231902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghosh MC, Tong WH, Zhang D, Ollivierre-Wilson H, Singh A, Krishna MC, Mitchell JB, Rouault TA. Tempol-mediated activationoflatent iron regulatory protein activity prevents symptoms of neurodegenerative disease in IRP2 knockout mice. Proc Natl Acad Sci U S A. 2008;105:12028–12033. doi: 10.1073/pnas.0805361105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang J, Chen G, Filebeen C, Pantopoulos K. Insights on regulation and function of the iron regulatory protein 1 (IRP1) Hemoglobin. 2008;32:109–115. doi: 10.1080/03630260701680326. [DOI] [PubMed] [Google Scholar]
- 77.Mueller S. Iron regulatory protein 1 as a sensor of reactive oxygen species. Biofactors. 2005;24:171–181. doi: 10.1002/biof.5520240121. [DOI] [PubMed] [Google Scholar]
- 78.Daba A, Koromilas AE, Pantopoulos K. Alternative ferritin mRNA translation via internal initiation. RNA. 2012;3:547–556. doi: 10.1261/rna.029322.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brown NM, Anderson SA, Steffen DW, Carpenter TB, Kennedy MC, Walden WE, Eisenstein RS. Novel role of phosphorylation in Fe–S cluster stability revealed by phosphomimetic mutations at Ser-138 of iron regulatory protein 1. Proc Natl Acad Sci U S A. 1998;95:15235–15240. doi: 10.1073/pnas.95.26.15235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Deck KM, Vasanthakumar A, Anderson SA, Goforth JB, Kennedy MC, Antholine WE, Eisenstein RS. Evidence that phosphorylation of iron regulatory protein 1 at Serine 138 destabilizes the [4Fe–4S] cluster in cytosolic aconitase by enhancing 4Fe–3Fe cycling. J Biol Chem. 2009;284:12701–12709. doi: 10.1074/jbc.M807717200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pantopoulos K, Hentze MW. Activation of iron regulatory protein-1 by oxidative stress in vitro. Proc Natl Acad Sci U S A. 1998;95:10559–10563. doi: 10.1073/pnas.95.18.10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Goessling LS, Mascotti DP, Bhattacharyya-Pakrasi M, Gang H, Thach RE. Irreversible steps in the ferritin synthesis induction pathway. J Biol Chem. 1994;269:4343–4348. [PubMed] [Google Scholar]
- 83.Haile DJ, Rouault TA, Harford JB, Kennedy MC, Blondin GA, Beinert H, Klausner RD. Cellular regulation of the iron-responsive element binding protein: disassembly of the cubane iron–sulfur cluster results in high-affinity RNA binding. Proc Natl Acad Sci U S A. 1992;89:11735–11739. doi: 10.1073/pnas.89.24.11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen OS, Schalinske KL, Eisenstein RS. Dietary iron intake modulates the activity of iron regulatory proteins (IRPs) and the abundance of ferritin and mitochondrial aconitase in rat liver. J Nutr. 1997;127:238–248. doi: 10.1093/jn/127.2.238. [DOI] [PubMed] [Google Scholar]
- 85.Meyron-Holz EG, Ghosh MC, Iwai K, LaVaute T, Brazzolotto X, Berger UV, Land W, Olivarerre-Wilson H, Grinberg A, Love P, Rouault TA. Genetic ablations of iron regulatory protein 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004;23:386–395. doi: 10.1038/sj.emboj.7600041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Neonaki M, Graham DC, White KN, Bomford A. Down-regulation of liver iron-regulatory protein 1 in haemochromatosis. Biochem Soc Trans. 2001;30:726–728. doi: 10.1042/bst0300726. [DOI] [PubMed] [Google Scholar]
- 87.Fillebeen C, Chahine D, Caltagirone A, Segal P, Pantopoulos K. A phosphomimetic mutation at Ser-138 renders iron regulatory protein 1 sensitive to iron-dependent degradation. Mol Cell Biol. 2003;23:6973–6981. doi: 10.1128/MCB.23.19.6973-6981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang J, Fillebeen C, Chen G, Biederbick A, Lill R, Pantopoulos K. Iron-dependent degradation of apo-IRP1 by the ubiquitin–proteasome pathway. Mol Cell Biol. 2007;27:2423–2430. doi: 10.1128/MCB.01111-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Salahudeen AA, Thompson JW, Ruiz JC, Ma HW, Kinch LN, Li Q, Grishin NV, Bruick RK. An E3 ligase possessing an iron responsive hemerythrin domain is a regulator of iron homeostasis. Science. 2009;326:718–721. doi: 10.1126/science.1176326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vashisht AA, Zumbrennen KB, Huang X, Powers DN, Durazo A, Sun D, Bhaskaran N, Persson A, Uhlen M, Sangfelt O, Spruck C, Leibold EA, Wohlschlegel JA. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science. 2009;326:718–721. doi: 10.1126/science.1176333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moroishi T, Nishiyama M, Takeda Y, Iwai K, Nakayama KI. The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo. Cell Metab. 2011;14:339–351. doi: 10.1016/j.cmet.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 92.Mochel F, Knight MA, Tong WH, Hernandez D, Ayyad K, Taivassalo T, Andersen PM, Singleton A, Rouault TA, Fischbeck KH, Haller RG. Splice mutation in the iron–sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am J Hum Genet. 2008;82:652–660. doi: 10.1016/j.ajhg.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Eisenstein RS, Tuazon PT, Schalinske KL, Anderson SA, Traugh JA. Iron responsive element binding protein: phosphorylation by protein kinase C. J Biol Chem. 1993;268:27363–27370. [PubMed] [Google Scholar]
- 94.Davis MR, Shawron KM, Rendina E, Peterson SK, Lucas EA, Smith BJ, Clarke SL. Hypoxia inducible factor-2 alpha is translationally repressed in response to dietary iron deficiency in Sprague–Dawley rats. J Nutr. 2011;141:1590–1596. doi: 10.3945/jn.111.144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zimmer M, Lamb J, Ebert BL, Lynch M, Neil C, Schmidt E, Golub TR, Iliopoulos O. The connectivity map links iron regulatory protein-1-mediated inhibition of hypoxia-inducible factor-2a translation to the anti-inflammatory 15-deoxy-delta12,14-prostaglandin J2. Cancer Res. 2010;70:3071–3079. doi: 10.1158/0008-5472.CAN-09-2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 97.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 2009;9:152–164. doi: 10.1016/j.cmet.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mastrogiannaki M, Matak P, Keith B, Simon MC, Vaulont S, Peyssonnaux C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest. 2009;119:1159–1166. doi: 10.1172/JCI38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Anderson ER, Xue X, Shah YM. Intestinal hypoxia-inducible factor-2alpha (HIF-2alpha) is critical for efficient erythropoiesis. J Biol Chem. 2011;286:19533–19540. doi: 10.1074/jbc.M111.238667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Taylor M, Qu A, Anderson ER, Matsubara T, Martin A, Gonzalez FJ, Shah YM. Hypoxia-inducible factor-2alpha mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology. 2011;140:2044–2055. doi: 10.1053/j.gastro.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schranzhofer M, Schifrer M, Cabrera JA, Kopp S, Chiba P, Beug H, Mullner EW. Remodeling the regulation of iron metabolism during erythroid differentiation to ensure efficient heme biosynthesis. Blood. 2006;107:4159–4167. doi: 10.1182/blood-2005-05-1809. [DOI] [PubMed] [Google Scholar]
- 103.Ke Y, Theil EC. An mRNA loop/bulge in the ferritin iron-responsive element forms in vivo and was detected by radical probing with Cu-1,10-phenanthroline and iron regulatory protein footprinting. J Biol Chem. 2002;277:2373–2376. doi: 10.1074/jbc.C100614200. [DOI] [PubMed] [Google Scholar]
- 104.Cooperman SS, Meyron-Holtz EG, Olivierre-Wilson H, Ghosh MC, McConnell JP, Rouault TA. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood. 2005;106:1084–1091. doi: 10.1182/blood-2004-12-4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Smith SR, Cooperman S, Lavaute T, Tresser N, Ghosh M, Meyron-Holtz E, Land W, Ollivierre H, Jortner B, Switzer III R, Messing A, Rouault TA. Severity of neurodegeneration correlates with compromise of iron metabolism in mice with iron regulatory protein deficiencies. Ann N Y Acad Sci. 2004;1012:65–83. doi: 10.1196/annals.1306.006. [DOI] [PubMed] [Google Scholar]
- 106.Kim HY, LaVaute T, Iwai K, Klausner RD, Rouault TA. Identification of a conserved and functional iron-responsive element in the 5′-untranslated region of mammalian mitochondrial aconitase. J Biol Chem. 1996;271:24226–24230. doi: 10.1074/jbc.271.39.24226. [DOI] [PubMed] [Google Scholar]
- 107.Schalinske KL, Chen OS, Eisenstein RS. Iron differentially stimulates translation of mitochondrial aconitase and ferritin mRNAs in mammalian cells. J Biol Chem. 1998;273:3740–3746. doi: 10.1074/jbc.273.6.3740. [DOI] [PubMed] [Google Scholar]
- 108.Goforth JB, Anderson SA, Nizzi CP, Eisenstein RS. Multiple determinants within iron-responsive elements dictate iron regulatory protein binding and regulatory hierarchy. RNA. 2010;16:154–169. doi: 10.1261/rna.1857210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Khan MA, Walden WE, Goss DJ, Theil EC. Direct Fe2+ sensingby iron-responsive messenger RNA:repressor complexes weakens binding. J Biol Chem. 2009;284:30122–30128. doi: 10.1074/jbc.M109.041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Galy B, Ferring-Appel D, Kaden S, Grone HJ, Hentze MW. Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell Metab. 2008;7:79–85. doi: 10.1016/j.cmet.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 111.Surdej P, Richman L, Kuhn LC. Differential translational regulation of IRE-containing mRNAs in Drosophila melanogaster by endogenous IRP anda constitutive human IRP1 mutant. Insect Biochem Mol Biol. 2008;38:891–894. doi: 10.1016/j.ibmb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 112.Burdon KP, Sharma S, Chen CS, Dimasi DP, Mackey DA, Craig JE. A novel deletion in the FTL gene causes hereditary hyperferritinemia cataract syndrome (HHCS) by alteration of the transcription start site. Hum Mutat. 2007;28:742. doi: 10.1002/humu.9501. [DOI] [PubMed] [Google Scholar]
- 113.Allerson CR, Cazzola M, Rouault TA. Clinical severity and thermodynamic effects of iron-responsive element mutations in hereditary hyperferritinemia–cataract syndrome. J Biol Chem. 1999;274:26439–26447. doi: 10.1074/jbc.274.37.26439. [DOI] [PubMed] [Google Scholar]
- 114.Walden WE, Selezneva A, Volz K. Accommodating variety in iron-responsive elements: crystal structure of transferrin receptor 1 B IRE bound to iron regulatory protein 1. FEBS Lett. 2012;586:32–35. doi: 10.1016/j.febslet.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 115.Zhang DL, Hughes RM, Ollivierre-Wilson H, Ghosh MC, Rouault TA. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009;9:461–473. doi: 10.1016/j.cmet.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leibold EA, Munro HN. Characterization and evolution of the expressed rat ferritin light subunit gene and its pseudogene family: conservation of sequences with non-coding regions of ferritin genes. J Biol Chem. 1987;262:7335–7341. [PubMed] [Google Scholar]
- 117.Aziz N, Munro HN. Iron regulates ferritin mRNA through a segment of its 5′ untranslated region. Proc Natl Acad Sci U S A. 1987;84:8478–8482. doi: 10.1073/pnas.84.23.8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hentze MW, Caughman SW, Rouault TA, Barriocanal JG, Dancis A, Harford JB, Klausner RD. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science. 1987;238:1570–1573. doi: 10.1126/science.3685996. [DOI] [PubMed] [Google Scholar]
- 119.Leibold EA, Munro HN. Cytoplasmic protein binds in vitro to a highly conserved sequence in the 5′ untranslated region of ferritin heavy- and light-subunit mRNAs. Proc Natl Acad Sci U S A. 1988;85:2171–2175. doi: 10.1073/pnas.85.7.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Casey JL, Hentze MW, Koeller DM, Caughman SW, Rouault TA, Klausner RD, Harford JB. Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation. Science. 1988;240:924–928. doi: 10.1126/science.2452485. [DOI] [PubMed] [Google Scholar]
- 121.Selezneva AI, Cavigiolio G, Theil EC, Walden WE, Volz K. Crystallization and preliminary X-ray diffraction analysis of iron regulatory protein 1 in complex with ferritin IRE RNA. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:249–252. doi: 10.1107/S1744309106004192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Harrell CM, McKenzie AR, Patino MM, Walden WE, Theil EC. Ferritin mRNA: interactions of iron regulatory element with translational regulator protein P-90 and the effect on base-paired flanking regions. Proc Natl Acad Sci U S A. 1991;88:4166–4170. doi: 10.1073/pnas.88.10.4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bettany AJ, Eisenstein RS, Munro HN. Mutagenesis of the iron-regulatory element further defines a role for RNA secondary structure in the regulation of ferritin and transferrin receptor expression. J Biol Chem. 1992;267:16531–16537. [PubMed] [Google Scholar]
- 124.Gdaniec Z, Sierputowska-Gracz H, Theil EC. Iron regulatory element and internal loop/bulge structure for ferritin mRNA studied by cobalt(III) hexamine binding, molecular modeling, and NMR spectroscopy. Biochemistry. 1998;37:1505–1512. doi: 10.1021/bi9719814. [DOI] [PubMed] [Google Scholar]
- 125.Henderson BR, Menotti E, Bonnard C, Kuhn LC. Optimal sequence and structure of iron-responsive elements. J Biol Chem. 1994;269:17481–17489. [PubMed] [Google Scholar]
- 126.Henderson BR, Menotti E, Kuhn LC. Iron regulatory proteins 1 and 2 bind distinct sets of RNA target sequences. J Biol Chem. 1996;271:4900–4908. doi: 10.1074/jbc.271.9.4900. [DOI] [PubMed] [Google Scholar]
- 127.Liang LG, Hall KB. A model of the iron responsive element RNA hairpin loop structure determined from NMR and thermodynamic data. Biochemistry. 1996;35:13586–13596. doi: 10.1021/bi961310q. [DOI] [PubMed] [Google Scholar]
- 128.Volz K. The functional duality of iron regulatory protein 1. Curr Opin Struct Biol. 2008;18:106–111. doi: 10.1016/j.sbi.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Stevens SG, Gardner PP, Brown C. Two covariance models for iron-responsive elements. RNA Biol. 2011;319:1047–1052. doi: 10.4161/rna.8.5.16037. [DOI] [PubMed] [Google Scholar]
- 130.Walden WE, Selezneva AI, Dupuy J, Volbeda A, Fontecilla-Camps JC, Theil EC, Volz K. Structure of dual function iron regulatory protein 1 complexed with ferritin IRE-RNA. Science. 2006;314:1903–1908. doi: 10.1126/science.1133116. [DOI] [PubMed] [Google Scholar]
- 131.Dix DJ, Lin PN, McKenzie AR, Walden WE, Theil EC. The influence of the base-paired flanking region on structure and function of the ferritin mRNA iron regulatory element. J Mol Biol. 1993;231:230–240. doi: 10.1006/jmbi.1993.1278. [DOI] [PubMed] [Google Scholar]
- 132.Dupuy J, Volbeda A, Carpentier P, Darnault C, Moulis JM, Fontecilla-Camps JC. Crystal structure of human iron regulatory protein1 as cytosolic aconitase. Structure. 2006;14:129–139. doi: 10.1016/j.str.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 133.Ke Y, Wu J, Leibold EA, Walden WE, Theil EC. Loops and bulge/loops in iron-responsive element isoforms influence iron regulatory protein binding. Fine-tuning of mRNA regulation? J Biol Chem. 1998;273:23637–23640. doi: 10.1074/jbc.273.37.23637. [DOI] [PubMed] [Google Scholar]
- 134.Piccinelli P, Samuelsson T. Evolution of the iron-responsive element. RNA. 2007;13:952–966. doi: 10.1261/rna.464807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ke Y, Sierzputowska-Gracz H, Gdaniec Z, Theil EC. Internal loop/bulge and hairpin loop of the iron-responsive element of ferritin mRNA contribute to maximal iron regulatory protein 2 binding and translational regulation in the iso-iron-responsive element/iso-iron regulatory protein family. Biochemistry. 2000;39:6235–6242. doi: 10.1021/bi9924765. [DOI] [PubMed] [Google Scholar]
- 136.Gunshin H, Allerson CR, Polycarpou-Schwarz M, Rofts A, Rogers JT, Kishi F, Hentze MW, Rouault TA, Andrews NC, Hediger MA. Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett. 2001;509:309–316. doi: 10.1016/s0014-5793(01)03189-1. [DOI] [PubMed] [Google Scholar]
- 137.Butt J, Kim HY, Basilion JP, Cohen S, Iwai K, Philpott CC, Altschul S, Klausner RD, Rouault TA. Differences in the RNA binding sites of iron regulatory proteins and potential target diversity. Proc Natl Acad Sci U S A. 1996;93:4345–4349. doi: 10.1073/pnas.93.9.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Meehan HA, Connell GJ. The hairpin loop but not the bulged C of the iron responsive element is essential for high affinity binding to the iron regulatory protein-1. J Biol Chem. 2001;276:14791–14796. doi: 10.1074/jbc.M010295200. [DOI] [PubMed] [Google Scholar]
- 139.Cho HH, Cahill CM, Vanderburg CR, Scherzer CR, Wang B, Huang X, Rogers JT. Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J Biol Chem. 2010;285:31217–31232. doi: 10.1074/jbc.M110.149161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.dos Santos CO, Dore LC, Valentine E, Shelat SG, Hardison RC, Ghosh M, Wang W, Eisenstein RS, Costa FF, Weiss MJ. An iron responsive element-like stem-loop regulates alpha-hemoglobin-stabilizing protein mRNA. J Biol Chem. 2008;283:26956–26964. doi: 10.1074/jbc.M802421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Guo B, Yu Y, Leibold EA. Iron regulates cytoplasmic levels of a novel iron-responsive element-binding protein without aconitase activity. J Biol Chem. 1994;269:24252–24260. [PubMed] [Google Scholar]
- 142.Samaniego F, Chin J, Iwai K, Rouault TA, Klausner RD. Molecular characterization of a second iron-responsive element binding protein, iron regulatory protein 2. J Biol Chem. 1994;49:30904–30910. [PubMed] [Google Scholar]
- 143.Guo B, Phillips JD, Yu Y, Leibold EA. Iron regulates the intracellular degradation of iron regulatory protein 2 by the proteasome. J Biol Chem. 1995;270:21645–21651. doi: 10.1074/jbc.270.37.21645. [DOI] [PubMed] [Google Scholar]
- 144.Iwai K, Klausner RD, Rouault TA. Requirements for iron-regulated degradation of the RNA binding protein, iron regulatory protein 2. EMBO J. 1995;14:5350–5357. doi: 10.1002/j.1460-2075.1995.tb00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kang DK, Jeong J, Drake SK, Wehr NB, Rouault TA, Levine RL. Iron regulatory protein 2 as iron sensor. Iron-dependent oxidative modification of cysteine. J Biol Chem. 2003;278:14857–14864. doi: 10.1074/jbc.M300616200. [DOI] [PubMed] [Google Scholar]
- 146.Jeong J, Rouault TA, Levine RL. Identification of a heme-sensing domain in iron regulatory protein 2. J Biol Chem. 2004;279:45450–45454. doi: 10.1074/jbc.M407562200. [DOI] [PubMed] [Google Scholar]
- 147.Yamanaka K, Ishikawa H, Megumi Y, Tokunaga F, Kanie M, Rouault TA, Morishima I, Minato N, Ishimori K, Iwai K. Identification of the ubiquitin-protein ligase that recognizes oxidized IRP2. Nat Cell Biol. 2003;5:336–340. doi: 10.1038/ncb952. [DOI] [PubMed] [Google Scholar]
- 148.Ishikawa H, Kato M, Hori H, Ishimori K, Kirisako T, Tokunaga F, Iwai K. Involvement of heme regulatory motif in heme-mediated ubiquitination and degradation of IRP2. Mol Cell. 2005;19:171–181. doi: 10.1016/j.molcel.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 149.Bourdon E, Kang DK, Ghosh MC, Drake SK, Wey J, Levine RL, Rouault TA. The role of endogenous heme synthesis and degradation domain cysteines in cellular iron-dependent degradation of IRP2. Blood Cells Mol Dis. 2003;31:247–255. doi: 10.1016/s1079-9796(03)00161-x. [DOI] [PubMed] [Google Scholar]
- 150.Hanson ES, Rawlins ML, Leibold EA. Oxygen and iron regulation of iron regulatory protein 2. J Biol Chem. 2003;278:40337–40342. doi: 10.1074/jbc.M302798200. [DOI] [PubMed] [Google Scholar]
- 151.Wang J, Chen G, Muckenthaler M, Galy B, Hentze MW, Pantopoulos K. Iron-mediated degradation of IRP2, an unexpected pathway involving a 2-oxoglutarate-dependent oxygenase activity. Mol Cell Biol. 2004;24:954–965. doi: 10.1128/MCB.24.3.954-965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zumbrennen KB, Hanson ES, Leibold EA. HOIL-1 is not required for iron-mediated IRP2 degradation in HEK293 cells. Biochim Biophys Acta. 2008;1783:246–252. doi: 10.1016/j.bbamcr.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- 154.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 155.Bailly X, Vanin S, Chabasse C, Mizuguchi K, Vinogradov SN. A phylogenomic profile of hemerythrins, the nonheme diiron binding respiratory proteins. BMC Evol Biol. 2008;8:244. doi: 10.1186/1471-2148-8-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kao WC, Wang VC, Huang YC, Yu SS, Chang TC, Chan SI. Isolation, purification and characterization of hemerythrin from Methylococcus capsulatus (Bath) J Inorg Biochem. 2008;102:1607–1614. doi: 10.1016/j.jinorgbio.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 157.Isaza CE, Silaghi-Dumitrescu R, Iyer RB, Kurtz DM, Jr, Chan MK. Structural basis for O2 sensing by the hemerythrin-like domain of a bacterial chemotaxis protein: substrate tunnel and fluxional N terminus. Biochemistry. 2006;45:9023–9031. doi: 10.1021/bi0607812. [DOI] [PubMed] [Google Scholar]
- 158.Thompson JW, Salahudeen AA, Chollangi S, Ruiz JC, Brautigam CA, Makris TM, Lipscomb JD, Tomchick DR, Bruick RK. Structural and molecular characterization of the iron-sensing hemerythrin-like domain within F-box and leucine-rich repeat protein 5 (FBXL5) J Biol Chem. 2012;287:7357–7365. doi: 10.1074/jbc.M111.308684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang J, Chen G, Lee J, Pantopoulos K. Iron-dependent degradation of IRP2 requires its C-terminal region and IRP structural integrity. BMC Mol Biol. 2008;9:15. doi: 10.1186/1471-2199-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Meyron-Holtz EG, Ghosh MC, Rouault TA. Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science. 2004;306:2087–2090. doi: 10.1126/science.1103786. [DOI] [PubMed] [Google Scholar]
- 161.Hausmann A, Lee J, Pantopoulos K. Redox control of iron regulatory protein 2 stability. FEBS Lett. 2011;585:687–692. doi: 10.1016/j.febslet.2011.01.036. [DOI] [PubMed] [Google Scholar]
- 162.Cairo G, Pietrangelo A. Nitric-oxide mediated activation of iron-regulatory protein controls hepatic iron metabolism during acute inflammation. Eur J Biochem. 1995;232:358. doi: 10.1111/j.1432-1033.1995.358zz.x. [DOI] [PubMed] [Google Scholar]
- 163.Tacchini L, Recalcati S, Bernelli-Zazzera A, Cairo G. Induction of ferritin synthesis in ischemic-reperfused rat liver: analysis of the molecular mechanisms. Gastroenterology. 1997;113:946–953. doi: 10.1016/s0016-5085(97)70191-4. [DOI] [PubMed] [Google Scholar]
- 164.Zumbrennen KB, Wallander ML, Romney SJ, Leibold EA. Cysteine oxidation regulates the RNA-binding activity of Iron Regulatory Protein 2. Mol Cell Biol. 2009;29:2219–2229. doi: 10.1128/MCB.00004-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ozer A, Bruick RK. Non-heme dioxygenases: cellular sensors and regulators jelly rolled into one? Nat Chem Biol. 2007;3:144–153. doi: 10.1038/nchembio863. [DOI] [PubMed] [Google Scholar]
- 166.Kim S, Wing SS, Ponka P. S-nitrosylation of IRP2 regulates its stability via the ubiquitin–proteasome pathway. Mol Cell Biol. 2004;24:330–337. doi: 10.1128/MCB.24.1.330-337.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chang AH, Jeong J, Levine RL. Iron regulatory protein 2 turnover through a nonproteasomal pathway. J Biol Chem. 2011;286:23698–23707. doi: 10.1074/jbc.M110.216788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dycke C, Charbonnier P, Pantopoulos K, Moulis JM. A role for lysosomes in the turnover of human iron regulatory protein 2. Int J Biochem Cell Biol. 2008;40:2826–2832. doi: 10.1016/j.biocel.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 169.Morgan EH. Inhibition of reticulocyte iron uptake by NH4Cl and CH3NH2. Biochim Biophys Acta. 1981;642:119–134. doi: 10.1016/0005-2736(81)90143-7. [DOI] [PubMed] [Google Scholar]
- 170.Torti SV, Torti FM. Ironing out cancer. Cancer Res. 2011;71:1511–1514. doi: 10.1158/0008-5472.CAN-10-3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Cozzi A, Corsi B, Levi S, Santambrogio P, Albertini A, Arosio P. Overexpression of wild type and mutated human ferritin H-chain in HeLa cells: in vivo role of ferritin ferroxidase activity. J Biol Chem. 2000;275:25122–25129. doi: 10.1074/jbc.M003797200. [DOI] [PubMed] [Google Scholar]
- 172.Kakhlon O, Gruenbaum Y, Cabantchik ZI. Repression of ferritin expression increases the labile iron pool, oxidative stress, and short-term growth of human erythroleukemia cells. Blood. 2001;97:2863–2871. doi: 10.1182/blood.v97.9.2863. [DOI] [PubMed] [Google Scholar]
- 173.Cozzi A, Corsi B, Levi S, Santambrogio P, Biasiotto G, Arosio P. Analysis of the biologic functions of H- and L-ferritins in HeLa cells by transfection with siRNAs and cDNAs: evidence for a proliferative role of L-ferritin. Blood. 2004;103:2377–2383. doi: 10.1182/blood-2003-06-1842. [DOI] [PubMed] [Google Scholar]
- 174.Baldi A, Lombardi D, Russo P, Palescandolo E, De Luca A, Santini D, Baldi F, Rossiello L, Dell’Anna ML, Mastrofrancesco A, Maresca V, Flori E, Natali PG, Picardo M, Paggi MG. Ferritin contributes to melanoma progression by modulating cell growth and sensitivity to oxidative stress. Clin Cancer Res. 2005;11:3175–3183. doi: 10.1158/1078-0432.CCR-04-0631. [DOI] [PubMed] [Google Scholar]
- 175.Pinnix ZK, Miller LD, Wang W, D’Agostino R, Jr, Kute T, Willingham MC, Hatcher H, Tesfay L, Sui G, Di X, Torti SV, Torti FM. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010;2:43ra56. doi: 10.1126/scisignal.3001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Maffettone C, Chen G, Drozdov I, Ouzounis C, Pantopoulos K. Tumorigenic properties of iron regulatory protein 2 (IRP2) mediated by its specific 73-amino acids insert. PLoS One. 2010;5:e10163. doi: 10.1371/journal.pone.0010163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chen G, Fillebeen C, Wang J, Pantopoulos K. Overexpression of iron regulatory protein 1 suppresses growth of tumor xenografts. Carcinogenesis. 2007;28:785–791. doi: 10.1093/carcin/bgl210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wallander ML, Zumbrennen KB, Rodansky ES, Romney SJ, Leibold EA. Iron-independent phosphorylation of iron regulatory protein 2 regulates ferritin during the cell cycle. J Biol Chem. 2008;283:23589–23598. doi: 10.1074/jbc.M803005200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Meyron-Holtz EG, Ghosh MC, Iwai K, LaVaute T, Brazzolotto X, Berger UV, Land W, Ollivierre-Wilson H, Grinberg A, Love P, Rouault TA. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004;23:386–395. doi: 10.1038/sj.emboj.7600041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Smith SR, Ghosh MC, Ollivierre-Wilson H, Hang Tong W, Rouault TA. Complete loss of iron regulatory proteins 1 and 2 prevents viability of murine zygotes beyond the blastocyst stage of embryonic development. Blood Cells Mol Dis. 2006;36:283–287. doi: 10.1016/j.bcmd.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 181.LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer Rr, Grinberg A, Love P, Tresser N, Rouault TA. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- 182.Galy B, Ferring D, Hentze MW. Generation of conditional alleles of the murine iron regulatory protein (Irp)-1 and -2 genes. Genesis. 2005;43:181–188. doi: 10.1002/gene.20169. [DOI] [PubMed] [Google Scholar]
- 183.Galy B, Ferring D, Minana B, Bell O, Janser HG, Muckenthaler M, Schumann K, Hentze MW. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2) Blood. 2005;106:2580–2589. doi: 10.1182/blood-2005-04-1365. [DOI] [PubMed] [Google Scholar]
- 184.Ferring-Appel D, Hentze MW, Galy B. Cell-autonomous and systemic context-dependent functions of iron regulatory protein 2 in mammalian iron metabolism. Blood. 2009;113:679–687. doi: 10.1182/blood-2008-05-155093. [DOI] [PubMed] [Google Scholar]
- 185.Jeong SY, Crooks DR, Wilson-Ollivierre H, Ghosh MC, Sougrat R, Lee J, Cooperman S, Mitchell JB, Beaumont C. TA Rouault, Iron insufficiency compromises motor neurons and their mitochondrial function in Irp2-null mice. PLoS One. 2011;6:e25404. doi: 10.1371/journal.pone.0025404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ferreira C, Santambrogio P, Martin ME, Andrieu V, Feldmann G, Henin D, Beaumont C. H ferritin knockout mice: a model of hyperferritinemia in the absence of iron overload. Blood. 2001;98:525–532. doi: 10.1182/blood.v98.3.525. [DOI] [PubMed] [Google Scholar]
- 187.Galy B, Holter SM, Klopstock T, Ferring D, Becker L, Kaden S, Wurst W, Grone HJ, Hentze MW. Iron homeostasis in the brain: complete iron regulatory protein 2 deficiency without symptomatic neurodegeneration in the mouse. Nat Genet. 2006;38:967–969. doi: 10.1038/ng0906-967. (discussion 969–970) [DOI] [PubMed] [Google Scholar]
- 188.De Domenico I, Vaughn MB, Paradkar PN, Lo E, Ward DM, Kaplan J. Decoupling ferritin synthesis from free cytosolic iron results in ferritin secretion. Cell Metab. 2011;13:57–67. doi: 10.1016/j.cmet.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 189.Vanoaica L, Darshan D, Richman L, Schumann K, Kuhn LC. Intestinal ferritin H is required for an accurate control of iron absorption. Cell Metab. 2010;12:273–282. doi: 10.1016/j.cmet.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 190.Galy B, Ferring-Appel D, Sauer SW, Kaden S, Lyoumi S, Puy H, Kolker S, Grone HJ, Hentze MW. Iron regulatory proteins secure mitochondrial iron sufficiency and function. Cell Metab. 2010;12:194–201. doi: 10.1016/j.cmet.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 191.Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14:804–810. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Dallman PR, Goodman JR. Enlargement of mitochondrial compartment in iron and copper deficiency. Blood. 1970;35:496–505. [PubMed] [Google Scholar]
- 193.Hintze KJ, Theil EC. DNA and mRNA elements with complementary responses to hemin, antioxidant inducers, and iron control ferritin-L expression. Proc Natl Acad Sci U S A. 2005;102:15048–15052. doi: 10.1073/pnas.0505148102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Beaumont C, Leneuve P, Devaux I, Scoazec JY, Berthier M, Loiseau MN, Grandchamp B, Bonneau D. Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinaemia and cataract. Nat Genet. 1995;11:444–446. doi: 10.1038/ng1295-444. [DOI] [PubMed] [Google Scholar]
- 195.Girelli D, Corrocher R, Bisceglia L, Olivieri O, De Franceschi L, Zelante L, Gasparini P. Molecular basis for the recently described hereditary hyperferritinemia-cataract syndrome: a mutation in the iron-responsive element of ferritin L-subunit gene (the “Verona mutation”) Blood. 1995;86:4050–4053. [PubMed] [Google Scholar]
- 196.Cazzola M, Foglieni B, Bergamaschi G, Levi S, Lazzarino M, Aosio P. A novel deletion of the L-ferritin iron-responsive element responsible for severe hereditary hyperferritinaemia-cataract syndrome. Br J Haematol. 2002;116:667–670. doi: 10.1046/j.0007-1048.2001.03310.x. [DOI] [PubMed] [Google Scholar]
- 197.Kato J, Fujikawa K, Kanda M, Fukuda N, Sasaki K, Takayama T, Kobune M, Takada K, Takimoto R, Hamada H, Ikeda T, Niitsu Y. A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am J Hum Genet. 2001;69:191–197. doi: 10.1086/321261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Curtis AR, Fey C, Morris CM, Bindoff LA, Ince PG, Chinnery PF, Coulthard A, Jackson MJ, Jackson AP, McHale DP, Hay D, Barker WA, Markham AF, Bates D, Curtis A, Burn J. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet. 2001;28:350–354. doi: 10.1038/ng571. [DOI] [PubMed] [Google Scholar]
- 199.Luscieti S, Santambrogio P, Langlois d’Estaintot B, Granier T, Cozzi A, Poli M, Gallois B, Finazzi D, Cattaneo A, Levi S, Aosio P. Mutant ferritin L-chains that cause neurodegeneration act in a dominant-negative manner to reduce ferritin iron incorporation. J Biol Chem. 2010;285:11948–11957. doi: 10.1074/jbc.M109.096404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Muhoberac BB, Baraibar MA, Vidal R. Iron loading-induced aggregation and reduction of iron incorporation in heteropolymeric ferritin containing a mutant light chain that causes neurodegeneration. Biochim Biophys Acta. 2011;1812:544–548. doi: 10.1016/j.bbadis.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Vidal R, Miravalle L, Gao X, Barbeito AG, Baraibar MA, Hekmatyar SK, Widel M, Bansal N, Delisle MB, Ghetti B. Expression of a mutant form of the ferritin light chain gene induces neurodegeneration and iron overload in transgenic mice. J Neurosci. 2008;28:60–67. doi: 10.1523/JNEUROSCI.3962-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kannengiesser C, Jouanolle AM, Hetet G, Mosser A, Muzeau F, Henry D, Bardou-Jacquet E, Mornet M, Brissot P, Deugnier Y, Grandchamp B, Beaumont C. A new missense mutation in the L ferritin coding sequence associated with elevated levels of glycosylated ferritin in serum and absence of iron overload. Haematologica. 2009;94:335–339. doi: 10.3324/haematol.2008.000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Cox TC, Bottomley SS, Wiley JS, Bawden MJ, Matthews CS, May BK. X-linked pyridoxine-responsive sideroblastic anemia due to a Thr388-to-Ser substitution in erythroid 5-aminolevulinate synthase. N Engl J Med. 1994;330:675–679. doi: 10.1056/NEJM199403103301004. [DOI] [PubMed] [Google Scholar]
- 204.Cotter PD, Baumann M, Bishop DF. Enzymatic defect in “X-linked” sideroblastic anemia: molecular evidence for erythroid delta-aminolevulinate synthase deficiency. Proc Natl Acad Sci U S A. 1992;89:4028–4032. doi: 10.1073/pnas.89.9.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Whatley SD, Ducamp S, Gouya L, Grandchamp B, Beaumont C, Badminton MN, Elder GH, Holme SA, Anstey AV, Parker M, Corrigall AV, Meissner PN, Hift RJ, Marsden JT, Ma Y, Mieli-Vergani G, Deybach JC, Puy H. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008;83:408–414. doi: 10.1016/j.ajhg.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.De Domenico I, Ward DM, Musci G, Kaplan J. Iron overload due to mutations in ferroportin. Haematologica. 2006;91:92–95. [PMC free article] [PubMed] [Google Scholar]
- 207.Fernandes A, Preza GC, Phung Y, De Domenico I, Kaplan J, Ganz T, Nemeth E. The molecular basis of hepcidin-resistant hereditary hemochromatosis. Blood. 2009;114:437–443. doi: 10.1182/blood-2008-03-146134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Shahidi NT, Nathan DG, Diamond LK. Iron deficiency anemia associated with an error of iron metabolism in two siblings. J Clin Invest. 1964;43:510–521. doi: 10.1172/JCI104937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mims MP, Guan Y, Pospisilova D, Priwitzerova M, Indrak K, Ponka P, Divoky V, Prchal JT. Identification of a human mutation of DMT1 in a patient with microcytic anemia and iron overload. Blood. 2005;105:1337–1342. doi: 10.1182/blood-2004-07-2966. [DOI] [PubMed] [Google Scholar]
- 210.Iolascon A, d’Apolito M, Servedio V, Cimmino F, Piga A, Camaschella C. Microcytic anemia and hepatic iron overload in a child with compound heterozygous mutations in DMT1 (SCL11A2) Blood. 2006;107:349–354. doi: 10.1182/blood-2005-06-2477. [DOI] [PubMed] [Google Scholar]
- 211.Coon KD, Siegel AM, Yee SJ, Dunckley TL, Mueller C, Nagra RM, Tourtellotte WW, Reiman EM, Papassotiropoulos A, Petersen FF, Stephan DA, Kirsch WM. Preliminary demonstration of an allelic association of the IREB2 gene with Alzheimer’s disease. J Alzheimers Dis. 2006;9:225–233. doi: 10.3233/jad-2006-9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Smith MA, Wehr K, Harris PL, Siedlak SL, Connor JR, Perry G. Abnormal localization of iron regulatory protein in Alzheimer’s disease. Brain Res. 1998;788:232–236. doi: 10.1016/s0006-8993(98)00002-x. [DOI] [PubMed] [Google Scholar]
- 213.Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K, Leong SL, Perez K, Johanssen T, Greenough MA, Cho HH, Galatis D, Moir RD, Masters CL, McLean C, Tanzi RE, Cappai R, Barnham KJ, Ciccotosto GD, Rogers JT, Bush AI. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell. 2010;142:857–867. doi: 10.1016/j.cell.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BX, Adlard PA, Cherny RA, Lam LQ, Roberts BR, Volitakis I, Egan GF, McLean CA, Cappai R, Duce JA, Bush AI. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18:291–295. doi: 10.1038/nm.2613. [DOI] [PubMed] [Google Scholar]
- 215.DeMeo DL, Mariani T, Bhattacharya S, Srisuma S, Lange C, Litonjua A, Bueno R, Pillai SG, Lomas DA, Sparrow D, Shapiro SD, Criner GJ, Kim HP, Chen Z, Choi AM, Reilly J, Silverman EK. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. Am J Hum Genet. 2009;85:493–502. doi: 10.1016/j.ajhg.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Chappell SL, Daly L, Lotya J, Alsaegh A, Guetta-Baranes T, Roca J, Rabinovich R, Morgan K, Millar AB, Donnelly SC, Keatings V, MacNee W, Stolk J, Hiemstra PS, Miniati M, Monti S, O’Connor CM, Kalsheker N. The role of IREB2 and transforming growth factor beta-1 genetic variants in COPD: a replication case-control study. BMC Med Genet. 2011;12:24. doi: 10.1186/1471-2350-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]