

Abstract
Cervical carcinomas are almost universally associated with high-risk human papillomavirus (HPV) infections, and are a leading cause of cancer death in women worldwide. HPV oncoproteins contribute to cancer initiation and progression and their expression is necessary for the maintenance of the transformed state. The fact that the initiating oncogenic insult, infection with a high-risk HPV and viral oncoprotein expression, is common to almost all cervical cancers offers unique opportunities for prevention, early detection, and therapy. The potential for prevention has been realized by introduction of prophylactic vaccines that are to prevent transmission of specific high-risk HPVs. Given, however, that these vaccines have no therapeutic efficacy and HPV-associated cervical cancers arise years if not decades after the initial infection, it has been estimated that there will be no measurable decline of HPV-associated tumors before 2040. Cervical cancer alone will be diagnosed in more than 375,000 US women between now and 2040. Other HPV-associated anogenital and head and neck cancers are predicted to afflict another 700,000 men and women over this time period. Hence, therapeutic efforts to combat high-risk HPV-associated disease remain of critical importance.
INTRODUCTION
Papillomaviruses contain double stranded, circular DNA genomes of approximately 8,000 basepairs packaged into nonenveloped icosahedral particles. The viral genome contains an early (E) region that consists of up to eight open reading frames (ORFs), which encode nonstructural proteins. The late (L) region contains the L1 and L2 ORFs, which encode the major and minor capsid proteins, respectively. Early and late ORFs are encoded on the same DNA strand. A third region of the viral genome is referred to as the upstream regulatory region, long control region (LCR) or noncoding region and contains DNA elements that regulate viral genome replication and transcription (Fig 1A).1
Fig 1.

Human papillomavirus (HPV) genome organization. (A) The HPV16 genome is shown as a black circle; arrows indicate the early (E; black) and late (L; red) promoters in the long control region (LCR). Early (E) and late (L) open reading frames (blue) are expressed at different stages during epithelial differentiation. (B) Schematic representation of the minimal HPV sequences integrated into a host chromosome. Integration is a frequent event during malignant progression of high-risk HPV-associated lesions. Expression of the HPV E6 and E7 oncoproteins from the viral LCR is universally maintained in HPV-associated cervical cancers, whereas other portions of the viral genomes, including the E5 oncoprotein, are generally no longer expressed after integration. (C) Schematic representation of the HPV16 E5 (top), E6 (middle), and E7 (bottom) oncoproteins. Sequences involved in targeting known cellular proteins that are relevant for cellular transformation are indicated as red boxes (C, cysteine; L, leucine; Y, tyrosine; E, glutamic acid; X, any amino acid). The cysteine-rich sequence in E5 (CXXC) aids in formation of homodimers. The cysteine-rich domains (CXXC-X29-CXXC) in E6 and E7 are evolutionarily related and function as zinc-binding domains. The amino terminal domain of the E7 oncoprotein that shares sequence similarity to the adenovirus early 1A protein (Ad E1A) and simian vacuolating virus 40 large tumor antigen (SV40 TAg), which contains the binding site for the retinoblastoma tumor suppressor protein (pRB), is also shown. See Introduction and Oncogenic Activities of High-Risk HPVs for details. TM, transmembrane domain.
The life cycle of human papillomaviruses (HPVs) is closely linked to the differentiation stage of the infected cells. Keratinocytes in the basal layer of squamous epithelia are the initial targets for HPV infection (Fig 2). After infection, HPV genomes are maintained at low copy numbers and can persist for years to decades in the nuclei of basal epithelial cells. HPV genome amplification and synthesis of viral progeny, however, is restricted to terminally differentiated cells. With the exception of the E1 helicase, HPVs depend on host cell DNA synthesis enzymes for genome replication. Hence, a key aspect of the viral replication strategy is to maintain differentiated keratinocytes in a DNA synthesis competent state.1 HPVs are nonlytic viruses and the newly synthesized infectious viral particles are retained within the denucleated epithelial squames when they are sloughed off, and they remain infectious over extended periods of time.2
Fig 2.
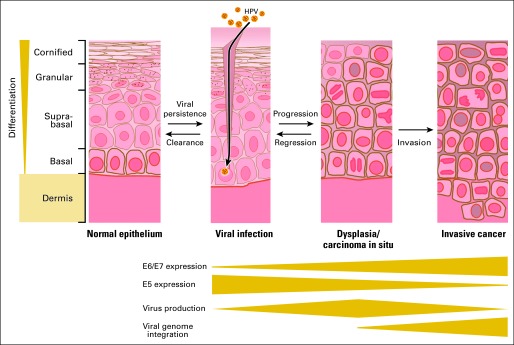
Human papillomavirus (HPV) infection of epithelial cells. HPVs infect basal cells of squamous epithelia through sites of mechanical trauma. Infections with high-risk HPVs can lead to dysplasia and carcinoma in situ and to invasive squamous cell carcinoma. Progression is a rare and slow process and many lesions regress spontaneously. Dysplasia is characterized by expansion of basal-like cells into the suprabasal layer and mitotic activity in these layers. Multipolar mitoses can occur as a consequence of E7 oncoprotein expression and contribute to genomic instability. Malignant progression is frequently accompanied by integration of portions of the HPV genome into a host cell chromosome. This leads to increased HPV E6/E7 oncoprotein expression, loss of E5 expression, and failure to produce viral progeny. See Epidemiology and Natural History of HPV Infections for details.
EPIDEMIOLOGY AND NATURAL HISTORY OF HPV INFECTIONS
Approximately 30 of the 200 HPV types that have been characterized specifically infect mucosal surfaces.3 While mucosal HPVs are mainly transmitted sexually through direct mucosal contact,4 penetrative sexual intercourse is not strictly required as these viruses can apparently be transferred from the introitus to the cervix.5 Although rare, there is also evidence for perinatal infection during passage through the birth canal, and auto- and hetero-inoculation through close contact and transmission via fomites have also been suggested.6,7
The most frequent HPV-positive site in males is the penile shaft, which often harbors several HPVs.8 Hence, multiple HPV types may be simultaneously transmitted during a single sexual contact. Circumcision in males may decrease transmission and persistent infection,9 but in most studies there was no correlation between male circumcision and cervical cancer incidence.10 Consistent condom use has been associated with decreased HPV-infection and disease.11 Animal model studies suggested that a common component of vaginal lubricants, the polysaccharide carrageenan, inhibits HPV transmission, whereas the spermicidal compound nonoxynol-9 might increase transmission rates.12,13
HPVs can gain access to basal epithelial cells through a traumatized epithelial barrier (Fig 2). Moreover, basal-like cells are accessible and vulnerable to HPV infection at the cervical squamocolumnar transformation zone. Reserve cells, which can give rise to columnar as well as squamous progeny, might be particularly significant targets of infection by high-risk HPVs.14–16 Since serology has only recently become available and HPV DNA is often only transiently detected, overall HPV exposure rates have been difficult to assess.17 However, it has been estimated that most women will have been infected with at least one mucosal HPV type during their reproductive life.18 Before widespread availability of prophylactic HPV vaccines, an overall HPV prevalence rate of 26.8% has been estimated for US females aged 14 to 59 years. The highest HPV prevalence, approximately 45%, was observed among women age 25 to 29 years.19
DISEASES ASSOCIATED WITH MUCOSAL HPV INFECTIONS
Clinically, mucosal HPVs are classified into low-risk and high-risk types according to the potential of malignant progression of the lesions they cause (Table 1).1,20,21 Infections with low-risk HPVs are associated with genital warts (condyloma acuminata). HPV6 and 11 are the most abundant low-risk HPVs and cause more than 90% of condylomata acuminata.22 Although these lesions are at an extremely low risk for malignant progression and frequently regress over time, patients generally insist on their removal by surgical options, such as laser coagulation or cryotherapy. The topical ointment Imiquimod (Aldara; Graceway Pharmaceuticals LLC, Bristol, TN), which stimulates the host's T-helper 1 immune response, notably the production of γ-interferon, may be used for treatment of recurrent disease.23 In rare cases, low-risk HPV infections can cause slow growing giant condyloma, named after their initial discoverers, Buschke and Lowenstein. These lesions are highly destructive to adjacent normal tissue and can metastasize.24
Table 1.
HPV Types and Associated Diseases
| HPV | Cutaneous | Mucosal |
|---|---|---|
| Benign lesions | Common skin warts: HPV27, 29, 37, 38, 46, 48, 49, 57 | Anogenital warts: HPV6, 11, 42, 44 |
| Plantar warts: HPV1, 2, 4, 63 | Oral warts: HPV2, 4, 6, 7, 11, 16, 32 | |
| Flat warts: HPV3, 10, 28, 41 | Oral focal epithelial hyperplasia: HPV13, 32 | |
| Lip lesions: HPV2 | Respiratory papillomatosis: HPV6, 11 | |
| Conjunctival papillomatosis: HPV6, 11 | ||
| Giant condyloma (Buschke-Lowenstein): HPV6, 11 | ||
| Cancer | Nonmelanoma skin cancers (mostly in patients with epidermodysplasia verruciformis or immunosuppression): HPV5, 8, and many others | Anogenital cancers: highest risk: HPV16, 18, 31, 45; other high risk: HPV33, 35, 39, 51, 52, 54, 56, 58, 59, 68; probably high risk: HPV26, 42, 43, 44, 53, 54, 55, 62, 66, 68, 73, 82 |
| Head and neck squamous cell carcinoma: HPV16 |
Recurrent respiratory papillomatosis is also caused by low-risk HPVs and is the most common benign neoplasm of the nasopharynx, trachea, and larynx among children suffering from wart-like growths in the aerodigestive tract. It has been suggested that these HPV infections may have been caused by vertical HPV transmission during vaginal delivery by an infected mother. Most lesions are cytologically benign, but they grow very rapidly and can spread locally. Progression to squamous cell carcinoma has been observed in some patients with recurrent respiratory papillomatosis.25,26
Harald zur Hausen's discovery that some HPVs are associated with premalignant cervical lesions and cancers27,28 was recognized with the 2008 Nobel Prize in Medicine. Epidemiologic studies have shown that almost all cervical cancers are caused by persistent infection with one of approximately 15 high-risk HPV types (Tables 1 and 229).21 HPV types 16 and 18 are the two most abundant high-risk HPVs and are present in approximately 50% of high-grade cervical intraepithelial neoplasias (CIN) and approximately 70% of cervical cancers.30 HPV16 is most frequently detected in squamous cell carcinomas, whereas HPV18 appears to be more frequently associated with adenocarcinomas.31
Table 2.
Estimated Contribution of High-Risk Mucosal Human Papillomaviruses to Various Cancers, Adapted From WHO/ICO Information Center on HPV and Cervical Cancer29
| Site | % |
|---|---|
| Cervix | > 99 |
| Anus | 84.2 |
| Vagina | 69.9 |
| Penis | 47.0 |
| Vulva | 44.4 |
| Oropharynx | 35.6 |
| Oral cavity | 23.5 |
Abbreviation: HPV, human papillomavirus.
Most high-risk HPV infections of the cervix are transient and asymptomatic and even when lesions arise, they are frequently cleared within 6 to 18 months postexposure presumably by elimination through the host's immune response (Fig 2).32,33 However, in some cases, there is no clearance and the risk for cervical cancer has been linked to long-term, persistent high-risk HPV infection.33 Women with persistent HPV16 infection are at an approximately 40% risk of developing premalignant lesions after 3 to 5 years (Fig 2).34–36 Overall, a high-risk HPV infection is a far more significant relative risk (RR) factor for cervical cancer development of (RR, approximately 50 to 100) than cigarette smoking is for developing lung cancer (RR, approximately 10).37 The average age of diagnosis of precancerous lesion ranges from 25 to 35 years, depending on two main factors, age at sexual debut and intensity of screening.31 An estimated 11,270 cases of cervical cancer have been diagnosed in 2009 in the United States and approximately 4,070 women have died from the disease.38 The burden of cervical cancer is far higher in countries with inadequate screening systems, where it remains one of the leading causes of cancer death in women.39 In these settings, cases are generally detected at advanced stages and the disease especially affects younger women.
Screening for high-risk HPV-associated precancerous cervical lesions utilizes the Papanicolaou (ie, Pap) test, an inexpensive and well-tolerated procedure, which, on implementation in the early 1960s, dramatically reduced the incidence and mortality rates of cervical carcinomas.40 Cytological abnormalities are classified accordingly to the Bethesda system as either atypical squamous cells, which designate a variety of cellular abnormalities that may or may not be indicative of neoplasia, or squamous intraepithelial lesions that are further classified as low grade and high grade. Abnormal cytology commonly requires a follow-up by colposcopy and, if necessary, a biopsy and endocervical curettage for histologic evaluation. Biopsied lesions are classified based on their location and severity of histologic abnormalities as cervical intraepithelial lesions (ie, CIN I, CIN II, CIN III), carcinoma in situ, and invasive cervical carcinoma. Standard therapeutic modalities for localized lesions typically involve tissue destructive procedures of the transformation zone, such as colposcopically guided laser treatment, loop electrosurgical excision, cone-shaped knife excision, and cryotherapy. Women generally test as HPV DNA negative after successful treatment. Invasive cervical cancer requires more aggressive surgery that typically involves complete removal of the uterus by hysterectomy. In young females with small cancers that are strictly limited to the cervix, a fertility-conserving surgery, such as trachelectomy, can be discussed.
High-risk HPV infections also contribute to some cancers of the vagina, vulva, penis, and anus (Table 2). The aggregate burden of these anogenital tract tumors is quite significant and the American Cancer Society estimates that in 2009, there will have been 3,580 women diagnosed with vulvar cancer, 2,160 women with vaginal and other genital cancers, 1,250 men with penile cancers, and 3,190 women and 2,100 men with anal cancer.41
Approximately 20% of head and neck cancers, particularly those that occur in patients who lack exposure to the classical risk factors tobacco and alcohol have been linked to high-risk HPV infections (Tables 1, 2).42–44 Data collected on sexual history suggest that infections of the oral cavity with high-risk HPVs may be through sexual contact, but oral-to-oral transmission cannot be ruled out.45,46 Oropharyngeal squamous cell carcinoma (OPSCC) have been most strongly associated with HPVs, and the HPV detection rate on OPSCC ranges between 20% to 50%.47–49 While HPV-associated OPSCC have more aggressive histopathologic features, these tumors appear to be more responsive to standard chemotherapy and radiation therapy regimens.42
HPVs have also been detected by some investigators in a variety of other carcinomas including breast, colon, lung, prostate, bladder, and esophageal cancers. Overall, these associations appear more tenuous and have not been confirmed by other researchers and/or extensive population-based studies.50
ONCOGENIC ACTIVITIES OF HIGH-RISK HPVs
High-risk HPVs encode three transforming proteins, E5, E6, and E7 (Figs 1C and 3). E5 is a small transmembrane protein that may function by inducing ligand-independent dimerization and activation of receptor protein tyrosine kinases including the epidermal growth factor receptor (Fig 1C).51,52 The E5 oncoprotein, together with E6, contributes to formation of koilocytes, a cell type that is used for diagnosis of HPV infections.53 Since malignant progression of precursor lesions is frequently associated with physical integration of portions of the viral genome into a host cell chromosome, E5 is generally not expressed in cervical carcinomas (Figs 1B, 2). Hence, whereas the HPV E5 oncoprotein may contribute to some early steps of viral transformation, it is not necessary for malignant progression and/or maintenance of the transformed phenotype. In contrast, the E6 and E7 oncoproteins are consistently expressed in cervical cancers (Fig 2). Several lines of evidence support the concept that expression of E6 and E7 is necessary for induction and maintenance of the transformed state of HPV-associated cancers.54 First, when HPV16 E6/E7 expressing primary human epithelial cells are grown under conditions that allow formation of a stratified skin like structure, they exhibit many of the histopathologic abnormalities of high-grade premalignant lesions.55 Second, HPV16 E6/E7 expression in basal epithelial cells causes cervical cancer in transgenic mice when the animals are treated with low doses of estrogen that mimic continuous estrus.56 Third, extinguishing expression of E6 and E7 expression in cervical carcinoma cell lines causes senescence, growth arrest, or cell death.57
Fig 3.

Transforming activities of high-risk human papillomavirus (HPV) oncoproteins. Major mechanisms utilized by the high-risk HPV E5, E6, and E7 oncoproteins to induce specific hallmarks of cellular transformation. See Oncogenic Activities of High-Risk HPVs for details.
HPV E6 and E7 are low molecular-weight cysteine-rich metal-binding proteins, which lack intrinsic enzymatic activities and specific DNA-binding properties (Fig 1C). They function by association with and functional modulation of host cellular regulatory protein complexes (Fig 3). The transforming mechanisms of high-risk HPVs represent strategies that these viruses employ to reprogram the infected host to establish a persistent infection and to ensure efficient production of viral progeny. During normal infection, these activities are exquisitely controlled, but in the context of a nonproductive infection, these control mechanisms are disturbed, causing cellular transformation (Figs 2, 3).58
High-risk HPV E7 proteins associate with and induce proteasome-mediated degradation of the retinoblastoma tumor suppressor (pRB; Fig 1C). Since pRB functions as a negative regulator of DNA synthesis, this allows for uncontrolled S phase entry. In a normal cell, aberrant DNA synthesis causes activation of the p53 tumor suppressor protein, which in turn triggers a cell death response. Presumably to short-circuit this cell abortive mechanism, high-risk HPV E6 oncoproteins target the p53 tumor suppressor protein for degradation by reprogramming a cellular ubiquitin ligase, E6AP (Fig 1C). To ensure infinite proliferative life span of infected cells, high-risk HPV E6 proteins activate transcription of hTERT, the catalytic protein subunit of telomerase.54 High-risk HPV E6 proteins also contain a binding site for cellular PDZ proteins, many of which are involved in signal transduction and regulate cell polarity. The integrity of the E6 PDZ binding site is essential for cellular transformation54 (Fig 1C).
High-risk HPV E6 and E7 oncoproteins also act as potent mitotic mutators and subvert genomic stability, thereby contributing to malignant progression.59 HPV E6 and E7 oncoproteins induce mitotic abnormalities through several mechanisms, most notably aberrant centrosome synthesis, which causes formation of multipolar mitoses.60 Tripolar mitoses have been described as a hallmark of high-risk HPV associated premalignant lesions and tumors (Fig 2).61 The HPV E6/E7 oncoproteins not only induce mitotic aberrations but may also subvert mitotic checkpoint activities that would normally be triggered by these alterations (Fig 3). This not only causes a higher incidence of mutations with every round of cell division but also enables aberrant cells to remain in the proliferative pool.62
HPV oncoproteins have also been reported to promote angiogenesis63 and induce some hallmarks of cancer associated epithelial to mesenchymal transition, a process that is important in invasion and metastasis.64 Moreover, HPV16 E7 expression causes the Warburg effect—a metabolic shift from an oxidative phosphorylation to a glycolysis based metabolism (Fig 3).65
SCREENING METHODS
As discussed earlier, introduction of the Pap smear has reduced cervical carcinoma rates by approximately 70%. Following recommendations by the American Cancer Society, the onset of an annual cervical cancer screening by Pap smear should be approximately 3 years after the first intercourse, but no later than age 21. With the newer liquid-based monolayer cytology techniques intervals between tests may be extended to 2 years. The liquid-based Pap test appears to have a lower rate of false-positive results, presumably as a result of standardized specimen preparation and fixation.66,67 Pap smears, however, still show significant deficits on diagnostic validity in terms of sensitivity and specificity.68 For women older than 30 years with three previous normal Pap tests, the American Cancer Society recently suggested a combination of Pap screening every 3 years in combination with an HPV DNA test. HPV DNA testing has a 25% to 35% higher sensitivity than cytology.69 The only HPV DNA screening test approved by the US Food and Drug Administration is the Hybrid capture II system (Qiagen, Valencia, CA). It is based on nucleic acid hybridization and distinguishes between the presence and absence of the most frequent high-risk and low-risk HPV types. Bearing a certain risk of cross-hybridization and inability of type specification, this method is cost intense and has a lower sensitivity than polymerase chain reaction–based methods that are frequently used in scientific studies.70 It has been suggested that HPV testing might eventually replace conventional Pap smears.71
In addition to HPV-encoded proteins, various cellular proteins are aberrantly expressed in (pre-) cancerous lesions and may thus be used as biomarkers. A promising biomarker for high-risk HPV associated neoplasias is p16INK4A, an inhibitor of cdk4/cdk6 cyclin D complexes. Increased p16INK4A expression represents a direct consequence of high-risk HPV E7 oncoprotein expression and is detected in premalignant lesions as well as cervical carcinomas.72,73 A number of other biomarkers have also been described but none of them is predictive for lesions that have a high propensity for malignant progression.74
PROPHYLACTIC VACCINATION
Prophylactic vaccines against a variety of human viral pathogens had a major impact on public health. The currently available prophylactic HPV vaccines are similar in concept to the Hepatitis B vaccine and consist of highly purified virus-like particles (VLPs) that are composed of the major capsid protein, L1, produced as recombinant proteins. L1 VLPs are morphologically and immunologically similar to naive HPV virions, but they do not contain a viral genome and, thus, they are not infectious.75 While such vaccines appear highly efficacious, the protection is specific to the vaccine HPV types. A quadrivalent vaccine containing L1 VLPs of the two most abundant high-risk (HPV16/18) and low-risk (HPV6/11) HPVs has been developed by Merck. VLPs are produced in yeast and formulated with an amorphous aluminum hydroxyphosphate sulfate based adjuvant. In 2006, the US Food and Drug Administration approved use of this vaccine in females and in October 2009 in males. GlaxoSmithKline (GSK) produces a bivalent vaccine that consists of HPV16 and HPV18 L1 VLPs. This vaccine was approved in the EU in 2007 and in the US in 2009. VLPs are produced in insect cells and are formulated with a proprietary adjuvant, which is claimed to yield particularly long lasting, robust immune responses.76 Immunogenicity, safety, and clinical efficacy have been proven for both of these vaccines in large clinical trials with high-grade CIN as an end point.77 Recently published follow-up data for 35 months (GSK) and 42 months (Merck) postvaccination indicate efficacies of 94% and 98%, respectively.78,79 It has been suggested that high and sustained serum levels of neutralizing antibodies are necessary to ensure efficient antibody transudation at the site of infection.80 A recent study with a mouse model of HPV transmission showed that humoral immunity induced by an HPV16 L1 VLP vaccine prevents virion binding to the basement membrane and/or transfer to epithelial cells.81 Postvaccination antibody titers reach serum levels that are approximately 10- to 100-fold higher than those observed after natural infection and remain high during a 5-year follow-up period80; an antibody decay model predicted that 99% of vaccinated women will develop almost life-long protection.82 An additional vaccine dose after 5 years induced a strong recall response with titers equal to peak titer after initial vaccination.83 Nonetheless, the real life long-term protective effect of these vaccines remains to be determined.
Some cross-protection against closely related high-risk, nonvaccine HPV types 31 (79%), 33 (46%), and 45 (76%) infections has been documented for the GSK vaccine.78,84 Conceivably, the Merck product may also offer some protection against a few nonvaccine HPV types. More detailed studies are required to determine if a broader protective immunity and prevention of HPV infection at other anatomic sites is elicited by the current vaccines.
Since these vaccines are designed to be purely prophylactic, they need to be administered before sexual activity. The US Government Advisory Committee on Immunization Practices recommends routine vaccination of females age 11 to 12 years. In addition, it can be given to girls as young as age 9 as well as to unvaccinated women age 13 to 26 years. Similar guidelines have been issued by the American Cancer Society and the American College of Obstetricians and Gynecologists. In October 2009, the US Food and Drug Administration approved Merck's vaccine in the US for prevention of genital warts in males age 9 to 26 years. In Europe, HPV vaccination recommendations vary from country to country but mostly follow American Cancer Society guidelines.
Concerns regarding vaccine safety arose shortly after vaccine distribution in larger populations of children and adolescents. The documented adverse events after immunization included mostly local site reaction, dizziness, nausea, headache, and hypersensitivity reactions including urticaria. Overall reporting rates were as expected for other vaccines, but disproportional reporting of syncope and venous thromboembolic events were noted.85 Even though the trials excluded pregnant women, many women became pregnant during the evaluation phase; based on these data, both vaccines show good safety profiles. So far, investigation of the few isolated deaths indicate these are unlikely to be related to vaccination.86
FUTURE DEVELOPMENTS IN HPV PROPHYLAXIS AND THERAPY
Second-Generation Prophylactic Vaccines
The current L1 VLP-based vaccines do not predictably offer protection from infection with nonvaccine HPVs. Vaccines based on the minor capsid protein, L2, however, elicit antibody responses that are neutralizing against a broad spectrum of HPVs.87,88 Since these antibody responses are directed against linear L2 epitopes, such vaccines can be peptide-based and hence may be much cheaper to produce and will have longer shelf lives. Studies in animal models have been very promising89 and it will be interesting to see whether such vaccine candidates show similarly encouraging results in clinical studies with human patients.
Mechanism-Based Therapeutic Opportunities
Given the importance of the E6 and E7 oncoproteins for HPV-associated carcinogenesis, these proteins are attractive therapeutic targets. The conceptually most straightforward strategy would be to interfere with E6/E7 oncoprotein expression. Proof of principle experiments using antisense90 and RNA interference approaches91 support the viability of such strategies. A second approach may be to identify small molecules that interfere with association of E6 and/or E7 oncoproteins proteins with critical cellular targets such as the p53 or pRB tumor suppressors, respectively. Thus far, such approaches have not been particularly fruitful. A third possibility may be to target cellular enzymes that are reprogrammed by E6 and E7 expression. Known enzymatic activities include protein kinases, histone modifying enzymes as well as components of the ubiquitin/proteasome system.58 The latter is particularly appealing since proteasome inhibitors are already in clinical use, and it will be interesting to determine whether HPV-associated tumors are sensitive to these compounds. A fourth possibility might be to inhibit cellular pathways that cooperate with HPV E6/E7 oncoprotein expression to accelerate carcinogenic progression. In a mouse model, estrogen is a necessary cofactor for induction of cervical cancers by HPV E6/E7.56 In this model, estrogen receptor antagonists were shown to prevent cervical cancer development and even caused regression of existing tumors.92 It remains to be determined whether such compounds have similar effects in human cervical carcinomas. Lastly, attractive therapeutic opportunities may also arise from cellular signal transduction network perturbations that are induced by viral oncoprotein expression. One expects that certain signal transduction pathways that are redundant in normal cells will become essential as a consequence of HPV oncoprotein expression. This phenomenon is referred to as synthetic lethality.93 One may discover such interactions by unbiased loss of function screens using small molecules or small inhibitory RNA libraries. An important proof of principle has been the identification of signal transductions pathways that become essential in K-ras oncogene expressing cells.94,95 Studies with HPV expressing cells have shown that HPV16 E7 expression alters kinase sensitivities in cells.96 A more recently published small hairpin RNA screen led to identification of two kinases that are essential for proliferation/survival of HPV-positive cervical carcinoma cell lines as well as HPV16 immortalized human epithelial cells but are dispensable in normal human epithelial cells. The sensitivity to these kinases was mapped to expression of the HPV E6 oncoprotein and its ability to induce degradation of the p53 tumor suppressor. Hence, PAK3 and SGK2 may be targets for therapy of HPV-associated lesions and cancers as well as other tumors that have lost functional p53 expression.97
Therapeutic Vaccines
As previously mentioned, the available prophylactic vaccines have no therapeutic effects and it will take decades before their use will markedly reduce the prevalence of HPV-associated cancers.98 In contrast, HPV-specific therapeutic vaccines would have an immediate impact on cervical cancer incidence. Prime targets for potential therapeutic vaccine strategies are the E6 and E7 oncoproteins, as they are expressed in premalignant lesions as well as cancers.54
A number of approaches are being evaluated. Studies with small HPV16 E7 peptide fragments linked to a nonspecific-helper peptide yielded variable clinical responses.99 HPV16 L2/E6/E7 fusion proteins induced E6/E7 specific cytotoxic T lymphocyte (CTL) responses in only eight of 40 healthy volunteers.100 Fusion of E7 to a heat-shock protein promoted a stronger immune response, but the clinical efficacy was limited at best.101,102 More promising results were obtained with HPV16 E6/E7–derived, synthetic, long peptide vaccines. When tested in a small group of patients with cervical cancer, they caused robust expansion of HPV16 specific CD4+ and CD8+ T-cells103 and a recent, nonplacebo controlled, phase II study conducted in women with HPV16 positive high-grade vulvar intraepithelial neoplasia was quite encouraging and induced a robust HPV16 specific immune response as well as very promising clinical responses.104
Other immunization strategies that are being considered include the application of naked viral DNA, viral RNA, vector-based systems or intranasal application of modified HPV protein expressing bacterial strains. Such strategies may have advantages as compared to traditional protein or peptide-based vaccines, such as the priming of stronger T-helper 1 and CTL responses, in addition to prolonged antigen exposure. Since the HPV E6 and E7 proteins are relatively weak immunogens, attempts have been made to increase immunogenicity of such vaccines by coexpressing E6 and E7 with immunomodulatory proteins, fusion of HPV proteins to ubiquitin or various other proteins.76,99,105
Vaccinia virus, adenovirus, or adenoassociated virus vectors infect a variety of cell types and efficiently express tumor antigens, which are presented as viral antigens. Hence, they may elicit strong humoral as well as T-cell responses. Several vaccinia virus vector–based vaccines designed to induce long-lasting CTL responses against HPV E2, E6, and E7 antigens are being tested in clinical studies.105 One vaccine of this type contained modified HPV16 and HPV18 E6/E7 sequences. A trial of patients with cervical cancer documented immunogenicity and safety,106 but failed to provide clear evidence for efficacy.107
Various strategies are being evaluated to generate a combined therapeutic and prophylactic vaccine. DNA vaccination with truncated versions of HPV16 L1 and E7 genes, induced L1-specific antibodies, but also L1- and E7-specific CTL responses.108 A VLP-based vaccine consisting of an L1/E7 chimera has also been tested in patients with HPV16-positive precursor lesions. It was safe, but only showed moderate therapeutic efficacy.109 Conceptually similar vaccines yielded comparable results.76
CONCLUDING REMARKS
Cancers that are caused by viral infections offer unique opportunities for prophylaxis, early detection, and therapy. For HPV-associated cancers, this has been partially realized by the introduction of Pap smear programs, which caused a 70% decrease in cervical cancer rates and the successful development of prophylactic vaccines, which have the potential to cause an additional 70% reduction. These vaccines, however, have no therapeutic efficacy in individuals who have already been infected, and it will take decades before these vaccines will have a measurable impact on the incidence rates of HPV-associated tumors. Moreover, it is not clear whether nonvaccine high-risk HPV types will fill the void left by elimination of the currently prevalent vaccine types, HPV16 and HPV18. The most significant problem, however, is that the high cost the current vaccines practically prohibits their use in low-resource settings, where, due to inadequate screening, HPV-associated tumors are particularly prevalent. Hence, basic research aimed at the development of therapeutic strategies for HPV-associated tumors should continue to be a high priority. Eradication of high-risk HPVs would require development of a crossprotective, needle-free, single-dose, long-duration, heat-stable, affordable vaccine that has both therapeutic and prophylactic efficacy and can be used in low-resource settings.
Acknowledgment
We are grateful to Christine Nguyen, PhD, for help with the preparation of the figures.
Footnotes
Supported by Public Health Services Grants No. CA066980, CA081135, HG004233, and CA141583 (K.M.), and postdoctoral fellowship HE5494/1-1 from the Deutsche Forschungsgemeinschaft (K.H.).
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Karl Münger, Arbor Vita (C), Merck (U) Stock Ownership: Karl Münger, Arbor Vita Honoraria: None Research Funding: None Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Karin Hellner, Karl Münger
Collection and assembly of data: Karin Hellner
Data analysis and interpretation: Karin Hellner, Karl Münger
Manuscript writing: Karin Hellner, Karl Münger
Final approval of manuscript: Karin Hellner, Karl Münger
REFERENCES
- 1.Howley PM, Lowy DR. Papillomaviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins; 2007. pp. 2299–2354. [Google Scholar]
- 2.Lee C, Laimins L. The differentiation-dependent life cycle of human papillomaviruses in keratinocytes. In: Garcea RL, DiMaio D, editors. The Papillomaviruses. New York, NY: Springer; 2007. pp. 45–68. [Google Scholar]
- 3.de Villiers EM, Fauquet C, Broker TR, et al. Classification of papillomaviruses. Virology. 2004;324:17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 4.Burchell AN, Winer RL, de Sanjose S, et al. Chapter 6: Epidemiology and transmission dynamics of genital HPV infection. Vaccine. 2006;24(suppl 3):S3/52–61. doi: 10.1016/j.vaccine.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 5.Winer RL, Lee SK, Hughes JP, et al. Genital human papillomavirus infection: Incidence and risk factors in a cohort of female university students. Am J Epidemiol. 2003;157:218–226. doi: 10.1093/aje/kwf180. [DOI] [PubMed] [Google Scholar]
- 6.Ferenczy A, Bergeron C, Richart RM. Human papillomavirus DNA in fomites on objects used for the management of patients with genital human papillomavirus infections. Obstet Gynecol. 1989;74:950–954. [PubMed] [Google Scholar]
- 7.Roden RB, Lowy DR, Schiller JT. Papillomavirus is resistant to desiccation. J Infect Dis. 1997;176:1076–1079. doi: 10.1086/516515. [DOI] [PubMed] [Google Scholar]
- 8.Nielson CM, Flores R, Harris RB, et al. Human papillomavirus prevalence and type distribution in male anogenital sites and semen. Cancer Epidemiol Biomarkers Prev. 2007;16:1107–1114. doi: 10.1158/1055-9965.EPI-06-0997. [DOI] [PubMed] [Google Scholar]
- 9.Gray RH, Serwadda D, Kong X, et al. Male circumcision decreases acquisition and increases clearance of high-risk human papillomavirus in HIV-negative men: A randomized trial in Rakai, Uganda. J Infect Dis. 2010;201:1455–1462. doi: 10.1086/652184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castellsague X, Bosch FX, Munoz N, et al. Male circumcision, penile human papillomavirus infection, and cervical cancer in female partners. N Engl J Med. 2002;346:1105–1112. doi: 10.1056/NEJMoa011688. [DOI] [PubMed] [Google Scholar]
- 11.Shields TS, Brinton LA, Burk RD, et al. A case-control study of risk factors for invasive cervical cancer among U.S. women exposed to oncogenic types of human papillomavirus. Cancer Epidemiol Biomarkers Prev. 2004;13:1574–1582. [PubMed] [Google Scholar]
- 12.Buck CB, Thompson CD, Roberts JN, et al. Carrageenan is a potent inhibitor of papillomavirus infection. PLoS Pathog. 2006;2:e69. doi: 10.1371/journal.ppat.0020069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts JN, Buck CB, Thompson CD, et al. Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat Med. 2007;13:857–861. doi: 10.1038/nm1598. [DOI] [PubMed] [Google Scholar]
- 14.Smedts F, Ramaekers F, Troyanovsky S, et al. Basal-cell keratins in cervical reserve cells and a comparison to their expression in cervical intraepithelial neoplasia. Am J Pathol. 1992;140:601–612. [PMC free article] [PubMed] [Google Scholar]
- 15.Tsutsumi K, Sun Q, Yasumoto S, et al. In vitro and in vivo analysis of cellular origin of cervical squamous metaplasia. Am J Pathol. 1993;143:1150–1158. [PMC free article] [PubMed] [Google Scholar]
- 16.Martens JE, Arends J, Van der Linden PJ, et al. Cytokeratin 17 and p63 are markers of the HPV target cell, the cervical stem cell. Anticancer Res. 2004;24:771–775. [PubMed] [Google Scholar]
- 17.Pagliusi SR, Dillner J, Pawlita M, et al. Chapter 23: International Standard reagents for harmonization of HPV serology and DNA assays: An update. Vaccine. 2006;24(suppl 3):S3/193–200. doi: 10.1016/j.vaccine.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 18.Baseman JG, Koutsky LA. The epidemiology of human papillomavirus infections. J Clin Virol. 2005;32(suppl 1):S16–24. doi: 10.1016/j.jcv.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 19.Dunne EF, Unger ER, Sternberg M, et al. Prevalence of HPV infection among females in the United States. JAMA. 2007;297:813–819. doi: 10.1001/jama.297.8.813. [DOI] [PubMed] [Google Scholar]
- 20.Munoz N, Castellsague X, de Gonzalez AB, et al. Chapter 1: HPV in the etiology of human cancer. Vaccine. 2006;24(suppl 3):S3/1–10. doi: 10.1016/j.vaccine.2006.05.115. [DOI] [PubMed] [Google Scholar]
- 21.Kumar V, Fausto N, Mitchell R. The female genital system and breast. In: Kumar V, Abbas AK, Fausto N, et al., editors. Robbins Basic Pathology. ed 8. Philadelphia, PA: Saunders; 2007. pp. 679–718. [Google Scholar]
- 22.Bosch X, Harper D. Prevention strategies of cervical cancer in the HPV vaccine era. Gynecol Oncol. 2006;103:21–24. doi: 10.1016/j.ygyno.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 23.Beutner KR, Ferenczy A. Therapeutic approaches to genital warts. Am J Med. 1997;102:28–37. doi: 10.1016/s0002-9343(97)00181-2. [DOI] [PubMed] [Google Scholar]
- 24.Frega A, Stentella P, Tinari A, et al. Giant condyloma acuminatum or buschke-Lowenstein tumor: Review of the literature and report of three cases treated by CO2 laser surgery: A long-term follow-up. Anticancer Res. 2002;22:1201–1204. [PubMed] [Google Scholar]
- 25.Rady PL, Schnadig VJ, Weiss RL, et al. Malignant transformation of recurrent respiratory papillomatosis associated with integrated human papillomavirus type 11 DNA and mutation of p53. Laryngoscope. 1998;108:735–740. doi: 10.1097/00005537-199805000-00021. [DOI] [PubMed] [Google Scholar]
- 26.Stamataki S, Nikolopoulos TP, Korres S, et al. Juvenile recurrent respiratory papillomatosis: Still a mystery disease with difficult management. Head Neck. 2007;29:155–162. doi: 10.1002/hed.20491. [DOI] [PubMed] [Google Scholar]
- 27.Durst M, Gissmann L, Ikenberg H, et al. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc Natl Acad Sci U S A. 1983;80:3812–3815. doi: 10.1073/pnas.80.12.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boshart M, Gissmann L, Ikenberg H, et al. A new type of papillomavirus DNA and its prevalence in genital cancer biopsies and in cell lines derived from cervical cancer. The EMBO Journal. 1984;3:1151–1157. doi: 10.1002/j.1460-2075.1984.tb01944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.WHO/ICO Information Center on HPV and Cervical Cancer. http://www.who.int/hpvcentre/statistics/en/
- 30.Smith JS, Lindsay L, Hoots B, et al. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: A meta-analysis update. Int J Cancer. 2007;121:621–632. doi: 10.1002/ijc.22527. [DOI] [PubMed] [Google Scholar]
- 31.Schiffman M, Castle PE, Jeronimo J, et al. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907. doi: 10.1016/S0140-6736(07)61416-0. [DOI] [PubMed] [Google Scholar]
- 32.Stanley M. Immune responses to human papillomavirus. Vaccine. 2006;24(suppl 1):S16–22. doi: 10.1016/j.vaccine.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 33.Plummer M, Schiffman M, Castle PE, et al. A 2-year prospective study of human papillomavirus persistence among women with a cytological diagnosis of atypical squamous cells of undetermined significance or low-grade squamous intraepithelial lesion. J Infect Dis. 2007;195:1582–1589. doi: 10.1086/516784. [DOI] [PubMed] [Google Scholar]
- 34.Schiffman M, Herrero R, Desalle R, et al. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology. 2005;337:76–84. doi: 10.1016/j.virol.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Khan MJ, Castle PE, Lorincz AT, et al. The elevated 10-year risk of cervical precancer and cancer in women with human papillomavirus (HPV) type 16 or 18 and the possible utility of type-specific HPV testing in clinical practice. J Natl Cancer Inst. 2005;97:1072–1079. doi: 10.1093/jnci/dji187. [DOI] [PubMed] [Google Scholar]
- 36.Castle PE, Solomon D, Schiffman M, et al. Human papillomavirus type 16 infections and 2-year absolute risk of cervical precancer in women with equivocal or mild cytologic abnormalities. J Natl Cancer Inst. 2005;97:1066–1071. doi: 10.1093/jnci/dji186. [DOI] [PubMed] [Google Scholar]
- 37.Franco EL, Harper DM. Vaccination against human papillomavirus infection: A new paradigm in cervical cancer control. Vaccine. 2005;23:2388–2394. doi: 10.1016/j.vaccine.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 38.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 39.Parkin DM, Bray F. Chapter 2: The burden of HPV-related cancers. Vaccine. 2006;24(suppl 3):S3/11–25. doi: 10.1016/j.vaccine.2006.05.111. [DOI] [PubMed] [Google Scholar]
- 40.Solomon D, Breen N, McNeel T. Cervical cancer screening rates in the United States and the potential impact of implementation of screening guidelines. CA Cancer J Clin. 2007;57:105–111. doi: 10.3322/canjclin.57.2.105. [DOI] [PubMed] [Google Scholar]
- 41.American Cancer Society: ACS Cancer Statistics, Cases and Deaths by Site, State 2009. http://www.cancer.org/docroot/PRO/content/PRO_1_1_Cancer_Facts_Figures_Suppl_Data_2009.asp.
- 42.Gillison ML. Human papillomavirus-associated head and neck cancer is a distinct epidemiologic, clinical, and molecular entity. Semin Oncol. 2004;31:744–754. doi: 10.1053/j.seminoncol.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 43.Gillison ML, D'Souza G, Westra W, et al. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J Natl Cancer Inst. 2008;100:407–420. doi: 10.1093/jnci/djn025. [DOI] [PubMed] [Google Scholar]
- 44.Gillison ML, Koch WM, Capone RB, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92:709–720. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- 45.D'Souza G, Kreimer AR, Viscidi R, et al. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med. 2007;356:1944–1956. doi: 10.1056/NEJMoa065497. [DOI] [PubMed] [Google Scholar]
- 46.D'Souza G, Agrawal Y, Halpern J, et al. Oral sexual behaviors associated with prevalent oral human papillomavirus infection. J Infect Dis. 2009;199:1263–1269. doi: 10.1086/597755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Syrjanen S. Human papillomavirus (HPV) in head and neck cancer. J Clin Virol. 2005;32(suppl 1):S59–66. doi: 10.1016/j.jcv.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 48.Andrews E, Seaman WT, Webster-Cyriaque J. Oropharyngeal carcinoma in non-smokers and non-drinkers: A role for HPV. Oral Oncol. 2009;45:486–491. doi: 10.1016/j.oraloncology.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 49.Hobbs CG, Sterne JA, Bailey M, et al. Human papillomavirus and head and neck cancer: A systematic review and meta-analysis. Clin Otolaryngol. 2006;31:259–266. doi: 10.1111/j.1749-4486.2006.01246.x. [DOI] [PubMed] [Google Scholar]
- 50.Gillison ML, Shah KV. Role of mucosal human papillomavirus in nongenital cancers. J Natl Cancer Inst Monogr. 2003:57–65. doi: 10.1093/oxfordjournals.jncimonographs.a003484. [DOI] [PubMed] [Google Scholar]
- 51.Talbert-Slagle K, DiMaio D. The bovine papillomavirus E5 protein and the PDGF beta receptor: It takes two to tango. Virology. 2009;384:345–351. doi: 10.1016/j.virol.2008.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DiMaio D, Mattoon D. Mechanisms of cell transformation by papillomavirus E5 proteins. Oncogene. 2001;20:7866–7873. doi: 10.1038/sj.onc.1204915. [DOI] [PubMed] [Google Scholar]
- 53.Krawczyk E, Suprynowicz FA, Liu X, et al. Koilocytosis: A cooperative interaction between the human papillomavirus E5 and E6 oncoproteins. Am J Pathol. 2008;173:682–688. doi: 10.2353/ajpath.2008.080280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Munger K, Howley PM, DiMaio D. Human papillomavirus E6 and E7 oncogenes. In: Garcea RL, DiMaio D, editors. The Papillomaviruses. New York, NY: Springer; 2007. pp. 195–251. [Google Scholar]
- 55.McCance DJ, Kopan R, Fuchs E, et al. Human papillomavirus type 16 alters human epithelial cell differentiation in vitro. Proc Natl Acad Sci U S A. 1988;85:7169–7173. doi: 10.1073/pnas.85.19.7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arbeit JM, Howley PM, Hanahan D. Chronic estrogen-induced cervical and vaginal squamous carcinogenesis in human papillomavirus type 16 transgenic mice. Proc Natl Acad Sci U S A. 1996;93:2930–2935. doi: 10.1073/pnas.93.7.2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thierry F, Yaniv M. The BPV1–E2 trans-acting protein can be either an activator or a repressor of the HPV18 regulatory region. Embo J. 1987;6:3391–3397. doi: 10.1002/j.1460-2075.1987.tb02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McLaughlin-Drubin ME, Munger K. The human papillomavirus E7 oncoprotein. Virology. 2009;384:335–344. doi: 10.1016/j.virol.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.White AE, Livanos EM, Tlsty TD. Differential disruption of genomic integrity and cell cycle regulation in normal human fibroblasts by the HPV Oncoproteins. Genes Dev. 1994;8:666–677. doi: 10.1101/gad.8.6.666. [DOI] [PubMed] [Google Scholar]
- 60.Duensing S, Lee LY, Duensing A, et al. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci U S A. 2000;97:10002–10007. doi: 10.1073/pnas.170093297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crum CP, Ikenberg H, Richart RM, et al. Human papillomavirus type 16 and early cervical neoplasia. N Engl J Med. 1984;310:880–883. doi: 10.1056/NEJM198404053101403. [DOI] [PubMed] [Google Scholar]
- 62.Nguyen CL, Hayakawa H, Munger K. Activities of human papillomavirus oncoproteins that contribute to genomic instability and malignant progression. In: Norrild B, editor. Human Paillomavirus Gene Regulation and Transformation. Trivandrum, India: Transworld Research Network; 2007. pp. 139–164. [Google Scholar]
- 63.Chen W, Li F, Mead L, et al. Human papillomavirus causes an angiogenic switch in keratinocytes which is sufficient to alter endothelial cell behavior. Virology. 2007;367:168–174. doi: 10.1016/j.virol.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hellner K, Mar J, Fang F, et al. HPV16 E7 oncogene expression in normal human epithelial cells causes molecular changes indicative of an epithelial to mesenchymal transition. Virology. 2009;391:57–63. doi: 10.1016/j.virol.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zwerschke W, Mazurek S, Massimi P, et al. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc Natl Acad Sci U S A. 1999;96:1291–1296. doi: 10.1073/pnas.96.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Uyar DS, Eltabbakh GH, Mount SL. Positive predictive value of liquid-based and conventional cervical Papanicolaou smears reported as malignant. Gynecol Oncol. 2003;89:227–232. doi: 10.1016/s0090-8258(02)00102-6. [DOI] [PubMed] [Google Scholar]
- 67.Bernstein SJ, Sanchez-Ramos L, Ndubisi B. Liquid-based cervical cytologic smear study and conventional Papanicolaou smears: A metaanalysis of prospective studies comparing cytologic diagnosis and sample adequacy. Am J Obstet Gynecol. 2001;185:308–317. doi: 10.1067/mob.2001.116736. [DOI] [PubMed] [Google Scholar]
- 68.Wright TC., Jr Cervical cancer screening in the 21st century: Is it time to retire the PAP smear? Clin Obstet Gynecol. 2007;50:313–323. doi: 10.1097/GRF.0b013e31804a8285. [DOI] [PubMed] [Google Scholar]
- 69.Clavel C, Masure M, Bory JP, et al. Human papillomavirus testing in primary screening for the detection of high-grade cervical lesions: A study of 7932 women. Br J Cancer. 2001;84:1616–1623. doi: 10.1054/bjoc.2001.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lie AK, Risberg B, Borge B, et al. DNA- versus RNA-based methods for human papillomavirus detection in cervical neoplasia. Gynecol Oncol. 2005;97:908–915. doi: 10.1016/j.ygyno.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 71.Mayrand MH, Duarte-Franco E, Rodrigues I, et al. Human papillomavirus DNA versus Papanicolaou screening tests for cervical cancer. N Engl J Med. 2007;357:1579–1588. doi: 10.1056/NEJMoa071430. [DOI] [PubMed] [Google Scholar]
- 72.Ordi J, Garcia S, del Pino M, et al. P16 INK4a immunostaining identifies occult CIN lesions in HPV-positive women. Int J Gynecol Pathol. 2009;28:90–97. doi: 10.1097/PGP.0b013e31817e9ac5. [DOI] [PubMed] [Google Scholar]
- 73.Klaes R, Friedrich T, Spitkovsky D, et al. Overexpression of p16(INK4A) as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int J Cancer. 2001;92:276–284. doi: 10.1002/ijc.1174. [DOI] [PubMed] [Google Scholar]
- 74.Khan AM, Singer A. Biomarkers in cervical precancer management: The new frontiers. Future Oncol. 2008;4:515–524. doi: 10.2217/14796694.4.4.515. [DOI] [PubMed] [Google Scholar]
- 75.Giannini SL, Hanon E, Moris P, et al. Enhanced humoral and memory B cellular immunity using HPV16/18 L1 VLP vaccine formulated with the MPL/aluminium salt combination (AS04) compared to aluminium salt only. Vaccine. 2006;24:5937–5949. doi: 10.1016/j.vaccine.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 76. Huh WK, Roden RB. The future of vaccines for cervical cancer Gynecol Oncol 2008. 109 S 48 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koutsky LA, Harper DM. Chapter 13: Current findings from prophylactic HPV vaccine trials. Vaccine. 2006;24(suppl 3):S3/114–121. doi: 10.1016/j.vaccine.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 78.Paavonen J, Naud P, Salmeron J, et al. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): Final analysis of a double-blind, randomised study in young women. Lancet. 2009;374:301–314. doi: 10.1016/S0140-6736(09)61248-4. [DOI] [PubMed] [Google Scholar]
- 79.Kjaer SK, Sigurdsson K, Iversen OE, et al. A pooled analysis of continued prophylactic efficacy of quadrivalent human papillomavirus (types 6/11/16/18) vaccine against high-grade cervical and external genital lesions. Cancer Prev Res. 2009;2:868–878. doi: 10.1158/1940-6207.CAPR-09-0031. [DOI] [PubMed] [Google Scholar]
- 80. Frazer IH. Measuring serum antibody to human papillomavirus following infection or vaccination Gynecol Oncol 2010. 118 S 8 11 [DOI] [PubMed] [Google Scholar]
- 81.Kines RC, Thompson CD, Lowy DR, et al. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc Natl Acad Sci U S A. 2009;106:20458–20463. doi: 10.1073/pnas.0908502106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fraser C, Tomassini JE, Xi L, et al. Modeling the long-term antibody response of a human papillomavirus (HPV) virus-like particle (VLP) type 16 prophylactic vaccine. Vaccine. 2007;25:4324–4333. doi: 10.1016/j.vaccine.2007.02.069. [DOI] [PubMed] [Google Scholar]
- 83.Olsson SE, Villa LL, Costa RL, et al. Induction of immune memory following administration of a prophylactic quadrivalent human papillomavirus (HPV) types 6/11/16/18 L1 virus-like particle (VLP) vaccine. Vaccine. 2007;25:4931–4939. doi: 10.1016/j.vaccine.2007.03.049. [DOI] [PubMed] [Google Scholar]
- 84.Smith JF, Brownlow M, Brown M, et al. Antibodies from women immunized with Gardasil cross-neutralize HPV 45 pseudovirions. Hum Vaccin. 2007;3:109–115. doi: 10.4161/hv.3.4.4058. [DOI] [PubMed] [Google Scholar]
- 85.Slade BA, Leidel L, Vellozzi C, et al. Postlicensure safety surveillance for quadrivalent human papillomavirus recombinant vaccine. JAMA. 2009;302:750–757. doi: 10.1001/jama.2009.1201. [DOI] [PubMed] [Google Scholar]
- 86.Agorastos T, Chatzigeorgiou K, Brotherton JM, et al. Safety of human papillomavirus (HPV) vaccines: A review of the international experience so far. Vaccine. 2009;27:7270–7281. doi: 10.1016/j.vaccine.2009.09.097. [DOI] [PubMed] [Google Scholar]
- 87.Slupetzky K, Gambhira R, Culp TD, et al. A papillomavirus-like particle (VLP) vaccine displaying HPV16 L2 epitopes induces cross-neutralizing antibodies to HPV11. Vaccine. 2007;25:2001–2010. doi: 10.1016/j.vaccine.2006.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jagu S, Karanam B, Gambhira R, et al. Concatenated multitype L2 fusion proteins as candidate prophylactic pan-human papillomavirus vaccines. J Natl Cancer Inst. 2009;101:782–792. doi: 10.1093/jnci/djp106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gambhira R, Karanam B, Jagu S, et al. A protective and broadly cross-neutralizing epitope of human papillomavirus L2. J Virol. 2007;81:13927–13931. doi: 10.1128/JVI.00936-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.von Knebel Doeberitz M, Rittmuller C, zur Hausen H, et al. Inhibition of tumorigenicity of cervical cancer cells in nude mice by HPV E6-E7 anti-sense RNA. Int J Cancer. 1992;51:831–834. doi: 10.1002/ijc.2910510527. [DOI] [PubMed] [Google Scholar]
- 91.Butz K, Ristriani T, Hengstermann A, et al. SiRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene. 2003;22:5938–5945. doi: 10.1038/sj.onc.1206894. [DOI] [PubMed] [Google Scholar]
- 92.Chung SH, Lambert PF. Prevention and treatment of cervical cancer in mice using estrogen receptor antagonists. Proc Natl Acad Sci U S A. 2009;106:19467–19472. doi: 10.1073/pnas.0911436106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kaelin WG., Jr Synthetic lethality: A framework for the development of wiser cancer therapeutics. Genome Med. 2009;1:99. doi: 10.1186/gm99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luo J, Emanuele MJ, Li D, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Scholl C, Frohling S, Dunn IF, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–834. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 96.Baldwin A, Li W, Grace M, et al. Kinase requirements in human cells: II. Genetic interaction screens identify kinase requirements following HPV16 E7 expression in cancer cells. Proc Natl Acad Sci U S A. 2008;105:16478–16483. doi: 10.1073/pnas.0806195105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baldwin A, Grueneberg DA, Hellner K, et al. Kinase requirements in human cells: V. Synthetic lethal interactions between p53 and the protein kinases SGK2 and PAK3. Proc Natl Acad Sci U S A. 2010;107:12463–12468. doi: 10.1073/pnas.1007462107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Frazer IH. Prevention of cervical cancer through papillomavirus vaccination. Nat Rev Immunol. 2004;4:46–54. doi: 10.1038/nri1260. [DOI] [PubMed] [Google Scholar]
- 99.Cid-Arregui A. Therapeutic vaccines against human papillomavirus and cervical cancer. Open Virol J. 2009;3:67–83. doi: 10.2174/1874357900903010067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.de Jong A, O'Neill T, Khan AY, et al. Enhancement of human papillomavirus (HPV) type 16 E6 and E7-specific T-cell immunity in healthy volunteers through vaccination with TA-CIN, an HPV16 L2E7E6 fusion protein vaccine. Vaccine. 2002;20:3456–3464. doi: 10.1016/s0264-410x(02)00350-x. [DOI] [PubMed] [Google Scholar]
- 101.Einstein MH, Kadish AS, Burk RD, et al. Heat shock fusion protein-based immunotherapy for treatment of cervical intraepithelial neoplasia III. Gynecol Oncol. 2007;106:453–460. doi: 10.1016/j.ygyno.2007.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Goldstone SE, Palefsky JM, Winnett MT, et al. Activity of HspE7, a novel immunotherapy, in patients with anogenital warts. Dis Colon Rectum. 2002;45:502–507. doi: 10.1007/s10350-004-6229-6. [DOI] [PubMed] [Google Scholar]
- 103.Welters MJ, Kenter GG, Piersma SJ, et al. Induction of tumor-specific CD4+ and CD8+ T-cell immunity in cervical cancer patients by a human papillomavirus type 16 E6 and E7 long peptides vaccine. Clin Cancer Res. 2008;14:178–187. doi: 10.1158/1078-0432.CCR-07-1880. [DOI] [PubMed] [Google Scholar]
- 104.Kenter GG, Welters MJ, Valentijn AR, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med 361:1838- 1847;2009 doi: 10.1056/NEJMoa0810097. [DOI] [PubMed] [Google Scholar]
- 105.Trimble CL, Frazer IH. Development of therapeutic HPV vaccines. Lancet Oncol. 2009;10:975–980. doi: 10.1016/S1470-2045(09)70227-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaufmann AM, Stern PL, Rankin EM, et al. Safety and immunogenicity of TA-HPV, a recombinant vaccinia virus expressing modified human papillomavirus (HPV)-16 and HPV-18 E6 and E7 genes, in women with progressive cervical cancer. Clin Cancer Res. 2002;8:3676–3685. [PubMed] [Google Scholar]
- 107.Fiander AN, Tristram AJ, Davidson EJ, et al. Prime-boost vaccination strategy in women with high-grade, noncervical anogenital intraepithelial neoplasia: Clinical results from a multicenter phase II trial. Int J Gynecol Cancer. 2006;16:1075–1081. doi: 10.1111/j.1525-1438.2006.00598.x. [DOI] [PubMed] [Google Scholar]
- 108.Kuck D, Leder C, Kern A, et al. Efficiency of HPV 16 L1/E7 DNA immunization: Influence of cellular localization and capsid assembly. Vaccine. 2006;24:2952–2965. doi: 10.1016/j.vaccine.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 109.Kaufmann AM, Nieland JD, Jochmus I, et al. Vaccination trial with HPV16 L1E7 chimeric virus-like particles in women suffering from high grade cervical intraepithelial neoplasia (CIN 2/3) Int J Cancer. 2007;121:2794–2800. doi: 10.1002/ijc.23022. [DOI] [PubMed] [Google Scholar]