

Abstract
New therapeutic approaches for castration-resistant prostate cancer (CRPC) introduce new treatment dilemmas: how best to sequence these options to maximally benefit patients, what tests to perform before and after treatment to assess disease status, and how to interpret the test results and use them to guide treatment. New and specific end points for different classes of drugs are needed to provide the information to guide these treatment decisions. In 2008, the Prostate Cancer Working Group 2 consensus criteria for early-phase clinical trials redefined clinical trial end points as first, to control, relieve, or eliminate disease manifestations present when treatment is started and second, to prevent or delay future disease manifestations. Disease manifestations include prostate-specific antigen (PSA), soft-tissue disease (nodes and/or viscera), bone disease (most common site of spread), and symptoms. Recent US Food and Drug Administration (FDA) approvals for CRPC therapies have been based on the prevent/delay end points that reflect unequivocal benefit to a patient: prolongation of life or reduction in skeletal-related events (SREs). For the practicing oncologist, the control/relieve/eliminate outcomes should serve primarily to inform the decision of whether to continue therapy. In this review, we consider individual end points such as PSA, imaging, and patient-reported outcomes in the context of the control/relieve/eliminate and prevent/delay framework. We address the time-to-event end points of metastasis prevention, SRE, time to progression, and overall survival in the context of regulatory approvals. We also discuss circulating tumor cells measured with the CellSearch assay, recently cleared by the FDA for monitoring CRPC.
INTRODUCTION
A physician treating a patient with progressive castration-resistant prostate cancer (CRPC) now has a range of new therapies with diverse mechanisms of action available in the clinical armamentarium. These new agents include biologic (sipuleucel-T [Provenge; Dendreon, Seattle, WA]),1 cytotoxic (cabazitaxel [Jevtana; sanofi-aventis, Bridgewater, NJ]),2 hormonal (abiraterone) agents,3 a bone-seeking radioisotope (apharadine; Bayer, Wayne, NJ, and Berlin, Germany) and an antibody that blocks the interaction between tumor and bone (denosumab [Xgeva; Amgen, Thousand Oaks, CA]).4 Each has been shown in definitive phase III trials to prolong life or prevent the development of bone metastases that can produce some of the most feared complications of the disease. The new issues facing the clinician are how best to sequence these options to maximally benefit patients, what tests to perform before and after treatment to assess disease status, and how to interpret the test results and use them to guide management. These issues are straightforward for many cancers. However, for prostate cancer, they are not because of the heterogeneity of the disease, difficulty in assessing disease in bone, and uncertainty of interpreting the clinical significance of post-therapy changes in prostate-specific antigen (PSA) in different contexts. Needed are new and specific end points for the different classes of drugs under evaluation in CRPC clinical trials to provide the information necessary to guide these treatment decisions.
To assist in interpreting clinical data generated by prostate cancer clinical trials, in 2008, the Prostate Cancer Working Group 2 (PCWG2) reported consensus guidelines on clinical trial conduct that included objectives, the evaluation of patients, and trial eligibility.5 A key outcome was to better align clinical research with clinical practice by developing outcomes more relevant to prostate cancer and its management than the Response Evaluation Criteria in Solid Tumors (RECIST) criteria previously in use.6 The PCWG2 paradigm considered post-treatment outcomes in two general categories: first, to control, relieve, or eliminate disease manifestations present when treatment is started, and second, to prevent or delay future disease manifestations. The result was a shift from a focus on early outcome measures used to categorize patients as responders or nonresponders to one that enables treatment to continue until it is clear that it is not working or no longer benefitting a patient and should be stopped. The global response categories of complete response, partial response, and stable disease were eliminated and replaced by the recommendation to report changes in each disease manifestation separately.
Although the PCWG2 guidelines have changed how we conduct, report, and interpret prostate cancer clinical trials, these guidelines should not be confused with the regulatory requirements for US Food and Drug Administration (FDA) approval. The regulatory approval criteria remain the same, requiring the demonstration of clinical benefit: an improvement in how a patient feels or functions or how long he or she survives. With the exception of the relief of pain, all the recent approvals for CRPC have been based on the prevent/delay end points that reflect unequivocal benefit to a patient: prolongation of life or a reduction in skeletal-related events (SREs). Thus, from the point of view of the practicing oncologist, the control/relieve/eliminate outcomes should serve primarily to inform the decision of whether to continue therapy. To do so requires that the outcome for each disease manifestation be interpreted carefully, because the significance of a change in any single measure, be it PSA or imaging, differs as a function of the mode of action of the drug being studied and the purpose for which it is being administered. This careful approach helps to ensure a drug is allowed sufficient time to work and is not discontinued prematurely. Premature discontinuations can occur if there are changes in an outcome measure that does not accurately reflect the favorable effects of drug on host nonmalignant tissues, such as a bone-targeting agent or immune modulator, which influence the disease trajectory favorably independent of a direct effect on the tumor.
Here, we consider individual end points such as PSA, imaging, and patient-reported outcomes in the context of the control/relieve/eliminate and prevent/delay framework. Next, we address the time-to-event end points of metastasis prevention, SREs, time to progression, and overall survival (OS) in the context of regulatory approvals. Separately, we discuss circulating tumor cells (CTCs) measured with the CellSearch assay (Veridex, Warren, NJ) recently cleared by the FDA as an aid to monitoring CRPC.7
CLINICAL PRACTICE AND CLINICAL RESEARCH: SHARED GOALS OF TREATMENT
Treatment is administered with therapeutic intent whether it is given off (clinical practice) or on protocol (clinical research). Off-protocol considerations before starting therapy include determining the reason therapy is needed and it is expected to do for the individual and his disease, disease manifestations now present or likely to occur in the future (and when), factors that might predict for the success or failure of one treatment versus another to guide treatment selection, and the specific intervention that will be used. Next, one must determine the testing and evaluation methods that will be used to assess the effects of therapy and, at each time point that the disease is reassessed, how the test information will be used to inform the decision of whether to continue treatment. On-protocol therapy applies much the same considerations to a patient population: the study objectives that address patients' unmet need(s); eligibility criteria that seek to homogenize the patient population with respect to prognosis and likelihood of response to the intervention; and end points used to assess the effect of the intervention, plus a statistical design based on the analysis of the outcome of each individual and the population as a whole, to conclude whether or not the trial was successful and, if so, what to do next. Individuals in whom the considerations align with a protocol can be considered for enrollment.
Disease
The PCWG2 criteria divides the disease continuum into milestones or states based on disease extent and measured level of androgens in the blood (Fig 1). CRPC states include patients whose sole manifestation of the disease is a rising PSA as well as those with detectable metastases who have not yet received chemotherapy, are about to start first-line cytotoxic therapy, or have received chemotherapy. As illustrated, the objectives, issues, and unmet needs are similar for an individual patient or patient population in a given clinical state: what manifestations are present, what is the prognosis, what is the best choice of treatment for this patient's cancer, and, once administered, is it working?
Fig 1.

Prostate cancer clinical states model; framework for patient management and drug development. PSA, prostate-specific antigen. Data adapted.6,8
Patient Assessment
As outlined by the PCWG2,5 a patient's pretreatment evaluation should include a complete history and physical examination; assessment of symptoms and attribution of each one to disease, prior therapy, or pre-existing comorbid conditions; and complete blood count, chemistry panel, PSA, and testosterone. Transaxial imaging with computed tomography or magnetic resonance imaging and a radionuclide bone scan are also advised. Increasingly, bone turnover markers are being ordered along with CTCs.
Prognosis is determined by the functional status of the patient, demographic characteristics, presence or absence of pain, and disease manifestations. Disease manifestations include a rising PSA, soft-tissue disease (nodes and/or viscera), bone disease (most common site of spread), and symptoms. These and other individual parameters (eg, performance status, site of disease, lactate dehydrogenase count, and PSA level) are often combined into statistical models called nomograms to predict a patient's clinical outcome or stratify a group of patients at trial entry. However, nomograms should not be used to assess efficacy or compare outcomes between trials, because the population used to develop the nomogram will almost surely differ from the patient cohort under study.
Factors that predict sensitivity to treatment include prior systemic therapy, which historically has largely focused on whether a patient has received a cytotoxic drug, whereas second-line (postdocetaxel) trials consider outcomes based on the amount of docetaxel received; time from last dose to progression, and prior response. Of particular interest is identifying agents showing efficacy in patients who had never responded to docetaxel or who responded and later progressed while receiving docetaxel. For example, in a trial of mitoxantrone/prednisone and ixabepilone, response rates were similar between patients who were primarily refractory and those who had shown a previous response.9 Similarly, the response rate to cabazitaxel was independent of the amount of prior docetaxel received, which also suggests a degree of noncross-resistance.2
Now, with a range of therapies targeting the androgen-receptor signaling axis in late-stage development, there is also a focus on the specific hormonal therapies a patient has received and the level of testosterone in the blood. The PCWG2 defines castrate as 50 ng/mL or less, but with the increasing study of CYP17 inhibitors that produce androgen levels in the range of 1 to 2 ng/mL,10 this may need to be redefined. Unknown at this point is whether response to docetaxel, the first-line cytotoxic standard, will vary as a function of androgen levels in blood.
Molecular determinants are also under study, such as receptor tyrosine kinase mutations in other tumor types.11 Receptor tyrosine kinase mutations are rare in prostate cancer.12 Gene expression profiles13 are also under study, but at this point, rebiopsy for profiling is not part of routine management.
Drug
Increased understanding of prostate cancer biology and extensive profiling efforts have led to the identification of a range of new targets and to the evaluation of a range of new approaches beyond traditional hormones and cytotoxics. These new approaches include biologics, bone-targeting agents, antiangiogenic agents, proapoptotic approaches, specific pathway inhibitors, differentiating agents, and inhibitors of other components of the metastatic process. Different outcome measures will be needed to assess different drugs. For a patient to make an informed decision, it is essential to discuss why a treatment is being offered and what it is anticipated to do for the patient relative to what it might do to him (ie, risk/reward ratio of intervention planned).
OUTCOMES BY DISEASE MANIFESTATION
When determining or planning the specific outcomes of success or failure of an individual's treatment, it is important to consider the mechanism of action of a drug and anticipated timing of benefit, if benefit does occur. For biologics, in particular, the effects may take several months, and in some cases, they may not be assessable using traditional outcome measures, as was the case with sipuleucel-T, which showed no effect on PSA or time to progression but did show a survival benefit.1 The PCWG2 recommends serially monitoring changes in disease manifestations present at baseline with the same modality used before treatment. If all parameters are improving, continuing is straightforward. Difficulties arise when parameters are discordant (eg, PSA rising while there is no change in other parameters, or PSA declining while imaging studies grow worse). The central question is whether the adverse change in a single parameter, or discordant parameters, is sufficient to stop treatment. Because early changes can be misleading, it is generally advised to treat a patient for a minimum of 12 weeks and to avoid using early changes in PSA or imaging during this interval as the sole decision-making criterion. In cases in which outcomes are equivocal or discordant, it is also advised that treatment be continued in the absence of clear-cut evidence of progression or worsening clinical status.
PSA
The use of post-therapy PSA measurements as an outcome was first proposed to screen for drug activity based on the hypothesis that a reduction in tumor burden would be mirrored by a decline in PSA, and an increase in burden by a rise.14 It seemed simple: a drug not producing a defined degree of decline would not be worthy of further study, whereas one that did would merit it, and this would be determined by a routine, quantitative, easily attainable blood test. Metrics were proposed for use in practice that included documenting the decline more than once and over time, along with drug class–specific metrics that recognized that not all drugs worked immediately15 (eg, up to 20% of patients treated with docetaxel do not show declines until 12 weeks or more).16 The use of a decline of 50% or more from baseline as a response measure was derived in part from prognostic factor analyses associating this degree of decline with survival,17–20 which unfortunately became misconstrued as an indication that the treatment was providing a direct clinical benefit to the patient and, going one step farther, that the PSA decline could serve as the basis for regulatory approval. Missing were analyses of the strength of association between PSA decline and survival. For example, in an analysis using every post-treatment PSA measurement rather than a cutoff, only 17% of the variability in OS was explained by the time-dependent PSA.21 Also missing were multiple phase III randomized trials with the proposed PSA outcome measure embedded to prospectively address the question.
By the Prentice22 criteria, a surrogate must first, influence the end point, and second, fully capture the effect of an intervention on the efficacy end point. In addition, third, the intervention must affect the surrogate and end point. TAX-327 showed a survival benefit for docetaxel once every 3 weeks relative to mitoxantrone, establishing a new FDA-approved standard of care.23 In the analysis, patients who showed a 50% or greater decline in PSA from baseline had 60% reduction in mortality relative to those lacking such a decline, which accounted for half of the treatment effect on OS. Unfortunately, this was not confirmed with the weekly docetaxel schedule, which did not show a statistically significant difference in survival despite having a similar PSA decline rate, leading to the appropriate conclusion that the 50% decline was not a surrogate for survival.16 In a second trial showing a survival benefit for a docetaxel-containing regimen (SWOG 99-16 [Southwest Oncology Group 99-16]),24 a post-treatment decline of 30% or more was proposed as a surrogate for survival based on the demonstration that the effect of treatment on outcome was not significant after adjusting for the 30% PSA decline.25 However, this demonstration of nonsignificance is not the same as proving the treatments are equivalent after adjusting for post-treatment PSA, which is the second point the Prentice criteria. The question of PSA surrogacy requires further prospective validation in multiple randomized trials.26
In practice, a therapy that produces any degree of PSA decline, in the absence of other signs of deterioration or progression, is continued. For an individual, is it better to have a 20% PSA decline for 1 year or more or a 90% decline for 2 months? No single degree of decline has been established as a surrogate, and to standardize reporting, the PCWG2 advises reporting PSA change data using waterfall plots,27 showing the maximal decline from baseline, and separately, the degree of decline at a fixed time point (12 weeks).5 Waterfall plots provide a simple but informative display of PSA change data.
For consistency of trial reporting, the PCWG2 defines PSA progression as the date that an increase of 25% or more and absolute increase of 2 ng/mL or more from the nadir are documented. For patients who had an initial PSA decline during treatment, this must be confirmed by a second value 3 or more weeks later. However, PSA progression alone is not necessarily an indication to stop treatment, because in some cases, PSA levels may rise slowly after an initial decline (Fig 2A) or stabilize after an initial rise with no other sign of clinical progression (Fig 2B). Our view is that stopping therapy in patients with apparent PSA progression alone is not appropriate, because there are cases in which additional years of disease control would not have been realized had therapy been stopped on the basis of PSA change alone (Fig 2B).5
Fig 2.

(A) Prostate-specific antigen drift: A slow rise in PSA after an initial rapid decline with no evidence of radiographic or clinical progression for 28 months while receiving MDV3100; (B) an initial rapid rise in PSA with subsequent decline above baseline on a second value in a patient who remained biochemically, radiographically, and clinically stable for 22 months on abiraterone acetate.
Soft Tissue
Control/relieve/eliminate outcomes for soft-tissue disease in nodes and/or viscera follow RECIST,28 with the addition that changes in size are reported as waterfall plots to facilitate comparison between studies. Prevent/delay end points (eg, progression in nodal or visceral site) are also defined using RECIST. However, it must be recognized that although imaging of soft-tissue disease is part of virtually every CRPC trial and used routinely in clinical practice, it may furnish misinformation because of the lack of standards for assessing disease at baseline and recording results. A central issue is that none of the outcome criteria used have been directly associated with clinical outcomes. Tumor shrinkage, although encouraging, does not necessarily equate to an improvement in survival, nor does an increase in size necessarily equate with a decrement in survival. For example, a modest increase in the size of a pelvic lymph node may be less significant than continued control of disease in bone. Lymphocytic infiltration of a tumor mass after biologic therapy is another example in which an apparent increase in size may not reflect tumor enlargement.
Bone
Although skeletal lesions are considered nontargets in RECIST, they comprise the most frequent site of distant metastasis in prostate cancer and are the primary source of morbidity and mortality related to the disease.5 This provides the clinician with a conundrum; the site of clinical interest is poorly assessable by standard imaging methods. The most common method of assessing bone lesions is the bone scan, yet bone scintigraphy does not directly assess the cancer itself but rather its derivative effects on bone. As a result, bone scans cannot be used to measure cancerous lesions, but they can detect bone trauma, degenerative changes, infection, and other inflammatory processes unrelated to the patient's cancer. Finally, bone scans have typically demonstrated disease progression far more quickly than they do disease regression, which can lead clinicians to discontinue therapy, because they do not observe a response.5 Further biasing clinicians in favor of terminating treatment is the flare phenomenon, in which bony lesions emerge or enlarge response to an effective therapy, a result of rapidly healing bone around regressing cancer (Fig 3).29
Fig 3.
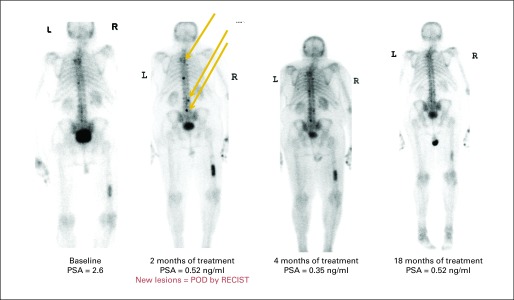
Flare on bone scan. Two new lesions at 8 weeks were not followed by subsequent additional lesions, so patient remained on study; bone scan markedly improved over 18 months. POD, progression of disease; PSA, prostate-specific antigen; RECIST, Response Evaluation Criteria in Solid Tumors.
To contend with the difficulties of bone scan use and prevent premature termination of treatment, the PCWG2 has proposed standards for bone scan interpretation, setting the principle that a patient should remain on study or treatment until two new bone lesions are observed. To accommodate early flare phenomena, the appearance of two new lesions on the first scan performed after treatment (usually 8 to 12 weeks after treatment start) has been deemed insufficient to warrant treatment discontinuation. Instead, to document progression, two additional lesions would need to be observed on a subsequent follow-up scan 6 weeks or longer after the first scan.5
Since the original publication of the PCWG2 guidelines, many investigators have extended this concept of verifying progression with an additional scan even after the period in which flare phenomena might be observed. As a result, many trials mandate not only that two new lesions be documented after the initial on-treatment scan before a patient is declared to have progressed but also that those two new lesions be observed in a confirmatory scan performed at least 6 weeks after the second of the two lesions is noted. Hence, contemporarily, the PCWG2 defines bone scan progression as either two new lesions noted on the first on-treatment scan followed by two additional lesions on the next scan or two new lesions seen on any scan after the first on-treatment scan that are confirmed on a subsequent scan. These criteria can be found in a series of simplified bone scan data capture forms available to clinicians and radiologists alike, which are being used to prospectively qualify the PCWG2-proposed end point in phase III trials.30
Metastasis prevention.
Preventing bone metastasis is another time-to-event end point that is a direct clinical benefit in its own right. In Trial 147, men with nonmetastatic CRPC were randomly assigned to the RANK ligand inhibitor denosumab versus placebo. The results showed that denosumab significantly improved both median bone metastasis–free survival by 4.2 months and time to first occurrence of bone metastasis. OS times were similar.31 Previous trials with atrasentan32 and zibotentan33 were unsuccessful. A challenge to conducting these trials is keeping patients on study until the detectable metastasis end point is achieved when the agent under study does not affect PSA, and levels continue to rise in all patients.
SREs.
An SRE is another standalone end point of clinical benefit, first used in trials of the bisphosphonate zoledronic acid, which led to full FDA approval for several tumor types, including prostate cancer.34 SREs were defined as pain requiring a change in oncolytic therapy, spinal cord compression, need for radiation therapy or surgical intervention to treat a bone complication, or development of hypercalcemia. Significantly, this trial established that a clearly demonstrated objective patient benefit reducing the morbidity of bone metastases could lead to full approval despite the drug failing to show a survival benefit or effect on PSA.34,35 This same end point was subsequently used in a phase III registration trial of the RANK ligand inhibitor denosumab, which proved superior to zoledronic acid in a head-to-head comparison.4
Patient-Reported Outcomes
Control, relief, and elimination of symptoms such as pain are important therapeutic objectives. They considered trial end points independent of other measures, for example, are because pain response and PSA response do not correlate well, and the presence of pain is an adverse prognostic factor for survival.36 In clinical trials, symptoms such as pain should be assessed using validated self-reporting questionnaires that enable intensity, and interference with usual activities.37 Confirmation of both favorable and unfavorable findings is also essential to optimize management.
Patient-reported outcomes.
The patient subjective experience is an essential dimension in the evaluation and management of men with CRPC. Pain, in particular, can be debilitating for patients, is associated with decreased survival, and can be an important clinical trial end point in defining treatment benefit.38 Mitoxantrone was labeled in 1996 as initial chemotherapy for the treatment of patients with pain related to advanced hormone-refractory prostate cancer based on evidence from two randomized controlled trials designed to show reduction in pain, without a demonstration of an OS benefit.39–41
The science of developing and administering patient-reported outcomes (PROs) in clinical trials and during routine practice has evolved substantially over the past decade, and approaches used in the past would likely not result in drug approval or labeling today. A 2009 FDA guidance document defined a PRO as any report of a patient's health status that comes directly from the patient, without interpretation of the patient's response by anyone else. The document also noted that PRO instruments are most appropriate for measuring treatment impact in a clinical trial when the outcome of interest is best known by the patient, such as pain intensity.42 The FDA acknowledges that capturing the patient's perspective through adequate PRO measures can be an important aspect of defining a treatment effect in a clinical trial. The PRO guidance delineates how the FDA determines the adequacy of PRO measures used in clinical trials to support labeling claims. As with other measures that support labeling claims, PRO instruments used for effectiveness end points should be well developed and must provide reliable assessments of what they are intended to measure within the context of use.
Although pain end points were included in pivotal trials of docetaxel, cabazitaxel, and abiraterone, these were exploratory end points and therefore could not be used as the basis for drug approval or labeling in the United States. For future trials, because of the importance of pain as an end point in prostate cancer and the complexity of assessing it in a trial, it is advisable that sponsors begin planning pain end points early in the product development cycle and initiate early discussions with PRO experts and the FDA. A dedicated pain study may be advisable and could be a path to drug approval (eg, if palliative outcomes were shown in a statistically and clinically convincing manner).43
In clinical practice, regular assessment of pain is also essential, because pain is prevalent in men with CRPC.44 Transient increases in pain may occur before improvement, and those occurring in the first 12 weeks should be ignored in the absence of other compelling evidence of disease progression. Products with demonstrated symptom palliation properties should be considered for men with pain. Mitoxantrone, docetaxel, and abiraterone have demonstrated modest reductions in pain intensity among subpopulations of patients, and substantial pain improvements may be seen after treatment with samarium. Cabazitaxel showed no significant difference in pain palliation rates compared with mitoxantrone in the TROPIC (Treatment of Hormone-Refractory Metastatic Prostate Cancer Previously Treated With a Taxotere-Containing Regimen) trial, but rates of pain palliation were low with either treatment, at fewer than 10% of patients in this docetaxel-pretreated population.2 However, the toxicities of these approaches and potential negative impact on quality of life must be weighed against potential pain benefits. All patients in pain should be offered narcotic analgesics, including a long-acting agent, with consideration of palliative radiation therapy as warranted.
Other important PROs for consideration in trials and practice include anorexia (decreased appetite), anxiety, constipation, diarrhea, sleep disturbance, mucositis, nausea, pain, peripheral sensory neuropathy, rash, vomiting, urinary symptoms, global health-related quality of life, and interference of symptoms with usual activities. The PCWG2 considers symptoms and health-related quality of life independently from other outcome measures based on the importance of symptoms for clinical benefit, limited correlations between pain response and post-therapy PSA decline, and independent status of pain and PSA as predictors of survival.36 Symptoms related to treatment benefit as well as to potential toxicity should be evaluated.45 Although it is rare for health-related quality of life to serve as the basis for drug labeling or approval in the United States, it serves a more prominent regulatory role in Europe, and its assessment is important to understand the patient subjective experience with disease and treatment.
CLINICAL PRACTICE VERSUS FDA REGULATORY APPROVAL: LEVEL OF EVIDENCE
Prolonging life—the delay or prevention of death from disease—is the gold standard for drug approval. Delay and prevention of SREs is also a standalone end point of clinical benefit that has led to the approval of several bone-targeted agents. Other composite time-to-event progression measures have not faired as well.
Time to Progression
Progression-free survival (PFS) is defined as the time from study entry or random assignment to disease progression in bone or soft tissue, symptoms, or death. It is an attractive end point, because progression events occur months to years before survival can be measured, and PFS is not confounded by the effect of subsequent treatments. Because the definition of PFS produces a higher number of events than that of OS, using PFS can also lead to trials with smaller sample sizes. Using PFS in a phase II study also has the advantage that a comparable end point can be used in a subsequent definitive randomized phase III comparative trial to test the same agent. This consistency in the end point of phase II and III studies reduces the risk of an ineffective finding in the large-scale, resource-intense phase III trial after the determination that the therapy was effective in the lead-in phase II study.46
Various PFS definitions have been used, which in practice include PSA alone, radiographic changes defined as a size increase of an existing lesion, new metastatic lesions, or disease-related symptoms.47,48 Although each of the changes outlined represents some form of disease reactivation, their clinical significance varies, and as such, it is not clear that each warrants a change in therapy. To date, definitions based on PSA and radiographic progression have been unsatisfactory in the CRPC population, in which recent studies of biomarker-derived progression end points have shown only modest associations with survival.47,48
CTCs
CTCs include those with stem-cell or stem cell–like properties or any degree of differentiation. They may originate from the primary tumor or metastatic sites and are estimated to represent at most one cell in 1 billion of the circulating cells in the blood.49 Presently, there are a range of assays and devices in various stages of development and clinical testing. These approaches generally include an enrichment step followed by different detection and characterization methods.49,50 Enrichment is achieved using the following methods: first, morphologic or physical methods; second, positive selection with antibodies to cell surface markers on CTCs; or third, negative selection with antibodies to the common leukocyte antigen CD45 to deplete mononuclear cells or those that eliminate RBCs and mononuclear cells. Detection and characterization methods use tumor- or tissue-specific markers or various cytometric methods.49,50 Importantly, there is no standardized definition of a CTC and no single feature that distinguishes CTCs from normal, nonmalignant blood elements. Consequently, different assays do not measure the same malignant cell population, and as such, each reports a different biomarker, not all of which are useful in a given context.
Our interest in CTCs in prostate cancer is based on promising early results showing that the presence of mRNA for PSA in the mononuclear cell fraction by real-time polymerase chain reaction could provide information distinct from PSA51 and serve as a non–PSA-based outcome measure.52 Presently, CellSearch (Veridex) is the only CTC assay cleared by the FDA for use in the clinic.7 With this assay, a CTC is defined as a cell that is morphologically intact; has a nucleus surrounded by cytoplasm when stained with 4′,6-diamidino-2-phenylindole; and expresses cytokeratin 8, 18, or 19 but does not express the mononuclear cell determinant CD-45.53,54 Assay results are reported as the number of cells meeting the analytically valid definition per 7.5 mL of blood, which represents only a proportion of cytokeratin-positive events.55 The cells isolated are confirmed to have molecular features of prostate cancer.54 CTC counts trend toward higher values in relation to PSA level and extent of disease in bone, but the association is modest, suggesting that each parameter provides independent information.56
Clearance for use was obtained first for breast followed by colorectal and later prostate cancers based on trials of similar design that enrolled patients who were about to start a new line of chemotherapy57–59 and that showed baseline CTC number was prognostic for survival in univariate and multivariate analyses before treatment and a range of times post-treatment including, 2 to 5, 6 to 8, 9 to 11, and 13 to 20 weeks. For prostate cancer, a cutoff of four or fewer cells per 7.5 mL of blood was considered favorable and five or more cells per 7.5 mL of blood unfavorable. At all time points evaluated, the post-therapy CTC number was more predictive than a decline of 50% or more in PSA.58 In separate analyses of CTC number as a continuous variable,60 it is noteworthy that a low count alone did not assure a long survival.57–59,61,62 In a multivariate analysis, only CTC count and lactate dehydrogenase were prognostic; PSA was no longer predictive.63
The FDA clearance states that “presence of CTC in the peripheral blood, as detected by the CellSearch Circulating Tumor Cell Kit, is associated with decreased progression free survival and decreased overall survival in patients treated for metastatic breast, colorectal or prostate cancer … Serial testing for CTC should be used in conjunction with other clinical methods for monitoring [these cancers].”7(p3) It has not been shown to be a surrogate for survival, nor can it replace other methods of assessing disease. It should be noted that many investigators are not yet using CTC enumeration testing in day-to-day practice; they are waiting until more data and more standardized methods of measuring CTCs become available.
One way to use CTC counts is to obtain a baseline sample before starting a new therapy. In our experience, the proportion of patients with unfavorable counts has been approximately 25% in the prechemotherapy population, 50% in patients receiving first-line chemotherapy, and 70% in those receiving second-line chemotherapy (Danila et al, unpublished data). If the counts are unfavorable at baseline, the approach of our group is to serially monitor them after each cycle of therapy. Conversions from unfavorable to favorable tend to occur early, and in one study, they were maximal at 8 weeks.63 Patients in whom CTC counts do not decline tend to do poorly. Trials are ongoing in breast cancer to address whether an early change in therapy for patients who do not show a CTC conversion will affect outcome. If the count is favorable after two baseline determinations, we repeat the assessment at the time of progression.
At this point, it is too early to rely solely on CTC enumeration results to guide patient management. Going forward, use of the CTC assay in specific contexts will require further qualification. These efforts are ongoing through a formal collaboration with the Center for Devices and Radiologic Health branch of the FDA. CTC enumeration was studied in the phase I/II trial of abiraterone, with favorable conversion rates.10,64 The trial subsequently showed a significant survival benefit, and analyses are ongoing to define a biomarker panel including CTCs that is prognostic for survival after treatment and which explains a sufficient portion of the survival benefit observed. Once determined, the panel will be prospectively studied in a phase III trial of similar design, with the CTC end point embedded, as it is in trials of MDV3100 (AFFIRM; NCT00974311), TAK-700 (NCT01193257), and ipilimumab (NCT01057810). There is also an ongoing effort to qualify CTCs as a surrogate outcome measure.
It is hoped that in the future, CTCs will provide a noninvasive, real-time liquid biopsy for just-in-time disease characterization to guide treatment selection. Several new technologies are in development that could detect cells in more patients and more cells overall with greater purity. Techniques include fluorescence-activated cell sorting,67 filtration,68,69 and microfluidics,70–72 although further analytic validation will be required before they can be considered for clearance or qualification.49,50,73 The specific assay needed will depend on the context of use and the biologic determinant being measured.
CONCLUSION
In CRPC, it is often difficult to determine the effectiveness of a new treatment or what metrics to use with an individual patient to choose the right treatment at the right time. This is because of the complexity of the disease overall, of assessing disease in bone, and of interpreting PSA results after therapy. Biomarkers continue to be refined in this disease, which should provide more specific end points and clinical guidance in the future.
Footnotes
Supported by the Sidney Kimmel Center for Prostate and Urologic Cancers and in part by the Memorial Sloan-Kettering Cancer Center Special Program of Research Excellence in Prostate Cancer (Grant No. P50 CA92629), Department of Defense Prostate Cancer Research Program (Grant No. PC051382), Research and Therapeutics Program for Prostate Cancer, and Prostate Cancer Foundation.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Howard I. Scher, Veridex (U), Aragon (U), Bristol-Meyers Squibb (U), Exelixis (U), Foundation Medicine (U), Genentech (U), Medivation (U), Amgen (C), Ortho Biotech Oncology Research and Development (C), Dendreon (C), Enzon (C), Millennium (C), Novartis (C), Roche (C), sanofi-aventis (U); Ethan Basch, Exelixis (U); Glenn Heller, Veridex (U); Michael Morris, Millennium (C) Stock Ownership: Howard I. Scher, Genta, previously owned stock in Johnson & Johnson Honorarium: None Research Funding: Howard I. Scher, Veridex, Bristol-Meyers Squibb, Ortho Biotech Oncology Research and Development, Medivation, Aragon; Michael Morris, Genta, Algeta, sanofi-aventis, GlaxoSmithKline, Agensys Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Administrative support: Howard I. Scher
Provision of study materials or patients: Howard I. Scher
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 2.de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet. 2010;376:1147–1154. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 3.de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone acetate and survival of patients with metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fizazi K, Carducci M, Smith M, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomised, double-blind study. Lancet. 2011;377:813–822. doi: 10.1016/S0140-6736(10)62344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scher HI, Halabi S, Tannock I, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: Recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26:1148–1159. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scher HI, Morris MJ, Kelly WK, et al. Prostate cancer clinical trial end points: “RECIST”ing a step backwards. Clin Cancer Res. 2005;11:5223–5232. doi: 10.1158/1078-0432.CCR-05-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veridex. FDA clearance document for CellSearch Circulating Tumor Cell Kit. http://www.accessdata.fda.gov/cdrh_docs/pdf7/K073338.pdf.
- 8.Scher HI, Heller G. Clinical states in prostate cancer: Towards a dynamic model of disease progression. Urology. 2000;55:323–327. doi: 10.1016/s0090-4295(99)00471-9. [DOI] [PubMed] [Google Scholar]
- 9.Harzstark AL, Rosenberg JE, Weinberg VK, et al. Ixabepilone, mitoxantrone, and prednisone for metastatic castration-resistant prostate cancer after docetaxel-based therapy: A phase 2 study of the department of defense prostate cancer clinical trials consortium. Cancer. doi: 10.1002/cncr.25810. [epub ahead of print on December 29, 2010] [DOI] [PubMed] [Google Scholar]
- 10.Attard G, Reid AH, A'Hern R, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27:3742–3748. doi: 10.1200/JCO.2008.20.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ilson DH, Kelsen D, Shah M, et al. A phase 2 trial of erlotinib in patients with previously treated squamous cell and adenocarcinoma of the esophagus. Cancer. 2011;117:1409–1414. doi: 10.1002/cncr.25602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Febbo PG. Epigenetic events highlight the challenge of validating prognostic biomarkers during the clinical and biologic evolution of prostate cancer. J Clin Oncol. 2009;27:3088–3090. doi: 10.1200/JCO.2008.20.9783. [DOI] [PubMed] [Google Scholar]
- 14.Scher HI, Curley T, Geller N, et al. Trimetrexate in prostatic cancer: Preliminary observations on the use of prostate-specific antigen and acid phosphatase as a marker in measurable hormone-refractory disease. J Clin Oncol. 1990;8:1830–1838. doi: 10.1200/JCO.1990.8.11.1830. [DOI] [PubMed] [Google Scholar]
- 15.Scher HI, Mazumdar M, Kelly WK. Clinical trials in relapsed prostate cancer: Defining the target. J Natl Cancer Inst. 1996;88:1623–1634. doi: 10.1093/jnci/88.22.1623. [DOI] [PubMed] [Google Scholar]
- 16.Berthold DR, Pond GR, Roessner M, et al. Treatment of hormone-refractory prostate cancer with docetaxel or mitoxantrone: Relationships between prostate-specific antigen, pain, and quality of life response and survival in the TAX-327 study. Clin Cancer Res. 2008;14:2763–2767. doi: 10.1158/1078-0432.CCR-07-0944. [DOI] [PubMed] [Google Scholar]
- 17.Kelly WK, Scher HI, Mazumdar M, et al. Prostate-specific antigen as a measure of disease outcome in metastatic hormone-refractory prostate cancer. J Clin Oncol. 1993;11:607–615. doi: 10.1200/JCO.1993.11.4.607. [DOI] [PubMed] [Google Scholar]
- 18.Scher HI, Kelly WM, Zhang ZF, et al. Post-therapy serum prostate-specific antigen level and survival in patients with androgen-independent prostate cancer. J Natl Cancer Inst. 1999;91:244–251. doi: 10.1093/jnci/91.3.244. [DOI] [PubMed] [Google Scholar]
- 19.Small EJ, Fratesi P, Reese DM, et al. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol. 2000;18:3894–3903. doi: 10.1200/JCO.2000.18.23.3894. [DOI] [PubMed] [Google Scholar]
- 20.Smith DC, Dunn RL, Strawderman MS, et al. Change in serum prostate-specific antigen as a marker of response to cytotoxic therapy for hormone-refractory prostate cancer. J Clin Oncol. 1998;16:1835–1843. doi: 10.1200/JCO.1998.16.5.1835. [DOI] [PubMed] [Google Scholar]
- 21.Verbel DA, Heller G, Kelly WK, et al. Quantifying the amount of variation in survival explained by prostate-specific antigen. Clin Cancer Res. 2002;8:2576–2579. [PubMed] [Google Scholar]
- 22.Prentice RL. Surrogate endpoints in clinical trials: Definition and operational criteria. Stat Med. 1989;8:431–440. doi: 10.1002/sim.4780080407. [DOI] [PubMed] [Google Scholar]
- 23.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 24.Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–1520. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 25.Petrylak DP, Ankerst DP, Jiang CS, et al. Evaluation of prostate-specific antigen declines for surrogacy in patients treated on SWOG 99-16. J Natl Cancer Inst. 2006;98:516–521. doi: 10.1093/jnci/djj129. [DOI] [PubMed] [Google Scholar]
- 26.Fleming MT, Morris MJ, Heller G, et al. Post-therapy changes in PSA as an outcome measure in prostate cancer clinical trials. Nature Clin Pract Oncol. 2006;3:658–667. doi: 10.1038/ncponc0664. [DOI] [PubMed] [Google Scholar]
- 27.Seidman AD, Scher HI, Petrylak D, et al. Estramustine and vinblastine: Use of prostate specific antigen as a clinical trial end point for hormone refractory prostatic cancer. J Urol. 1992;147:931–934. doi: 10.1016/s0022-5347(17)37426-8. [DOI] [PubMed] [Google Scholar]
- 28.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 29.Shah SK, Ryan CJ, Molina A, et al. Bone scan flare in patients (pts) receiving abiraterone acetate (AA) for metastatic androgen deprivation-resistant prostate cancer (mADRPC): Analysis of data from a phase II study of the Department of Defense Prostate Cancer Clinical Trials Consortium. Presented at the American Society of Clinical Oncology Genitourinary Cancers Symposium; March 5-7, 2010; San Francisco, CA. abstr 4. [Google Scholar]
- 30.Morris MJ, Farrelly JS, Fox JJ, et al. The Prostate Cancer Clinical Trials Consortium (PCCTC) bone scan data capture tool for clinical trials using Prostate Cancer Working Group 2 (PCWG2) criteria: Effect on data accuracy and workload. Presented at the American Society of Clinical Oncology Genitourinary Cancers Symposium; February 17-19, 2011; Orlando, FL. abstr 121. [Google Scholar]
- 31.Bioresearch Online. XGEVA (denosumab) significantly improved bone metastasis-free survival in men with prostate cancer. http://www.bioresearchonline.com/article.mvc/XGEVA-Denosumab-Significantly-Improved-0001.
- 32.Carducci MA, Padley RJ, Breul J, et al. Effect of endothelin-A receptor blockade with atrasentan on tumor progression in men with hormone-refractory prostate cancer: A randomized, phase II, placebo-controlled trial. J Clin Oncol. 2003;21:679–689. doi: 10.1200/JCO.2003.04.176. [DOI] [PubMed] [Google Scholar]
- 33.AstraZeneca. AstraZeneca halts phase III trial of zibotentan in non-metastatic castrate resistant prostate cancer. http://www.astrazeneca.com/Media/Press-releases/Article/0022011AstraZeneca-halts-phase-III-trial-of-ZIBOTENTAN.
- 34.Saad F, Gleason DM, Murray R, et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst. 2002;94:1458–1468. doi: 10.1093/jnci/94.19.1458. [DOI] [PubMed] [Google Scholar]
- 35.Saad F, Gleason DM, Murray R, et al. Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J Natl Cancer Inst. 2004;96:879–882. doi: 10.1093/jnci/djh141. [DOI] [PubMed] [Google Scholar]
- 36.Berthold DR, Pond G, de Wit R, et al. Association of pain and quality of life (QOL) response and survival of patients (pts) with metastatic hormone refractory prostate cancer (mHRPC) treated with docetaxel or mitoxantrone in the TAX-327 study. Presented at the American Society of Clinical Oncology Prostate Cancer Symposium; February 24-26, 2006; San Francisco, CA. abstr 140. [Google Scholar]
- 37.Turk DC, Dworkin RH, Burke LB, et al. Developing patient-reported outcome measures for pain clinical trials: IMMPACT recommendations. Pain. 2006;125:208–215. doi: 10.1016/j.pain.2006.09.028. [DOI] [PubMed] [Google Scholar]
- 38.Halabi S, Vogelzang NJ, Kornblith AB, et al. Pain predicts overall survival in men with metastatic castration-refractory prostate cancer. J Clin Oncol. 2008;26:2544–2549. doi: 10.1200/JCO.2007.15.0367. [DOI] [PubMed] [Google Scholar]
- 39.Hospira Worldwide. Mitoxantrone prescribing information. http://www.drugs.com/pro/mitoxantrone.html.
- 40.Kantoff PW, Halabi S, Conaway M, et al. Hydrocortisone with or without mitoxantrone in men with hormone-refractory prostate cancer: Results of the cancer and leukemia group B 9182 study. J Clin Oncol. 1999;17:2506–2513. doi: 10.1200/JCO.1999.17.8.2506. [DOI] [PubMed] [Google Scholar]
- 41.Tannock IF, Osoba D, Stockler MR, et al. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: A Canadian randomized trial with palliative end points. J Clin Oncol. 1996;14:1756–1764. doi: 10.1200/JCO.1996.14.6.1756. [DOI] [PubMed] [Google Scholar]
- 42.US Food and Drug Administration. Guidance for industry: Patient-reported outcomes measures—Use in medical product development to support labeling claims. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM193282.pdf. [DOI] [PMC free article] [PubMed]
- 43.Basch E, Trentacosti AM, Burke L, et al. Measurement of pain palliation in metastatic prostate cancer: The FDA perspective. Presented at the American Society of Clinical Oncology Genitourinary Cancers Symposium; March 5-7, 2010; San Francisco, CA. abstr 79. [Google Scholar]
- 44.Basch EM, Burke L, Kane R, et al. Prevalence and severity of pain in men with metastatic prostate cancer. Presented at the American Society of Clinical Oncology Genitourinary Cancers Symposium; February 26-28, 2009; Orlando, FL. abstr 205. [Google Scholar]
- 45.Basch E. The missing voice of patients in drug-safety reporting. N Engl J Med. 2010;362:865–869. doi: 10.1056/NEJMp0911494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fazzari M, Heller G, Scher HI. The phase II/III transition: Toward the proof of efficacy in cancer clinical trials. Control Clin Trials. 2000;21:360–368. doi: 10.1016/s0197-2456(00)00056-8. [DOI] [PubMed] [Google Scholar]
- 47.Halabi S, Vogelzang NJ, Ou SS, et al. Progression-free survival as a predictor of overall survival in men with castrate-resistant prostate cancer. J Clin Oncol. 2009;27:2766–2771. doi: 10.1200/JCO.2008.18.9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scher HI, Warren M, Heller G. The association between measures of progression and survival in castrate-metastatic prostate cancer. Clin Cancer Res. 2007;13:1488–1492. doi: 10.1158/1078-0432.CCR-06-1885. [DOI] [PubMed] [Google Scholar]
- 49.Pantel K, Alix-Panabières C. Circulating tumour cells in cancer patients: Challenges and perspectives. Trends Mol Med. 2010;16:398–406. doi: 10.1016/j.molmed.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 50.Yu M, Stott S, Toner M, et al. Circulating tumor cells: Approaches to isolation and characterization. J Cell Biol. 2011;192:373–382. doi: 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghossein RA, Scher HI, Gerald WL, et al. Detection of circulating tumor cells in patients with localized and metastatic prostatic carcinoma: Clinical implications. J Clin Oncol. 1995;13:1195–1200. doi: 10.1200/JCO.1995.13.5.1195. [DOI] [PubMed] [Google Scholar]
- 52.Kantoff PW, Halabi S, Farmer DA, et al. Prognostic significance of reverse transcriptase polymerase chain reaction for prostate-specific antigen in men with hormone-refractory prostate cancer. J Clin Oncol. 2001;19:3025–3028. doi: 10.1200/JCO.2001.19.12.3025. [DOI] [PubMed] [Google Scholar]
- 53.Cristofanilli M, Budd GT, Ellis MJ, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351:781–791. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 54.Shaffer DR, Leversha MA, Danila DC, et al. Circulating tumor cell analysis in patients with progressive castration-resistant prostate cancer. Clin Cancer Res. 2007;13:2023–2029. doi: 10.1158/1078-0432.CCR-06-2701. [DOI] [PubMed] [Google Scholar]
- 55.Coumans FA, Doggen CJ, Attard G, et al. All circulating EpCAM+CK+CD45− objects predict overall survival in castration-resistant prostate cancer. Ann Oncol. 2010;21:1851–1857. doi: 10.1093/annonc/mdq030. [DOI] [PubMed] [Google Scholar]
- 56.Imbriaco M, Larson SM, Yeung HW, et al. A new parameter for measuring metastatic bone involvement by prostate cancer: The bone scan index. Clin Cancer Res. 1998;4:1765–1772. [PubMed] [Google Scholar]
- 57.Cohen SJ, Punt CJ, Iannotti N, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:3213–3221. doi: 10.1200/JCO.2007.15.8923. [DOI] [PubMed] [Google Scholar]
- 58.de Bono JS, Scher HI, Montgomery RB, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14:6302–6309. doi: 10.1158/1078-0432.CCR-08-0872. [DOI] [PubMed] [Google Scholar]
- 59.Hayes DF, Cristofanilli M, Budd GT, et al. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin Cancer Res. 2006;12:4218–4224. doi: 10.1158/1078-0432.CCR-05-2821. [DOI] [PubMed] [Google Scholar]
- 60.Danila DC, Heller G, Gignac GA, et al. Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin Cancer Res. 2007;13:7053–7058. doi: 10.1158/1078-0432.CCR-07-1506. [DOI] [PubMed] [Google Scholar]
- 61.Cohen SJ, Punt CJ, Iannotti N, et al. Prognostic significance of circulating tumor cells in patients with metastatic colorectal cancer. Ann Oncol. 2009;20:1223–1229. doi: 10.1093/annonc/mdn786. [DOI] [PubMed] [Google Scholar]
- 62.Iinuma H, Watanabe T, Mimori K, et al. Clinical significance of circulating tumor cells, including cancer stem-like cells, in peripheral blood for recurrence and prognosis in patients with Dukes' Stage B and C colorectal cancer. J Clin Oncol. 2011;29:1547–1555. doi: 10.1200/JCO.2010.30.5151. [DOI] [PubMed] [Google Scholar]
- 63.Scher HI, Jia X, de Bono JS, et al. Circulating tumour cells as prognostic markers in progressive, castration-resistant prostate cancer: A reanalysis of IMMC38 trial data. Lancet Oncol. 2009;10:233–239. doi: 10.1016/S1470-2045(08)70340-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reid AH, Attard G, Danila DC, et al. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28:1489–1495. doi: 10.1200/JCO.2009.24.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scher HI, Heller G, Molina A, et al. Evaluation of circulating tumor cell (CTC) enumeration as an efficicacy response biomarker of overall survival (OS) in metastatic castration-resistant prostate cancer (mCRPC): Planned final analysis (FA) of COU-AA-301, a randomized double-blind, placebo-controlled phase III study of abiraterone acetate (AA) plus low-dose prednisone (P) post doxetaxel. J Clin Oncol. 2011;29(suppl):293s. abstr LBA4517. [Google Scholar]
- 67.Racila E, Euhus D, Weiss A, et al. Detection and characterization of carcinoma cells in the blood. Proc Natl Acad Sci U S A. 1998;95:4589–4594. doi: 10.1073/pnas.95.8.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zheng S, Lin H, Liu JQ, et al. Membrane microfilter device for selective capture, electrolysis and genomic analysis of human circulating tumor cells. J Chromatogr A. 2007;1162:154–161. doi: 10.1016/j.chroma.2007.05.064. [DOI] [PubMed] [Google Scholar]
- 69.Lin HK, Zheng S, Williams AJ, et al. Portable filter-based microdevice for detection and characterization of circulating tumor cells. Clin Cancer Res. 2010;16:5011–5018. doi: 10.1158/1078-0432.CCR-10-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nagrath S, Sequist LV, Maheswaran S, et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450:1235–1239. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stott SL, Hsu CH, Tsukrov DI, et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci U S A. 2010;107:18392–18397. doi: 10.1073/pnas.1012539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gleghorn JP, Pratt ED, Denning D, et al. Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody. Lab Chip. 2010;10:27–29. doi: 10.1039/b917959c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pantel K, Brakenhoff RH, Brandt B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat Rev Cancer. 2008;8:329–340. doi: 10.1038/nrc2375. [DOI] [PubMed] [Google Scholar]