

Abstract
In the kidney, human organic cation transporters (OCTs) and multidrug and toxin extrusion proteins (MATEs) are the major transporters for the secretion of cationic drugs into the urine. In the human kidney, OCT2 mediates the uptake of drugs from the blood at the basolateral membrane of tubular epithelial cells, and MATE1 and MATE2-K secrete drugs from cells into the lumen of proximal tubules. However, the expression of these transporters depends on the species of the animal. In the rodent kidney, OCT1 and OCT2 are expressed at the basolateral membrane, and MATE1 localizes at the brush-border membrane. Together, these transporters recognize various compounds and have overlapping, but somewhat different, substrate specificities. OCTs and MATEs can transport important drugs, such as metformin and cisplatin. Therefore, functional variation in OCTs and MATEs, including genetic polymorphisms or inter-individual variation, may seriously affect the pharmacokinetics and/or pharmacodynamics of cationic drugs. In this review, we summarize the recent findings and clinical importance of these transporters.
KEY WORDS: cation, kidney, MATE, OCT
INTRODUCTION (ORGANIC CATION TRANSPORT IN THE KIDNEY)
The kidney is one of the main organs responsible for the excretion of drugs and xenobiotics. Renal excretion consists of three steps: glomerular filtration, tubular secretion, and re-absorption. Drugs are actively secreted through a two-step membrane transport process that includes distinct systems at the brush-border and basolateral membranes of epithelial cells. The characterization of the transport systems for organic cations was performed using isolated membrane vesicles (1). The electrogenic basolateral transport of organic cations is driven by an internal negative membrane potential, while electroneutral brush-border transport is dominated by a H+/organic cation antiport process. Renal drug transporters have been identified and characterized at the molecular level (2,3). There are two SLC protein families involved in cationic drug secretion, including the SLC22 family of organic cation transporters (OCTs) at the basolateral membrane, and the SLC47 multidrug and toxin extrusion proteins (MATEs) at the brush-border membrane (Fig. 1; Table I). OCTs and MATEs can transport various, structurally unrelated organic cations.
Fig. 1.
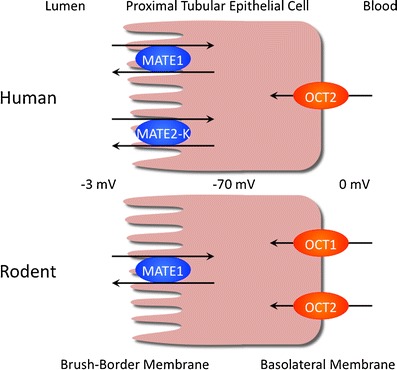
Physiological significance of OCTs and MATEs in the human and rodent kidney. In the human kidney, MATE1 and MATE2-K excrete cationic compounds via epithelial cells in cooperation with basolateral OCT2. In the rodent kidney, MATE1 is expressed at the brush-border membrane of epithelial cells, and both OCT1 and OCT2 are located at the basolateral membrane
Table I.
| Amino acids | TMD | Tissue distributiona | Membrane localization | |
|---|---|---|---|---|
| OCT1/SLC22A1 | 554 | 12 | Liver | Sinusoidal membrane (liver) |
| OCT2/SLC22A2 | 555 | 12 | Kidney | Basolateral membrane (kidney) |
| OCT3/SLC22A3 | 556 | 12 | Placenta, testis, brain, etc. | Basolateral membrane (placenta and lung) |
| Apical membrane (Intestine) | ||||
| OCTN1/SLC22A4 | 551 | 12 | Kidney, skeletal muscle, etc. | Brush-border membrane (kidney) |
| OCTN2/SLC22A5 | 557 | 12 | Kidney, liver, heart, etc. | Brush-border membrane (kidney) |
| MATE1/SLC47A1 | 570 | 13 | Kidney, liver, and muscle | Brush-border membrane (kidney) |
| Canalicular membrane (liver) | ||||
| MATE2-K/SLC47A2 | 566 | 13 | Kidney | Brush-border membrane (kidney) |
TMD trans-membrane domain
aTissue distribution of transporter proteins
ORGANIC CATION TRANSPORTERS (OCTs, SLC22A1-3) AND ORGANIC CATION AND CARNITINE TRANSPORTERS (OCTNs, SLC22A4-5) IN THE KIDNEY
Identification of OCTs in the Kidney
In 1994, the first OCT isoform, OCT1, was found to be highly expressed in the rat kidney and play a role in cationic drug secretion (4). Subsequently, mammalian orthologs of OCT1 were identified in humans, mice and rabbits. Using knockout mice (5), it was demonstrated that OCT1 plays an important role in the rodent kidney. In humans, OCT1 is expressed at extremely low levels in the kidney and is mainly found in the liver (6). These data suggest that there is a species difference for the role of OCT1 in renal drug secretion.
OCT2 was isolated from the rat kidney through homology cloning of the OCT1 sequence (7). Subsequently, human orthologs of OCT2 were identified (8). To investigate the pharmacological and physiological roles of OCT1 and OCT2, Jonker et al. (9) generated OCT1/2 single-knockout and OCT1/2 double-knockout mice. Accumulation of tetraethylammonium (TEA) in the liver was 4- to 6-fold lower in OCT1(−/−) mice, compared with their wild-type counterparts. In addition, excretion of TEA into the urine over 1 h was increased from 53% to 80% in the OCT1 knockout mouse. These data appear to be inconsistent with the purported role of OCT1 in the kidney. Jonker et al. (5) speculated that the decreased TEA uptake by the liver indirectly increased the urinary excretion of TEA. Absence of OCT2 alone had little effect on the pharmacokinetics of (TEA); however, the renal secretion of this compound was completely abolished in OCT1/2(−/−) mice. As a consequence, plasma concentrations of TEA were substantially increased in OCT1/2(−/−) mice. Sugawara-Yokoo et al. (10) reported that OCT1 is concentrated in the proximal tubules of the renal cortex, and OCT2 is abundant in the proximal tubules of the outer stripe of the outer medulla. Thus, OCT1 and OCT2 are differentially distributed along the proximal tubules. Both isoforms are considered essential for the renal secretion of organic cations; on the other hand, the expression of OCT2, but not OCT1, is much higher than levels of other renal drug transporters in the human kidney (6). In the human kidney, the OCT2 protein is located at the basolateral membrane of whole segments of the proximal tubular epithelia. Based on the low expression of OCT1 in the human kidney, OCT2 is the dominant OCT at the basolateral membrane of proximal epithelial cells. OCTs are part of the transport systems that mediate electrogenic cation uniport in both directions across the plasma membrane in vitro. The substrate concentration and membrane potentials between −50 and −100 mV provide the driving force for cation translocation from the blood to epithelial cells.
The third member of the OCT family, OCT3/SLC22A3, was identified as an extraneuronal monoamine transporter. A study using OCT3 knock-out mice (11) indicated that OCT3 plays an important role in the heart rather than the kidney. Homozygous mutant mice were viable and fertile, with no obvious physiological defects. However, OCT3(−/−) mice showed impaired uptake activity into the heart as measured by the accumulation of intravenously administered tritiated 1-methyl-4-phenylpyridinium ([3H]MPP+). While a 72% reduction in MPP+ levels was measured in hearts of OCT3(−/−) mice, no significant differences were found in any other organs or in plasma when comparing wild-type and mutant mice. The importance of OCT3 in the renal secretion of cationic drugs is unknown.
Organic cation and carnitine tranporters (OCTN1 and OCTN2) were identified as novel OCTs (SLC22A4 and SLC22A5) (12,13) in 1997 and 1998. These transporters mediate the trafficking of l-carnitine, TEA, and acetyl-l-carnitine in a Na+-dependent manner. In addition, it was reported that ergothioneine is transported by OCTN1 (14). Ergothioneine is associated with chronic inflammatory conditions, neurodegeneration, and cardiovascular diseases. However, the roles of OCTN1 in the kidney are not clear. Although OCTN2 is ubiquitously expressed, it is highly expressed specifically in the liver and kidney. The OCTN2 protein localizes at the brush-border membrane of cortical proximal tubular epithelia. It is possible that OCTN2 plays a role in organic cation transport at the apical membrane.
Urakami et al. (15) identified differences in the affinity of OCT1 and OCT2 for various substrates. The substrate specificity of these proteins was well summarized in a review by Fujita et al. in 2006 (16). Urakami et al. (15) reported that the inhibitory patterns of n-tetraalkylammonium compounds were closely related with the length of their alkyl side chain. In general, an increase in the length of alkyl side chains is associated with an increase in hydrophobicity. Recently, substrate features were investigated by a quantitative structure-activity relationship (QSAR) analysis (17). Computationally derived QSAR models established human OCT2 selectivity. A 2D-QSAR emphasized the importance of hydrophobicity, structural bulk, and molecular flexibility as important determinants in the binding of substrates to OCT2. A 3D-QSAR displayed better predictive power and served to emphasize the fact that molecular size and shape play a significant role in defining the interaction of substrates with OCT2. In contrast, hydrophobicity was not a major factor in determining the affinity of OCT for guanidine compounds (18). It was determined that the specificity of OCT1 and OCT2 is not simply determined by guanidino groups. Among guanidine compounds, aminoguanidine was identified as a new superior substrate for OCT2. Recently, Kido et al. (19) screened a prescription drug library against OCT2. Of the 910 compounds screened, 244 inhibited OCT2. Computational analyses revealed key properties of inhibitors versus noninhibitors, including differences in overall molecular charge. Based on the classification of OCT2 inhibitory ligands by Kido et al., Harper and Wright (20) determined the kinetic basis of inhibition by these ligands using CHO cells expressing human OCT2. In their report, TEA, diphenidol, and phenyltoloxamine were mixed-type inhibitors, and adrenosterone and carvediol displayed noncompetitive inhibitory profiles. Harper and Wright underscored the caution required for the development of predictive models of ligand interactions with multidrug transporters.
Recently, Koepsell (21) summarized the substrate recognition and translocation by OCTs. A detailed characterization of OCT1 and OCT2, in combination with homology modeling based on lactose permease (LacY) structures and extensive mutagenesis, provided the basic understanding of how these transporters work; furthermore, it was determined that several amino acids are important for substrate recognition and translocation. During substrate translocation, OCT1 and OCT2 may perform a series of conformational changes involving an outward-facing conformation, an occluded state, and an inward-facing conformation. However, Koepsell states that detailed crystal structures in different conformations with bound ligands should be elucidated in future investigations. In addition, ligand binding and ligand-induced allosteric effects should be characterized by multiple experimental methods.
One important substrate of OCT2 is creatinine (22). To estimate renal function, the glomerular filtration rate (GFR) is determined through the analysis of serum concentration of creatinine and creatine clearance. However, creatinine clearance usually exceeds the GFR because of the tubular secretion of creatinine. In addition, overestimates of the GFR based on creatinine clearance have been made in patients with renal diseases, especially those with glomerular disorders. Urakami et al. (22) found that only OCT2 mediated creatinine transport out of the several organic ion transporters assessed, such as OCT1, organic anion transporter (OAT)1, and OAT3. OCT2 regulates creatinine uptake at the basolateral membranes of renal proximal tubules. Indeed, the fluoroquinolone antibacterial agent DX-619 inhibits OCT2-mediated transport of creatinine (23). Human studies with healthy volunteers also suggested that DX-619 increased the serum creatinine concentration by inhibiting excretory tubular transporters (24). In addition, DX-619 was able to inhibit OCT2, MATE1, and MATE2-K at its therapeutic dose, leading to a significant inhibition of the tubular secretion of creatinine and an elevation of serum creatinine levels (25).
It has been determined that OCTs are regulated by various factors in the kidney, such as PKA, the Ca2+/calmodulin pathway and steroid hormones. This was well summarized in the review by Koepsell et al. (26). In addition, the expression levels of OCTs are changed in multiple pathological conditions, including animal models of partial nephrectomy, streptozotocin-induced diabetes, and hyperuricemia.
MULTIDRUG AND TOXIN EXTRUSION (MATE and SLC47)
Identification of MATE Transporters
The transporter protein involved in the excretion step at tubular epithelia was identified as MATE1, a mammalian ortholog of the MATE family conferring multidrug resistance in bacteria (27). The MATE transporters are primarily expressed in the kidney and liver, and they are localized at the apical membranes of the renal tubules and bile canaliculi. MATE1 mediates the H+-coupled electroneutral exchange of TEA and MPP, which are typical substrates for renal and hepatic H+/organic cation exchangers. Thus, MATE1 appears to be a polyspecific antiporter that directly transports organic cations into the urine and bile. Originally, human MATE2 was also identified as an ortholog of the MATE family, and the mRNA of MATE2 was detected predominantly in the kidney; however, the transport activity of MATE2 was not examined. Subsequent to the identification of MATE1 and MATE2, another H+/organic cation antiporter, human kidney-specific MATE 2 (MATE2-K), was isolated from the human kidney as an active splice variant of human MATE2 (28). MATE2-K mRNA was found to be expressed predominantly in the kidney. In contrast, no signal for MATE2 mRNA was detected in the human kidney. MATE2-K plays a role in the H+ gradient-dependent antiport of TEA, and it is the second member of the H+/organic cation antiporters responsible for the tubular secretion of cationic drugs across the brush-border membranes. The regulation of MATEs were summarized in the review of Yonezawa and Inui (29). Sp1 and AP-1 may regulate the transcription of MATE1 mRNA by interacting with the promoter region of the MATE1 gene.
Mouse Mate2 was identified by Otsuka et al. (27), and its deduced amino acid sequence was 38.1% identical to its human counterpart. In addition, the tissue distribution of MATE2 mRNA was found to be significantly different between the human and the mouse. Hiasa et al. (30) reported that rodent MATE2 was a third member of the SLC47 family. Mouse MATE2 mRNA was detected specifically in the testes. Therefore, the function of this MATE in the kidney has not been determined. In contrast to rodent MATE2, rabbit MATE2-K has a 74% sequence identity its human counterpart (31). Thus, care is needed when discussing the nomenclature and classification of MATE2.
Substrate Specificity of MATE1 and MATE2-K
Tanihara et al. (32) examined the substrate specificity of MATE1 and MATE2-K to find functional differences between these two transporters. Using a combined in vitro and computational approach, Astorga et al. (33) found that MATE1 and MATE2-K had markedly overlapping selectivity for a broad range of cationic compounds. They identified several physicochemical parameters that influence ligand-binding to MATE1, such as the presence and location of multiple hydrophobic moieties, hydrogen donors, and ionizable features.
Although MATE1 and MATE2-K have similar substrate specificities, the zwitterions cephalexin and cephradine were revealed to be specific substrates of MATE1, but not MATE2-K. In 1985, Inui et al. (34) indicated that cephalexin should share a common carrier transport system with organic cations in renal brush-border membranes through use of membrane vesicles. Watanabe et al. (35) carried out experiments on the transport of cephalexin by MATE1 and pharmacokinetic analyses of cephalexin in MATE1(−/−) mice. Cephalexin was transported by human MATE1 with a bell-shaped pH profile. After intravenous administration of cephalexin, the urinary excretion of the compound was significantly reduced, and the renal concentration was markedly increased in MATE1(−/−) mice, compared with MATE1(+/+) mice. The renal clearance of cephalexin in MATE1(−/−) mice was approximately 60% of that in Mate1(+/+) mice and seemed to be similar to the clearance of creatinine. Overall, it was demonstrated that MATE1 is responsible for the renal tubular secretion of the zwitterionic substrate cephalexin in vivo. At the basolateral membrane, the OAT3 plays an important role in the metabolism of cephalosporin antibiotics (36). For the renal secretion of some zwitterionic drugs, MATEs may cooperate with the basolateral OAT family.
THE CLINICAL IMPORTANCE OF OCT2, MATE1, AND MATE2-K IN THE KIDNEY
Transcellular Transport of Cationic Drugs by OCTs and MATEs
The substrate specificity, membrane localization and driving forces suggest that OCT2 and MATE1 or MATE2-K mediate the tubular secretion of cationic drugs from the blood to urine. Sato et al. (37) established double-transfected Madin–Darby canine kidney cells as an in vitro model that mimics the vectorial transport of cationic drugs across human epithelial cells. Indeed, TEA was transported unidirectionally from the basolateral to apical side of the membrane in these double transfectants. An important observation by Sato et al. (37) was the clear directional transport of procainamide and quinidine in double transfectants. In humans, procainamide and quinidine are actively secreted via the renal tubules into the urine. However, it was difficult to detect the uptake of procainamide and quinidine by OCT- or MATE-expressing HEK293 cells because these compounds are lipophilic cations. The double-transfected MDCK-hOCT1/hMATE1 and MDCK-hOCT2/hMATE1 cells clearly solved the technical limitations of previous uptake experiments, and they would be useful in vitro tools to examine the renal tubular secretion of cationic drugs in humans.
OCTs and MATEs are the sites for drug-drug interactions between cationic drugs (Table II). Therefore, Tsuda et al. (38) assessed the drug interactions between cimetidine and metformin through use of double-transfected Madin–Darby canine kidney cells stably expressing both OCT2 and MATE1 as an in vitro model of proximal tubular epithelial cells. The results of their investigation suggested that apical MATE1 is involved in drug interactions between cimetidine and cationic compounds in proximal tubular epithelial cells. Similar results were obtained by Kusuhara et al. (39) and Ito et al. (40). Kusuhara et al. (39) found that the renal clearance of metformin was significantly decreased by pyrimethamine at microdoses and therapeutic doses. They considered the drug–drug interaction to be attributable to the inhibition of MATE proteins by pyrimethamine. Recently, Ito et al. (40) concluded that competitive inhibition of OCT2 is unlikely to underlie the drug-drug interaction caused by cimetidine in the renal elimination of cationic drugs and that competitive inhibition of MATEs by cimetidine is likely to be important in vivo at clinical doses.
Table II.
Comparison of Affinity for Human MATE1, MATE2-K, and OCT2 of Organic Cations and Various Drugs
| Compound | Affinity (Ki) for transporters (μM) | Plasma concentration (μM) | ||
|---|---|---|---|---|
| MATE1 | MATE2-K | OCT2 | ||
| Organic cation | ||||
| TEA | 380 (km)a | 760 (km)a | 48–270 | |
| MPP | 100 (km)a | 110 (km)a | 19–78 | |
| Guanidine | 2,100 | 4,200 | 3,030 | |
| Antidiabetic drug | ||||
| Metformin | 667 | 6,516 | 339–1,700 | 4.1 |
| Phenformin | 16 (IC50)b | 73 (IC50)b | 65 | Not used |
| Antiarrhythmic drug | ||||
| Disopyamide | 84 | 292 | 64 | >4.4 |
| Procainamide | 217 | 178 | 50–58 | 11–52 |
| Quinidine | 29 (IC50)b | 23 (IC50)b | 19 | 6–18 |
| Verapamil | 27.5 | 32.1 | 206 | 0.6 |
| Histamin H1 blocker | ||||
| Chlorpheniramine | 87.6 | 191.2 | 26 | 0.26 |
| Diphenhydramine | 87 | 266.5 | 32 | 0.9 |
| Histamin H2 blocker | ||||
| Cimetidine | 1.1 | 7.3 | 8.6–73 | 2.0–3.6 |
| Famotidine | 0.6 | 9.7 | 111–204 | 0.04 |
| Ranitidine | 25 | 25 | 40–265 | 0.28 |
| Antiparkinsonian drug | ||||
| Amantadine | 112 | 1,167 | 20–28 | 1.6 |
| Pramipexole | 141 | 24 | 17 | 0.001 |
| Antimalarial drug | ||||
| Quinine | 5 | 42 | 23–34 | 34 |
| Anticancer drug | ||||
| Cisplatin | 3,500 | >5,000 | 6,000 | 11 |
| Oxaliplatin | 4,000 | 1,000 | >5,000 | 0.83 |
Table II was modified from Tsuda et al. (38). The affinities (K m, K i, or IC50) for OCT2 were obtained from four different references (15,18,26,53). The affinities for MATE1 and MATE2-K (K m, K i, or IC50) were obtained from three references (32,33,38). The plasma concentrations were obtained from Thummel et al. (54)
a K m values were obtained from Tanihara et al. (32)
bIC50 values from Astorga et al. (33). Other K i values for MATE1 and MATE2-K were obtained from Tsuda et al. (38)
Pharmacokinetics and Pharmacodynamics of Metformin
Metformin is widely used in the treatment of type II diabetes mellitus. It is almost entirely excreted into the urine in an unmodified form. Lactic acidosis is a fatal adverse effect of metformin and can occur in patients without any risk factors. Metformin is a substrate for OCT2, MATE1, and MATE2-K (29,41). Choi et al. (42) characterized genetic variants of MATE2-K, determined their association with the response to metformin, and elucidated their impact. Four nonsynonymous variants and four variants in the MATE2-K basal promoter region were identified from ethnically diverse populations. Two nonsynonymous variants, including c.485C>T (Pro162Leu) and c.1177G>A (Gly393Arg), were shown to be associated with significantly lower metformin uptake and a reduction in protein levels when the variants were expressed in HEK293 cells. MATE2-K basal promoter haplotypes containing the most common variant, g.-130G>A (>26% allele frequency), were associated with a significant increase in promoter activity levels and reduced binding to the transcriptional repressor myeloid zinc finger 1. Patients with diabetes who were homozygous for g.-130A had a significantly poorer response to metformin treatment than carriers of the reference allele, g.-130G, when assessed for the relative change in glycated hemoglobin (HbA1c). Choi et al. (42) suggested that MATE2-K plays a role in the antidiabetic response to metformin and that the next challenge in pharmacogenomic research is to improve the outcome for patients through this pathway.
To clarify the pharmacokinetic role of MATE1 in vivo, Tsuda et al. (43) carried out the targeted disruption of the murine MATE1 gene. After a single intravenous administration of metformin, the area under the blood concentration-time curve of metformin in MATE1(−/−) mice showed a 2-fold increase. The urinary excretion of metformin after intravenous administration was significantly decreased in MATE1(−/−) mice, compared with MATE1(+/+) mice. The renal secretory clearance of metformin in MATE1(−/−) mice was approximately 14% of that in MATE1(+/+) mice. The report of Tsuda et al. (43) was the first to demonstrate an essential role for MATE1 in the systemic clearance of metformin. Recently, Toyama et al. (44) reported that significantly higher blood lactate levels and lower pH and HCO3− levels were observed in MATE1(−/−) mice 7 days after metformin administration in the drinking water. Using knockout mice, Toyama et al. (44,45) also reported that dysfunctional MATE1 caused a remarkable elevation in the concentration of metformin in the liver and led to lactic acidosis, suggesting that homozygous MATE1 variants, but not heterozygous variants, are risk factors for metformin-induced lactic acidosis (Fig. 2).
Fig. 2.
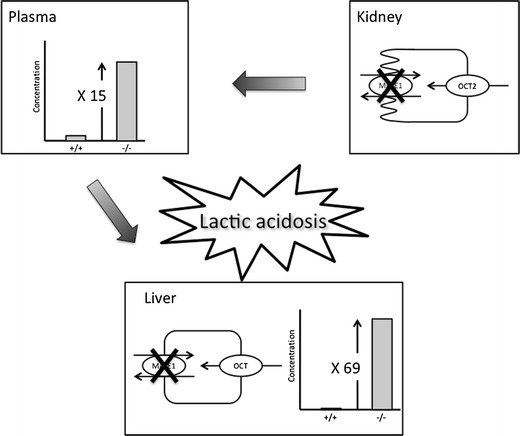
The influence of dysfunctional MATE1 on pharmacokinetics and toxicodynamics of metformin. Plasma concentrations of metformin are increased in Slc47a1 knockout mice, compared with their wild-type counterparts. This increased concentration is due to the loss of urinary and biliary efflux of metformin. Therefore, impaired functioning of MATEs synergistically increases metformin accumulation in the liver and blood lactate levels resulting in the development of lactic acidosis
Tzvetkov et al. (46) examined the effects of genetic polymorphisms in OCT1, OCT2, OCT3, OCTN1, and MATE1 on the pharmacokinetics of metformin in healthy male Caucasians. Low-activity genotypes of OCT1 were related to an increase in the renal clearance of metformin. The result was similar to the aforementioned OCT(−/−) mice. It is possible that dysfunction of OCT1 indirectly affects the renal clearance of organic cations in humans.
Nephrotoxicity of Cisplatin
Members of the SLC22 organic ion transporter family mediate the renal excretion of both endogenous and exogenous substances. Thus, the functional and molecular variations of renal SLC22 transporters under acute kidney injury have an impact on the systemic clearance of substrate drugs and can result in altered pharmacokinetics or unexpected adverse events caused by the accumulation of drugs (47). On the other hand, renal drug transporters are involved in drug-induced nephrotoxicity. In 2011, Yonezawa and Inui (48) summarized the roles of OCT and MATE in the nephrotoxicity caused by platinum agents. Briefly, it was reported that OCTs and MATEs play a role in the pharmacokinetics of platinum agents. Interestingly, only cisplatin induces nephrotoxicity, and the toxicity is kidney specific. Kidney-specific OCT2 mediates the transport of cisplatin and is the determinant for cisplatin-induced nephrotoxicity. In addition, cisplatin and oxaliplatin are substrates for these transporters, but carboplatin and nedaplatin are not. Substrate specificity could regulate the nephrotoxic features of platinum agents (Fig. 3). Nakamura et al. (49) examined the role of MATE1 in the nephrotoxicity of cisplatin in vivo and in vitro. When cisplatin was administered intraperitoneally, the lifespan was significantly shorter in MATE1(−/−) mice than MATE1(+/+) mice. Three days after the administration of cisplatin, plasma creatinine, and blood urea nitrogen (BUN) levels were increased and creatinine clearance was decreased in both MATE1(+/+) and MATE1(−/−) mice, compared with vehicle-treated controls. Moreover, a significant rise in creatinine and BUN levels was observed in cisplatin-treated MATE1(−/−) mice, compared with MATE1(+/+) mice. A pharmacokinetic analysis revealed the plasma concentration and renal accumulation of cisplatin to be higher in MATE1(−/−) mice than MATE1(+/+) mice. Furthermore, the combination of a selective MATE inhibitor, pyrimethamine, with cisplatin also elevated creatinine and BUN levels, compared with cisplatin alone. In vitro, mouse MATE1 transported cisplatin as well as OCT1 and OCT2. In conclusion, MATE1 mediates the efflux of cisplatin and is involved in cisplatin-induced nephrotoxicity.
Fig. 3.
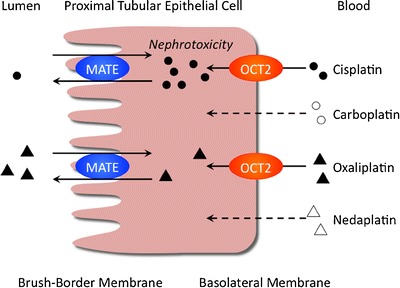
Renal handling of cisplatin, carboplatin, oxaliplatin, and nedaplatin by OCT2 and MATE. Cisplatin is transported by OCT2 and, to a much lesser extent, by MATE. Cisplatin accumulates at higher levels than other platinum agents in the kidney, which causes nephrotoxicity. Carboplatin and nedaplatin are not transported by OCT or MATE. Oxaliplatin is a substrate for OCT2 and MATE but does not induce nephrotoxicity
Susceptibility to cisplatin (CDDP) nephrotoxicity is known to vary between individuals, but the basis of this variation has not been fully elucidated. Recently, Iwata et al. (50) reported that the 808G>T SNP in OCT2 ameliorated CDDP-induced nephrotoxicity without altering the disposition of CDDP; whereas, the rs2289669 G>A SNP in MATE1 had no effect on CDDP toxicity. A total of 27% of patients with 808GG experienced over grade 2 Scr elevation, but patients with 808GT did not show any apparent nephrotoxicity. Song et al. (51) reported that the c.808G>T variant caused a significant decrease in transport activity without a change in protein expression. These results were consistent with the conclusions of Yonezawa et al. (48). It is speculated that the inter-individual variation in cisplatin-induced kidney injury is affected by variations in OCT2 activity.
CONCLUSIONS AND PERSPECTIVES
The kidney plays an important role in drug excretion. Various transporters at the brush-border and basolateral membranes mediate vectorial transport into the urine. OCTs and MATEs are responsible for transporting cationic drugs at the basolateral membrane and brush-border membrane, respectively (Table II). The variation in transporter functions may affect the pharmacokinetics and pharmacodynamics of cationic drugs, such as metformin and cisplatin. The serious adverse effects of metformin and cisplatin were partially but significantly affected by the transporter function.
As OCTs and MATEs are important sites for drug-drug interactions, strategic inhibition of these transporters could help to prevent the adverse effects of drugs. For example, Tanihara et al. (52) reported that the renal accumulation and subsequent nephrotoxicity of cisplatin were significantly decreased by the oral administration of imatinib. The concomitant administration of imatinib clearly protected against severe renal impairment induced by cisplatin as determined by histological examination. Therefore, the administration of imatinib with cisplatin prevents nephrotoxicity by inhibiting the OCT2-mediated renal accumulation of cisplatin in rats. Clinical application of this compound to block drug–drug interactions should improve the effectiveness and safety of cisplatin. In addition, inter-individual variation in the pharmacokinetics of cationic drugs may be caused by changes in transporter function. Using animal models and other experimental methods, changes in OCTs or MATEs are currently being investigated by several groups. In future studies, it is expected that renal drug excretion will be estimated based on transporter information.
ACKNOWLEDGMENTS
This work was supported in part by a grant-in-aid for Scientific Research (KAKENHI) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Conflict of Interest
The authors declare no conflict of interests.
REFERENCES
- 1.Inui K, Okuda M. Cellular and molecular mechanisms of renal tubular secretion of organic anions and cations. Clin Exp Nephrol. 1998;2:100–108. doi: 10.1007/BF02479930. [DOI] [Google Scholar]
- 2.Burckhardt G, Wolff NA. Structure of renal organic anion and cation transporters. Am J Physiol Renal Physiol. 2000;278:F853–F866. doi: 10.1152/ajprenal.2000.278.6.F853. [DOI] [PubMed] [Google Scholar]
- 3.Inui K, Masuda S, Saito H. Cellular and molecular aspects of drug transport in the kidney. Kidney Int. 2000;58:944–958. doi: 10.1046/j.1523-1755.2000.00251.x. [DOI] [PubMed] [Google Scholar]
- 4.Grundemann D, Gorboulev V, Gambaryan S, Veyhl M, Koepsell H. Drug excretion mediated by a new prototype of polyspecific transporter. Nature. 1994;372:549–552. doi: 10.1038/372549a0. [DOI] [PubMed] [Google Scholar]
- 5.Jonker JW, Wagenaar E, Mol CA, Buitelaar M, Koepsell H, Smit JW, et al. Reduced hepatic uptake and intestinal excretion of organic cations in mice with a targeted disruption of the organic cation transporter 1 (OCT1 [Slc22a1]) gene. Mol Cell Biol. 2001;21:5471–5477. doi: 10.1128/MCB.21.16.5471-5477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Motohashi H, Sakurai Y, Saito H, Masuda S, Urakami Y, Goto M, et al. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J Am Soc Nephrol. 2002;13:866–874. doi: 10.1681/ASN.V134866. [DOI] [PubMed] [Google Scholar]
- 7.Okuda M, Saito H, Urakami Y, Takano M, Inui K. cDNA cloning and functional expression of a novel rat kidney organic cation transporter, OCT2. Biochem Biophys Res Commun. 1996;224:500–507. doi: 10.1006/bbrc.1996.1056. [DOI] [PubMed] [Google Scholar]
- 8.Gorboulev V, Ulzheimer JC, Akhoundova A, Ulzheimer-Teuber I, Karbach U, Quester S, et al. Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol. 1997;16:871–881. doi: 10.1089/dna.1997.16.871. [DOI] [PubMed] [Google Scholar]
- 9.Jonker JW, Wagenaar E, Van Eijl S, Schinkel AH. Deficiency in the organic cation transporters 1 and 2 (OCT1/OCT2 [Slc22a1/Slc22a2]) in mice abolishes renal secretion of organic cations. Mol Cell Biol. 2003;23:7902–7908. doi: 10.1128/MCB.23.21.7902-7908.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugawara-Yokoo M, Urakami Y, Koyama H, Fujikura K, Masuda S, Saito H, et al. Differential localization of organic cation transporters rOCT1 and rOCT2 in the basolateral membrane of rat kidney proximal tubules. Histochem Cell Biol. 2000;114:175–180. doi: 10.1007/s004180000186. [DOI] [PubMed] [Google Scholar]
- 11.Zwart R, Verhaagh S, Buitelaar M, Popp-Snijders C, Barlow DP. Impaired activity of the extraneuronal monoamine transporter system known as uptake-2 in Orct3/Slc22a3-deficient mice. Mol Cell Biol. 2001;21:4188–4196. doi: 10.1128/MCB.21.13.4188-4196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamai I, Yabuuchi H, Nezu J, Sai Y, Oku A, Shimane M, et al. Cloning and characterization of a novel human pH-dependent organic cation transporter, OCTN1. FEBS Lett. 1997;419:107–111. doi: 10.1016/S0014-5793(97)01441-5. [DOI] [PubMed] [Google Scholar]
- 13.Wu X, Prasad PD, Leibach FH, Ganapathy V. cDNA sequence, transport function, and genomic organization of human OCTN2, a new member of the organic cation transporter family. Biochem Biophys Res Commun. 1998;246:589–595. doi: 10.1006/bbrc.1998.8669. [DOI] [PubMed] [Google Scholar]
- 14.Grundemann D. The ergothioneine transporter controls and indicates ergothioneine activity—a review. Prev Med. 2012;54(Suppl):S71–S74. doi: 10.1016/j.ypmed.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 15.Urakami Y, Okuda M, Masuda S, Akazawa M, Saito H, Inui K. Distinct characteristics of organic cation transporters, OCT1 and OCT2, in the basolateral membrane of renal tubules. Pharm Res. 2001;18:1528–1534. doi: 10.1023/A:1013070128668. [DOI] [PubMed] [Google Scholar]
- 16.Fujita T, Urban TJ, Leabman MK, Fujita K, Giacomini KM. Transport of drugs in the kidney by the human organic cation transporter, OCT2 and its genetic variants. J Pharm Sci. 2006;95:25–36. doi: 10.1002/jps.20536. [DOI] [PubMed] [Google Scholar]
- 17.Suhre WM, Ekins S, Chang C, Swaan PW, Wright SH. Molecular determinants of substrate/inhibitor binding to the human and rabbit renal organic cation transporters hOCT2 and rbOCT2. Mol Pharmacol. 2005;67:1067–1077. doi: 10.1124/mol.104.004713. [DOI] [PubMed] [Google Scholar]
- 18.Kimura N, Masuda S, Katsura T, Inui K. Transport of guanidine compounds by human organic cation transporters, hOCT1 and hOCT2. Biochem Pharmacol. 2009;77:1429–1436. doi: 10.1016/j.bcp.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 19.Kido Y, Matsson P, Giacomini KM. Profiling of a prescription drug library for potential renal drug-drug interactions mediated by the organic cation transporter 2. J Med Chem. 2011;54:4548–4558. doi: 10.1021/jm2001629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harper JN, Wright SH. Multiple mechanisms of ligand interaction with the human organic cation transporter, OCT2. Am J Physiol Renal Physiol. 2013;304:F56–F67. doi: 10.1152/ajprenal.00486.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koepsell H. Substrate recognition and translocation by polyspecific organic cation transporters. Biol Chem. 2011;392:95–101. doi: 10.1515/bc.2011.009. [DOI] [PubMed] [Google Scholar]
- 22.Urakami Y, Kimura N, Okuda M, Inui K. Creatinine transport by basolateral organic cation transporter hOCT2 in the human kidney. Pharm Res. 2004;21:976–981. doi: 10.1023/B:PHAM.0000029286.45788.ad. [DOI] [PubMed] [Google Scholar]
- 23.Okuda M, Kimura N, Inui K. Interactions of fluoroquinolone antibacterials, DX-619 and levofloxacin, with creatinine transport by renal organic cation transporter hOCT2. Drug Metab Pharmacokinet. 2006;21:432–436. doi: 10.2133/dmpk.21.432. [DOI] [PubMed] [Google Scholar]
- 24.Sarapa N, Wickremasingha P, Ge N, Weitzman R, Fuellhart M, Yen C, et al. Lack of effect of DX-619, a novel des-fluoro(6)-quinolone, on glomerular filtration rate measured by serum clearance of cold iohexol. Antimicrob Agents Chemother. 2007;51:1912–1917. doi: 10.1128/AAC.01223-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imamura Y, Murayama N, Okudaira N, Kurihara A, Okazaki O, Izumi T, et al. Prediction of fluoroquinolone-induced elevation in serum creatinine levels: a case of drug-endogenous substance interaction involving the inhibition of renal secretion. Clin Pharmacol Ther. 2011;89:81–88. doi: 10.1038/clpt.2010.232. [DOI] [PubMed] [Google Scholar]
- 26.Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res. 2007;24:1227–1251. doi: 10.1007/s11095-007-9254-z. [DOI] [PubMed] [Google Scholar]
- 27.Otsuka M, Matsumoto T, Morimoto R, Arioka S, Omote H, Moriyama Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci U S A. 2005;102:17923–17928. doi: 10.1073/pnas.0506483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masuda S, Terada T, Yonezawa A, Tanihara Y, Kishimoto K, Katsura T, et al. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J Am Soc Nephrol. 2006;17:2127–2135. doi: 10.1681/ASN.2006030205. [DOI] [PubMed] [Google Scholar]
- 29.Yonezawa A, Inui K. Importance of the multidrug and toxin extrusion MATE/SLC47A family to pharmacokinetics, pharmacodynamics/toxicodynamics and pharmacogenomics. Br J Pharmacol. 2011;164:1817–1825. doi: 10.1111/j.1476-5381.2011.01394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hiasa M, Matsumoto T, Komatsu T, Omote H, Moriyama Y. Functional characterization of testis-specific rodent multidrug and toxic compound extrusion 2, a class III MATE-type polyspecific H+/organic cation exporter. Am J Physiol Cell Physiol. 2007;293:C1437–C1444. doi: 10.1152/ajpcell.00280.2007. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Cherrington NJ, Wright SH. Molecular identification and functional characterization of rabbit MATE1 and MATE2-K. Am J Physiol Renal Physiol. 2007;293:F360–F370. doi: 10.1152/ajprenal.00102.2007. [DOI] [PubMed] [Google Scholar]
- 32.Tanihara Y, Masuda S, Sato T, Katsura T, Ogawa O, Inui K. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H+-organic cation antiporters. Biochem Pharmacol. 2007;74:359–371. doi: 10.1016/j.bcp.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 33.Astorga B, Ekins S, Morales M, Wright SH. Molecular determinants of ligand selectivity for the human multidrug and toxin extruder proteins MATE1 and MATE2-K. J Pharmacol Exp Ther. 2012;341:743–755. doi: 10.1124/jpet.112.191577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inui K, Takano M, Okano T, Hori R. H+ gradient-dependent transport of aminocephalosporins in rat renal brush border membrane vesicles: role of H+/organic cation antiport system. J Pharmacol Exp Ther. 1985;233:181–185. [PubMed] [Google Scholar]
- 35.Watanabe S, Tsuda M, Terada T, Katsura T, Inui K. Reduced renal clearance of a zwitterionic substrate cephalexin in MATE1-deficient mice. J Pharmacol Exp Ther. 2010;334:651–656. doi: 10.1124/jpet.110.169433. [DOI] [PubMed] [Google Scholar]
- 36.Ueo H, Motohashi H, Katsura T, Inui K. Human organic anion transporter hOAT3 is a potent transporter of cephalosporin antibiotics, in comparison with hOAT1. Biochem Pharmacol. 2005;70:1104–1113. doi: 10.1016/j.bcp.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 37.Sato T, Masuda S, Yonezawa A, Tanihara Y, Katsura T, Inui K. Transcellular transport of organic cations in double-transfected MDCK cells expressing human organic cation transporters hOCT1/hMATE1 and hOCT2/hMATE1. Biochem Pharmacol. 2008;76:894–903. doi: 10.1016/j.bcp.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 38.Tsuda M, Terada T, Ueba M, Sato T, Masuda S, Katsura T, et al. Involvement of human multidrug and toxin extrusion 1 in the drug interaction between cimetidine and metformin in renal epithelial cells. J Pharmacol Exp Ther. 2009;329:185–191. doi: 10.1124/jpet.108.147918. [DOI] [PubMed] [Google Scholar]
- 39.Kusuhara H, Ito S, Kumagai Y, Jiang M, Shiroshita T, Moriyama Y, et al. Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin Pharmacol Ther. 2011;89:837–844. doi: 10.1038/clpt.2011.36. [DOI] [PubMed] [Google Scholar]
- 40.Ito S, Kusuhara H, Yokochi M, Toyoshima J, Inoue K, Yuasa H, et al. Competitive inhibition of the luminal efflux by multidrug and toxin extrusions, but not basolateral uptake by organic cation transporter 2, is the likely mechanism underlying the pharmacokinetic drug–drug interactions caused by cimetidine in the kidney. J Pharmacol Exp Ther. 2012;340:393–403. doi: 10.1124/jpet.111.184986. [DOI] [PubMed] [Google Scholar]
- 41.Kimura N, Masuda S, Tanihara Y, Ueo H, Okuda M, Katsura T, et al. Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab Pharmacokinet. 2005;20:379–386. doi: 10.2133/dmpk.20.379. [DOI] [PubMed] [Google Scholar]
- 42.Choi JH, Yee SW, Ramirez AH, Morrissey KM, Jang GH, Joski PJ, et al. A common 5′-UTR variant in MATE2-K is associated with poor response to metformin. Clin Pharmacol Ther. 2011;90:674–684. doi: 10.1038/clpt.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsuda M, Terada T, Mizuno T, Katsura T, Shimakura J, Inui K. Targeted disruption of the multidrug and toxin extrusion 1 (MATE1) gene in mice reduces renal secretion of metformin. Mol Pharmacol. 2009;75:1280–1286. doi: 10.1124/mol.109.056242. [DOI] [PubMed] [Google Scholar]
- 44.Toyama K, Yonezawa A, Masuda S, Osawa R, Hosokawa M, Fujimoto S, et al. Loss of multidrug and toxin extrusion 1 (MATE1) is associated with metformin-induced lactic acidosis. Br J Pharmacol. 2012;166:1183–1191. doi: 10.1111/j.1476-5381.2012.01853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toyama K, Yonezawa A, Tsuda M, Masuda S, Yano I, Terada T, et al. Heterozygous variants of multidrug and toxin extrusions (MATE1 and MATE2-K) have little influence on the disposition of metformin in diabetic patients. Pharmacogenet Genomics. 2010;20:135–138. doi: 10.1097/FPC.0b013e328335639f. [DOI] [PubMed] [Google Scholar]
- 46.Tzvetkov MV, Vormfelde SV, Balen D, Meineke I, Schmidt T, Sehrt D, et al. The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin Pharmacol Ther. 2009;86:299–306. doi: 10.1038/clpt.2009.92. [DOI] [PubMed] [Google Scholar]
- 47.Saito H. Pathophysiological regulation of renal SLC22A organic ion transporters in acute kidney injury: pharmacological and toxicological implications. Pharmacol Ther. 2010;125:79–91. doi: 10.1016/j.pharmthera.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 48.Yonezawa A, Inui K. Organic cation transporter OCT/SLC22A and H+/organic cation antiporter MATE/SLC47A are key molecules for nephrotoxicity of platinum agents. Biochem Pharmacol. 2011;81:563–568. doi: 10.1016/j.bcp.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 49.Nakamura T, Yonezawa A, Hashimoto S, Katsura T, Inui K. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochem Pharmacol. 2010;80:1762–1767. doi: 10.1016/j.bcp.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 50.Iwata K, Aizawa K, Kamitsu S, Jingami S, Fukunaga E, Yoshida M, et al. Effects of genetic variants in SLC22A2 organic cation transporter 2 and SLC47A1 multidrug and toxin extrusion 1 transporter on cisplatin-induced adverse events. Clin Exp Nephrol. 2012;16:843–851. doi: 10.1007/s10157-012-0638-y. [DOI] [PubMed] [Google Scholar]
- 51.Song IS, Shin HJ, Shim EJ, Jung IS, Kim WY, Shon JH, et al. Genetic variants of the organic cation transporter 2 influence the disposition of metformin. Clin Pharmacol Ther. 2008;84:559–562. doi: 10.1038/clpt.2008.61. [DOI] [PubMed] [Google Scholar]
- 52.Tanihara Y, Masuda S, Katsura T, Inui K. Protective effect of concomitant administration of imatinib on cisplatin-induced nephrotoxicity focusing on renal organic cation transporter OCT2. Biochem Pharmacol. 2009;78:1263–1271. doi: 10.1016/j.bcp.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 53.Ishiguro N, Saito A, Yokoyama K, Morikawa M, Igarashi T, Tamai I. Transport of the dopamine D2 agonist pramipexole by rat organic cation transporters OCT1 and OCT2 in kidney. Drug Metab Dispos. 2005;33:495–499. doi: 10.1124/dmd.104.002519. [DOI] [PubMed] [Google Scholar]
- 54.Thummel KE, Shen DD, Isoherranen N. Design and optimization of dosage regimens: pharmacokinetic data. In: Brunton LL, Chabner BA, Knollmann BC, editors. Goodman and Gilman’s the pharmacological basis of therapeutics. 12. New York: McGraw-Hill; 2011. pp. 1891–1990. [Google Scholar]