

Abstract
Target identification of the known bioactive compounds and novel synthetic analogs is a very important research field in medicinal chemistry, biochemistry, and pharmacology. It is also a challenging and costly step towards chemical biology and phenotypic screening. In silico identification of potential biological targets for chemical compounds offers an alternative avenue for the exploration of ligand–target interactions and biochemical mechanisms, as well as for investigation of drug repurposing. Computational target fishing mines biologically annotated chemical databases and then maps compound structures into chemogenomical space in order to predict the biological targets. We summarize the recent advances and applications in computational target fishing, such as chemical similarity searching, data mining/machine learning, panel docking, and the bioactivity spectral analysis for target identification. We then described in detail a new web-based target prediction tool, TargetHunter (http://www.cbligand.org/TargetHunter). This web portal implements a novel in silico target prediction algorithm, the Targets Associated with its MOst SImilar Counterparts, by exploring the largest chemogenomical databases, ChEMBL. Prediction accuracy reached 91.1% from the top 3 guesses on a subset of high-potency compounds from the ChEMBL database, which outperformed a published algorithm, multiple-category models. TargetHunter also features an embedded geography tool, BioassayGeoMap, developed to allow the user easily to search for potential collaborators that can experimentally validate the predicted biological target(s) or off target(s). TargetHunter therefore provides a promising alternative to bridge the knowledge gap between biology and chemistry, and significantly boost the productivity of chemogenomics researchers for in silico drug design and discovery.
Key words: ChEMBL, chemogenomics, machine learning, target identification, TargetHunter
INTRODUCTION
High-throughput screening (HTS) and high-content screening (HCS) are technologies, for rapid identification of potent compounds, producing an explosive amount of annotated biological data in the chemogenomics databases. For example, using “high-content screening” as a keyword, 239 bioassays were retrieved in PubChem and 5,900 bioassays were recorded with the keyword “HTS” on the date of our study (Sep 30, 2012), which contained 1,295 compounds with activity (IC50, etc.) ≤1 μM. Current methods of cellular screening and pharmacogenetic profiling can rapidly reveal phenotypic responses to chemicals, but cannot immediately pinpoint their molecular targets (1). Meanwhile, the methodology of modern combinatorial chemistry has generated thousands or even millions of organic compounds for medicinal chemistry research. PubChem has archived 35.6 million of unique chemicals. Among them, 25.3 million satisfy the rule of five (2), 1.85 million have been tested in at least one bioassay, and 0.8 million have been reported as active.
How to explore the therapeutic potential of this huge number of compounds is a major challenge. The bioactivity records from HTS/HCS together with decades of published, patented, and proprietary research on protein–ligand interactions contain a wealth of data on biological activities. By combining data-mining methods and diverse chemogenomics libraries, such as PubChem (3–5) and ChEMBL databases (6), it may be possible to map the known “chemistry space” onto the known “biological activity space” in the form of models that enable prediction of the targets, pathways, or therapeutic relevance (7). Target fishing (8), target identification, or ligand profiling is used to investigate and validate biological targets that interact with small organic molecules. The target can be a therapeutic protein or a cancer cell line. The small organic molecules can be newly or to-be-synthesized chemicals, or agents that are active in phenotypic screening (9–11) whose target or mechanism of action remains unknown. Target identification can also include a search for potential off-target effects of therapeutic compounds and can be useful for the repurposing of drugs (12).
Various methods for computational target prediction have been developed with the help of advances in molecular descriptors and data-mining algorithms. The technologies involved have been summarized in comprehensive reviews (8,13,14). Bender et al., the pioneer researchers on target prediction, stated that “current approaches to predicting targets of small molecules can be broadly grouped into four classes: chemical similarity searching, data mining/machine learning, panel docking, and the analysis of bioactivity spectra” (15). The basic principle, with the advantages and disadvantages of these methods, are listed below.
Chemical similarity as a criterion for in silico target identification is based on the well-established medicinal chemistry concept that structurally similar compounds have similar physicochemical properties and possibly similar biological profiles (16–22). With such methods, the small molecules usually are represented as chemical fingerprints, and the similarity between two molecules is measured by the Tanimoto similarity metric (23). Meanwhile, a pharmacophore-based similarity matrix, such as SHED-based approach (24), has been developed for target identification. In addition, statistical analysis has been added to traditional chemical similarity scores in order to assess the statistical significance of similarity. For example, fitting with extreme value distributions, the similarity ensemble approach models the possibility of the occurrence of higher scores when comparing two ligand sets. This method has been successfully used in drug repurpose (25) and side-effect prediction (26). If a target has few known bioactive ligands, the prediction for this target may be not feasible by similarity search.
Another method for in silico target identification is based on algorithms of data mining and machine learning. This method tries to generate a statistical model by mining or analyzing the properties of active compounds for a target. The derived model predicts the probability of a query compound being associated with this target. An example of this method was reported by Nidhi et al. (7). The authors used the World of Molecular Bioactivity dataset (27) to build multiple-category models (MCM) for predicting protein targets and therapeutic activities on MDL Drug Database Report (MDDR; Molecular Design Ltd., San Leandro, CA, USA) (28). This algorithm subsequently has been used for predicting adverse drug reactions and off-target effects (29). Such models can extract the important target-specific information. Because one target may have thousands of the structurally diverse ligands, one unique model may not recover all the features, and the prediction performance may not be satisfying. Extensive discussion and comparison studies will accompany the description of TargetHunter prediction.
Panel docking methods are also used for in silico target identification. A compound is docked to a wide panel of proteins to determine its potential partners. TarFisDock (30) is an online web service that implements panel docking for target prediction. In TarFisDock, a query compound is docked to 698 protein structures to explore the potential interactions with these candidate proteins. TarFisDock identified the protein target of two anti-Helicobacter pylori probes (31). Another example of panel docking is INVDOCK; the early work is conducted by Chen et al. (32). Recently, this method has been applied to the analysis of pharmacodynamics mechanism of Chinese medicinal plants for chronic kidney disease (33). Obviously, the application of such methods is limited by availability of high-quality protein structures and accuracy of docking programs, also by the necessity of high-computational power.
Methods based on bioactivity spectra are more complicated for in silico target identification. Bioactivity spectra are the response of a compound to a series of cell lines, DNA microarrays, or proteins. If two compounds target the same signaling pathway or protein, they generate similar bioactivity spectra. They induce similar patterns of either gene expression or phenotypic responses. The bioactivity spectra-based computational method predicts potential targets of a chemical by profiling similarity measurements and hierarchical clustering (34,35). For example, Cheng et al. have developed a target identification algorithm by combining the search for similarity bioactivity profile with mining public databases (36). These methods require many expensive and time consuming wet experiments to produce the bioactivity spectra.
While the foregoing algorithms or methods have advantages of mining the potential biological targets of active compounds, a public accessible, user friendly, and reliable tool for target identification is still not available to meet the demand of broad scientific research communities. Here, we introduce a new target identification tool, TargetHunter, as a method to fulfill this requirement.
TargetHunter is an online program for predicting the biotargets of chemical compounds. It is built on biologically annotated chemical genomic (chemogenomic) databases with millions of bioactivity records, such as the ChEMBL database. It predicts the biological targets of a query compound by the Targets Associated with its MOst SImilar Counterparts (TAMOSIC) algorithm, which assigns the targets associated with the most similar compounds of a query chemical as the predicted targets. As illustrated in Fig. 1, a graphic diagram of the TargetHunter program shows that a query compound is input on a web interface (Fig. 1a), and the TAMOSIC algorithm at the server is executed by generating molecular fingerprints (Fig. 1b) and comparing with compounds in a chemogenomics database, e.g., ChEMBL library (Fig. 1c). Then, the targets associated with the top N most similar compounds of the query compound are output as the potential targets of the queried compound with ranked similarity scores (Fig. 1d). TargetHunter program has the following five unique features. (a) Easy to operate. The user can draw a structure or upload the query compound library (such as sdf file), and click a button to retrieve the prediction results for single molecular query or batch compound library query. (b) Query data retrieval. The query results can be stored for later retrieval in the web cloud computing environments. (c) Choice of desired fingerprints and databases. It provides query functions with the user’s choice of various molecular fingerprints and different chemical databases. (d) High accuracy. The program has been validated on a subset of known high-potency compounds and also can remove false positives and calculate confidence rating. These functions reduce the risk of the predictions. (e) Integrated BioassayGeoMap or bioassay finder. The TargetHunter program allows users to find easily the potential collaborators within a defined distance who may already have the bioassays established to validate the predicted biological targets or off-targets experimentally. More details are described in the next section.
Fig. 1.

Schematic overview of TargetHunter for the target identification of organic compounds, showing from query window to prediction output (a–d)
MATERIALS AND METHODS
Databases
We use ChEMBL (6) as an example. ChEMBL is a chemogenomics database maintained by the European Bioinformatics Institute. It serves as the knowledge database and testing set in this study. As a manually curated chemical database of bioactive molecules with drug-like properties (37), ChEMBL version 11 was the largest publically available compound-target database when the project was initiated. This database contains 1,060,258 distinct compounds, 8,603 targets, and 5,479,146 bioactivity entries from 42,516 publications and PubChem bioassays. It also provides a hierarchical scheme, including the compounds, biological targets, information on bioactivity type, and sourcing references. The request for data retrieval is supported by the Structured Query Language (SQL). To assess the prediction accuracy of our TAMOSIC and the reported MCM algorithms, a subset of high-potency compounds from the ChEMBL database was extracted. This subset was generated according to two selection criteria. (a) For each target in ChEMBL, compounds were used only if they had an activity value (IC50/EC50/Ki/ED50) less than 10 μM at high confidence level (ChEMBL level 9) for direct interactions. (b) Targets were considered only if there were more than 30 qualified compounds.
On the basis of these selection standards, a total of 117,535 unique compounds from 794 targets were retrieved. The number of compounds associated with each target ranged from 30 to 2,180, with an average of 216.6 compounds per target. These targets included a wide variety of proteins, such as enzymes, kinase, and receptors. A compound might have more than one target listed in this subset. All these pairs of compound and target were considered in this study.
Molecular Descriptors
Recently, Heikamp et al. (38) conducted a large-scale similarity search on 266 well-defined compound activity classes extracted from the ChEMBL database using the Extended Connectivity Fingerprint (ECFP; 39). Inspired by their results, we adopted the ECFPs implemented in the ChemAxon software as molecular descriptors, either to calculate the similarity between two compounds for the TAMOSIC algorithm or to train the MCM (7). To be consistent with a previous MCM study, ECFPs with a neighborhood size of six (ECFP6) were selected. For TAMOSIC, the ECFP fingerprints of ChEMBL compounds were represented as 1,024-bit strings for simplicity. For MCM, the ECFP fingerprints were represented in sparse arrays for consistency. In addition, two other types of fingerprints, the ECFP4 fingerprint from ChemAxon and the FP2 fingerprint (40) from Openbabel, were used in our online web service as alternative options.
TAMOSIC Algorithm
The TAMOSIC algorithm was developed and implemented in TargetHunter for target identification. The algorithm assigns the targets associated with the most similar compounds of a queried chemical as its predicted targets. The prediction is based on the known medicinal chemistry concept that structurally similar compounds have similar physicochemical properties and probably similar biological profiles.
To validate the performance of the TAMOSIC algorithm, 16,790 (1/7) of these unique compounds, along with their ChEMBL IDs and SMILES structures, were randomly selected as the testing set. The remaining was left as the training set. The reason for using this division is that the ratio of 1/7 is very close to the 15%/85% splitting method in the MCM literature. For each target, its active compounds in the training and testing sets were identified by ChEMBL IDs. These testing pairs of compound and target were predicted by comparing the testing compounds with all compounds in the training set, including the actives from this target and from other targets. The similarity scores were recorded and ranked. For comparison studies, we adopted the same strategy as the MCM algorithm; i.e., only the top N most similar compounds were considered to predict the possible targets. Thus, if the most similar compound from the training set has an association with the target, which is also associated with this query compound from the testing set, this is named as the correct prediction by the first guess. If the second most similar compound associates with the same target, it is classified as a correct prediction by the second guess. Finally, the prediction accuracy for testing compounds from ith target and the whole testing set can be estimated by Formula F1 and F2, respectively.
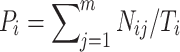 |
1 |
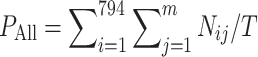 |
2 |
where m is the number of guesses; m is set to 3 in this study. Nij is the number of correctly predicted pairs of compound and target from target i by jth guess. T is the number of pairs of testing compound and target. Ti is the number of pairs of testing compound and target from target i. Because some compounds are associated with multiple targets in our study, T is the total number of pairs of testing compound and target. This number is not exactly identical to the total number of testing compounds.
MCM Algorithm
For comparison, the MCM algorithm was tested on the identical data set. The MCM algorithm was used by Nidhi et al. to categorize the possible targets of a query compound (7). The concept of MCM is based on the naive Bayes algorithm but with a Laplacian-corrected estimator. Given a compound that possesses n features, the score for this compound being active against a target is estimated as the sum of the contribution of each individual feature.
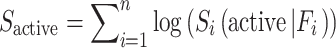 |
3 |
Where 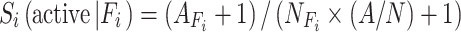 is the contribution of feature Fi, which is calculated from the active compounds of this target having this feature Fi, AFi is the number of active compounds of this target that have the feature Fi, is the total number of compounds that possess feature Fi, including both active and inactive ones. A is the number of active compounds. N is the total number of compounds. “1” is the Laplacian-corrected estimator. Intuitively, if a query compound contains more features favored by most of the active compounds of a target, the query compound has a higher probability of being active against this target.
is the contribution of feature Fi, which is calculated from the active compounds of this target having this feature Fi, AFi is the number of active compounds of this target that have the feature Fi, is the total number of compounds that possess feature Fi, including both active and inactive ones. A is the number of active compounds. N is the total number of compounds. “1” is the Laplacian-corrected estimator. Intuitively, if a query compound contains more features favored by most of the active compounds of a target, the query compound has a higher probability of being active against this target.
As currently implemented, independent scoring models are created separately for each ChEMBL target according to Formula F3. When building a multiple-category model for one target, training compounds associated with this target are defined as active, while other training compounds are defined as inactive. A total of 794 models are generated.
To predict the target of a query compound from the testing set, the molecular fingerprint of the query is generated and relative estimator scores are evaluated by all the 794 MCM models. The target with the highest score is assumed to be the most likely one for the query compound. Similarly, the target with the next highest score can be assigned as the second most likely and so on. To be consistent with the previous MCM study, only the top 3 plausible target predictions are considered.
To assess how the results of these two algorithms will generalize to an independent dataset, sevenfold cross-validations were also performed for both TAMOSIC and MCM. As described in the above paragraphs, these 117,535 unique compounds were assigned to seven subsets of equal size. In each round, one of the subsets was used as the test set and the remaining subsets were used as the training set. This process was repeated seven times using the different possible test sets. The resulting accuracies were averaged for each method. Differences in the prediction accuracies of these two approaches were tested for statistical significance using a paired t test implemented in the R package. Significance was accepted at the 0.05 level of probability (p < 0.05).
Parameters for TAMOSIC
An important parameter of TAMOSIC is the Tanimoto threshold that excludes irrelevant targets. If all of the Tanimoto similarities between a query compound and any of the annotated ChEMBL compounds are less than this threshold, TargetHunter considers this query as inactive to any of the archived targets in ChEMBL. The Tanimoto threshold is determined by analyzing the relationship between Tanimoto similarity and the prediction accuracy. First, the similarity scores between the testing compounds and their most similar counterparts in the training database are plotted to obtain the primary estimation of this threshold. Then, the Tanimoto similarities with different ranges and the corresponding accuracies are explored to determine inactivity. The range of Tanimoto coefficients from 0 to 1.0 is split into 11 bins. Each bin contains the percentage of correct predictions within the first three guesses if the Tanimoto of the first guess is in the range of this bin.
The foregoing steps aim at revealing an appropriate Tanimoto threshold score in order to remove potential false positives. If a query compound has more than one similar compound with a Tanimoto coefficient higher than this threshold, ranking is needed to identify the targets with top priority. The targets can be ranked according to Tanimoto scores between the ligands of these targets and the query compound. However, in order to make confidence-rated predictions and effectively remove false positives, the conditional probability of a compound being active against a target (T) is estimated using logistic regression. The target-specific similarity model is based on the hypothesis that targets require different structural patterns for ligands to bind. The divergent ligand–receptor interaction modes may result in different thresholds of similarity scores that have identical sensitivity and precision for ligand screening. Based on these assumptions, the logistic regression is conditioned on specific targets and the similarity score between the compound and its nearest neighbor associated with the targets.
The probability of a compound being active given T and score is
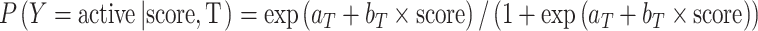 |
4 |
where T is the target; score is the highest Tanimoto coefficient between the compound and its nearest neighbor associated with T; Y is the categorical label. In addition, 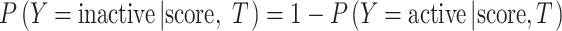 . aT and bT are intercept and slope, and must be estimated from observations. Note that aT and bT are constant regarding T. To “learn” aT and bT, both active and inactive compounds are randomly sampled from the whole ChEMBL dataset for each target T. For a given target with index k, i.e., Tk, let CTk represent the active compound set associated with Tk.
. aT and bT are intercept and slope, and must be estimated from observations. Note that aT and bT are constant regarding T. To “learn” aT and bT, both active and inactive compounds are randomly sampled from the whole ChEMBL dataset for each target T. For a given target with index k, i.e., Tk, let CTk represent the active compound set associated with Tk.
To study the similarity threshold for Tk, calculate the following scores for each active compound 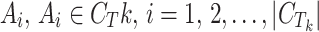 , and create a score set:
, and create a score set:
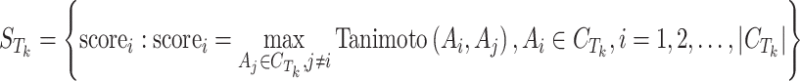 |
5 |
On the other hand, a set of negative observations are required to estimate the parameters in logistic regression. To create the negative score set, the following protocol is conducted:
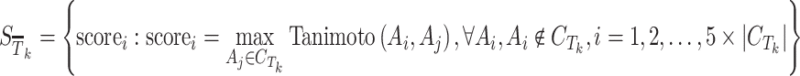 |
6 |
In this step, we assume compound 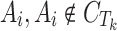 is not active against Tk, which is not always true. The intercept and slope aTk, bTk, for Tk are estimated by a maximum likelihood estimator:
is not active against Tk, which is not always true. The intercept and slope aTk, bTk, for Tk are estimated by a maximum likelihood estimator:
 |
7 |
The aTk and bTk have to be solved numerically. The default similarity threshold is chosen for Tk when 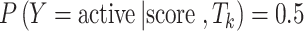 . Therefore, a ligand is predicted to be active against Tk if the score is greater than
. Therefore, a ligand is predicted to be active against Tk if the score is greater than 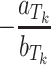 , or inactive otherwise. The training error rate is calculated as the ratio of correct predictions with default threshold to the total number of sampled compounds.
, or inactive otherwise. The training error rate is calculated as the ratio of correct predictions with default threshold to the total number of sampled compounds.
Web Service
An online service, TargetHunter, is built on the TAMOSIC algorithm to automate the target prediction calculation and make it accessible to academic and industrial researchers. The server is constructed on the LAMPP (Linux, Apache, MySQL, PHP, and Python) platform (http://www.cbligand.org/TargetHunter). It also embeds a key function, a geographical bioassay locator that can assist users to find suitable bioassays available nearby in order to validate the target prediction. If compounds are predicted to interact with a particular target by TargetHunter, the next likely step is finding a collaborator to perform biological experiments and validate the hypothesis. A researcher usually can find a candidate laboratory nearby, for example on the same campus. Accordingly, GeoMap of Bioassay service in TargetHunter is designed to facilitate the search for possible collaborators in the geographic vicinity. This function helps to identify laboratories with established bioassays against a particular target through Google Map technology. Our rationale is that if a bioactivity record of a target for any compound is reported, the technology and protocols for testing the bioactivity of a new sample usually are easy to apply. Based on this assumption, the affiliation information was collected from the references in the ChEMBL database and was then converted into geographic coordinates through the Geocoding technology in Google MAP. This geographic coordinate-affiliation information was then stored in the TargetHunter database, allowing a search for nearby available bioassays.
RESULTS
Performance of TAMOSIC Algorithm and its Comparison with MCM Algorithm
According to the previously mentioned protocols, the prediction performances of TAMOSIC and MCM were assessed with a subset of high-potency compounds from ChEMBL. The average accuracy of sevenfold cross-validations of TAMOSIC is 90.9% (±0.2%), while that of MCM is 74.8% (±0.2%) by the top 3 guesses. The differences in prediction accuracy for these two methods are statistically significant (paired t test, p value = 6.41e-13 < 0.05). The variations of prediction accuracies on different subsets are small because the dataset size is large. As such, only the first round of calculations was analyzed in the following steps. Figure 2 compares the prediction accuracy (PAll) of the first round of calculations using TAMOSIC and MCM. The TAMOSIC algorithm correctly identifies 85.0% of all testing compound-target pairs from 794 targets by the most similar compound or first guess, 4.4% by the second guess, and 1.7% by the third guess (Fig. 2). Overall, 91.1% of the compounds are correctly assigned to their known targets within three guesses. The MCM algorithm only reaches an overall accuracy of 74.8% with the top three guesses, e.g., 50.9% by the first guess, 16.3% by the second guess, and 7.6% by the third guess.
Fig. 2.

The plots of the prediction results of TAMOSIC and MCM algorithms on the testing compounds from the ChEMBL subset. The x-axis denotes the number of top N guesses by TAMOSIC and MCM, and the y-axis is the prediction accuracy. N ranges from 1 to 100 in the left figure and ranges from 1 to 10 in the right figure. TAMOSIC Targets Associated with MOst SImilar Counterparts, MCM multiple-category models
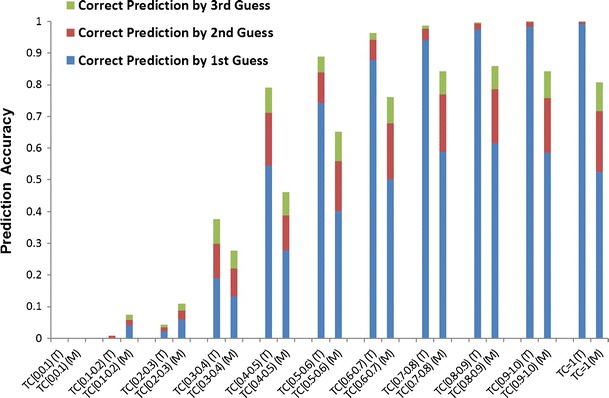
To assess further the two algorithms, 10 individual targets along with their ligands were selected to illustrate the prediction capabilities of TAMOSIC and MCM. These 10 targets are representative for popular drug targets, such as hydrolases, kinases, and GPCRs. Table I lists the names of these targets, the corresponding ChEMBL IDs, and the quantity of associated testing samples. In all ten cases, TAMOSIC outperforms MCM, as shown in Fig. 3.
Table I.
Ten Showcases of Protein Targets and Qualified Compounds that were Used for Evaluating the Performance of TAMOSIC in Comparison with MCM
| CHEMBL ID | Target name | Qualified compounds |
|---|---|---|
| CHEMBL220 | Acetylcholinesterase | 657 |
| CHEMBL221 | Cyclooxygenase-1 | 228 |
| CHEMBL230 | Cyclooxygenase-2 | 607 |
| CHEMBL243 | Human immunodeficiency virus type 1 protease | 2,180 |
| CHEMBL247 | Human immunodeficiency virus type 1 reverse transcriptase | 1,418 |
| CHEMBL203 | Epidermal growth factor receptor erbB1 | 704 |
| CHEMBL204 | Thrombin | 1,098 |
| CHEMBL2056 | Dopamine D1 receptor | 290 |
| CHEMBL260 | MAP kinase p38 alpha | 850 |
| CHEMBL235 | Peroxisome proliferator-activated receptor gamma | 991 |
Fig. 3.

Comparison of prediction accuracies of ten showcases of protein targets by TAMOSIC and MCM algorithms. TAMOSIC Targets Associated with MOst SImilar Counterparts, MCM multiple-category models
Threshold of Tanimoto Coefficient for TargetHunter
An important advantage of TargetHunter is the application of a Tanimoto threshold for excluding irrelevant targets or false positives. These exclusions are deficient in MCM and other established methodologies. If the similarities (Tanimoto) between a query compound and any ChEMBL compounds fall below the defined threshold, TargetHunter will regard the query as inactive to the targets associated with these compounds, regardless of the ranking of the database compounds in the score list. To determine the threshold, the similarity scores from each compound in the testing set and its closest neighbor in the training set are plotted out in Fig. 4, showing the distribution of these calculated Tanimoto coefficient (Tc) values. As illustrated in Fig. 4, specifically, 81.6% of the testing compounds have similarity scores larger than a Tc of 0.60 to the most similar ones in the training compounds, and 89.4% have similarity scores larger than a Tc of 0.50. These percentages indicate how to select a proper threshold.
Fig. 4.

The distribution of the Tanimoto coefficient values comparing any pair of a compound from the testing set with its most similar counterpart from the training set
To estimate the threshold further, the prediction accuracies for both TAMOSIC and MCM in these 11 bins are calculated. Figure 5 shows that the performance of TAMOSIC is better than MCM when Tc values are larger than 0.3. Each of these Tc values is calculated between one compound in the testing set and its most similar compound in the training set. For TAMOSIC, the prediction accuracy increases when the Tc values rise. For example, when the Tc values range from 0.5 to 0.6, the prediction accuracies by the first, second, and third guesses are 74.2%, 9.6%, and 5.0%, respectively. An expected accuracy of 88.8% therefore can be obtained by guesses from the top three most similar compounds; when Tanimoto falls into a range of 0.3–0.4, the accuracy of the top 3 guesses is only 37.5%. A prediction based on low Tc therefore is not satisfactory. From the foregoing analysis, we suggest a default Tanimoto threshold of 0.5 to eliminate ambiguous predictions. However, users have the option to adjust it according to their knowledge and preferences.
Fig. 5.
The plot of Tanimoto coefficient ranges and prediction accuracies of TAMOSIC (T) and MCM (M). The Tanimoto scores are calculated from the pairwise comparisons of each query compound in the testing set with its most similar counterpart in the training set. The range of Tanimoto coefficients is split into 11 bins. In each bin, the percentages of correct target prediction by first, second and third guesses are calculated separately for all testing compounds with Tanimoto coefficients falling into this bin. TAMOSIC Targets Associated with MOst SImilar Counterparts, MCM Multiple-Category Models
Confidence-Rated Prediction for Individual Targets
Logistic regression was applied to model the probability of a ligand being active against a certain target. The probabilistic model forms a sigmoidal curve regarding similarity score and determines a decision threshold value correspondingly. Intuitively, a decision function can be formulated as 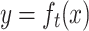 , where x is the similarity score, t is a given threshold value, and y represents a categorical label (0, inactive; 1, active), y = 1 if x ≥ t, y = 0 o/w. Logistic regression yields a relaxed form of this function by calculating P(y|x,t).
, where x is the similarity score, t is a given threshold value, and y represents a categorical label (0, inactive; 1, active), y = 1 if x ≥ t, y = 0 o/w. Logistic regression yields a relaxed form of this function by calculating P(y|x,t).
The basic assumption of the target-conditioned model is the discrepancy in the thresholds of similarity scores for different targets. To examine the rigor of this hypothesis, the distribution of the similarity thresholds and the corresponding logistic functions are plotted in Fig. 6. As shown in Fig. 6a, the solved threshold values span mostly from 0.2 to 0.5. The average and standard deviation are 0.364 ± 0.086. Figure 6b displays the 794 solved logistic functions that express the fitted probability of a ligand being active against the 794 corresponding targets. The threshold value depends both on the definition of ECFP and the occurrence of common structural patterns observed in active compounds. Figure 6 demonstrates that the structural boundary between active and inactive compounds greatly depends on the targets and that target-dependent models can provide more informative suggestions for target prediction.
Fig. 6.
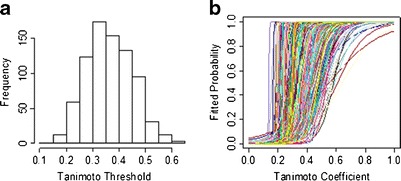
a Histogram plots of similarity threshold values according to 794 solved logistic functions. b Fitted probability plots of being active against 794 targets as functions of the Tanimoto coefficients
Case Study of the use of TargetHunter for Target Prediction
Cell-based phenotypic screening has generated a huge amount of bioactivity data and tools integrated with knowledge databases would help to interpret these results and guide further experimental validations. For example, Cheng et al. (36) have reported a target identification by bioactivity profile similarity search. Here, we take a test compound from PubChem bioassays to illustrate how TargetHunter can be used to identify a target and also explore the mechanism of action for small organic molecules.
As shown in Fig. 7, compound CID 46907796 can induce cellular apoptosis with an activity concentration (AC50) of 0.4136 and 4.908 μM, reported in PubChem by cell-based bioassay with assay IDs (AIDs) of 488848 and 488931, respectively. However, the mechanism of action for this bioactivity is not known. Queried with CID 46907796, TargetHunter retrieved two related compounds (ChEMBL IDs 1724922 and 1711746, with Tanimoto scores of 0.78 and 0.63, respectively). Both of these compounds target the nuclear factor erythroid 2-related factor 2 (Nrf2, tested in PubChem bioassay AID: 504444). Nrf2 is known to act as an anti-apoptosis protein (41). Therefore, inhibition of Nrf2 is very likely the cause of the cellular apoptosis induced by compound CID 46907796.
Fig. 7.
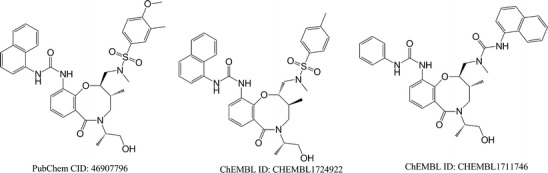
Structure of compound CID 46907796 (AC50 = 0.4136 and 4.908 μM) and its similar compounds, ChEMBL 1724922 and ChEMBL1711746 (two Nrf2 inhibitors)
Case Study for the Application of TargetHunter in Drug Repurposing
Drug repurposing (12) discovers novel therapies from already approved drugs. It has advantages (12,25) over the discovery of new chemical entities, which is often lengthy and costly. TargetHunter can help in the data mining of the chemogenomics database through querying commercial drugs and predicting drug repurposing. Figure 8 shows two interesting examples. Darifenacin from Novartis, a selective antagonist of the muscarinic M3 receptor, is used to treat urinary incontinence (42). TargetHunter found that the most similar compound is UK-201844 (with ECFP6 fingerprints, the Tanimoto coefficient is 0.83), which can inhibit the gp160 process of human immunodeficiency virus type 1 as reported by Pfizer (43). Although these two compounds have different chiral centers and ring size, they share multiple features. Darifenacin, therefore, is predicted to be an inhibitor of HIV-1 gp160. Another example shows that an antihypertensive drug from Pfizer, Polythiazide, is very similar to a compound named CHEMBL1577 (with ECFP6 fingerprints, the Tanimoto coefficient is 0.64), which is an inhibitor of Nrf2 reported in PubChem bioassay AID 504444. Since Nrf2 is a transcription factor that maintains cellular redox homoestasis and protects cells from xenobiotics (44,45), inhibition of Nrf2 would represent a novel therapeutic method that could improve survival of patients undergoing chemotherapy or radiotherapy. The prediction implies that Polythiazide may be an Nrf2 modulator with therapeutic potential for cancer treatment.
Fig. 8.
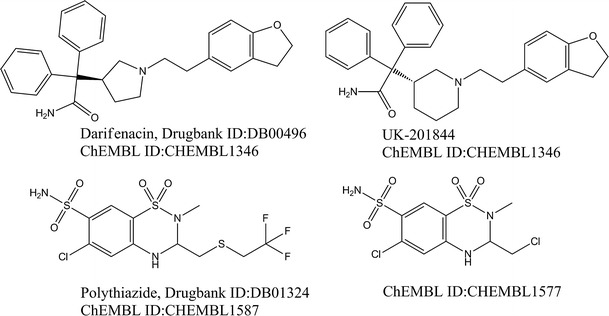
Case study of new target prediction by TargetHunter for known drugs on the market
Web Service and BioassayGeoMap
TargetHunter is available online (http://www.cbligand.org/TargetHunter) and the TAMOSIC algorithm is also implemented on the server side. Through a web browser, users can either sketch or upload a file containing a compound structure and submit it as a query. Upon receipt of the query compound, the server searches the ChEMBL database to identify the most similar compounds based on Tanimoto similarity. Those compounds, together with the associated protein targets, bioactivity information, and references are retrieved and displayed in the web browser. TargetHunter also lists compounds with lower bioactivity, if they are reported in the ChEMBL database, because these records also provide valuable information for the prediction of improbable targets. Another important feature is that the implemented function of GeoMap of Bioassay or BioassayGeoMap (www.cbligand.org/TargetHunter/bioassay_geomap.php) can help users easily to identify the reported bioassays for possible collaborations in order to validate the target prediction. Figure 9 shows an example of the target prediction of Darifenacin together with the GeoMap function for searching potential collaborators. We previously stated that Darifenacin is predicted to interact with human immunodeficiency virus 1. Our TargetHunter program also identifies three different groups at the University of Pittsburgh that have already reported technologies for testing anti-HIV compounds. We believe that TargetHunter can help to originate collaborations across different scientific communities. For example, a chemist can find a biological laboratory nearby to test compounds that are predicted to be active by TargetHunter.
Fig. 9.

An illustration of target prediction for Darifenacin and the assistant hyperlink by BioassayGeoMap for finding potential collaborators
DISCUSSION
The in silico identification of the biological targets of small organic molecules has attracted substantial attention from biologists, chemists and pharmacologists. Computational “target hunter” demonstrates many advantages over traditional experimental methods, including high throughput nature, the wide coverage of candidate targets, and the capability of processing virtual compounds. More importantly, it assists the discovery of off-targets and identification of mechanisms of action (46), thereby playing a crucial role in many scientific projects. In silico methods have been successfully applied in many chemogenomics tasks, but are still far from perfect. For example, docking-based methods depend on precise scoring functions, crystal structures of target proteins, and extensive computational resources. Methods based on the bioactivity profile rely on large amounts of bioactivity data tested experimentally, such as Cerep Bioprint database (34), which is usually proprietary in large pharmaceutical companies. All these requirements limit their usage and availability to the scientific communities. A new approach, TargetHunter, therefore is presented here for target identification, and its performance is evaluated by comparison with the published method MCM.
Our pilot study shows that TargetHunter outperforms MCM on the high-potency subset of the ChEMBL dataset. Despite the simplicity of TargetHunter, its nearest neighbor search effectively retrieves the targets of 91.1% of testing compounds within the first three guesses. Moreover, mechanisms for the removal of false positives and confidence-rated predictions, which are not well addressed in MCM, are built into TargetHunter. The removal of false positives reduces the risk of yielding predictions that could incorrectly affect the downstream experiments for drug discovery.
It is very surprising that a simple algorithm could achieve such high accuracy comparable with MCM. As we discussed before, for MCM, modeling tens to thousands of compounds with one unique formula is a challenging mission. For our TAMOSIC, “While any single similarity score used in a chemoinformatics method may, like a firefly, cast only a small point of light, even fireflies are bright-when collected in the billions.” (47) With the increasing size of the ChEMBL database and also by incorporating other chemogenomics databases, we believe that TargetHunter will provide more powerful service for target prediction.
A limitation of the use of the TAMOSIC algorithm is the subjective definition of structural similarity. It is difficult for TargetHunter to make predictions for compounds having structural features that are not represented in the current database. Of course, such limitation is also true for other ligand-based predictions, such as MCM models. For example, as shown in Fig. 5, MCM-based prediction is only a little better than TAMOSIC-based when Tc values fall in ranges of 0.1–0.3, which means that MCM gives better prediction than TAMOSIC only if query compounds have fewer features common to target-annotated compounds. However, the prediction accuracies for both of these methods are less than 15% in this range. For 2D fingerprints, the bits are usually from the structural features, not from pharmacophores, such as hydrogen bond donors and hydrogen bond receptors. The 3D molecular descriptors have proven superior for probes with low structural similarity to other compounds in the database (1). Accordingly, the introduction of 3D similarity may improve the prediction accuracy for targets with fewer available ligands.
TargetHunter relies on the huge amount of high quality chemogenomics data in the current literature. TargetHunter can easily incorporate, although currently does not, other commercial databases. Most of the commercial databases obtain a better integration of biological activity spectrum information, which may be more useful to link chemical samples to therapeutic usage. The prediction of therapeutic activities (7) and ADME properties (34) may be feasible for TargetHunter by the integration of other systematically annotated databases such as MDDR (28).
Meanwhile, many sources of bioactivity data are now open to academic users. How to integrate such data in the target prediction is still a challenge. The prediction of targets with higher accuracy will be enabled by the integration of different algorithms (48), more chemogenomics data, such as proteomics and transcriptome information (49,50), new methods, such as combining information on target sequences and on ligands (51,52), new technologies, such as GPU for the acceleration of Tanimoto calculation (53), and even clinical data (54,55). In addition, all the target prediction relies heavily on the experimental validation. In most case, the testing assays for the predicted targets are not available in the researcher’s laboratory. Collaboration with other laboratories that have well-established assays therefore is essential. Our BioassayGeoMap will meet this need and could boost the cooperation of chemogenomics researchers who study ligand-target interactions and explore biochemical mechanisms in addition to drug repurposing.
Currently, only bioassays from the literature archived in the ChEMBL database are considered by BioassayGeoMap, which represents only portions of all the reported ones. The static literature reference neglects updates of the bioactivity data from various laboratories, for example, the relocation of a laboratory and bioassays that have become obsolete. To remedy this shortcoming, we plan to provide an option to select the most recent research literature in order to highlight more accurate information. We also plan to create a self-sustaining data curation platform to allow researchers to report their published bioassays and data to TargetHunter and amend them at any time.
CONCLUSION
This article presents a web-interfaced target identification program, TargetHunter with a built-in powerful data-mining algorithm (TAMOSIC) for predicting potential biological targets of query compounds. The variables used in the TargetHunter program have been thoroughly evaluated and discussed. The performance of TargetHunter has been compared with the reported MCM method. As a practical tool introduced to interdisciplinary researchers, TargetHunter also provides a convenient BioassayGeoMap service. The case studies demonstrate that TargetHunter is a promising technique for new target identification or repurposing drugs. Its BioassayGeoMap service can help to locate potential collaborators who are capable of performing relevant bioassays for validation of the predictions. The developed TargetHunter web portal with its implemented data mining algorithm can facilitate the study of biological mechanisms and discovery of new drugs by scientists in academic institutes and pharmaceutical industries.
ACKNOWLEDGMENTS
The authors gratefully acknowledge financial support from NIH Grants(NIH R01DA025612, NIGMS P50-GM067082, and NIH R21HL109654) and the National Natural Science Foundation of China (NSFC 81090410 and NSFC 90913018). We thank Dr. Herbert Barry III for improvements of the manuscript and Dr. Qin Ouyang for preparation of the figures.
Footnotes
Lirong Wang and Chao Ma equally contributed to the paper.
REFERENCES
- 1.Nettles JH, Jenkins JL, Bender A, Deng Z, Davies JW, Glick M. Bridging chemical and biological space: “target fishing” using 2D and 3D molecular descriptors. J Med Chem. 2006;49(23):6802–6810. doi: 10.1021/jm060902w. [DOI] [PubMed] [Google Scholar]
- 2.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37(suppl 2):W623–W633. doi: 10.1093/nar/gkp456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolton EE, Wang Y, Thiessen PA, Bryant SH. PubChem: integrated platform of small molecules and biological activities. Annu Rep Comput Chem. 2008;4:217–241. doi: 10.1016/S1574-1400(08)00012-1. [DOI] [Google Scholar]
- 5.Xie X-QS. Exploiting PubChem for virtual screening. Expert Opin Drug Discov. 2010;5(12):1205–1220. doi: 10.1517/17460441.2010.524924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40(1):D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nidhi, Glick M, Davies JW, Jenkins JL. Prediction of biological targets for compounds using multiple-category Bayesian models trained on chemogenomics databases. J Chem Inf Model. 2006;46(3):1124–1133. doi: 10.1021/ci060003g. [DOI] [PubMed] [Google Scholar]
- 8.Jenkins JL, Bender A, Davies JW. In silico target fishing: predicting biological targets from chemical structure. Drug Discov Today Technol. 2007;3(4):413–421. doi: 10.1016/j.ddtec.2006.12.008. [DOI] [Google Scholar]
- 9.Abraham VC, Taylor DL, Haskins JR. High content screening applied to large-scale cell biology. Trends Biotechnol. 2004;22(1):15–22. doi: 10.1016/j.tibtech.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 10.Bleicher KH, Bohm HJ, Muller K, Alanine AI. Hit and lead generation: beyond high-throughput screening. Nat Rev Drug Discov. 2003;2(5):369–378. doi: 10.1038/nrd1086. [DOI] [PubMed] [Google Scholar]
- 11.Carpenter AE, Sabatini DM. Systematic genome-wide screens of gene function. Nat Rev Genet. 2004;5(1):11–22. doi: 10.1038/nrg1248. [DOI] [PubMed] [Google Scholar]
- 12.Oprea TI, Bauman JE, Bologa CG, Buranda T, Chigaev A, Edwards BS, et al. Drug repurposing from an academic perspective. Drug Discov Today Ther Strateg. 2011. doi:10.1016/j.ddstr.2011.10.002. [DOI] [PMC free article] [PubMed]
- 13.Rognan D. Structure‐based approaches to target fishing and ligand profiling. Mol Inf. 2010;29(3):176–187. doi: 10.1002/minf.200900081. [DOI] [PubMed] [Google Scholar]
- 14.Rognan D, editors. Computational approaches to target fishing and ligand profiling. Theory and applications in computational chemistry: the first decade of the second millennium: International Congress TACC-2012; 2012.
- 15.Bender A, Young DW, Jenkins JL, Serrano M, Mikhailov D, Clemons PA, et al. Chemogenomic data analysis: prediction of small-molecule targets and the advent of biological fingerprint. Comb Chem High Throughput Screen. 2007;10(8):719–731. doi: 10.2174/138620707782507313. [DOI] [PubMed] [Google Scholar]
- 16.Martin YC, Kofron JL, Traphagen LM. Do structurally similar molecules have similar biological activity? J Med Chem. 2002;45(19):4350–4358. doi: 10.1021/jm020155c. [DOI] [PubMed] [Google Scholar]
- 17.Schuffenhauer A, Floersheim P, Acklin P, Jacoby E. Similarity metrics for ligands reflecting the similarity of the target proteins. J Chem Inf Comput Sci. 2003;43(2):391–405. doi: 10.1021/ci025569t. [DOI] [PubMed] [Google Scholar]
- 18.Mitchell JBO. The relationship between the sequence identities of alpha helical proteins in the PDB and the molecular similarities of their ligands. J Chem Inf Comput Sci. 2001;41(6):1617–1622. doi: 10.1021/ci010364q. [DOI] [PubMed] [Google Scholar]
- 19.Brown RD, Martin YC. The information content of 2D and 3D structural descriptors relevant to ligand-receptor binding. J Chem Inf Comput Sci. 1997;37(1):1–9. doi: 10.1021/ci960373c. [DOI] [Google Scholar]
- 20.Johnson M. Concepts and applications of molecular similarity. J Med Chem. 1991;34(12):3409. [Google Scholar]
- 21.Bender A, Glen RC. Molecular similarity: a key technique in molecular informatics. Org Biomol Chem. 2004;2(22):3204–3218. doi: 10.1039/b409813g. [DOI] [PubMed] [Google Scholar]
- 22.Patterson DE, Cramer RD, Ferguson AM, Clark RD, Weinberger LE. Neighborhood behavior: a useful concept for validation of “molecular diversity” descriptors. J Med Chem. 1996;39(16):3049–3059. doi: 10.1021/jm960290n. [DOI] [PubMed] [Google Scholar]
- 23.Tanimoto TT. IBM Internal Report. November 17, 1957.
- 24.Gregori-Puigjané E, Mestres J. SHED: Shannon entropy descriptors from topological feature distributions. J Chem Inf Model. 2006;46(4):1615–1622. doi: 10.1021/ci0600509. [DOI] [PubMed] [Google Scholar]
- 25.Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, et al. Predicting new molecular targets for known drugs. Nature. 2009;462(7270):175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lounkine E, Keiser MJ, Whitebread S, Mikhailov D, Hamon J, Jenkins JL, et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature. 2012;486(7403):361–367. doi: 10.1038/nature11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olah M, Mracec M, Ostopovici L, Rad R, Bora A, Hadaruga N, et al. WOMBAT: world of molecular bioactivity. Chemoinformatics in Drug Discov. 2005. doi:10.1002/3527603743.ch9.
- 28.MDDR. 2012. http://accelrys.com/products/databases/bioactivity/mddr.html. Accessed Oct 12 2012.
- 29.Bender A, Scheiber J, Glick M, Davies JW, Azzaoui K, Hamon J, et al. Analysis of pharmacology data and the prediction of adverse drug reactions and off‐target effects from chemical structure. ChemMedChem. 2007;2(6):861–873. doi: 10.1002/cmdc.200700026. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Gao Z, Kang L, Zhang H, Yang K, Yu K, et al. TarFisDock: a web server for identifying drug targets with docking approach. Nucleic Acids Res. 2006;34(Web Server issue):W219–W224. doi: 10.1093/nar/gkl114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cai J, Han C, Hu T, Zhang J, Wu D, Wang F, et al. Peptide deformylase is a potential target for anti‐Helicobacter pylori drugs: reverse docking, enzymatic assay, and X‐ray crystallography validation. Protein Sci. 2006;15(9):2071–2081. doi: 10.1110/ps.062238406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen YZ, Zhi DG. Ligand-protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins. 2001;43(2):217–226. doi: 10.1002/1097-0134(20010501)43:2<217::AID-PROT1032>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 33.Zhu W, Qiu XH, Xu XJ, Lu CJ. Computational network pharmacological research of Chinese medicinal plants for chronic kidney disease. SCIENCE CHINA Chem. 2010;53(11):2337–2342. doi: 10.1007/s11426-010-4082-0. [DOI] [Google Scholar]
- 34.Krejsa CM, Horvath D, Rogalski SL, Penzotti JE, Mao B, Barbosa F, et al. Predicting ADME properties and side effects: the BioPrint approach. Curr Opin Drug Discov Dev. 2003;6(4):470–480. [PubMed] [Google Scholar]
- 35.Fliri AF, Loging WT, Thadeio PF, Volkmann RA. Biological spectra analysis: linking biological activity profiles to molecular structure. Proc Natl Acad Sci U S A. 2005;102(2):261–266. doi: 10.1073/pnas.0407790101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng T, Li Q, Wang Y, Bryant SH. Identifying compound-target associations by combining bioactivity profile similarity search and public databases mining. J Chem Inf Model. 2011;51(9):2440–2448. doi: 10.1021/ci200192v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bender A. Databases: compound bioactivities go public. Nat Chem Biol. 2010;6(5):309. doi: 10.1038/nchembio.354. [DOI] [Google Scholar]
- 38.Heikamp K, Bajorath J. Large-scale similarity search profiling of ChEMBL compound data sets. J Chem Inf Model. 2011;51(8):1831–1839. doi: 10.1021/ci200199u. [DOI] [PubMed] [Google Scholar]
- 39.Rogers D, Hahn M. Extended-connectivity fingerprints. J Chem Inf Model. 2010;50(5):742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 40.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan KP, Yang M, Ito S. Activation of nuclear factor (erythroid-2 like) factor 2 by toxic bile acids provokes adaptive defense responses to enhance cell survival at the emergence of oxidative stress. Mol Pharmacol. 2007;72(5):1380. doi: 10.1124/mol.107.039370. [DOI] [PubMed] [Google Scholar]
- 42.Bozkurt TE, Sahin-Erdemli I. M1 and M3 muscarinic receptors are involved in the release of urinary bladder-derived relaxant factor. Pharmacol Res. 2009;59(5):300–305. doi: 10.1016/j.phrs.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 43.Blair WS, Cao J, Jackson L, Jimenez J, Peng Q, Wu H, et al. Identification and characterization of UK-201844, a novel inhibitor that interferes with human immunodeficiency virus type 1 gp160 processing. Antimicrob Agents Chemother. 2007;51(10):3554–3561. doi: 10.1128/AAC.00643-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 45.Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008. doi:10.1053/j.gastro.2008.06.082. [DOI] [PubMed]
- 46.Gregori-Puigjané E, Setola V, Hert J, Crews BA, Irwin JJ, Lounkine E, et al. Identifying mechanism-of-action targets for drugs and probes. Proc Natl Acad Sci U S A. 2012;109(28):11178–11183. doi: 10.1073/pnas.1204524109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gregori-Puigjané E, Keiser MJ. Chemoinformatic approaches to target identification. In: Harris CJ, Morphy JR, editors. Designing multi-target drugs. London: Royal Society of Chemistry; 2012. [Google Scholar]
- 48.Brummond KM, Goodell J, LaPorte M, Wang L, Xie X-Q. Synthesis and in silico screening of a library of carboline-containing compounds. Beilstein J Org Chem. 2012;8:1048–1058. doi: 10.3762/bjoc.8.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanseau P, Agarwal P, Barnes MR, Pastinen T, Richards JB, Cardon LR, et al. Use of genome-wide association studies for drug repositioning. Nat Biotechnol. 2012;30(4):317–320. doi: 10.1038/nbt.2151. [DOI] [PubMed] [Google Scholar]
- 50.Hu G, Agarwal P. Human disease–drug network based on genomic expression profiles. PLoS One. 2009;4(8):e6536. doi: 10.1371/journal.pone.0006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang L, Ma C, Xie X-Q. Linear and non-linear support vector machine for the classification of human 5-HT1A ligand functionality. Mol Inf. 2012;31(1):85–95. doi: 10.1002/minf.201100126. [DOI] [PubMed] [Google Scholar]
- 52.Geppert H, Humrich J, Stumpfe D, Gärtner T, Bajorath J. Ligand prediction from protein sequence and small molecule information using support vector machines and fingerprint descriptors. J Chem Inf Model. 2009;49(4):767–779. doi: 10.1021/ci900004a. [DOI] [PubMed] [Google Scholar]
- 53.Ma C, Wang L, Xie XQ. GPU accelerated chemical similarity calculation for compound library comparison. J Chem Inf Model. 2011;51(7):1521–1527. doi: 10.1021/ci1004948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang L, Agarwal P. Systematic drug repositioning based on clinical side-effects. PLoS One. 2011;6(12):e28025. doi: 10.1371/journal.pone.0028025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yao L, Zhang Y, Li Y, Sanseau P, Agarwal P. Electronic health records: implications for drug discovery. Drug Discov Today. 2011;16(13–14):594–599. doi: 10.1016/j.drudis.2011.05.009. [DOI] [PubMed] [Google Scholar]