

Abstract
Nicotine, the main tobacco alkaloid leading to smoking dependence, rapidly crosses the blood–brain barrier (BBB) to become concentrated in the brain. Recently, it has been shown that nicotine interacts with some organic cation transporters (OCT), but their influence at the BBB has not yet been assessed in vivo. In this study, we characterized the transport of nicotine at the mouse luminal BBB by in situ brain perfusion. Its influx was saturable and followed the Michaelis–Menten kinetics (Km = 2.60 mM, Vmax = 37.60 nmol/s/g at pH 7.40). At its usual micromolar concentrations in the plasma, most (79%) of the net transport of nicotine at the BBB was carrier-mediated, while passive diffusion accounted for 21%. Studies on knockout mice showed that the OCT Oct1–3, P-gp, and Bcrp did not alter [3H]-nicotine transport at the BBB. Neither did inhibiting the transporters Mate1, Octn, or Pmat. The in vivo manipulation of intracellular and/or extracellular pH, the chemical inhibition profile, and the trans-stimulation experiments demonstrated that the nicotine transporter at the BBB shared the properties of the clonidine/proton antiporter. The molecular features of this proton-coupled antiporter have not yet been identified, but it also transports diphenhydramine and tramadol and helps nicotine cross the BBB at a faster rate and to a greater extent. The pharmacological inhibition of this nicotine/proton antiporter could represent a new strategy to reduce nicotine uptake by the brain and thus help curb addiction to smoking.
KEY WORDS: blood–brain barrier, nicotine, organic cation, proton antiporter, transporter
INTRODUCTION
Nicotine is the predominant tobacco alkaloid that binds specifically to the neuronal acetylcholine receptor. An acute increase in dopamine output in the nucleus accumbens following nicotine intake is believed to mediate the addictive properties of smoking (1). Nicotine is most addictive when inhaled, whereas nicotine delivered orally or through the skin rarely leads to addiction (1,2). The rapid transport of nicotine from the blood to the brain seems to be critical for its neurobiological effects (1,3). A positron emission tomography study of human subjects showed that the nicotine concentration in the brain reached 50% of its maximum within 15 s, illustrating how a rapid rise contributes to nicotine dependence in smokers (4). Nicotine must cross the blood–brain barrier (BBB) to reach the brain parenchyma. This barrier is formed by endothelial cells linked together by tight junctions that impede paracellular permeation. Therefore, solutes like nicotine cross the BBB by the transcellular route, which is a rate-limiting step. Although our knowledge of how xenobiotics are transported across the BBB is still incomplete, at least some of the biochemical properties of the BBB (e.g., transporters) that affect the movement of drugs across it have been identified. BBB transporters lying on the luminal (blood side) and/or abluminal membranes can increase and/or decrease the transport of solutes into the brain parenchyma by unidirectional or bidirectional processes (5,6). These carrier-mediated processes at the BBB are still being investigated.
We have previously reported that a proton-coupled antiporter, whose molecular nature is still unknown, is involved in the transport of the organic drugs clonidine and diphenhydramine in vivo. Nicotine inhibits/interacts with this transporter, as do some other psychotropic drugs like opiates (7). Earlier transport experiments using Caco-2 cells and slices of rat kidney have shown that the movement of nicotine is due to both passive and facilitated antiporter-mediated fluxes (8,9).
Based on these findings, we developed a working hypothesis that the transport of nicotine across the BBB would involve both passive and facilitated components. This transport pattern could support the idea that the more rapidly nicotine reaches the brain, the greater its potential for addiction.
Here, we determined the functional characteristics of nicotine transport at the BBB by in situ brain perfusion, which is a sensitive method for studying the intrinsic transport properties of the luminal side of the BBB (10,11). In this in situ method, “blood” composition is controlled by using an artificial perfusion fluid whose composition, in terms of solutes or ions, can be varied, which is not possible with other in vivo models (12). The in situ brain perfusion method is well-suited to studies on high-capacity, “low-affinity” transport systems at the BBB. Assessing the function of carrier-mediated systems requires concentrations of substrates or inhibitors that are high enough to produce nonlinear saturation kinetics, thereby revealing the involvement of facilitated/active transport systems.
Our results suggest that the transport of nicotine across the mouse BBB involves a passive component and a distinct “proton/amine” antiporter that also transports tramadol and diphenhydramine (7). The features of this reversible “proton/amine” antiporter at the mouse BBB highlight its importance in facilitating the rate and extent of the delivery of nicotine to the brain.
MATERIALS AND METHODS
Drugs and Chemicals
[3H]-Nicotine (75 Ci/mmol) and [14C]-sucrose (565 mCi/mmol) were purchased from PerkinElmer (Courtaboeuf, France). GF120918 and PSC833 were gifts from GlaxoSmithKline (Uxbridge, UK) and Novartis (Basel, Switzerland), respectively. 3,4-Methylenedioxymethamphetamine (MDMA, ecstasy) was synthesized (purity >99%) in the laboratory of Prof. Galons (Faculté de Pharmacie, Université Paris Descartes). Other chemicals were obtained as reported by André et al. (2009).
Animals
Male Swiss mice (8–10 weeks old) were obtained from Janvier (Genest, France). Male Oct1 and Oct2 double knockout Fvb mice [Oct1,2(−/−)], Oct3 single knockout Fvb mice [Oct3(−/−)], and Mdr1a, Mdr1b, and breast cancer resistance protein (Bcrp) triple knockout [Mdr1a,Mdr1b,Bcrp(−/−)] Fvb mice were bred in-house from progenitors obtained from Dr. A.H. Schinkel’s laboratory (Netherlands Cancer Institute, Amsterdam, The Netherlands); wild-type Fvb mice were used as controls in some experiments. All mice were housed in a controlled environment (19 ± 2°C, 55 ± 10% relative humidity) with a 12-h light/dark cycle and access to food and tap water ad libitum. The experimental procedures used complied with the ethical rules of the French national agency for experimentation with laboratory animals and were approved by the University IMTCE-IFR71 ethics committee (no. 86-23, Faculté de Pharmacie, Université Paris Descartes).
In Situ Brain Perfusion
Surgical Procedure and Perfusion
We assessed the function and transport properties of carrier-mediated systems by measuring the transport of [3H]-nicotine at the BBB of mice by in situ brain perfusion, in which blood is completely replaced by an artificial perfusion fluid for a very short time (10,12,13). Mice were anesthetized with ketamine–xylazine (140–8 mg/kg, i.p.). A catheter was inserted into the right carotid artery after ligation of the appropriate vessels. The perfusion liquid was connected to the catheter. Before perfusion, the thorax was opened and the heart was removed to ensure the replacement of all the blood by the artificial fluid. Perfusion was started immediately at a flow rate of 2.5 ml/min. Each mouse was perfused with [3H]-nicotine (0.3 μCi/ml) and [14C]-sucrose (0.1 μCi/ml) as a vascular marker for 60 s, unless otherwise specified. The experiment was terminated by decapitating the mouse. The brain was removed from the skull and dissected out on a freezer pack. The right brain hemisphere and aliquots of perfusion fluid were placed in tared glass vials and weighed, digested with 2 ml of Solvable® solubilizer (PerkinElmer), and mixed with 10 ml of Ultima Gold XR® liquid scintillation cocktail (PerkinElmer). Labeled compounds were counted on a Tri-Carb® (PerkinElmer) counter with the appropriate dual (14C/3H) quenching curves and standards to measure disintegration per minute in samples.
Perfusion Fluid
The perfusion fluid was Krebs carbonate-buffered physiological saline (in millimoles per liter: 128, NaCl; 24, NaHCO3; 4.2, KCl; 2.4, NaH2PO4; 1.5, CaCl2; 0.9, MgSO4; and 9, d-glucose, unless otherwise specified) warmed to 37°C in a water bath and gassed with 95% O2/5% CO2 to bring the pH to 7.40, unless otherwise specified. Hydrochloric acid was added in some experiments to bring the pH to 5.40 or 6.40. The ionic composition of the Krebs carbonate perfusate was changed to remove Na+ and/or Cl− by iso-osmotic replacement. Sodium was replaced by lithium in “Li+” fluid (Li+, 128 mM). Sodium was removed and chloride was depleted in the “mannitol” fluid (mannitol, 256 mM; Cl−, 3 mM). Mice were also perfused with carbonate-free HEPES-buffered saline (in millimoles per liter: 152, NaCl; 4.2, KCl; 1.5, CaCl2; 0.9, MgSO4; 10, HEPES; and 9, d-glucose) warmed to 37°C, gassed with O2, and brought to pH 7.40 with sodium hydroxide. The perfusion fluid, which determines the extracellular pH (pHe), i.e., the pH of the blood/vascular fluid, was always adjusted and checked with a digital pH meter (±0.05 pH units) immediately before perfusion.
[3H]Nicotine Transport Study Conditions
Initial transport rates were measured under trans-influx zero conditions by perfusing [3H]-nicotine (~8 nM) and [14C]-sucrose for 60 s in the Krebs carbonate fluid (pHe 7.40), unless otherwise specified. Cis-inhibition was performed by perfusing [3H]-nicotine and [14C]-sucrose plus an unlabelled compound. Trans-stimulation of the distribution of [3H]-nicotine in the brain was performed using two syringes, each with its own infusion pump and connected to the carotid catheter by a four-way valve. This enabled immediate switching from one syringe to the other. The tissue was first “loaded” by perfusion with [3H]-nicotine in Krebs carbonate buffer (pHe 7.40) for 60 s. The second 60 s perfusion (pHe 6.40) was with [14C]-sucrose (0.1 μCi/ml), with or without the unlabelled test compound.
Apparent Tissue Distribution Volume and Initial Transport Kinetic Parameters
Calculations were done as previously described (10–12). The tissue “vascular” volume was estimated using the [14C]-sucrose distribution volume (Vv, in microliters per gram):
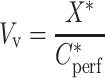 |
1 |
where X* (in disintegration per minute per gram) is the amount of [14C]-sucrose in the tissue sample and 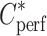 (in disintegration per minute per microliter) is the [14C]-sucrose concentration in the perfusion fluid. The data for any mouse whose Vv was above the normal value (13) were excluded from the study.
(in disintegration per minute per microliter) is the [14C]-sucrose concentration in the perfusion fluid. The data for any mouse whose Vv was above the normal value (13) were excluded from the study.
The apparent brain tissue distribution volume (Vbrain, in microliters per gram) was calculated as follows:
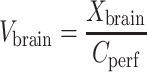 |
2 |
where Xbrain (in disintegration per minute per gram) is the amount of [3H]-nicotine in the right brain hemisphere and Cperf (in disintegration per minute per microliter) is the [3H]-nicotine concentration in the perfusion fluid. This total activity was corrected for “vascular” contamination using the Vv subtraction:
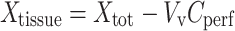 |
3 |
where Xtot (in disintegration per minute per gram) is the total quantity of [3H]-nicotine measured in the right brain tissue. The amount of [3H]-nicotine in the vascular “[14C]-sucrose” space (VvCperf) was calculated and subtracted from the total (Xtot) (Eq. 3).
As the vascular space was “washed” in the final perfusion with [3H]-nicotine-free fluid in the trans-stimulation experiments, there was no need to subtract the vascular [3H]-nicotine content. The [14C]-sucrose perfused during the final washout procedure was only used to estimate Vv in order to verify vascular integrity during these experiments.
The initial transport parameter or brain clearance, Kin (in microliters per second per gram), is calculated as follows:
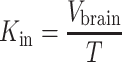 |
4 |
where T is the perfusion time (in seconds).
PS (permeability–surface area product; in microliters per second per gram) was calculated from Kin using the Crone–Renkin equation as follows:
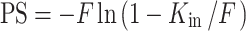 |
5 |
where F (vascular flow rate; in microliters per second per gram) has been previously calculated to be 42.3 μl/s/g for the brain (11).
The flux (Jin; in nanomoles per second per gram) is given by:
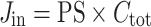 |
6 |
and
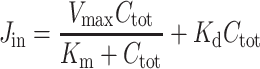 |
7 |
where Ctot is the total nicotine concentration in the perfusate, Vmax (in nanomoles per second per gram) is the maximal velocity of transport, Km (in millimolars) is the apparent Michaelis–Menten transport parameter, which represents the concentration at the half-maximal carrier velocity, and Kd (in microliters per second per gram) is an unsaturable flux component. The data were fitted using nonlinear regression analysis.
A relative value for the permeability of a tissue for a given compound was obtained from the extraction parameter (Ebrain; in percent), calculated with F (in microliters per second per gram), the perfusion liquid flow rate measured using [3H]-diazepam, a freely, passively diffusible, and completely extracted compound:
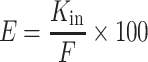 |
8 |
Data Analysis
The transport parameters represent the means ± standard deviation (SD) of four to eight mice. A Student’s two-tailed unpaired t test or one-way analysis of variance with post hoc test were used to identify significant differences between groups and the threshold for statistical significance set at p < 0.05. The transport parameters (Km, Vmax, and Kd) were estimated by plotting brain flux against total concentration using Eq. 7 and performing nonlinear regression with the WinNonlin® software. The errors associated with these parameters are asymptotic standard errors returned by the nonlinear regression routine and are a measure of the best-fit value.
RESULTS
Physical Integrity of the BBB
The [14C]-sucrose Vv was always measured for each mouse and was below 20 μl/g for the brain, in agreement with the normal Vv for [14C]-sucrose (13). Hence, the integrity of the vascular compartment was unaltered in all of our diverse experimental conditions. These results suggest that nicotine (up to 30 mM) had no acute effect on the physical integrity of the BBB during perfusion.
Time Course of [3H]-Nicotine Distribution in the Brain
The accumulation of [3H]-nicotine in the brain (Fig. 1) was measured under trans-influx zero conditions to determine the kinetic conditions required to measure transport solely across the membrane separating the sucrose (vascular) space from the nonsucrose (brain parenchyma) space. The perfusion time adopted ensured that the tissue distribution of the drug was that of the initial linear part of the distribution kinetics. The brain apparent distribution did not reach (pseudo)equilibrium during this initial linear phase (30–120 s) (Fig. 1). At these time periods, abluminal modulation had no effect on the redistribution of the labeled drug from the nonsucrose compartment into the sucrose (vascular) space through the exposed luminal (blood side) surface of the membrane and that the first membrane (luminal) delimiting the sucrose space was the only kinetic interface that affected the distribution of [3H]-nicotine. Thus, we used a perfusion time of 60 s for subsequent single time-point [3H]-nicotine experiments.
Fig. 1.
Time course of [3H]-nicotine transport by the Swiss mouse brain (solid line) expressed as apparent distribution volume (V brain, in microliters per gram), determined by in situ brain perfusion for 30, 60, 90, and 120 s. This line indicates a linear regression with r 2 = 0.988. Data represent the means ± SD of four to five animals
Effects of the ABC Transporters P-gp and Bcrp on [3H]-Nicotine Transport into the Brain
P-glycoprotein (P-gp/mdr1a/Abcb1a) and Bcrp (Abcg2), two ATP-binding cassette transporters that allow the unidirectional efflux of many xenobiotics at the BBB, were not involved in [3H]-nicotine transport (8 nM), since its transport rate measured by in situ brain perfusion experiments and expressed as a percentage of the rate in control Fvb mice (100 ± 15.0%; n = 5) was not significantly modified by the co-perfusion of the specific P-gp inhibitor PSC833 (5 μM) (102.5 ± 5.1%; n = 5) or the P-gp and Bcrp inhibitor GF120918 (3 μM) (103.9 ± 5.9%; n = 5). These conclusions were also supported by the lack of any significant difference in [3H]-nicotine transport in Mdr1a,Mdr1b,Bcrp(−/−) Fvb mice, which lack both P-gp and Bcrp (97.2 ± 12.1%; n = 6) and wild-type control Fvb mice under similar conditions.
Effects of Organic Cation Transporters on [3H]-Nicotine Transport into the Brain
We assessed the functional role of some carrier-mediated systems known to be involved in the transport of xenobiotics and endogenous cationic compounds belonging to the solute carrier transporter (SLC) superfamily. We measured the transport of [3H]-nicotine (8 nM) at the BBB by in situ perfusion in Fvb mice lacking both Oct1 and Oct2 or Oct3 (also called Slc22a1–3). The transport of [3H]-nicotine, expressed as a percentage of the rate in control Fvb mice (100 ± 15.0%; n = 5), did not differ significantly from that measured in Oct1,Oct2(−/−) (97.3 ± 12.2%; n = 5) or Oct3(−/−) mice (103.8 ± 6.1%; n = 5).
The involvement of organic cation transporters (OCT) was also assessed in cis-inhibition experiments in Swiss mice (Table I). We used both broad-range inhibitors such as tetraethylammonium that inhibit both Oct and Mate transporters and more specific inhibitors, such as choline, carnitine, the amphoteric substrate of Octn transporters (Slc22a4–5), agmatine (a polyamine transporter), and biogenic amine neurotransmitters (histamine, dopamine, serotonin), which inhibit Pmat (Slc29a4) and/or Oct transporters. These compounds, used at concentrations at which they are normally effective, did not significantly modulate the transport of [3H]-nicotine, suggesting that these SLC transporters are not involved in its transport at the BBB (Table I).
Table I.
Effects of Organic Cation Transporter Modulators on [3H]-nicotine Transport at the Mouse BBB
| Compound | Concentration (mM) | Relative transport rate (%) |
|---|---|---|
| Agmatine | 10 | 98 ± 14 |
| Carnitine | 15 | 96 ± 17 |
| Choline | 15 | 94 ± 16 |
| Dopamine | 10 | 99 ± 15 |
| Histamine | 10 | 97 ± 8 |
| Serotonin | 10 | 99 ± 9 |
| Tetraethylammonium | 15 | 105 ± 13 |
Cis-inhibition of [3H]-nicotine (8 nM) transport at the BBB by an unlabelled compound was evaluated by in situ brain perfusion with Krebs carbonate perfusion fluid (pHe 7.40) for 60 s in Swiss mice. Brain transport (K in) for [3H]-nicotine (8 nM) in controls, set at 100%, was 17.9 ± 1.1 μl/s/g (100 ± 6%). Data represent the means ± SD (n = 4–6)
Passive and Carrier-Mediated Nicotine Transport Components at the BBB
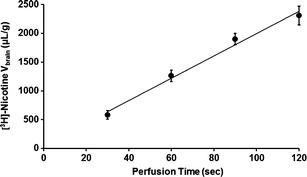
The brain influx (Jin) of nicotine was measured by perfusing the brain with Krebs carbonate buffer (pHe 7.40) for 60 s at diverse nicotine concentrations: 0.8 × 10−5, 10−4, 10−3, 5 × 10−3, 10−2, 5 × 10−2, 10−1, 1, 2, 5, 10, 20, and 30 mM (Fig. 2a, b). The concentrations used were chosen to reflect both unsaturated conditions (in the nanomolar range), including the pharmacological concentration range of nicotine, which is reported to be lower than 10 μM (Fig. 2b), and to exceed the maximal velocity of a carrier-mediated system in order to allow its characterization in terms of maximal and half-maximal transport velocity (Fig. 2a). The experimental determination of PS (see the “Materials and Methods” section) at the diverse nicotine concentrations allowed the measurement of the global or net flux (Jin), according to Eq. 6, which was then plotted against the nicotine concentration (Fig. 2a, b). The Jin was fitted to Eq. 7 (dashed line), which yielded both a saturable (Michaelis–Menten) and an unsaturable nicotine flux, as suggested by the shape of the Jin curve (Fig. 2a). Once the total flux, as indicated by Jin (dashed line), reached 10 mM, at which the carrier-mediated flux was presumably at a constant maximum velocity, the rate associated with the unsaturable component (Kd) could be estimated. Regression analysis of the passive diffusion component of nicotine into the brain at pHe 7.40 yielded a Kd of 3.67 ± 0.14 μl/s/g, equivalent to an extraction parameter, Epassive,brain, of 8.8% (see the “Materials and Methods” section). The unsaturated flux (dotted line) was subtracted from the total measured nicotine flux (Jin) to obtain the carrier-mediated nicotine component (solid line), which was plotted against the total (unbound) nicotine concentration. Modeling this carrier-mediated brain nicotine flux yielded an apparent Km of 2.60 ± 0.19 mM and a Vmax of 37.60 ± 2.88 nmol/s/g (solid line; Fig. 2a); this was best fitted with a Hill coefficient of 1. The carrier-mediated flux was higher than the passive diffusion flux for nicotine concentrations below 7.65 mM (the concentration at which the fluxes were equal (Eq. 7), as also shown graphically by the curve intercept; Fig. 2a). The global brain transport rate (Kin) for nicotine under unsaturated conditions was 17.8 ± 0.9 μl/s/g, which corresponds to an extraction parameter, Etotal,brain, of 42.2%.
Fig. 2.
Passive and carrier-mediated BBB fluxes of nicotine. a Total flux (J in; in nanomoles per second per gram; dashed line) measured in the right brain hemispheres of Swiss mice and plotted against total nicotine concentration in the Krebs carbonate perfusion fluid at pHe 7.40. The straight dotted line represents the passive diffusion of nicotine (K d of 3.67 ± 0.14 μl/s/g; for both cationic and neutral forms at pHe 7.40). The solid line represents the curve obtained by subtracting the passive flux from the total flux and fitted to the carrier-mediated Michaelis–Menten equation by nonlinear least squares regression. The estimated parameters are a K m of 2.60 ± 0.19 mM and a V max of 37.60 ± 2.88 nmol/s/g for the brain transport of nicotine. b Total (J in; dashed line), calculated according Eq. 6, and individual passive (dotted line) and carrier-mediated (solid line) nicotine fluxes at the BBB fitted according to each component of Eq. 7 and according to the physiological nicotine concentrations. Data represent the means ± SD of four to eight animals
Effects of Extracellular and/or Intracellular pH on [3H]-Nicotine Transport
Extracellular/Vascular pHe
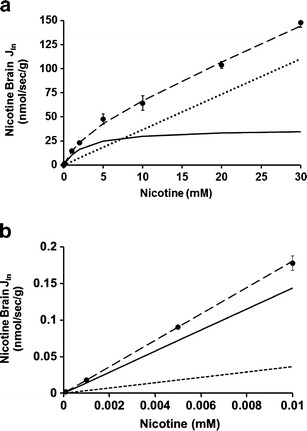
The fractions of neutral and cationic species of nicotine depend on the pHe, which can be estimated in the perfusate using pKa values for nicotine in water (pKa2, 8.02; pKa1, 3.12). Nicotine is 99% cationic at pH 5.40 and 81% cationic at pHe 7.40. The brain transport of [3H]-nicotine (8 nM) was significantly increased by vascular alkalinization (Fig. 3a). This increase could be related to the higher neutral nicotine fraction in the perfusate at pHe 7.40 and/or to a change in the transport rate according to the extracellular proton concentration. Adding unlabeled nicotine (10 mM) led to a significant 2.8-fold decrease in [3H]-nicotine transport at the BBB at pHe 7.40 and a 1.2-fold decrease at pHe 5.40, suggesting that a carrier-mediated system was involved (Fig. 3a). The smaller effect of nicotine saturation at pHe 5.40 could have been related to the higher extracellular proton concentration and the resulting inverted or inward proton gradient, whereas at the physiological pHe of 7.40, the gradient is outward, since the BBB intracellular pH (pHi) is more acidic, around 6.9–7.1 (14). Thus, the control experiments performed at a pHe of 5.40 could reflect a state in which the transporter is “inactive,” as the proton gradient at this pHe does not favor nicotine exchange by a hypothetical nicotine/proton antiporter. Experiments at this pHe, unlike those at pHe 7.40, would thus reflect only the cationic form of nicotine. The Kin for cationic nicotine transport across the BBB was thus estimated at this low pH (2.4 ± 0.1 μl/s/g). However, the confounding effects of both the increase in the cationic form of nicotine and the modulation of the transporter flux rate by the proton concentration make it impossible to assess the respective transporter rates for the neutral and cationic forms by comparing data obtained at the two pHe values.
Fig. 3.
Effects of changes in vascular perfusion fluid pHe, intracellular endothelial pHi, and the presence of sodium on [3H]-nicotine transport at the Swiss mouse BBB. a Effects of perfusion with Krebs carbonate fluid pHe at 5.40 or 7.40 on the brain transport (K in; in microliters per second per gram) of [3H]-nicotine with (black column) or without (white column) co-perfusion of unlabelled nicotine (10 mM) measured by in situ mouse carotid perfusion for 60 s. Data are presented as the means ± SD of five to seven animals. **p < 0.01, ***p < 0.001 for the experimental groups compared to the control group at pHe 5.4; ‡ p < 0.05, ‡‡ p < 0.01 for comparisons between data at the same pH with or without added unlabelled nicotine (10 mM). b Effect of altering pHi (empty column) and removing sodium (grey column). The vascular perfusion fluids were Krebs carbonate buffer plus NH4Cl (30 mM; “NH4”), HEPES-buffered fluid without or with ACTZ (10 mM) and “mannitol” (sodium-free and chloride-free) Krebs carbonate buffer used to alter pHi (empty column). The effect of a Na+-free carbonate perfusion fluid (pHe 7.40) was studied by replacing sodium with lithium (Li+; grey column). Data represent the means ± SD of four to seven animals. *p < 0.05, **p < 0.01, ***p < 0.001 for comparisons between experimental and control groups; † p < 0.05 for comparisons between the presence and absence of ACTZ (10 mM)
Intracellular pH
Alkalinization of BBB Endothelial Cells
Adding NH4Cl to the perfusate affects the pHi of cells by releasing freely diffusible NH3, which enters cells down a concentration gradient. Mice were perfused with Krebs bicarbonate fluid containing [3H]-nicotine with or without NH4Cl (30 mM) adjusted to pHe 7.40. This significantly reduced by 1.5-fold (p < 0.01) the transport of [3H]-nicotine at the BBB (Fig. 3b).
Physiologically, vascular barrier cells, i.e., endothelial cells, are exposed to the blood, which acts as a carbonate buffer. Replacing this vascular carbonate buffer with a carbonate-free fluid acutely affects the cellular pHi as a function of the cells’ enzymatic and/or transporter components. In situ brain perfusion with a carbonate-free buffer (e.g., HEPES fluid) can rapidly produce a transient increase in pHi that is dependent on carbonic anhydrase (CA) and/or a carbonate-coupled proton transporter. The carbonate-free perfusion fluid causes the intracellular CO2 to diffuse out of barrier cells to establish a new equilibrium between the intracellular and the extracellular or vascular medium. [3H]-nicotine transport into the brain after perfusion with carbonate-free HEPES fluid (pHe 7.40) was significantly lower than the transport measured after perfusion with Krebs carbonate fluid (pHe 7.40; Fig. 3b). This “carbonate/no carbonate” change was partially reduced when acetazolamide (ACTZ; 10 mM), a permeating CA inhibitor, was added to the HEPES-buffered fluid (pHe 7.40; Fig. 3b). The effect of ACTZ showed that the effects of the lack of carbonate were in part associated with a CA-sensitive process known to be present at the BBB.
Acidification of BBB Endothelial Cells
We perfused the brains of mice with [3H]-nicotine in Na+-free/Cl−-free carbonate buffer (mannitol) for 60 s at pHe 7.40 and compared the transport parameters measured with those of mice perfused with regular Krebs carbonate fluid (control; pHe 7.40; Fig. 3b). The lack of sodium with or without chloride depletion in the vascular compartment, which is known to acidify the BBB (7,14), increased the brain transport of [3H]-nicotine by 1.72-fold (p < 0.01).
Influence of Sodium on [3H]-Nicotine Transport
It can be difficult to study the influence of ions, such as sodium, on a putative H+-coupled transport system because exposure to a Na+-free extracellular fluid can alter the Na+-dependent transporters involved in controlling the pHi and thus indirectly alter the activity of the transporter. We perfused the brains of mice with [3H]-nicotine in Na+-free carbonate buffer (pHe 7.40) containing lithium, which does not significantly affect the pHi at the BBB (Fig. 3b) (7) and its transport with the parameters obtained after perfusion with [3H]-nicotine in regular Krebs carbonate fluid (control; pHe 7.40). There was no significant change in the brain transport of [3H]-nicotine, suggesting that sodium is not involved in nicotine transport at the BBB (Fig. 3b).
Effect of the Cis-inhibition of Selected Organic Compounds on [3H]-Nicotine Transport at the BBB
We performed cis-inhibition experiments on the brain transport of [3H]-nicotine to discriminate between the effects of known transporters and to obtain a molecular profile of the drugs that interacts with nicotine transport. The concentrations used were selected to modulate a high-capacity/low-affinity system and were, therefore, relatively high, in accordance with the Km value in the millimolar range. They do not reflect in vivo drug interactions, as these unbound concentrations can never result from systemic administration. Mice were perfused with Krebs carbonate buffer (pHe 7.40) with or without the co-perfusion of unlabeled xenobiotics (Table II). Experiments performed with nicotine at 10 μM with or without the co-perfusion of diphenhydramine (10 mM) revealed the significant inhibition of the nicotine transport rate in diphenhydramine-perfused mice (32 ± 2%; n = 5; p < 0.001) as compared to control mice (100 ± 4%).
Table II.
Effects of Drugs on [3H]-nicotine Transport at the Mouse BBB
| Compound | Concentration (mM) | Relative transport rate (%) |
|---|---|---|
| Codeine | 10 | 71 ± 5*** |
| Clonidine | 10 | 69 ± 5*** |
| Diphenhydramine | 5 | 41 ± 6*** |
| Diphenhydramine | 10 | 30 ± 2*** |
| MDMA | 10 | 35 ± 7*** |
| Naloxone | 3 | 74 ± 5*** |
| N-methyl naloxone | 3 | 102 ± 8 |
| Nicotine | 20 | 27 ± 1*** |
| Oxycodone | 10 | 77 ± 5*** |
| Tramadol | 10 | 39 ± 8*** |
Cis-inhibition of the BBB transport of [3H]-nicotine (8 nM) by an unlabeled compound was evaluated by in situ brain perfusion in Krebs carbonate perfusion fluid (pHe 7.40) for 60 s in Swiss mice. Brain transport (K in) for [3H]-nicotine (8 nM) in controls, set at 100%, was 18.1 ± 0.9 μl/s/g (100 ± 4%). Data represent the means ± SD (n = 4–7)
***p < 0.001 for the experimental group compared to the control group
Effect of the Trans-stimulation of Organic Compounds on [3H]-Nicotine Brain Transport
We performed trans-stimulation experiments despite the fact that no kinetic equilibrium was reached for [3H]-nicotine. We assessed the ability of some compounds present in the vascular compartment to further stimulate the exit of the accumulated [3H]-nicotine. The trans-efflux zero or exit of [3H]-nicotine was significantly stimulated (trans-stimulation) by some drugs (naloxone, tramadol, diphenhydramine) (Fig. 4), suggesting that they are also substrates of the hypothetical transporter. However, it cannot be definitely concluded that drugs whose trans-stimulation was not statistically significant were not also substrates. The trans-stimulation study also suggests that the transporter carries nicotine in both directions.
Fig. 4.
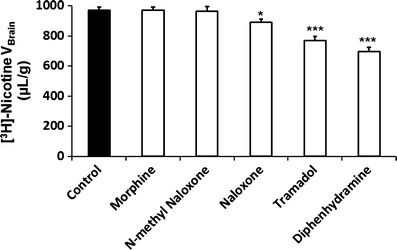
Trans-stimulation of [3H]-nicotine transport at the BBB. [3H]-nicotine brain distribution volume (V brain; in microliter per gram) was measured after in situ mouse brain perfusion with Krebs carbonate buffer (pHe 7.40) for 60 s, followed by a 60-s washout step without [3H]-nicotine. The last 60 s perfusion was at pHe 6.40 without (Control) or with 10 mM of an unlabelled test compound (diphenhydramine, N-methyl naloxone, naloxone, morphine, or tramadol). Data represent the means ± SD of six to eight animals. *p < 0.05, ***p < 0.001 for comparisons between experimental and control groups
DISCUSSION
Passive diffusion is the primary route by which solutes cross membranes. It was originally believed that central nervous system (CNS)-active (CNS+) compounds crossed the BBB and any tight barriers solely by this mechanism. Unlike benzodiazepines, some psychotropic substances are amines (pKa >8) that are mainly cationic at neutral pH. The relative “sufficient” lipophilicity of the neutral form is often used to predict/correlate with barrier permeability. As 19% of nicotine is uncharged (cLogP, 0.57; cLogD7.4, −0.47 (15)) at pH 7.4, it was assumed to cross the BBB by passive diffusion fast enough to produce its CNS effects, whereas 81% of positively charged nicotine did not (16–18). Although the pH partition theory is physiochemically relevant, it suffers from the misinterpretation of the CNS effects of many psychotropic drugs, such as amphetamines and MDMA, which are 99% cationic at neutral pH. This has led to speculation that there is a “pKa shift,” such that a cationic form loses some of its charge when it interacts with a membrane due to a decrease or “shift” in its pKa, enabling it to cross the membrane (19). A second hypothesis is that active or facilitated transport processes, capable of carrying the cationic form of some psychotropic substances across the BBB, are also involved.
Our in situ brain perfusion studies reveal that nicotine rapidly crosses the BBB, with an uptake clearance, Kin, of about 18 μl/s/g. This value can be considered to be intermediate in the Kin scale, which ranges from 0.2 μl/s/g for morphine to 60 μl/s/g for imipramine. When compared to the vascular flow marker diazepam, the extraction of nicotine by the BBB represents about 42%. The small fraction of non-ionized nicotine can diffuse passively across the BBB, as the binding of nicotine to plasma proteins (fu, 0.80 to 0.95 (20)) is not restrictive, and it has been suggested that all neuropharmacodynamic events result solely from this non-ionized fraction. However, pharmacokinetic studies in rodents have suggested that a carrier-mediated nicotine influx process could be involved at the BBB, which could explain its higher concentration in the brain than in plasma (9,21,22). In this study, we examined the permeability of the BBB to nicotine to see if more than one mechanism was involved, including the possible presence of an active efflux and/or influx transport processes.
Several new pharmacological and biological tools have been used to demonstrate that many CNS+ drugs are substrates of unidirectional efflux ABC transporters like P-gp, which affect their ability to cross the BBB and, consequently, their brain concentrations (6,23,24). Nicotine has been shown to be a potent inhibitor of the P-gp-mediated efflux of saquinavir at the rodent BBB (25). This may be how nicotine interacts with drugs that are P-gp substrates, as the blood concentration of unbound nicotine needed to inhibit P-gp can be reached by smoking tobacco (25). However, the role of P-gp in nicotine transport is controversial. Our studies suggest that P-gp and Bcrp, two major ABC transporters involved in the unidirectional efflux of many neutral and/or cationic drugs, are not involved in the nicotine flux at the mouse BBB. This agrees with experiments on Caco-2 cells showing that human P-gp is not involved in the transport of nicotine (8).
The carrier-mediated influx of nicotine at the BBB could result from its ability to interact with several SLC transporters (26). Previous studies have demonstrated that nicotine inhibits OCT, such as OCT1–3 (SLC22A1–3) and OCTN1–2, in vitro (8,26) and may be a substrate of PMAT and MATE1 (27,28). There is conflicting evidence for the expression and function of Oct1 and/or Oct2 at the BBB in vitro (29,30). Moreover, nicotine has been suggested to interact in vivo and in vitro with Oct transporters at the BBB (29). Our experiments found no evidence that these SLC carriers were involved in the BBB transport of nicotine, as it was not altered either by the deletion of Oct1–3 or by the inhibition of Oct, Mate1, Pmat, Octn, and choline transporters. This absence of an interaction of nicotine with known SLC transporters for organic cations at the BBB is reinforced by a recent study showing that Oct1–3, Pmat, and Mate1 transporters are not expressed or functional at the mouse BBB (31).
Here, we found that the in vivo mouse brain transport of nicotine is saturable, which indicates the critical role of a transport process in its transport at the BBB. This transport has an apparent Km of 2.6 mM at pHe 7.40, which is in accordance with the in vitro Km value obtained with Caco-2 cells (0.9 mM (8)). These relatively high apparent Km values suggest that nicotine is transported by a “low-affinity”, high-capacity polyspecific xenobiotic transporter, unlike the more specific endogenous substrate transporters with lower apparent Km (32). High apparent Km values are unlikely to produce substrate saturation. This would also prevent such a transport system from being readily detected by conventional in vivo pharmacokinetic approaches, which use lower/physiological concentrations, and in the absence of transporter-deficient mice.
Nicotine concentrations in the blood plasma of humans and rodents are reported to be <10 μM, which is many times lower than these Km values (22,25). The carrier-mediated BBB flux in vivo is unsaturated and increases linearly with the concentration of unbound nicotine in the vascular compartment. A comparison of the passive (~3.8 μl/s/g) and carrier-mediated (~14.0 μl/s/g) transport rates suggest that the carrier-mediated influx of nicotine is 3.7 times greater than its passive diffusion when concentrations are below the apparent carrier-mediated Km. Thus, the carrier-mediated flux may provide a faster nicotine BBB influx than does passive diffusion. This facilitated component could be responsible for the faster rise in nicotine concentration in the brain parenchyma. Quantitative analysis of the two transport components reveals that passive diffusion accounts for only 21% of the nicotine entering the brain, while active influx is responsible for 79% when blood concentrations are in the usual micromolar range. This makes it easier to understand why it rapidly enters the brain and indicates that cationic nicotine in the plasma also contributes to brain levels.
We exploited the great potential of the in situ method to investigate the molecular characteristics of nicotine transport in more detail. Modifying the vascular or intracellular proton concentration by diverse physical or biochemical protocols altered the transport of nicotine at the BBB in a way that suggests the presence of a sodium-independent nicotine/proton antiporter with features similar to those of the diphenhydramine and clonidine/proton antiporter at the mouse BBB (7). A similar transporter has also been found in BBB cell lines, where it transports oxycodone, diphenhydramine, and codeine (33–35). An analogous nicotine/proton antiporter has also been characterized in kidney tubules, Caco-2, JAR, and LLC-PK1 cell lines (8,36,37). Many cationic organic drugs, such as clonidine, diphenhydramine, and MDMA, are similarly transported by a proton antiporter in Caco-2 cells (38–40). The trans-stimulation of the BBB transport of nicotine by diphenhydramine, naloxone, or tramadol suggests that they share the same transporter. An analysis of the brain kinetics of tramadol suggests that its BBB influx transporter could also be the nicotine/proton antiporter (41). Although oxycodone and naloxone, which are structurally related to morphine, are carried by a similar proton antiporter (33,42), our trans-stimulation experiments could not determine whether morphine was a substrate of our hypothetical antiporter.
The data from our studies, including cis-inhibition and trans-stimulation experiments, suggest that nicotine transport at the BBB is bidirectional and oriented according to the nicotine concentration gradient, which can be extremely high in the case of cigarette smoke inhalation. As a consequence, modulating the rate at which a CNS+ drug crosses the BBB will probably affect its CNS distribution and pharmacodynamics. The pharmacokinetic BBB transport rate has been a major concern in comparing the sedation velocities of general anesthetic agents. Neurobiological studies have recently illustrated that, in addition to its concentration, the rate at which nicotine or cocaine enters the brain influences the neurobehavioral plasticity involved in addiction (3). The nicotine/proton antiporter could be a new pharmacological target whose inhibition could be used to limit addiction and help abstinence. We now need to determine whether the modulation/inhibition of this transporter can influence the CNS effects of nicotine. This is possible only if specific inhibitors of this transport system in vivo can be developed. Knowledge of the molecular nature of the new amine antiporter is also essential, as it might be possible to modulate its transport properties and design new CNS drugs based on pharmacophore determination. This new BBB feature adds a new and key element that must be considered when analyzing the pharmacological variability in response to exposure to CNS+ substances.
In conclusion, nicotine transport at the mouse BBB involves a proton antiporter flux that is kinetically much more important than its passive diffusion. This carrier-mediated system could be responsible for the faster uptake of nicotine by the brain, which may promote nicotine addiction.
ACKNOWLEDGMENTS
We thank Dr. Alfred H. Schinkel for supplying the knockout mice, GSK for generously supplying GF120918, and Novartis for generously supplying PSC833. We thank Dr. S. Rasika for editing the English text.
Conflict of Interest
No conflict of interest.
REFERENCES
- 1.Samaha AN, Yau WY, Yang P, Robinson TE. Rapid delivery of nicotine promotes behavioral sensitization and alters its neurobiological impact. Biol Psychiatry. 2005;57(4):351–360. doi: 10.1016/j.biopsych.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 2.West R, Hajek P, Foulds J, Nilsson F, May S, Meadows A. A comparison of the abuse liability and dependence potential of nicotine patch, gum, spray and inhaler. Psychopharmacology (Berl) 2000;149(3):198–202. doi: 10.1007/s002130000382. [DOI] [PubMed] [Google Scholar]
- 3.Samaha AN, Robinson TE. Why does the rapid delivery of drugs to the brain promote addiction? Trends Pharmacol Sci. 2005;26(2):82–87. doi: 10.1016/j.tips.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Berridge MS, Apana SM, Nagano KK, Berridge CE, Leisure GP, Boswell MV. Smoking produces rapid rise of [11C]nicotine in human brain. Psychopharmacology (Berl) 2010;209(4):383–394. doi: 10.1007/s00213-010-1809-8. [DOI] [PubMed] [Google Scholar]
- 5.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37(1):13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 6.Tournier N, Decleves X, Saubamea B, Scherrmann JM, Cisternino S. Opioid transport by ATP-binding cassette transporters at the blood–brain barrier: implications for neuropsychopharmacology. Curr Pharm Des. 2011;17(26):2829–2842. doi: 10.2174/138161211797440203. [DOI] [PubMed] [Google Scholar]
- 7.Andre P, Debray M, Scherrmann JM, Cisternino S. Clonidine transport at the mouse blood–brain barrier by a new H+ antiporter that interacts with addictive drugs. J Cereb Blood Flow Metab. 2009;29(7):1293–1304. doi: 10.1038/jcbfm.2009.54. [DOI] [PubMed] [Google Scholar]
- 8.Fukada A, Saito H, Inui K. Transport mechanisms of nicotine across the human intestinal epithelial cell line Caco-2. J Pharmacol Exp Ther. 2002;302(2):532–538. doi: 10.1124/jpet.102.034629. [DOI] [PubMed] [Google Scholar]
- 9.Fukada A, Saito H, Urakami Y, Okuda M, Inui K. Involvement of specific transport system of renal basolateral membranes in distribution of nicotine in rats. Drug Metab Pharmacokinet. 2002;17(6):554–560. doi: 10.2133/dmpk.17.554. [DOI] [PubMed] [Google Scholar]
- 10.Takasato Y, Rapoport SI, Smith QR. An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am J Physiol. 1984;247(3 Pt 2):H484–H493. doi: 10.1152/ajpheart.1984.247.3.H484. [DOI] [PubMed] [Google Scholar]
- 11.Dagenais C, Rousselle C, Pollack GM, Scherrmann JM. Development of an in situ mouse brain perfusion model and its application to mdr1a P-glycoprotein-deficient mice. J Cereb Blood Flow Metab. 2000;20(2):381–386. doi: 10.1097/00004647-200002000-00020. [DOI] [PubMed] [Google Scholar]
- 12.Smith QR. Brain perfusion systems for studies of drug uptake and metabolism in the central nervous system. Pharm Biotechnol. 1996;8:285–307. doi: 10.1007/978-1-4899-1863-5_15. [DOI] [PubMed] [Google Scholar]
- 13.Cattelotte J, Andre P, Ouellet M, Bourasset F, Scherrmann JM, Cisternino S. In situ mouse carotid perfusion model: glucose and cholesterol transport in the eye and brain. J Cereb Blood Flow Metab. 2008;28(8):1449–1459. doi: 10.1038/jcbfm.2008.34. [DOI] [PubMed] [Google Scholar]
- 14.Taylor CJ, Nicola PA, Wang S, Barrand MA, Hladky SB. Transporters involved in regulation of intracellular pH in primary cultured rat brain endothelial cells. J Physiol. 2006;576(Pt 3):769–785. doi: 10.1113/jphysiol.2006.117374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ChemSpider. The free chemical database. http://www.chemspider.com/Chemical-Structure.917.html. Accessed 16 June 2012.
- 16.Oldendorf W, Braun L, Cornford E. pH dependence of blood–brain barrier permeability to lactate and nicotine. Stroke. 1979;10(5):577–581. doi: 10.1161/01.STR.10.5.577. [DOI] [PubMed] [Google Scholar]
- 17.Putney JW, Jr, Borzelleca JF. On the mechanisms of 14C-nicotine distribution in rat submaxillary gland in vitro. J Pharmacol Exp Ther. 1971;178(1):180–191. [PubMed] [Google Scholar]
- 18.Seelig A, Gottschlich R, Devant RM. A method to determine the ability of drugs to diffuse through the blood–brain barrier. Proc Natl Acad Sci USA. 1994;91(1):68–72. doi: 10.1073/pnas.91.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beschiaschvili G, Seelig J. Peptide binding to lipid bilayers. Nonclassical hydrophobic effect and membrane-induced pK shifts. Biochemistry (Mosc) 1992;31(41):10044–10053. doi: 10.1021/bi00156a026. [DOI] [PubMed] [Google Scholar]
- 20.Busto U, Bendayan R, Sellers EM. Clinical pharmacokinetics of non-opiate abused drugs. Clin Pharmacokinet. 1989;16(1):1–26. doi: 10.2165/00003088-198916010-00001. [DOI] [PubMed] [Google Scholar]
- 21.Aceto MD, Awaya H, Martin BR, May EL. Antinociceptive action of nicotine and its methiodide derivatives in mice and rats. Br J Pharmacol. 1983;79(4):869–876. doi: 10.1111/j.1476-5381.1983.tb10531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosheh OA, Dwoskin LP, Miller DK, Crooks PA. Accumulation of nicotine and its metabolites in rat brain after intermittent or continuous peripheral administration of [2′-(14)C]nicotine. Drug Metab Dispos. 2001;29(5):645–651. [PubMed] [Google Scholar]
- 23.Doran A, Obach RS, Smith BJ, Hosea NA, Becker S, Callegari E, et al. The impact of P-glycoprotein on the disposition of drugs targeted for indications of the central nervous system: evaluation using the MDR1A/1B knockout mouse model. Drug Metab Dispos. 2005;33(1):165–174. doi: 10.1124/dmd.104.001230. [DOI] [PubMed] [Google Scholar]
- 24.Linnet K, Ejsing TB. A review on the impact of P-glycoprotein on the penetration of drugs into the brain. Focus on psychotropic drugs. Eur Neuropsychopharmacol. 2008;18(3):157–169. doi: 10.1016/j.euroneuro.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 25.Manda VK, Mittapalli RK, Bohn KA, Adkins CE, Lockman PR. Nicotine and cotinine increases the brain penetration of saquinavir in rat. J Neurochem. 2010;115(6):1495–1507. doi: 10.1111/j.1471-4159.2010.07054.x. [DOI] [PubMed] [Google Scholar]
- 26.Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res. 2007;24(7):1227–1251. doi: 10.1007/s11095-007-9254-z. [DOI] [PubMed] [Google Scholar]
- 27.Itagaki S, Ganapathy V, Ho HT, Zhou M, Babu E, Wang J. Electrophysiological characterization of the polyspecific organic cation transporter plasma membrane monoamine transporter. Drug Metab Dispos. 2012;40(6):1138–1143. doi: 10.1124/dmd.111.042432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsuda M, Terada T, Asaka J, Ueba M, Katsura T, Inui K. Oppositely directed H+ gradient functions as a driving force of rat H+/organic cation antiporter MATE1. Am J Physiol Renal Physiol. 2007;292(2):F593–F598. doi: 10.1152/ajprenal.00312.2006. [DOI] [PubMed] [Google Scholar]
- 29.Liou HH, Hsu HJ, Tsai YF, Shih CY, Chang YC, Lin CJ. Interaction between nicotine and MPTP/MPP+ in rat brain endothelial cells. Life Sci. 2007;81(8):664–672. doi: 10.1016/j.lfs.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 30.Dickens D, Owen A, Alfirevic A, Giannoudis A, Davies A, Weksler B, et al. Lamotrigine is a substrate for OCT1 in brain endothelial cells. Biochem Pharmacol. 2012;83(6):805–814. doi: 10.1016/j.bcp.2011.12.032. [DOI] [PubMed] [Google Scholar]
- 31.André P, Saubaméa B, Cochois-Guégan V, Marie-Claire C, Cattelotte J, Smirnova M, et al. Transport of biogenic amine neurotransmitters at the mouse blood–retina and blood–brain barriers by uptake1 and uptake2. J Cereb Blood Flow Metab. 2012;1989–2001. [DOI] [PMC free article] [PubMed]
- 32.Smith QR. Transport of glutamate and other amino acids at the blood–brain barrier. J Nutr. 2000;130(4S Suppl):1016S–1022S. doi: 10.1093/jn/130.4.1016S. [DOI] [PubMed] [Google Scholar]
- 33.Okura T, Hattori A, Takano Y, Sato T, Hammarlund-Udenaes M, Terasaki T, et al. Involvement of the pyrilamine transporter, a putative organic cation transporter, in blood–brain barrier transport of oxycodone. Drug Metab Dispos. 2008;36(10):2005–2013. doi: 10.1124/dmd.108.022087. [DOI] [PubMed] [Google Scholar]
- 34.Fischer W, Bernhagen J, Neubert RH, Brandsch M. Uptake of codeine into intestinal epithelial (Caco-2) and brain endothelial (RBE4) cells. Eur J Pharm Sci. 2010;41(1):31–42. doi: 10.1016/j.ejps.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 35.Sadiq MW, Borgs A, Okura T, Shimomura K, Kato S, Deguchi Y, et al. Diphenhydramine active uptake at the blood–brain barrier and its interaction with oxycodone in vitro and in vivo. J Pharm Sci. 2011;100(9):3912–3923. doi: 10.1002/jps.22567. [DOI] [PubMed] [Google Scholar]
- 36.Takami K, Saito H, Okuda M, Takano M, Inui KI. Distinct characteristics of transcellular transport between nicotine and tetraethylammonium in LLC-PK1 cells. J Pharmacol Exp Ther. 1998;286(2):676–680. [PubMed] [Google Scholar]
- 37.Zevin S, Schaner ME, Giacomini KM. Nicotine transport in a human choriocarcinoma cell line (JAR) J Pharm Sci. 1998;87(6):702–706. doi: 10.1021/js970455v. [DOI] [PubMed] [Google Scholar]
- 38.Fischer W, Metzner L, Hoffmann K, Neubert RH, Brandsch M. Substrate specificity and mechanism of the intestinal clonidine uptake by Caco-2 cells. Pharm Res. 2006;23(1):131–137. doi: 10.1007/s11095-005-8925-x. [DOI] [PubMed] [Google Scholar]
- 39.Kuwayama K, Inoue H, Kanamori T, Tsujikawa K, Miyaguchi H, Iwata Y, et al. Uptake of 3,4-methylenedioxymethamphetamine and its related compounds by a proton-coupled transport system in Caco-2 cells. Biochim Biophys Acta. 2008;1778(1):42–50. doi: 10.1016/j.bbamem.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 40.Mizuuchi H, Katsura T, Ashida K, Hashimoto Y, Inui K. Diphenhydramine transport by pH-dependent tertiary amine transport system in Caco-2 cells. Am J Physiol Gastrointest Liver Physiol. 2000;278(4):G563–G569. doi: 10.1152/ajpgi.2000.278.4.G563. [DOI] [PubMed] [Google Scholar]
- 41.Kanaan M, Daali Y, Dayer P, Desmeules J. Uptake/efflux transport of tramadol enantiomers and O-desmethyl-tramadol: focus on P-glycoprotein. Basic Clin Pharmacol Toxicol. 2009;105(3):199–206. doi: 10.1111/j.1742-7843.2009.00428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki T, Ohmuro A, Miyata M, Furuishi T, Hidaka S, Kugawa F, et al. Involvement of an influx transporter in the blood–brain barrier transport of naloxone. Biopharm Drug Dispos. 2010;31(4):243–252. doi: 10.1002/bdd.707. [DOI] [PubMed] [Google Scholar]