

Abstract
Motor complications of Parkinson’s disease (PD) are a consequence of pulsatile dopaminergic stimulation from standard oral levodopa therapy. Levodopa–carbidopa intestinal gel (LCIG) is infused continuously via an intrajejunal percutaneous gastrostomy tube. This was the first study designed to characterize the full pharmacokinetic profiles of levodopa, carbidopa, and levodopa metabolite, 3-O-methyldopa (3-OMD) with 16-h LCIG infusion. Nineteen advanced PD patients (mean age, 65 years) who were on LCIG therapy for ≥30 days were enrolled. Patients received their individualized LCIG infusion doses, and serial pharmacokinetic samples were collected. Eighteen patients completed the study; 19 were assessed for safety. Mean (SD) total levodopa and carbidopa doses were 1,580 (403) and 395 (101) mg, respectively. Mean (SD) Cavg (μg/mL) were 2.9 (0.84) for levodopa, 17.1 (4.99) for 3-OMD, and 0.22 (0.08) for carbidopa. The degree of fluctuation [defined as (Cmax − Cmin)/Cavg] in levodopa, 3-OMD, and carbidopa plasma concentrations was very low (0.52, 0.21, and 0.96, respectively) during hours 2–16 of infusion. Accordingly, the within-subject coefficients of variation in levodopa, 3-OMD, and carbidopa concentrations were low (13%, 6%, and 19%, respectively). Three patients (16%) reported ≥1 treatment-emergent adverse event; none were considered severe. Continuous intrajejunal LCIG infusion maintained stable plasma levodopa levels over 16 h. Consistent exposure has been shown to reduce motor and nonmotor complications associated with oral medications. LCIG was well tolerated, consistent with previous reports.
Keywords: Duodopa, LCIG, Levodopa-carbidopa intestinal gel, Parkinson’s disease, Pharmacokinetics
INTRODUCTION
Levodopa has been the mainstay of treatment for Parkinson’s disease (PD) since the 1960s (1,2). Unfortunately, many of those who initially respond positively to levodopa eventually develop motor complications, including “Off” periods (when medication has worn off and parkinsonian symptoms re-emerge) and levodopa-induced dyskinesias (1). These complications, arising from the narrowing of the therapeutic window (3,4) can be a major source of distress and disability for patients. As such, an important goal of PD therapy development has been to reduce “Off” time without inducing the development of dyskinesias.
The development of levodopa-associated motor complications may be related to the pulsatile dopaminergic stimulation characteristic of conventional oral levodopa regimens (1,4,5). Levodopa is rapidly metabolized and has a short plasma half-life of approximately 90 min (when administered with carbidopa), thus requiring frequent, repeated dosing (1,6). As levodopa is absorbed mainly in the proximal small intestine, gastric emptying plays an important role in determining plasma levodopa levels after intake of conventional oral formulations (7). Erratic gastric emptying is common in PD and likely contributes to fluctuations in levodopa plasma levels and the unpredictable motor responses observed with orally dosed levodopa (1,6,8,9).
Several studies have demonstrated the clinical utility of alternative methods for levodopa administration. Shoulson et al. (10) and Quinn et al. (11) demonstrated that continuous intravenous administration of levodopa produced stable plasma levels of levodopa and significantly reduced the “On”/”Off” phenomenon typical of the PD patients on oral therapies. Levodopa–carbidopa intestinal gel (LCIG) is a carboxymethylcellulose aqueous gel delivered directly to the proximal jejunum via a percutaneous endoscopic gastrojejunostomy (PEG-J) tube connected to a portable infusion pump (12–15). Infusion of LCIG in the jejunum bypasses gastric emptying, helping to avoid the fluctuation in plasma levodopa levels (16,17). Daytime infusion of LCIG has demonstrated significantly improved motor symptoms and quality-of-life measures in advanced PD patients compared to standard oral therapy (13).
Although not yet approved in the USA, LCIG is approved for clinical use in more than 40 countries (under the name Duodopa®) and has been used in over 2,800 patients worldwide. The present study was the first study to characterize the plasma levels of levodopa, carbidopa, and the levodopa metabolite, 3-O-methyldopa (3-OMD), during the entire time course of the 16-h LCIG infusion; therefore, the present study provides the most thorough pharmacokinetic characterization of LCIG in advanced PD patients to date.
METHODS
Subjects
Male and female subjects at least 30 years of age who were undergoing treatment with LCIG for at least 30 days were enrolled in the study. Subjects had previously undergone a PEG-J procedure for intrajejunal administration of LCIG. PEG (15 Fr Freka®, Fresenius Kabi, Bad Homburg, Germany) and J-tubes (9 Fr J-extension) had been placed in a single procedure under local anesthesia using endoscopic and/or fluoroscopic guidance. Screening took place 21 days prior to pharmacokinetic assessment. Subjects were in general good health, as determined by vital signs, medical history, physical examination, electrocardiogram (ECG), and laboratory tests, with body mass indices ranging from 18 to 30 kg/m2. Major exclusion criteria included the following: current diagnosis or history of drug or alcohol abuse within 12 months of baseline; other psychiatric, neurological, or behavioral disorders that may have interfered with the conduct or interpretation of the study, including a Mini-Mental State Examination score < 24; history of, or current, seizure disorders and subjects requiring treatment with anticonvulsants; or a history or presence of any condition that might interfere with absorption, distribution, metabolism, or excretion of study drug. The study protocol was approved by each respective institution’s ethics committee, the ethical committee of the University Hospital of Uppsala and the ethical committee of the State of Bremen, respectively. All subjects provided written informed consent prior to any procedures being performed.
Study Design
This was a multicenter, multiple-dose, open-label study in subjects with advanced PD. Subjects were confined in the clinic for 2 days (see Fig. 1). Subjects reported to the clinic on the day prior to the pharmacokinetic sample collection for baseline assessment, including a radiological check of intrajejunal tube placement. The tube was repositioned as necessary prior to the pharmacokinetic day. LCIG (Abbott Laboratories, Abbott Park, Illinois, USA), supplied in cassettes containing 100 mL of gel (20 mg/mL levodopa and 5 mg/mL carbidopa), was administered with a portable infusion pump (CADD-Legacy® Duodopa, Smiths Medical, Minnesota, USA). Individually optimized dosing of LCIG was delivered over a 16-h period, administered as a morning bolus followed by continuous infusion and, if needed, intermittent extra doses (patient-initiated based on symptom experience). Subjects who were on approximately 16- or 24-h LCIG infusion regimens were enrolled. Those subjects who received infusion for more than 16 h/day prior to study start had their pumps turned off after 16 h of infusion (the typically prescribed infusion duration) on the day prior to pharmacokinetic assessment. To compensate for the 8 h without LCIG infusion, oral levodopa–carbidopa tablets (Sinemet®) were allowed to be given for up to 3 h prior to the start of the pump on the pharmacokinetic day. The oral dosing regimen was determined by the investigator based on the patient’s motor symptoms. Patients on a 16-h infusion remained on their typical dosing regimen. No significant dose adjustment was allowed during the study or 14 days prior to screening. On the pharmacokinetic day, the patient’s usual morning dose was administered to rapidly achieve the therapeutic dose level and was expected to be 5–10 mL, corresponding to 100–200 mg levodopa and 25–50 mg carbidopa. The subsequent continuous infusion rate was kept within a range of 1–10 mL/h. Extra doses were given if the patient became hypokinetic during the day, but were discouraged during the pharmacokinetic sampling day. Oral levodopa–carbidopa IR was not allowed until the last pharmacokinetic sample was collected. If oral medication was required prior to the last pharmacokinetic sample, a final blood sample was taken prior to administration of oral drug. After collection of the last pharmacokinetic sample, subjects resumed their original levodopa–carbidopa regimens.
Fig. 1.
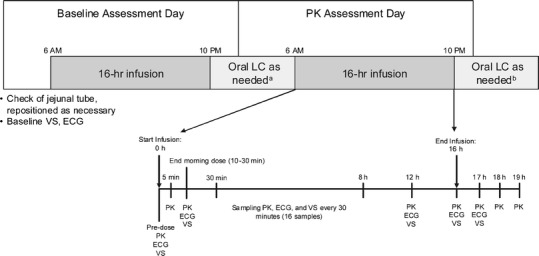
Study design. Superscript a Oral medication not allowed within 3 h of the start of infusion on the pharmacokinetic assessment day. Superscript b If oral medication was needed, final pharmacokinetic sample was taken prior to oral treatment. ECG electrocardiogram, LC levodopa/carbidopa, PK pharmacokinetic assessment, VS vital sign assessment
Diet
Prior to the pharmacokinetic sampling, subjects fasted overnight (starting from 10:00 pm on the previous day). On the pharmacokinetic assessment day, subjects received standardized, low protein meals at the following approximate times relative to starting LCIG infusion: breakfast (2–3 h), lunch (6 h), and dinner (12 h). Water was taken ad libitum. Subjects did not use or consume: any alcohol, products containing methylxanthine, grapefruit and grapefruit juice within the 48-h period prior to and during confinement; excessive amounts of caffeine or caffeine-containing products from 48 h prior to the confinement period; and caffeine on the day of pharmacokinetic sampling.
Concomitant Medication
Other than LCIG, any product containing levodopa or carbidopa was not allowed during the pharmacokinetic sampling day. Allowed medications (if stable for >30 days) included other anti-Parkinson drugs except those prohibited, oral antihypoglycemics, insulin, antihypertensives, anxiolytics, tricyclic antidepressants (excluding amitriptyline and desipramine), selective serotonin reuptake inhibitors (excluding fluoxetine and venlafaxine), and thyroid replacement or antithyroid therapy (if stable >90 days). Forbidden medications included the following: monoamine oxidase-A inhibitors and alpha-methyldopa (within last 60 days); selegiline, catechol-O-methyltransferase (COMT) inhibitors, dopamine, parenteral ergots, methylphenidate, amphetamine, beta blockers for treating tremor, isoprenaline, adrenaline, dobutamide, reserpine, flunarizine or cinnarizine, isoniazid, metoclopramide, and anticholinergics (within last 30 days); and iron salts (within last 7 days). Other forbidden medications included use of botulinum toxin type A or B (within at least 4 months), antineoplastic and immunosuppressants (within the last 5 years), and drugs known to increase risks for cardiac toxicity, Torsade de Pointes, sudden death or prolong QT interval (within five elimination half-lives before baseline and for the duration of the study).
Sample Collection and Analysis
Whole blood samples were collected via an indwelling catheter or by direct venous puncture on the pharmacokinetic sampling day immediately prior to the initiation of LCIG infusion in the morning and at the following time points after the initiation of infusion: 5 min, immediately after the end of the morning dose (if the end of the morning dose was at 5 or 30 min, only one sample was taken at the specified time point); every 30 min up to 8 h (0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 6.5, 7, 7.5, 8 h), 12, 16 (immediately after flushing the tube), 17, 18, and 19 h. The blood samples were collected in 6 mL potassium edetate anticoagulant-containing collection tubes and were immediately cooled to 0°C in ice water. Samples were centrifuged within 60 min of collection, and plasma was immediately transferred to polypropylene tubes containing sodium metabisulfite as an antioxidant. Tubes were stored at −20°C until shipment.
Plasma concentrations of levodopa, 3-OMD, and carbidopa were determined using a validated high performance liquid chromatography (HPLC) method with tandem mass spectrometric (MS/MS) detection (PPD, Middleton, Wisconsin, USA). Each 200 μL plasma sample was fortified with 20 μL of internal standard solution, and 100 μL of 2–8°C 10% perchloric acid solution was added. Samples were vortexed and stored at 2–8°C for 30 min. Samples were then vortexed and centrifuged. Supernatant (100 μL) was transferred to a 96-well plate, and 100 μL filtered water was added. Samples (50 μL) were injected and analyzed via HPLC with MS/MS. Spectrometric data were acquired in batch mode by running Applied Biosystem’s Analyst software (v 1.4.2, AB SCIEX, Framingham, Massachusetts, USA). MassLynx software (Waters, Milford, Massachusetts, USA) was used to integrate the peak area ratios of the analytes and internal standards.
Levodopa concentrations were determined using levodopa-d3 as an internal standard. The method was validated over the concentration range of 10–5,000 ng/mL (r2 ≥ 0.996), with interrun variability [percent coefficient of variation (%CV)] of <10.5% and mean bias between −6.4% and 1.4%. 3-OMD concentrations were determined using 3-OMD-d4 as an internal standard. The 3-OMD quantification method was validated over the concentration range of 25–25,000 ng/mL (r2 ≥ 0.996), with interrun variability (%CV) of <8.4% and mean bias between −4.8% and 0.88%. Carbidopa concentrations were established using carbidopa-d3 as an internal standard. The carbidopa quantification method was validated over the concentration range of 0.5–250 ng/mL (r2 ≥ 0.998), with interrun variability (%CV) of <5.6% and mean bias between −5.2% and 10.2%.
Samples with concentrations higher than the upper limit of quantitation were diluted and reanalyzed.
Pharmacokinetic Variables
Pharmacokinetic parameters for levodopa, carbidopa, and 3-OMD were estimated using noncompartmental analyses. The maximum observed plasma concentration (Cmax), the time to Cmax (Tmax), and the minimum observed plasma concentration (Cmin) over the 16-h infusion interval were determined directly from the plasma concentration–time data. Additionally, the Cmax and Cmin values were calculated for the interval of hours 2–16 of infusion. The terminal phase elimination rate constant (β, lambda z) for levodopa was determined from the slope of the least squares linear regression of the logarithms of the plasma concentration versus time data from the terminal log-linear phase of the profile. A minimum of three concentration–time data points was used to determine β. The terminal phase elimination half-life (t1/2) was calculated as ln(2)/β.
The area under the plasma concentration–time curve (AUC) from hour 0 to 16 (AUC0−16), AUC from hour 0 to the time of the last measurable concentration (AUCt), and AUC from hour 2 to 16 of infusion (AUC2−16) were calculated according to the linear-up log-down trapezoidal rule. The average plasma concentration (Cavg) for the 16 h of infusion as well as for the hour 2–16 infusion interval were calculated by dividing the AUC by the respective duration. The AUC from hour 0 to 24 (AUC0−24) was also calculated for levodopa by extrapolation from the last measurable concentration (Clast) based on the characterized values of β. A value for AUC0−24 could not be determined for 3-OMD or for carbidopa because the blood sampling schedule did not permit determination of a value for β. For levodopa, the apparent oral clearance value (CL/F, where F is the bioavailability) was calculated by dividing the total daily dose of levodopa by AUC0−24. For 3-OMD, the metabolite to parent ratios (M/P) for Cmax and AUC0−16 were calculated. The degree of fluctuation was calculated as 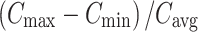 using parameters calculated for the hour 2–16 infusion interval.
using parameters calculated for the hour 2–16 infusion interval.
The mean and standard deviation was calculated for the pharmacokinetic variables, except t1/2, for which the harmonic mean and pseudo standard deviation are reported. The inter- and intrasubject CV for concentrations of each analyte were calculated for the interval of hours 2–16 of the infusion using a linear mixed model for log concentration, with time as a fixed and repeated effect with compound symmetric covariance structure.
Safety
Adverse events (AEs) were coded using the Medical Dictionary for Regulatory Activities (MedDRA), Version 13.0 (18). The number and percentage of subjects having a treatment-emergent adverse event were tabulated by MedDRA system organ class and preferred term. Study investigators recorded the start and stop date of each event, or designated it as “ongoing” at the end of the study. Investigators rated each AE as mild, moderate, or severe, and judged the potential relationship with study treatment. Laboratory data, vital signs, and ECGs were evaluated by site physicians, and abnormal values were assessed for their clinical significance. Values judged to be clinically significant were recorded as AEs. Descriptive statistics were provided for each scheduled time of measurement for vital signs and ECG.
RESULTS
Patient Disposition
Nineteen patients were enrolled (11 male and 8 female) and 18 subjects completed the study. One male patient was prematurely discontinued from the study prior to the pharmacokinetic sampling day for meeting an exclusion criterion (changed LCIG dose within 2 weeks of screening). Baseline demographics are shown in Table I.
Table I.
Baseline Demographic Characteristics
| Parameter | Value | |
|---|---|---|
| N | 19 | |
| Age (years), mean (SD) | 65.4 (7.9) | |
| Sex, n (%) | Male | 11 (58) |
| Female | 8 (42) | |
| Race, n (%) | White | 19 (100) |
| Weight (kg), mean (SD) | 66.2 (12.6) | |
| BMI (kg/m2), mean (SD) | 22.4 (3.3) |
Prior to the study, 5 of the 18 subjects were on 24-h LCIG infusion. These subjects were converted to 16-h infusion on the baseline assessment day as described under “Study Design.” Thirteen subjects were using nighttime oral levodopa/carbidopa or oral levodopa/benserazide prior to the study, and two subjects only were using other anti-PD medications: one subject was using amantadine, and one subject was using pramipexole.
Pharmacokinetics
On the day prior to pharmacokinetic assessment (i.e., baseline assessment), the total mean (SD) LCIG dose of levodopa and carbidopa was 1,770 (605) mg and 443 (151) mg, respectively. Following infusion termination on the night prior to the pharmacokinetic assessment day, 15 of the 18 subjects received single oral doses (100/25 mg for eight subjects and 200/50 mg for seven subjects) of levodopa/carbidopa. These nighttime doses were received between 10 and 11 pm, which corresponded to approximately 8 h prior to starting the LCIG infusion (at 6 or 7 am) on the pharmacokinetic assessment day. On the pharmacokinetic assessment day, the total mean (SD) LCIG dose of levodopa and carbidopa was 1,580 (403) mg and 395 (101) mg, respectively.
The total dose per day of LCIG was the sum of three components: morning dose, continuous (maintenance) dose, and extra doses. In the study, the morning dose ranged from 4 to 11.5 mL (infused at a rate of 40 mL/h), corresponding to 80/20 to 230/57.5 mg levodopa/carbidopa. The continuous dose infusion rate ranged from 2.7 to 6.1 mL/h (54/13.5 to 122/30.5 mg levodopa/carbidopa per hour). Extra doses were given if the patient became hypokinetic during the day. In the study, 13 subjects received extra doses, ranging from 1 to 3 mL (20/5 to 60/15 mg levodopa/carbidopa) for all subjects except one, who received 5 mL (100/25 mg levodopa/carbidopa). Use of extra doses of LCIG was discouraged during the pharmacokinetic sampling day, on which only two subjects received extra doses [two 2 mL (40/10 mg levodopa/carbidopa) extra doses for one subject and one 5 mL (100/25 mg levodopa/carbidopa) extra dose for another].
The pharmacokinetic parameters for levodopa, 3-OMD, and carbidopa are presented in Table II. The estimated elimination half-life, AUC0−24, and CL/F of levodopa were 1.5 ± 0.19 h, 53.8 ± 17.2 μg·h/mL, and 30.7 ± 7.52 L/h, respectively.
Table II.
Pharmacokinetic Parameters (Mean ± SD) of Levodopa, 3-OMD, and Carbidopa
| Parameter (units) | Analyte | ||
|---|---|---|---|
| Levodopa (n = 18) | 3-OMD (n = 18) | Carbidopa (n = 18) | |
| Baseline assessment day total LCIG dose (mg) | 1770 ± 605 | – | 443 ± 151 |
| PK assessment day total LCIG dose (mg) | 1580 ± 403 | – | 395 ± 101 |
| T max (h) | 2.85 ± 2.31 | 8.38 ± 5.77 | 5.70 ± 5.22 |
| C max (μg/mL) | 4.21 ± 1.36 | 19.0 ± 5.66 | 0.371 ± 0.149 |
| C avg (μg/mL) | 2.91 ± 0.836 | 17.1 ± 4.99 | 0.221 ± 0.083 |
| C min (μg/mL)a | 0.447 ± 0.282 | 15.1 ± 4.85 | 0.103 ± 0.067 |
| C min during hours 2–16 of infusion | 2.32 ± 0.582 | 15.4 ± 4.72 | 0.167 ± 0.073 |
| Degree of fluctuation during hours 2–16 of infusion | 0.52 ± 0.20 | 0.21 ± 0.11 | 0.96 ± 0.49 |
| AUC0−16 (μg·h/mL) | 46.5 ± 13.3 | 273 ± 79.8 | 3.54 ± 1.33 |
| AUCt (μg·h/mL) | 51.2 ± 14.9 | 316 ± 90.3 | 4.05 ± 1.65 |
a C min values during the 16 h of infusion were observed either at time 0 or 5 min after start of the infusion and were a result of drug washout prior to establishment of infusion.
The plasma concentration versus time profiles for levodopa, 3-OMD, and carbidopa are presented in Fig. 2. Levodopa concentrations rose quickly after the start of infusion and showed relatively low fluctuation during the 16 h of infusion. It is noteworthy that the Cmin values during the 16 h of infusion were observed either at time 0 or 5 min after start of the infusion, and these low concentrations were a result of drug washout prior to establishment of infusion (Fig. 2). During hours 2–16 of the infusion, the degree of fluctuation (mean ± SD) in plasma concentrations was very low for levodopa (0.52 ± 0.20) and 3-OMD (0.21 ± 0.11). The degree of fluctuation in plasma concentration was slightly higher for carbidopa (0.96 ± 0.49).
Fig. 2.
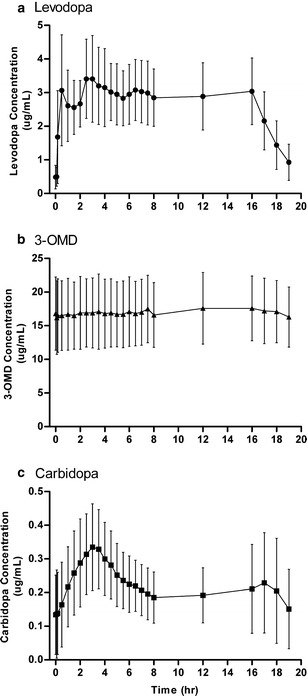
Plasma concentrations over time for a levodopa, b 3-OMD, and c carbidopa on the pharmacokinetic assessment day (16-h infusion). Symbols represent mean, and error bars represent SD; N = 18 for each analyte
The inter- and intrasubject %CVs for levodopa, 3-OMD, and carbidopa are presented in Table III. For levodopa, 3-OMD, and carbidopa, respectively, the intersubject %CVs were 32%, 33%, and 40% from hour 2–16, and intrasubject %CVs were low, 13%, 6%, and 19% over the same time interval. The 3-OMD to levodopa plasma exposure ratio was 462 ± 82% for Cmax and 597 ± 109% for AUC.
Table III.
Inter- and Intrasubject Coefficients of Variation for Levodopa, 3-OMD, and Carbidopa
| Analyte | N | Intersubject | Intrasubject | ||
|---|---|---|---|---|---|
| CV (%) | 95% CI | CV (%) | 95% CI | ||
| Levodopa | 18 | 32 | 18 − 42 | 13 | 12 − 15 |
| 3-OMD | 18 | 33 | 18 − 43 | 6 | 5 − 6 |
| Carbidopa | 18 | 40 | 22 − 53 | 19 | 17 − 21 |
Time interval is from hour 2−16 of LCIG infusion
3-OMD 3-O-methyldopa, CV coefficient of variation, CI confidence interval
Safety
The safety population included all 19 subjects enrolled. Of these, three (15.8%) reported at least one treatment-emergent AE. These included headache (n = 2), migraine, dizziness, and vomiting (n = 1 each). Headache, migraine, and vomiting were judged by the investigator as unrelated to study drug, and dizziness was judged as unlikely related to study drug. No AEs were considered severe, and none resulted in study discontinuation.
Four out of 19 (21%) subjects reported other adverse advents not considered as treatment-emergent since these events had onset prior to the start of the study drug on the pharmacokinetic assessment day: dystonia, spasmodic dysphonia, headache, excessive granulation tissue, and decrease in blood thyroid stimulating hormone. These adverse events were assessed by the investigator as possibly related or unrelated to study drug and ranged from moderate to severe.
Systolic and diastolic supine blood pressure (BP) decreased from a mean value of 152/86 mmHg at baseline to a mean of 120/66 mmHg at 3.5 h after starting the infusion. This was the maximal reduction from baseline. The mean systolic supine BP values were above 120 mmHg for the remainder of the pharmacokinetic day, and at hour 24 (in the morning following the pharmacokinetic day, corresponding to baseline), the mean supine BP was 128/76 mmHg. Only small mean differences from baseline in orthostatic change pressures were observed.
Discussion
This study is the first study to characterize the full plasma concentration versus time profiles of levodopa, carbidopa, and the levodopa metabolite, 3-OMD, with 16-h LCIG jejunal infusion in patients with advanced PD, a treatment modality approved for clinical use in more than 40 countries worldwide and approval of which is currently being sought in the USA. Given the mode of administration, interventional nature of treatment (direct delivery to the proximal jejunum via PEG-J), and the high total daily doses of levodopa and carbidopa typically needed in patients with advanced PD, this phase I study was conducted in the target patient population and recruited patients already on LCIG.
Intrajejunal administration of LCIG rapidly achieved therapeutic plasma levels of levodopa and maintained consistent levodopa levels over the course of infusion (Fig. 2). These results extend findings seen in previous investigations examining levodopa concentrations for up to 9 h (17,19,20). In the present study, the pharmacokinetic profile for levodopa revealed some relatively small peaks mainly during the first 3 h of infusion: one immediately after the high-infusion-rate morning dose (∼30 min after start of morning dose) and another around the 3-h time point (Fig. 2a). The first peak was a result of the change in infusion rate from the morning high-flow-rate infusion to the slower maintenance infusion, before the new steady-state plasma levels were established during the maintenance infusion. The second peak may be due to the slower absorption of carbidopa relative to levodopa (Fig. 2a, c). Given that breakfast and lunch were served 2–3 and 6 h, respectively, following the start of LCIG infusion, meal intake may also have contributed to the small fluctuations in levodopa concentrations observed around these times (Fig. 2a). The fast absorption and the short elimination half-life of levodopa allow these peaks to be discernible in this short time frame.
Over the majority of infusion duration, the degree of fluctuation in levodopa plasma concentrations was very low (0.52 ± 0.20 during hours 2–16). As a reference, the degree of fluctuation in levodopa plasma concentrations with immediate-release or controlled-release Sinemet administered every 8 h is reported to be 4.3 ± 0.9 and 1.9 ± 0.6, respectively (21), which correspond to 8.3- and 3.7-fold higher fluctuation than LCIG. The reported levodopa fluctuations with gastroretentive devices (22,23) and other novel extended release formulations (24) of levodopa–carbidopa, or levodopa–carbidopa formulated with COMT inhibitor (Stavelo®) (18), are also several folds higher than LCIG. Since the degree of fluctuation depends on the interdose interval and the half-life, patients with advanced PD take oral levodopa/carbidopa very frequently (every 1 or 2 h) in order to reduce these swings in concentrations with oral therapy (18). However, even with frequent oral dosing, patients still experience high intrasubject variability in levodopa concentrations due to erratic gastric emptying (17). With LCIG administration, the intrasubject variability in levodopa concentrations from hour 2–16 of infusion was low (13%, Table III), in agreement with findings from previous studies (13–14%) (17,19). Following termination of infusion, levodopa levels declined rapidly (Fig. 2a), highlighting further the need for constant daytime levodopa infusion to maintain effective symptom control in advanced PD patients.
When coadministered with a decarboxylase inhibitor (such as carbidopa), levodopa is known to be rapidly and completely absorbed from the small intestine in humans (9,25,26). The high small intestinal permeability of levodopa is a consequence of efficient transepithelial transport by the amino acid carrier for large neutral amino acids (LNAAs) (26–28). The bioavailability of orally administered standard IR levodopa–carbidopa (4:1 ratio) tablets is reported to be 84–99% (21). A recent pharmacokinetic analysis, which pooled data from three LCIG studies and fixed levodopa disposition parameters to values reported in the literature for intravenous infusion, suggested that LCIG has an absolute bioavailability of 88% (29). Therefore, the absolute bioavailability of LCIG is comparable to oral levodopa–carbidopa (4:1 ratio). This is in agreement with delivery of LCIG to the proximal small intestine, where the LNAA carrier density is highest. Detailed population pharmacokinetic analyses that estimate the bioavailability of LCIG relative to oral levodopa–carbidopa (4:1 ratio) tablets using pharmacokinetic data from head-to-head phase III efficacy trials of LCIG are warranted.
Levodopa is mainly metabolized by the aromatic amino acid decarboxylase (AADC; also referred to as dopa decarboxylase) and the COMT enzymes. Other metabolic routes include transamination and oxidation (30). When levodopa is administered alone, the decarboxylation of levodopa to dopamine by AADC is the major metabolic pathway. In the presence of the AADC inhibitor carbidopa, the main metabolic route for levodopa is the formation of 3-OMD by COMT (31). This metabolite is not active and has a half-life that is approximately ten times longer than levodopa itself (∼15 h), resulting in significantly higher plasma concentration of 3-OMD after chronic dosing (32,33). It has been suggested that 3-OMD may reduce brain uptake of levodopa by competing for active transport across the blood–brain barrier. However, studies have shown that 3-OMD levels associated with oral levodopa/carbidopa administration make only a small contribution to the LNAA pool circulating in plasma, and 3-OMD does not alter levodopa blood–brain barrier transport at clinical exposures (34–36). In the present study, 3-OMD showed a very flat pharmacokinetic profile with 16-h LCIG infusion (Fig. 2b) and very low intrasubject variability (6%) and degree of fluctuation (0.21) over hours 2–16 of infusion, in agreement with the long t1/2 of 3-OMD. The average 3-OMD concentration was approximately six-fold higher than that of levodopa at steady state (Table II), consistent with the ratio reported following repeated oral administration of levodopa/carbidopa in elderly subjects (6.5-fold) (21). 3-OMD/levodopa concentration ratios of 5–10 have been reported with long-term levodopa/carbidopa therapy (37). Taken together, these results suggest that 16-h LCIG infusion does not alter the 3-OMD/levodopa metabolite/parent exposure ratio at steady state and, therefore, may not adversely impact the brain uptake of levodopa compared to intermittent oral administration.
Carbidopa is coadministered with levodopa to reduce levodopa’s peripheral decarboxylation. Carbidopa has slower and more variable absorption and longer half-life than levodopa (21,38), and carbidopa is believed to have a different transport mechanism from levodopa (38). LCIG infusion produced a slow elevation in plasma carbidopa levels with only one discernible peak approximately 3 h post dose (Fig. 2c). The slow absorption of carbidopa and the slightly longer half-life compared to levodopa are likely responsible for this single peak, under which the peaks from the morning dose and the re-establishment of steady-state with the maintenance dose are likely merged. The slower absorption and longer half-life of carbidopa are also responsible for the longer duration until carbidopa plasma concentrations are stabilized (Fig. 2c).
The characterized exposures of levodopa, carbidopa, and 3-OMD in this study represent steady-state exposure with LCIG 16-h infusion and nighttime oral supplemental treatment. In the study, the nighttime doses of levodopa/carbidopa were received shortly after terminating the prior day’s infusion, approximately 8 h prior to starting the infusion on the pharmacokinetic assessment day. The observed carry-over concentrations of levodopa, carbidopa, and 3-OMD (Fig. 2, time 0) were expected based on the respective half-lives of these compounds.
A recent report by Elia et al. (39) compared the motor features and response to oral levodopa in PD patients being administered jejunal LCIG, subcutaneous apomorphine, or subthalamic nucleus deep brain stimulation (STN DBS). Their results indicated that while jejunal levodopa was found to be as efficacious as STN DBS in reducing “Off” state epochs and increasing “On” state epochs (both were significantly improved compared to apomorphine), the time to best motor “On” was significantly longer in the jejunal levodopa group. This delay to best motor “On” could arise from the absence of an initial high-flow-rate morning dose of jejunal levodopa as was used in the current work, resulting in a longer time required to reach therapeutic levodopa plasma levels (29). This possibility is supported by the fact that the time to best motor “On” was improved when a morning oral dose of levodopa was added in the study of Elia et al. (39). Our study is limited in that the effects of LCIG on motor symptoms were not assessed.
LCIG was found to be safe and well tolerated. None of the reported adverse events were considered severe or resulted in early study termination. The low number of AEs in the study may be due to the short duration of the study and the enrollment of patients who had been receiving LCIG treatment for at least 30 days. However, the safety results are generally in line with previous reports of LCIG. One safety finding of note was the decrease in mean systolic and diastolic BP upon LCIG dosing. This effect was greatest at hour 3.5 of infusion. The relatively high baseline BP (152/86 mmHg) allows the possibility that the observed decrease after LCIG treatment was due to the patients returning to a lower, more typical baseline value. The elevated baseline BP may have been due to the anticipation of study procedures or the deviation from a normal daily dosing routine. LCIG could not be started on the pharmacokinetic assessment day until completion of all baseline procedures per the protocol, and oral levodopa–carbidopa was not taken for approximately 8 h prior to the start of infusion. Hence, patients may have been in “Off” state when baseline BP was taken. The fact that the mean BP at hour 24 (a time corresponding to the baseline measure on the pharmacokinetic day) was 128/76 suggests that the change in daily routine prior to the pharmacokinetic day may have led to the higher BP at baseline. The lack of an untreated control group prevents the discrimination of a possible LCIG effect from fluctuations due to study procedures or circadian rhythm in this patient population.
CONCLUSION
The results of the present study demonstrate that jejunal infusion of LCIG results in low fluctuations in levodopa concentrations over the majority of the 16-h infusion duration in advanced Parkinson’s disease patients. Currently available PD medications often result in significant variations in plasma levodopa levels (40–42), which may contribute to the motor fluctuations observed in advanced disease (43). Stable plasma levodopa concentrations during LCIG administration may result in more consistent dopaminergic stimulation in the brains of PD patients and subsequent improvement in clinical symptoms.
Acknowledgments
This study was sponsored by Abbott. The study was conducted and clinical laboratory tests were performed by Quintiles. The authors thank Dr. Holger Honig and Research Nurses Anne Rüssmann and Sandra Leimbach of the Neurology clinic at Klinikum-Bremerhaven, and the staff of the Neurology clinic at Uppsala University Hospital for their assistance in the completion of the study. Nathan R. Rustay and Michelle M. Tangredi, of Abbott, provided medical writing support in the development of the manuscript.
Financial Disclosure/Conflict of Interest Concerning the Research Related to the Manuscript
Dr. Nyholm has been a study investigator in Abbott-sponsored studies and has received compensation from Abbott for serving as a consultant. Prof. Odin has been a study investigator in Abbott-sponsored studies and has received compensation from Abbott for serving as a consultant and lecturer. Dr. Johansson has been a study investigator in Abbott-sponsored studies and has received compensation from Abbott for serving as a lecturer. Drs Chatamra, Locke, Dutta, and Othman are employees of Abbott and receive compensation including salary, stock, and/or stock options from Abbott.
Funding source
Abbott funded this study and provided the study drug.
References
- 1.Hauser RA. Levodopa: past, present, and future. Eur Neurol. 2009;62(1):1–8. doi: 10.1159/000215875. [DOI] [PubMed] [Google Scholar]
- 2.Olanow CW, Agid Y, Mizuno Y, Albanese A, Bonuccelli U, Damier P, et al. Levodopa in the treatment of Parkinson’s disease: current controversies. Mov Disord. 2004;19(9):997–1005. doi: 10.1002/mds.20243. [DOI] [PubMed] [Google Scholar]
- 3.Nyholm D. The rationale for continuous dopaminergic stimulation in advanced Parkinson’s disease. Parkinsonism Relat disord. 2007;13(suppl):S13–S17. doi: 10.1016/j.parkreldis.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Stocchi F. The hypothesis of the genesis of motor complications and continuous dopaminergic stimulation in the treatment of Parkinson’s disease. Parkinsonism Relat disord. 2009;15(suppl 1):S9–S15. doi: 10.1016/S1353-8020(09)70005-7. [DOI] [PubMed] [Google Scholar]
- 5.Olanow CW, Obeso JA, Stocchi F. Continuous dopamine-receptor treatment of Parkinson’s disease: scientific rationale and clinical implications. Lancet Neurol. 2006;5(8):677–687. doi: 10.1016/S1474-4422(06)70521-X. [DOI] [PubMed] [Google Scholar]
- 6.Chase TN. The significance of continuous dopaminergic stimulation in the treatment of Parkinson’s disease. Drugs. 1998;55(suppl 1):1–9. doi: 10.2165/00003495-199855001-00001. [DOI] [PubMed] [Google Scholar]
- 7.Nyholm D, Lennernas H. Irregular gastrointestinal drug absorption in Parkinson’s disease. Expert Opin Drug Metabol Toxicol. 2008;4(2):193–203. doi: 10.1517/17425255.4.2.193. [DOI] [PubMed] [Google Scholar]
- 8.Kurlan R, Rothfield KP, Woodward WR, Nutt JG, Miller C, Lichter D, et al. Erratic gastric emptying of levodopa may cause “random” fluctuations of parkinsonian mobility. Neurology. 1988;38(3):419–421. doi: 10.1212/WNL.38.3.419. [DOI] [PubMed] [Google Scholar]
- 9.Nutt JG, Woodward WR, Hammerstad JP, Carter JH, Anderson JL. The “on-off” phenomenon in Parkinson’s disease. Relation to levodopa absorption and transport. N Engl J Med. 1984;310(8):483–488. doi: 10.1056/NEJM198402233100802. [DOI] [PubMed] [Google Scholar]
- 10.Shoulson I, Glaubiger GA, Chase TN. On–off response. Clinical and biochemical correlations during oral and intravenous levodopa administration in parkinsonian patients. Neurology. 1975;25(12):1144–1148. doi: 10.1212/WNL.25.12.1144. [DOI] [PubMed] [Google Scholar]
- 11.Quinn N, Parkes JD, Marsden CD. Control of on/off phenomenon by continuous intravenous infusion of levodopa. Neurology. 1984;34(9):1131–1136. doi: 10.1212/WNL.34.9.1131. [DOI] [PubMed] [Google Scholar]
- 12.Nyholm D, Aquilonius SM. Levodopa infusion therapy in Parkinson disease: state of the art in 2004. Clin Neuropharmacol. 2004;27(5):245–256. doi: 10.1097/01.wnf.0000144041.28224.b7. [DOI] [PubMed] [Google Scholar]
- 13.Nyholm D, Nilsson Remahl AI, Dizdar N, Constantinescu R, Holmberg B, Jansson R, et al. Duodenal levodopa infusion monotherapy vs oral polypharmacy in advanced Parkinson disease. Neurology. 2005;64(2):216–223. doi: 10.1212/01.WNL.0000149637.70961.4C. [DOI] [PubMed] [Google Scholar]
- 14.Honig H, Antonini A, Martinez-Martin P, Forgacs I, Faye GC, Fox T, et al. Intrajejunal levodopa infusion in Parkinson’s disease: a pilot multicenter study of effects on nonmotor symptoms and quality of life. Mov Disord. 2009;24(10):1468–1474. doi: 10.1002/mds.22596. [DOI] [PubMed] [Google Scholar]
- 15.Antonini A, Tolosa E. Apomorphine and levodopa infusion therapies for advanced Parkinson’s disease: selection criteria and patient management. Expert Rev Neurother. 2009;9(6):859–867. doi: 10.1586/ern.09.48. [DOI] [PubMed] [Google Scholar]
- 16.Antonini A, Odin P. Pros and cons of apomorphine and L-dopa continuous infusion in advanced Parkinson’s disease. Parkinsonism Relat disord. 2009;15(suppl 4):S97–S100. doi: 10.1016/S1353-8020(09)70844-2. [DOI] [PubMed] [Google Scholar]
- 17.Nyholm D, Askmark H, Gomes-Trolin C, Knutson T, Lennernas H, Nystrom C, et al. Optimizing levodopa pharmacokinetics: intestinal infusion versus oral sustained-release tablets. Clin Neuropharmacol. 2003;26(3):156–163. doi: 10.1097/00002826-200305000-00010. [DOI] [PubMed] [Google Scholar]
- 18.Nyholm D, Ehrnebo M, Lewander T, Trolin CG, Backstrom T, Panagiotidis G, et al. Frequent administration of levodopa/carbidopa microtablets vs levodopa/carbidopa/entacapone in healthy volunteers. Acta Neurol Scand. doi:10.1111/j.1600-0404.2012.01700.x. © 2012 John Wiley & Sons A/S. [DOI] [PubMed]
- 19.Nyholm D, Johansson A, Aquilonius SM, Hellquist E, Lennernas H, Askmark H. Complexity of motor response to different doses of duodenal levodopa infusion in Parkinson disease. Clin Neuropharmacol. 2012;35(1):6–14. doi: 10.1097/WNF.0b013e31823b1ffd. [DOI] [PubMed] [Google Scholar]
- 20.Nyholm D, Johansson A, Lennernas H, Askmark H. Levodopa infusion combined with entacapone or tolcapone in Parkinson disease: a pilot trial. Eur J Neurol. 2012;19(6):820–826. doi: 10.1111/j.1468-1331.2011.03614.x. [DOI] [PubMed] [Google Scholar]
- 21.Yeh KC, August TF, Bush DF, Lasseter KC, Musson DG, Schwartz S, et al. Pharmacokinetics and bioavailability of Sinemet CR: a summary of human studies. Neurology. 1989;39(11)(Suppl 2):25–38. [PubMed]
- 22.Chen C, Cowles VE, Sweeney M, Stolyarov ID, Illarioshkin SN. Pharmacokinetics and pharmacodynamics of gastroretentive delivery of levodopa/carbidopa in patients with Parkinson disease. Clin Neuropharmacol. 2012;35(2):67–72. doi: 10.1097/WNF.0b013e31824523de. [DOI] [PubMed] [Google Scholar]
- 23.Chen C, Cowles VE, Sweeney M, Stolyarov ID, Illarioshkin SN. Pharmacokinetics of levodopa/carbidopa delivered from gastric-retentive extended-release formulations in patients with Parkinson’s disease. J Clin Pharmacol. 2012;52(7):1069–1077. doi: 10.1177/0091270011409232. [DOI] [PubMed] [Google Scholar]
- 24.Hauser RA, Ellenbogen AL, Metman LV, Hsu A, O’Connell MJ, Modi NB, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord. 2011;26(12):2246–2252. doi: 10.1002/mds.23861. [DOI] [PubMed] [Google Scholar]
- 25.Bredberg E, Tedroff J, Aquilonius SM, Paalzow L. Pharmacokinetics and effects of levodopa in advanced Parkinson’s disease. Eur J Clin Pharmacol. 1990;39(4):385–389. doi: 10.1007/BF00315415. [DOI] [PubMed] [Google Scholar]
- 26.Lennernas H, Nilsson D, Aquilonius SM, Ahrenstedt O, Knutson L, Paalzow LK. The effect of L-leucine on the absorption of levodopa, studied by regional jejunal perfusion in man. Br J Clin Pharmacol. 1993;35(3):243–250. doi: 10.1111/j.1365-2125.1993.tb05691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wade DN, Mearrick PT, Morris JL. Active transport of L-dopa in the intestine. Nature. 1973;242(5398):463–465. doi: 10.1038/242463a0. [DOI] [PubMed] [Google Scholar]
- 28.Hidalgo IJ, Borchardt RT. Transport of a large neutral amino acid (phenylalanine) in a human intestinal epithelial cell line: Caco-2. Biochim Biophys Acta. 1990;1028(1):25–30. doi: 10.1016/0005-2736(90)90261-L. [DOI] [PubMed] [Google Scholar]
- 29.Westin J, Nyholm D, Palhagen S, Willows T, Groth T, Dougherty M, et al. A pharmacokinetic-pharmacodynamic model for duodenal levodopa infusion. Clin Neuropharmacol. 2011;34(2):61–65. doi: 10.1097/WNF.0b013e31820b570a. [DOI] [PubMed] [Google Scholar]
- 30.Nutt JG, Fellman JH. Pharmacokinetics of levodopa. Clin Neuropharmacol. 1984;7(1):35–49. doi: 10.1097/00002826-198403000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Nutt JG, Woodward WR, Anderson JL. The effect of carbidopa on the pharmacokinetics of intravenously administered levodopa: the mechanism of action in the treatment of parkinsonism. Ann Neurol. 1985;18(5):537–543. doi: 10.1002/ana.410180505. [DOI] [PubMed] [Google Scholar]
- 32.Contin M, Riva R, Martinelli P, Albani F, Baruzzi A. Effect of meal timing on the kinetic-dynamic profile of levodopa/carbidopa controlled release [corrected] in parkinsonian patients. Eur J Clin Pharmacol. 1998;54(4):303–308. doi: 10.1007/s002280050464. [DOI] [PubMed] [Google Scholar]
- 33.Sharpless NS, Muenter MD, Tyce GM, Owen CA., Jr 3-methoxy-4-hydroxyphenylalanine (3-O-methyldopa) in plasma during oral L-dopa therapy of patients with Parkinson’s disease. Clin chim Acta Int J Clin Chem. 1972;37:359–369. doi: 10.1016/0009-8981(72)90456-1. [DOI] [PubMed] [Google Scholar]
- 34.Contin M, Martinelli P. Pharmacokinetics of levodopa. J Neurol. 2010;257(suppl 2):S253–S261. doi: 10.1007/s00415-010-5728-8. [DOI] [PubMed] [Google Scholar]
- 35.Guttman M, Leger G, Cedarbaum JM, Reches A, Woodward W, Evans A, et al. 3-O-methyldopa administration does not alter fluorodopa transport into the brain. Ann Neurol. 1992;31(6):638–643. doi: 10.1002/ana.410310611. [DOI] [PubMed] [Google Scholar]
- 36.Nutt JG, Woodward WR, Gancher ST, Merrick D. 3-O-methyldopa and the response to levodopa in Parkinson’s disease. Ann Neurol. 1987;21(6):584–588. doi: 10.1002/ana.410210610. [DOI] [PubMed] [Google Scholar]
- 37.Hardie RJ, Malcolm SL, Lees AJ, Stern GM, Allen JG. The pharmacokinetics of intravenous and oral levodopa in patients with Parkinson’s disease who exhibit on-off fluctuations. Br J Clin Pharmacol. 1986;22(4):429–436. doi: 10.1111/j.1365-2125.1986.tb02913.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Durso R, Evans JE, Josephs E, Szabo G, Evans B, Fernandez HH, et al. Variable absorption of carbidopa affects both peripheral and central levodopa metabolism. J Clin Pharmacol. 2000;40(8):854–860. doi: 10.1177/00912700022009585. [DOI] [PubMed] [Google Scholar]
- 39.Elia AE, Dollenz C, Soliveri P, Albanese A. Motor features and response to oral levodopa in patients with Parkinson’s disease under continuous dopaminergic infusion or deep brain stimulation. Eur J Neurol. 2012;19(1):76–83. doi: 10.1111/j.1468-1331.2011.03437.x. [DOI] [PubMed] [Google Scholar]
- 40.Kuoppamaki M, Korpela K, Marttila R, Kaasinen V, Hartikainen P, Lyytinen J, et al. Comparison of pharmacokinetic profile of levodopa throughout the day between levodopa/carbidopa/entacapone and levodopa/carbidopa when administered four or five times daily. Eur J Clin Pharmacol. 2009;65(5):443–455. doi: 10.1007/s00228-009-0622-y. [DOI] [PubMed] [Google Scholar]
- 41.Stocchi F, Vacca L, Ruggieri S, Olanow CW. Intermittent vs continuous levodopa administration in patients with advanced Parkinson disease: a clinical and pharmacokinetic study. Arch Neurol. 2005;62(6):905–910. doi: 10.1001/archneur.62.6.905. [DOI] [PubMed] [Google Scholar]
- 42.Nyholm D, Lennernas H, Gomes-Trolin C, Aquilonius SM. Levodopa pharmacokinetics and motor performance during activities of daily living in patients with Parkinson’s disease on individual drug combinations. Clin Neuropharmacol. 2002;25(2):89–96. doi: 10.1097/00002826-200203000-00006. [DOI] [PubMed] [Google Scholar]
- 43.Stocchi F, Rascol O, Kieburtz K, Poewe W, Jankovic J, Tolosa E, et al. Initiating levodopa/carbidopa therapy with and without entacapone in early Parkinson disease: the STRIDE-PD study. Ann Neurol. 2010;68(1):18–27. doi: 10.1002/ana.22060. [DOI] [PubMed] [Google Scholar]