

Abstract
Physiologically based pharmacokinetic (PBPK) models are built using differential equations to describe the physiology/anatomy of different biological systems. Readily available in vitro and in vivo preclinical data can be incorporated into these models to not only estimate pharmacokinetic (PK) parameters and plasma concentration–time profiles, but also to gain mechanistic insight into compound properties. They provide a mechanistic framework to understand and extrapolate PK and dose across in vitro and in vivo systems and across different species, populations and disease states. Using small molecule and large molecule examples from the literature and our own company, we have shown how PBPK techniques can be utilised for human PK and dose prediction. Such approaches have the potential to increase efficiency, reduce the need for animal studies, replace clinical trials and increase PK understanding. Given the mechanistic nature of these models, the future use of PBPK modelling in drug discovery and development is promising, however some limitations need to be addressed to realise its application and utility more broadly.
KEY WORDS: absorption, clearance, distribution, dose prediction, physiologically based pharmacokinetic modelling
INTRODUCTION
Physiologically based pharmacokinetic (PBPK) models are tools that can be used to predict the pharmacokinetics (PK) and in combination with pharmacokinetic–pharmacodynamic (PKPD) models, predict the dose of new drug entities. These models are constructed using a series of differential equations that are parameterised using known physiology and represent a quantitative mechanistic framework by which the absorption, distribution, metabolism and excretion of new drugs can be explored. For this reason, they provide a useful starting point to understand and extrapolate PK and dose across in vitro and in vivo systems and across different species, populations and disease states.
Early reports of PBPK modelling use in the pharmaceutical industry are available but the application has been limited due to the mathematical complexity of the models and the large amounts of in vivo animal tissue concentration data required (1,2). However advances in the prediction of key parameters particularly tissue distribution such as the tissue to plasma partition coefficients (Kp values) (3–6) from in vitro and in silico data have made these models more attractive. Commercial PBPK packages have become available and strategies for the application of PBPK in the drug discovery setting have been published and recently evaluated (7,8). For this reason, the methodology has received an increase in attention in the last few years with several reports of its application for the prediction of animal and human PK for small molecules in discovery (7–13) and development (14–19).
Much of the literature describing the application of PBPK models for human PK and/or dose prediction has focused on small molecules. However a number of PBPK models for large molecules have also been reported (20–25). In general, these models require large amounts of in vivo tissue distribution data to describe how the large molecule distributes throughout the body. The absence of in vitro methods for the prediction of tissue distribution has limited their application in the pharmaceutical industry.
PBPK models provide the opportunity to integrate key input parameters from different sources to not only estimate PK parameters and predict plasma and tissue concentration–time profiles, but also to gain mechanistic insight into compound properties. The focus of this manuscript is on the application of PBPK models for both human PK prediction and human dose prediction. Examples are taken from the literature and on-going projects within Pfizer and include both small molecule and large molecule applications. Although many PBPK models have been published to predict drug–drug interactions, this is not the focus of this manuscript.
PBPK MODEL METHODOLOGIES
Small Molecule PBPK Models
PBPK models are made up of compartments corresponding to different tissues in the body, connected by the circulating blood system. Each compartment is defined by a tissue volume and tissue blood flow rate which is specific to the species of interest. These parameters have been reported in a number of publications (26,27). Each tissue can be described by perfusion rate limited or permeability rate limited assumptions. For small lipophilic molecules, blood flow to the tissue is the rate-limiting process and perfusion rate limited models are used. In contrast, for larger polar molecules, permeability across the cell membrane can become the rate-limiting process and permeability rate limited models are used. Generic PBPK models used in drug discovery usually assume perfusion rate limited kinetics with the liver and kidney being the only sites of clearance.
A schematic of a PBPK model is shown in Fig. 1. The mass balance differential equations used in these models have been described previously (8,10,28) and follow the principles shown below.
- Non-Eliminating Tissues:
where Q = blood flow (litre per hour), C = concentration (milligramme per litre), V = volume (litre), T = tissues, A = arterial, V = venous, CVT = CT / (Kp / B:P), Kp = tissue to plasma partition coefficient of the compound, and B:P = blood to plasma ratio.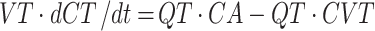
1 - Eliminating Tissues:
where CLint = the intrinsic clearance of the compound (litres/hour) and u = unbound.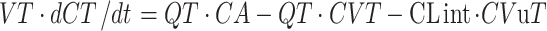
2
Fig. 1.
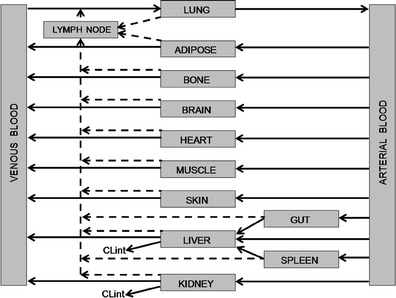
Schematic of a PBPK model. Blood flow (arrow), lymph flow (broken arrow)
As well as physiological parameters in the species of interest (e.g. rat, dog, human), to describe the intravenous (i.v.) and oral plasma concentration–time profile for a small molecule, the model requires compound-specific information (i.e. clearance, Kp values and the rate and extent of absorption). The model provides a physiological framework facilitating the incorporation of other mechanisms, e.g. active transport processes, as appropriate (29–31).
In vivo intrinsic organ clearance is a key parameter for PBPK models and can be scaled from a variety of in vitro systems (e.g. recombinant enzymes, microsomes and hepatocytes) using scaling factors such as inter-system extrapolation factors, microsomal recovery, hepatocellularity. These values can then be used directly in PBPK models (32) or can be used together with blood binding data and liver blood flow within liver models, e.g. well-stirred and parallel-tube models to predict hepatic CL (33). These approaches to predict hepatic clearance have been described in detail by a number of authors (34,35) and have been extensively validated in human (33,34,36–40). For other clearance mechanisms such as renal/biliary excretion of unchanged parent compound, other approaches can be used to predict in vivo intrinsic organ clearance. These methods may include allometry-based approaches (41–43) or scaling from in vitro systems if available (31,44,45).
A number of mechanistic, semi-mechanistic, and empirical methods have been described in the literature for the prediction of Kp values. In this publication, we will focus primarily on the mechanistic approaches. The development of mechanistic tissue composition-based equations for the prediction of Kp values and hence distribution in rat and human have enabled the routine application of PBPK methods in early drug discovery. These models dispense with the need for in vivo data and estimate the extent of tissue distribution from the physicochemical and in vitro binding characteristics to lipids and proteins of the compound. The equations developed by Poulin and coworkers (3,4,6) assume that the drug distributes homogenously into the tissue and plasma by passive diffusion accounting for non-specific binding to lipids estimated from drug lipophilicity data and specific reversible binding to proteins present in plasma and tissue estimated from plasma protein binding. Rodgers and coworker (5) extended these equations by incorporating ionisation/charge considerations. These equations account for partitioning of unionised drug into neutral lipids and neutral phospholipids, dissolution of ionised and unionised drug in tissue water, electrostatic interactions between ionised drug and acidic phospholipids for strong ionised bases and interactions with extracellular protein for neutrals, weak bases and acids. The tissue composition-based equations described above have been designed to describe specific interactions. Recently, unified algorithms combining these different processes have been developed to facilitate their application (6,46–48). Several studies have been performed to explore predictability of the different mechanistic approaches for Kp prediction using diverse drug datasets, with varying degrees of accuracy (8,10,11,48–53).
In order to predict oral PK, a mechanistic absorption model is required (54–56). These models rely on a variety of in vitro and/or in silico input data such as solubility, permeability, particle size, lipophilicity, pKa and dose, together with a series of differential equations to model the kinetics associated with dissolution, precipitation, uptake and absorption of a compound as it transits through the different segments of the digestive tract. Mechanistic absorption models are available in a number of commercial software. The absorption models developed by GastroPlus (‘Advanced Compartmental Absorption Transit’ model), SimCYP (‘Advanced, Dissolution, Absorption and Metabolism’ model) and PKSIM have been described in detail in the literature (57–59).
Overall, the integration of predictive in vitro systems for clearance, distribution and absorption together with the availability of commercial packages containing mechanistic PBPK models has revolutionised the application of PBPK modelling techniques throughout the industry.
Large Molecule PBPK Models
The major class of large molecules is the monoclonal antibodies (mAbs); for this reason, this section will focus on mAbs. The PK properties of different mAb molecules share similarities due to the IgG backbone. Following i.v. dosing, mAbs typically exhibit bi-exponential concentration decline in plasma, with a short alpha-phase (half-life of ∼3 days) followed by a long beta-phase (half-life of ∼3 weeks) in human (60). MAbs exhibit negligible urinary excretion because their size (molecular weight ∼150 kDa) restricts them from glomerular filtration. Instead, mAbs are eliminated by fluid phase endocytosis followed by lysosomal degradation (61–63). The long beta-phase is conferred by FcRn-mediated recycling which reduces clearance by fluid phase endocytosis (64). In vivo radioactive bio-distribution studies in mice have measured level of IgGs in different tissues (20,21,65,66). Experimental data in mice show tissue levels of ∼5–30% compared to plasma level for most tissues (an exception is the brain at <1%) (65). These studies have allowed for the development of PBPK models to describe the elimination and distributional properties of mAbs.
In a similar manner to the PBPK models described earlier in this manuscript for small molecules, mAb PBPK models are built up of a number of tissue compartments that are linked together by the blood flow. However in addition, lymph flow from all the tissues is delivered to the blood compartment via a lymph node compartment (Fig. 1). The models have been built to incorporate permeability limited distribution, with each tissue compartment being divided into vascular and interstitium compartments (20,21). These models were later extended to include an endosomal compartment to mathematically represent the IgG–FcRn interaction (66). Urva et al. (22) further added another compartment to represent cells expressing the target receptor in the tissue.
To mechanistically represent the transport of mAbs to tissues, PBPK models have decoupled overall IgG exchange between vascular and interstitium compartments in terms of passive (diffusion and convection) and active (FcRn-mediated transcytosis) transport processes. The passive transport flux from the vascular to the interstitium compartment can be described as shown in Eq. 3 (20,21,67,68).
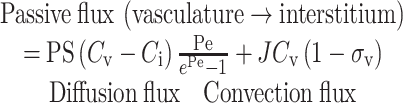 |
3 |
where, Cv and Ci are antibody concentrations in vascular and interstitium compartments, PS is permeability-surface area product, Pe is Peclet number, J is the fluid flow rate from vascular to interstitium compartment, and σv is vascular reflection coefficient (0 ≤ σv ≤ 1) representing the level of resistance provided to the mAb convection by the vascular endothelial barrier. The convective transport occurs by fluid exchange through gaps in the endothelial lining of capillary walls. Variations in gap sizes across different organs is captured by σv, e.g. σv for brain is close to 1 because endothelial cells at the blood brain barrier form tight junctions that do not allow convective transport of IgGs. The fluid from the interstitium returns to the circulation by the venous system (recirculation from interstitium) and the lymphatic system (21). Another set of reflection coefficients is associated with the convective transport via returning venous and lymphatic flow. The reflection coefficients are important determinants of Kp values for mAbs. For the diffusive transport term, PS captures the diffusivity and cross-sectional area of all the gaps. The Peclet number is a measure of the relative importance of convection to diffusion (Pe = J(1 − σv )/PS). Baxter et al. (21) concluded that diffusive flux contributes <2% to the transcapillary flux of IgGs based on PBPK model fitting to mouse distribution data. Hence, the diffusive flux has been neglected in many PBPK models. However, diffusion cannot be neglected for all mAb derivatives. In the same study, diffusion contributed ∼92% to the transcapillary exchange of Fabs (21).
Only recent models have included FcRn-mediated transport of IgG to tissues and its subsequent recycling (22–24,66,69). The role of FcRn in the uptake and protection of IgG was proposed by Brambell (70) and later confirmed by FcRn knockout mice studies. Mechanistic representation and parameterisation of this transport is challenging because the underlying FcRn biology is yet to be well characterised. Garg et al. (66) have modelled active FcRn-mediated transcytosis flux including the IgG uptake by fluid phase endocytosis; reversible binding to FcRn in the acidic environment of the endosomes and recycling of FcRn bound IgG back into the circulation and degradation of free IgG by a first-order process. Chabot et al. (24) have also allowed recycling of free IgG. Those models that have not incorporated FcRn have used a simple first-order elimination expression (20,21).
APPLICATION OF PBPK METHODOLOGIES FOR PK AND DOSE PREDICTION
Small Molecule PBPK Models
PBPK modelling can be applied for the purpose of PK and dose prediction across drug discovery and development from the early stages prior to lead development where limited data are available, during candidate selection and in early and late drug development where an expanse of data are available. The application of PBPK modelling for human PK and hence dose prediction has been more widespread in recent years with a number of publications from the Pharmaceutical Industry (7–13,16,17). In drug discovery and development, PBPK models can be iteratively refined to incorporate additional information on drug disposition as it becomes available from preclinical and clinical studies. A well-constructed PBPK model can play an important role in designing preclinical and clinical pharmacology studies by projecting drug PK profiles and doses under various scenarios. Table I gives a summary of some of these key applications. Jones et al. (8) proposed and validated a strategy for the use of PBPK for human PK simulations. Initially, the PBPK simulation is performed in animals using animal PBPK models, animal in vitro data and compound-specific physicochemical data. The animal simulation is compared with the in vivo data; if this simulation in animals is reasonable, then the human simulation is performed using a human PBPK model, human in vitro data and compound specific physicochemical data. If the simulation in animals is inaccurate, this would indicate a violation of one or more of the model assumptions, in this case, further experiments may be performed to understand the mismatch. This approach has been validated by a number of different authors using diverse drug datasets (8,10,11,71). Specific examples of successful prospective predictions of human PK using PBPK modelling can be found in the literature (72–74). These publications provide examples of where PBPK modelling has been used to design first-in-human clinical trials. In our laboratories, such predictions are successful in approximately 70% of cases.
Table I.
Summary of Some Key Examples of PBPK Application in Drug Discovery and Development for Simulation of Human PK and Dose in Different Populations
| Application | Reference |
|---|---|
| Prospective prediction of first-in-human PK in healthy volunteers | (72–74) |
| Retrospective prediction of first-in-human PK in healthy volunteers | (8–11,16,71) |
| Prediction of PK in paediatric populations | (78–82) |
| Prediction of PK in patients with hepatic impairment/liver cirrhosis | (75,76) |
| Prediction of PK in patients with renal impairment | (77,86,87) |
| Predictions of PK in other patient populations | (83–85) |
| Predictions of PK in polymorphic populations | (88) |
Given the mechanistic nature of these models, it is possible to incorporate physiological and mechanistic features to predict PK and dose in specific disease states and population groups. Incorporating known changes in hepatic blood flow, cytochrome P450, liver volume, hematocrit and liver/renal function as a function of disease or age has enabled reasonably accurate prediction of human PK for a number of compounds (75–77). In this regard, the strategy outlined by Jones et al. (8) can be extended to include these human populations (Fig. 2). The first workshop dedicated to exploring PBPK in drug development took place in 2002 (14). With the increased regulatory acceptance of such techniques (19), the use of PBPK models for this purpose is increasing. Indeed, current regulatory guidelines on PK in hepatically impaired patients recommend the development of PBPK models (15). Furthermore, the requirement of companies to submit a paediatric investigation plan before completion of the phase I trial has led many companies to use PBPK techniques to predict PK and set doses in children. Phase I PK data is used as a benchmark and differences in cytochrome P450 abundance, blood flows and tissue volumes can be incorporated to extrapolate to children using software such as SimCYP (78). In this context, between July 2008 and June 2010, the Office of Clinical Pharmacology at CDER, FDA, reviewed several IND and NDA submissions that incorporated PBPK simulations (18). Examples where PBPK approaches have been used to simulate PK in different populations and/or to justify dose adjustments/selection, include simulation of paediatric PK (78–82) and patient PK (83–85) from healthy volunteer data and prediction of the effects of organ impairment on PK (75–77,86,87).
Fig. 2.
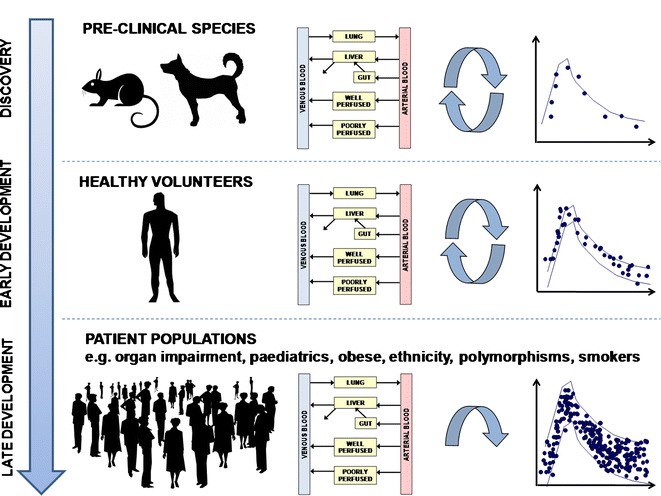
PBPK strategy through discovery to early- and late-stage development
Some in-house examples of where Pfizer have used PBPK approaches towards informed and model-based dose selection have been described in detail previously (88). These examples are described briefly below: (1) use of a high throughput dose optimisation PBPK tool in very early project stages to predict human dose for large number of compounds using in vitro or in silico inputs; (2) use of PBPK simulation techniques to help select the best compounds, doses and formulations to take forward to preclinical toxicological studies; (3) use of PBPK modelling at candidate selection to compare in-house and competitor substrate in terms of human PK and dose projection; (4) human PK and dose projections to help design first-in-human clinical trials; (5) food effect simulations to predict dose adjustments in the presence and absence of food; and (6) prediction of human PK mediated by polymorphic cytochrome P450 enzymes to estimate dose adjustments in these populations.
Large Molecule PBPK Models
One of the primary applications of PBPK models is to mechanistically translate preclinical data for predicting efficacious clinical dose and clinical PK (89). A compartmental model lumps the transport between plasma and tissue into a reversible first-order process. While such a model can fit data, its lumped parameters are not amenable for mechanistic translation. In contrast, PBPK models decouple vascular-interstitium exchange into fundamental physiological processes. Such decoupling allows for mechanistic translation across different species using species-specific (or animal model specific) physiological parameters for diffusion, convection and FcRn-mediated transport (21). Our discussions thus far have focussed on whole body PBPK models; however, the application of such models in the pharmaceutical industry for therapeutics is limited. In many applications the site of disease manifestation or target expression requires understanding of more detailed physiology than captured in the whole body PBPK models. Fit-for-purpose PBPK models offer a practical alternative. A fit-for-purpose PBPK model incorporates knowledge of biology around the therapeutic target, and of physiology around the target site of interest. In early stages, these models, assuming typical mAb properties, can guide affinity maturation by providing a quantitative link between affinity and dosing. In later drug development stages, the model can be updated with animal data specific to the therapeutic mAb to enable translation from preclinical species to human populations. We will illustrate fit-for-purpose PBPK models using one example from literature and one in-house example.
Adalimumab is an anti-TNF-alpha mAb approved for the treatment of rheumatoid arthritis (RA), an autoimmune disease which leads to inflammation of joints. To simulate adalimumab in human, Stepensky (90) developed a three-compartment model with central (vascular space), peripheral (extravascular space) and synovial fluid compartments. The terminal half-life of adalimumab is ∼2 weeks and the volumes for the central and peripheral compartments were ∼3 L each, similar to other reported mAbs (60). The synovial fluid compartment was modelled using physiological information for the synovial fluid volume and plasma–synovial fluid exchange rate. The rate of exchange at the plasma–synovium barrier has been experimentally reported by measuring the disappearance kinetics after intra-articular injection in RA patients (91). Using this model to simulate TNF-alpha knockdown via different dosing routes, the modelling indicates that intra-articular injections provide no benefit over i.v./subcutaneous injections. These results are expected based on the high levels of adalimumab in synovial fluid of RA patients (31–96% of plasma concentration). While these simulations are dependent on the model assumptions and parameters, our purpose to showcase this work is to provide an example of a fit-for-purpose PBPK model application. If model parameters and assumptions are reasonable, expensive clinical trials can be guided with such models.
In an early-stage project at Pfizer, we developed a fit-for-purpose PBPK model of a kidney target to explore mAb distribution and target suppression in the proximal tubule. While the specific details of the target biology are proprietary, we will explore a hypothetical target (cell surface target with a concentration of 100 pM and internalisation-mediated degradation half-life of 1 h−1). A schematic of our model is shown in Fig. 3. In our model, we have considered IgG transport between plasma and the proximal tubule through two transport routes: 1. glomerular filtration across the basement membrane, 2. Plasma <—(endothelial barrier)—> kidney interstitium <−−(epithelial barrier)—> proximal tubule. In a normal kidney, IgG transfer into the proximal tubule occurs predominantly via the first route. The rate of transport into the proximal tubule is characterised by the glomerular filtration rate (180 L/day) and the sieving coefficient (ratio of albumin concentration in filtrate to plasma). Excretion of very low amounts of endogenous IgG (urine ∼0.5–3.7 mg/day; plasma ∼10 g/L) in urine suggests low glomerular filtration (92). However, as a result of reabsorption, excreted IgG could be an underestimate of filtration. While direct measurement of IgG in the proximal tubule is not available, data is available for a comparable protein, albumin (∼2-fold lower molecular weight). A challenge in using the sieving coefficient is the large variation in these numbers reported in the literature. Based on recent debate in the field, we used a sieving coefficient range of 0.01–0.1% (93). Uptake from proximal tubule into plasma was set at 99% of filtered IgG, leaving 1% to be excreted by urine. For simplicity, we represent the proximal tubule as one compartment (volume = 30 mL) neglecting the concentration gradient along the nephron length (94). As the parameters in this model are linked directly to physiological processes, they can be altered to assess mAb distribution at different stages of the disease as well as treatment. To model the severe stage of nephritic kidney disease we included a 5-fold reduction in GFR with a high sieving coefficient of 1%, and to represent a compromised epithelial cell barrier we assumed instantaneously fast transport of IgG from the interstitium to the proximal tubule. Assuming typical mAb PK properties (60) and using i.v. dosing, Fig. 4 shows the simulated target suppression of mAbs with different affinities under the diseased condition. To achieve >95% target suppression, high dosing of 10 mg/kg every 2 weeks and high mAb affinity of 100 pM is required. Such simulations can be used to explore the utility of the target and to set expectations with respect to dosing and affinity requirements at early stages in the project life-cycle. With the improvement of kidney function as treatment progresses, it may become harder for the mAb to reach the proximal tubule due to improvements in the integrity of glomerulus and epithelial cell barriers. Dependent on the availability of data, treatment progression can be modelled by modifying the physiological parameters.
Fig. 3.
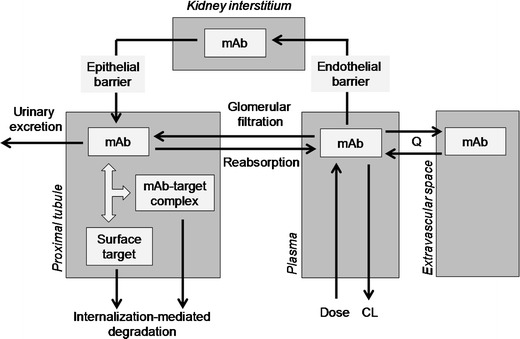
Graphical representation of mAb kidney model (see text for details of parameters)
Fig. 4.
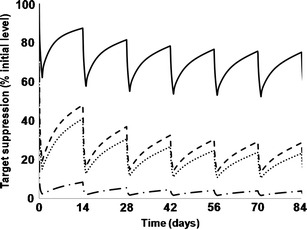
Model predictions of target suppression. Different curves correspond to different combinations of mAb affinities and dosing. MAb distribution parameters: Inter-compartmental clearance = 0.6 L/day, clearance = 0.25 L/day. Volumes of different compartments are: plasma = extravascular space accessed by mAb = 3 L; kidney interstitium = 100 mL; proximal tubule = 30 mL. Rate of fluid transport to kidney interstitium is set at 4.5 L/day, i.e. 0.5% of plasma flow rate to kidney; 85% of which is recirculated and remaining 15% ends up as lymphatic fluid. Reflection coefficients associated with endothelial barrier are 0.9 and 0.2 (24). Target parameters: expression = 100 pM; internalisation of free target = 1 h−1; internalisation of target–mAb complex = 5 h−1. Solid line represents KD = 1 nM, dose = 1 mg/kg; dotted line represents KD = 1 nM, dose = 10 mg/kg; dashed line represents KD = 100pM, dose = 1 mg/kg; dash-dot line represents KD = 100pM, dose = 10 mg/kg
CHALLENGES AND FUTURE DIRECTION
Small Molecule PBPK Models
The integration of physicochemical, in vitro and in vivo data into a PBPK framework to simulate PK profiles of small molecules in humans has become widespread within the Pharmaceutical Industry with numerous groups publishing on their experience with this approach (7–13,16–19). Although PBPK methods have shown utility in predicting human PK a few major challenges remain that need to be addressed to further increase the success of this approach.
Most publications show good predictions of distribution and absorption. One of the biggest challenges is to improve the accuracy of prediction methodologies for human clearance. Although a number of methods have been described in the literature, the prediction accuracy is still limited to only 50–80% of compounds being within 2-fold of actual observed clearance, with significant mis-predictions being observed for some compounds. These mis-predictions can become magnified when incorporated into a PBPK model to predict oral exposure. Knowing which method will work for the compound of interest before the human data is generated is a major hurdle, as no single method can predict human clearance for all compounds. Obviously an understanding of the physicochemistry and clearance mechanisms of compounds can help to narrow down the choice. An analysis of 15 compounds cleared via P450 metabolism, indicates that in 80% of cases a good/poor prediction of clearance in preclinical species resulted in a good/poor prediction of clearance in human respectively (calculated using data from (10)). However there are pitfalls, for low clearance compounds, some of the more widely used in vitro methods (i.e. depletion of parent compound in human liver microsomes and hepatocytes) show limitations as the disappearance from the assay over the time course of the incubation is too slow to accurately measure a depletion rate. Improvements in these methodologies to measure turnover over longer time periods will allow application of these techniques to a wider selection of compounds. A recent publication from Di et al. (95) shows promise in this regard.
With efforts to reduce clearance in in vitro systems during drug discovery, transporter-mediated PK is becoming more prevalent. Such processes provide additional challenges to clearance prediction. Several groups have described recently their PBPK modelling strategies to incorporate the influence of transporters in to the prediction of human clearance from in vitro systems (29–31). However these approaches require large relative activity factors to account for the differences in activity in vitro and activity in vivo. At Pfizer, we have shown success in predicting the human PK for a set of seven compounds that are substrates for both uptake transporters and metabolic enzymes by scaling data generated in vitro from sandwich cultured human hepatocytes (31). For these compounds, this approach was more successful than predicting clearance from either scaling in vitro metabolic data alone or from scaling preclinical PK data. The basis for these scaling factors is unclear. An evaluation of organic anion-transporting polypeptide (OATP) expression in sandwich cultured human hepatocytes, showed that, while OATP1B3/ OATP2B1 expression were reduced to ∼50% of that in suspension, OATP1B1 expression was increased to ∼150% (96). However, the difference between OATP expression in culture and in vivo is unknown and is likely to contribute to the scaling factors. As well as the liver, PBPK models can be used to simulate transporter processes other tissues, e.g. kidney and intestine. As our understanding of transporter mechanisms is enhanced and the abundance of these transporters in different tissues is known, these applications are likely to increase.
A particular strength of PBPK modelling is the mechanistic framework which allows the extrapolation of PK across different in vitro systems and species/populations by simply updating the physiological/anatomical parameters (e.g. tissue volumes, blood flows, transit times, expression levels). Knowledge of the variability of these different parameters enables the use of Monte Carlo methods, to predict variability in PK and to anticipate individuals whose attributes combine to give extreme PK risk (35,97,98). Further extensions of the approach taken by SimCYP to incorporate individual characteristics within the PBPK simulations will continue to evolve and be more widely applied. Indeed, recently a number of cases have been published where PBPK models have been developed for healthy volunteers and updated with patient physiology to predict PK in patients successfully (75–77,86,87). This approach has the potential to make personalised medicine a reality where the drug PK, efficacy and side effects can be predicted in a given individual with a known genotypic make-up for relevant ADME and pharmacology proteins. For this to be successful, different academic centres, pharmaceutical companies and regulatory authorities need to collaborate to ensure that that the missing system parameters in different disease populations are measured.
Large Molecule PBPK Models
The application of PBPK modelling for mAbs in the pharmaceutical industry has been limited. Most models have required large amounts of in vivo data for fitting. While published PBPK models can fit the experimental data, different groups have used not only different physiological parameters but also different representations of physiology to fit the data (Table II). This poses a challenge for their application for dose projections within the pharmaceutical industry, particularly with respect to the relevance of the different components and assumptions within the model. Reaching a consensus on the values of relevant systems parameters within these models and the model structure is required for its application. Some examples of these inconsistencies across models are described below.
Table II.
Similarities and Differences Between Published Large Molecule Whole Body PBPK Models
| Model description | Reference | ||
|---|---|---|---|
| Model structure | Multiple compartments with each tissue compartment being divided into vascular and interstitium compartments | (20,21) | |
| Multiple compartments with each tissue compartment being divided into vascular and interstitium compartments and endosomal compartment | (22–24,66,69) | ||
| IgG exchange between vascular and interstitium compartments | Diffusive transport | Peclet number dependency excluded | (20,100) |
| Peclet number dependency included | (21,24,25,69) | ||
| Convective transport | J defined as lymphatic flow rate | (20,22,23,66) | |
| J defined as venous recirculation + lymphatic flow rates | (21,24,25,69) | ||
| FcRn-mediated transcytosis | (22–25,66) | ||
| IgG elimination | First-order process | (20,21) | |
| FcRn-mediated recycling | (22–25,66,69) | ||
Lymphatic flow rates covering a 200-fold range (0.02–4%) have been used in the PBPK models published in the literature for the same organ (20,21,23,24,66). Despite large differences in lymphatic flow rates across different PBPK models, all models fit the experimental data reasonably well. However, such large differences in lymphatic flows across different models raise a fundamental question about their uniqueness and over-parameterisation (99). PBPK models are inconsistent in defining J in Eq. 3. Several models (20,22,23,66) have modelled J as the lymphatic flow. However, not all the fluid that drains from arterial capillaries ends up as lymphatic fluid. In a human approximately 0.5% of the plasma flow rate enters tissue interstitium of which the majority is recirculated back at the venous end of capillaries and the remaining (0.07% of blood flow) returns to circulation via lymphatic flow. In accordance with this physiological description, some models (21,24,25) have included venous recirculation in addition to the lymphatic flow. Using the human flow rates, different interpretations of J alone can lead to ∼7-fold difference in the rate of convective transport flux. This difference in convective flux can lead to changes in ability of the mAb to modulate (agonist or antagonist) the target influencing dose prediction. Another less prevalent inconsistency is in the description of diffusive transport flux. Some authors (20,100) did not include the Peclet number dependency in the diffusion term (see Eq. 3). Such a simplification can affect relative contribution of diffusive transport.
Another major difference across different mAb PBPK models is in the inclusion (or not) of IgG–FcRn interaction. Early PBPK models captured IgG distribution and clearance data without explicit representation of IgG–FcRn interaction (21). The beta-phase of a typical therapeutic mAb can be reasonably captured by first-order elimination process. Ferl et al. (69) and Garg et al. (66) published some of the first papers including the IgG–FcRn interaction in whole body PBPK models. While the FcRn knockout mice study from Garg et al. (66) has provided data for all of these models, more extensive data is needed to model FcRn-mediated processes. As with small molecule PBPK models describing active transport processes, the limitation for modelling the IgG–FcRn interaction is the lack of expression data measuring the number of FcRn copies on endothelial cells. In terms of utility, one of the main applications of an IgG–FcRn mechanistic model would be to predict changes in pharmacokinetics and distribution as a result of engineered modulations in mAb-FcRn affinity. There has been a significant therapeutic interest in increasing mAb-FcRn affinity to lower dose (or dosing frequency). However, these efforts have achieved limited success. For example, a Genentech study (101) demonstrated that a 4-fold increase in IgG1–FcRn affinity leads to a ∼2-fold improvement in clearance for one mAb, but a ∼80-fold increase in affinity did not significantly improve clearance for another mAb. One possible explanation for these observations is that the positive effect of improved rescue due to tighter IgG1–FcRn binding at acidic pH could be cancelled by reduced release of recycled IgG1 at neutral pH, if the modification also improves IgG1-FcRn binding at pH 7.4 (64). Existing PBPK models do not include any IgG–FcRn interaction at pH 7.4 so they cannot capture these data.
CONCLUSION
The application of PBPK methodology to predict human PK and/or dose of small molecules within the pharmaceutical industry has increased over the last decade, particularly with the development of in vitro to in vivo extrapolation techniques of key processes and the advent of commercially available ‘easy to use’ PBPK tools. In the pharmaceutical industry, the incomplete knowledge of FcRn biology has limited the ‘generic use’ of whole-body PBPK modelling for large molecules. Using examples from within Pfizer, as well as literature data, we have shown how PBPK techniques (whole body and fit-for-purpose) can be used throughout the stages of drug discovery and development to select dose in animals and different human populations. Given the mechanistic nature of these models, the future for PBPK modelling is promising with many potential opportunities to be explored. However several limitations need to be addressed, particularly around the incorporation of active transport processes (OATP, P-gp and FcRN mediated), to realise its application and utility more broadly.
ACKNOWLEDGMENTS
The authors would like to acknowledge the contribution of all colleagues in Pfizer Worldwide Research and Development that were involved in the projects described in this manuscript.
REFERENCES
- 1.Tanaka C, Kawai R, Rowland M. Dose-dependent pharmacokinetics of cyclosporin A in rats: events in tissues. Drug Metab Dispos. 2000;28(5):582–589. [PubMed] [Google Scholar]
- 2.Nestorov I. Whole body pharmacokinetic models. Clin Pharmacokinet. 2003;42(10):883–908. doi: 10.2165/00003088-200342100-00002. [DOI] [PubMed] [Google Scholar]
- 3.Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J Pharm Sci. 2002;91(1):129–156. doi: 10.1002/jps.10005. [DOI] [PubMed] [Google Scholar]
- 4.Berezhkovskiy LM. Determination of volume of distribution at steady state with complete consideration of the kinetics of protein and tissue binding in linear pharmacokinetics. J Pharm Sci. 2004;93(2):364–374. doi: 10.1002/jps.10539. [DOI] [PubMed] [Google Scholar]
- 5.Rodgers T, Rowland M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm Res. 2007;24(5):918–933. doi: 10.1007/s11095-006-9210-3. [DOI] [PubMed] [Google Scholar]
- 6.Poulin P, Haddad S. Advancing prediction of tissue distribution and volume of distribution of highly lipophilic compounds from a simplified tissue-composition-based model as a mechanistic animal alternative method. J Pharm Sci. 2012;101(6):2250–2261. doi: 10.1002/jps.23090. [DOI] [PubMed] [Google Scholar]
- 7.Theil FP, Guentert TW, Haddad S, Poulin P. Utility of physiologically based pharmacokinetic models to drug development and rational drug discovery candidate selection. Toxicol Lett. 2003;138(1–2):29–49. doi: 10.1016/S0378-4274(02)00374-0. [DOI] [PubMed] [Google Scholar]
- 8.Jones HM, Parrott N, Jorga K, Lave T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin Pharmacokinet. 2006;45(5):511–542. doi: 10.2165/00003088-200645050-00006. [DOI] [PubMed] [Google Scholar]
- 9.Brightman FA, Leahy DE, Searle GE, Thomas S. Application of a generic physiologically-based pharmacokinetic model to the estimation of xenobiotic levels in human plasma. Drug Metab Dispos. 2006;34(1):94–101. doi: 10.1124/dmd.105.004838. [DOI] [PubMed] [Google Scholar]
- 10.Jones HM, Gardner IB, Collard WT, Stanley PJ, Oxley P, Hosea NA, et al. Simulation of human intravenous and oral pharmacokinetics of 21 diverse compounds using physiologically based pharmacokinetic modelling. Clin Pharmacokinet. 2011;50(5):331–347. doi: 10.2165/11539680-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 11.De Buck SS, Sinha VK, Fenu LA, Nijsen MJ, Mackie CE, Gilissen RA. Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs. Drug Metab Dispos. 2007;35(10):1766–1780. doi: 10.1124/dmd.107.015644. [DOI] [PubMed] [Google Scholar]
- 12.Sinha VK, Snoeys J, Osselaer NV, Peer AV, Mackie C, Heald D. From preclinical to human–prediction of oral absorption and drug-drug interaction potential using physiologically based pharmacokinetic (PBPK) modeling approach in an industrial setting: a workflow by using case example. Biopharm Drug Dispos. 2012;33(2):111–121. doi: 10.1002/bdd.1782. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y, Jin JY, Mukadam S, Malhi V, Kenny JR. Application of IVIVE and PBPK modeling in prospective prediction of clinical pharmacokinetics: strategy and approach during the drug discovery phase with four case studies. Biopharm Drug Dispos. 2012;33(2):85–98. doi: 10.1002/bdd.1769. [DOI] [PubMed] [Google Scholar]
- 14.Rowland M, Balant L, Peck C. Physiologically based pharmacokinetics in drug development and regulatory science: a workshop report (Georgetown University, Washington, DC, May 29–30, 2002) AAPS PharmSci. 2004;6(1):E6. doi: 10.1208/ps060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rowland M, Peck C, Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45–73. doi: 10.1146/annurev-pharmtox-010510-100540. [DOI] [PubMed] [Google Scholar]
- 16.Peters SA. Evaluation of a generic physiologically based pharmacokinetic model for lineshape analysis. Clin Pharmacokinet. 2008;47(4):261–275. doi: 10.2165/00003088-200847040-00004. [DOI] [PubMed] [Google Scholar]
- 17.Edginton AN, Theil FP, Schmitt W, Willmann S. Whole body physiologically-based pharmacokinetic models: their use in clinical drug development. Expert Opin Drug Metab Toxicol. 2008;4(9):1143–1152. doi: 10.1517/17425255.4.9.1143. [DOI] [PubMed] [Google Scholar]
- 18.Zhao P, Zhang L, Grillo JA, Liu Q, Bullock JM, Moon YJ, et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther. 2011;89(2):259–267. doi: 10.1038/clpt.2010.298. [DOI] [PubMed] [Google Scholar]
- 19.Huang SM, Rowland M. The role of physiologically based pharmacokinetic modeling in regulatory review. Clin Pharmacol Ther. 2012;91(3):542–549. doi: 10.1038/clpt.2011.320. [DOI] [PubMed] [Google Scholar]
- 20.Covell DG, Barbet J, Holton OD, Black CD, Parker RJ, Weinstein JN. Pharmacokinetics of monoclonal immunoglobulin G1, F(ab’)2, and Fab’ in mice. Cancer Res. 1986;46(8):3969–3978. [PubMed] [Google Scholar]
- 21.Baxter LT, Zhu H, Mackensen DG, Jain RK. Physiologically based pharmacokinetic model for specific and nonspecific monoclonal antibodies and fragments in normal tissues and human tumor xenografts in nude mice. Cancer Res. 1994;54(6):1517–1528. [PubMed] [Google Scholar]
- 22.Urva SR, Yang VC, Balthasar JP. Physiologically based pharmacokinetic model for T84.66: a monoclonal anti-CEA antibody. J Pharm Sci. 2010;99(3):1582–1600. doi: 10.1002/jps.21918. [DOI] [PubMed] [Google Scholar]
- 23.Shah DK, Betts AM. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 2012;39(1):67–86. doi: 10.1007/s10928-011-9232-2. [DOI] [PubMed] [Google Scholar]
- 24.Chabot JR, Dettling DE, Jasper PJ, Gomes BC. Comprehensive mechanism-based antibody pharmacokinetic modeling. Conf Proc IEEE Eng Med Biol Soc. 2011;2011:4318–4323. doi: 10.1109/IEMBS.2011.6091072. [DOI] [PubMed] [Google Scholar]
- 25.Davda JP, Jain M, Batra SK, Gwilt PR, Robinson DH. A physiologically based pharmacokinetic (PBPK) model to characterize and predict the disposition of monoclonal antibody CC49 and its single chain Fv constructs. Int Immunopharmacol. 2008;8(3):401–413. doi: 10.1016/j.intimp.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Heal. 1997;13(4):407–484. doi: 10.1177/074823379701300401. [DOI] [PubMed] [Google Scholar]
- 27.Thompson CM, Johns DO, Sonawane B, Barton HA, Hattis D, Tardif R, et al. Database for physiologically based pharmacokinetic (PBPK) modeling: physiological data for healthy and health-impaired elderly. J Toxicol Environ Health B Crit Rev. 2009;12(1):1–24. doi: 10.1080/10937400802545060. [DOI] [PubMed] [Google Scholar]
- 28.Pang KS, Durk MR. Physiologically-based pharmacokinetic modeling for absorption, transport, metabolism and excretion. J Pharmacokinet Pharmacodyn. 2010;37(6):591–615. doi: 10.1007/s10928-010-9185-x. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe T, Kusuhara H, Maeda K, Shitara Y, Sugiyama Y. Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J Pharmacol Exp Ther. 2009;328(2):652–662. doi: 10.1124/jpet.108.146647. [DOI] [PubMed] [Google Scholar]
- 30.Poirier A, Cascais AC, Funk C, Lave T. Prediction of pharmacokinetic profile of valsartan in humans based on in vitro uptake-transport data. Chem Biodivers. 2009;6(11):1975–1987. doi: 10.1002/cbdv.200900116. [DOI] [PubMed] [Google Scholar]
- 31.Jones HM, Barton HA, Lai Y, Bi YA, Kimoto E, Kempshall S, et al. Mechanistic pharmacokinetic modeling for the prediction of transporter-mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab Dispos. 2012;40(5):1007–1017. doi: 10.1124/dmd.111.042994. [DOI] [PubMed] [Google Scholar]
- 32.Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, Rostami-Hodjegan A. The Simcyp population-based ADME simulator. Expert Opin Drug Metab Toxicol. 2009;5(2):211–223. doi: 10.1517/17425250802691074. [DOI] [PubMed] [Google Scholar]
- 33.Hallifax D, Foster JA, Houston JB. Prediction of human metabolic clearance from in vitro systems: retrospective analysis and prospective view. Pharm Res. 2010;27(10):2150–2161. doi: 10.1007/s11095-010-0218-3. [DOI] [PubMed] [Google Scholar]
- 34.Houston JB. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol. 1994;47(9):1469–1479. doi: 10.1016/0006-2952(94)90520-7. [DOI] [PubMed] [Google Scholar]
- 35.Rostami-Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov. 2007;6(2):140–148. doi: 10.1038/nrd2173. [DOI] [PubMed] [Google Scholar]
- 36.Iwatsubo T, Hirota N, Ooie T, Suzuki H, Shimada N, Chiba K, et al. Prediction of in vivo drug metabolism in the human liver from in vitro metabolism data. Pharmacol Ther. 1997;73(2):147–171. doi: 10.1016/S0163-7258(96)00184-2. [DOI] [PubMed] [Google Scholar]
- 37.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27(11):1350–1359. [PubMed] [Google Scholar]
- 38.Shiran MR, Proctor NJ, Howgate EM, Rowland-Yeo K, Tucker GT, Rostami-Hodjegan A. Prediction of metabolic drug clearance in humans: in vitro-in vivo extrapolation vs allometric scaling. Xenobiotica. 2006;36(7):567–580. doi: 10.1080/00498250600761662. [DOI] [PubMed] [Google Scholar]
- 39.Ring BJ, Chien JY, Adkison KK, Jones HM, Rowland M, Jones RD, et al. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 3: comparative assessement of prediction methods of human clearance. J Pharm Sci. 2011 May 3. [DOI] [PubMed]
- 40.Poulin P, Hop CE, Ho Q, Halladay JS, Haddad S, Kenny JR. Comparative assessment of in vitro-in vivo extrapolation methods used for predicting hepatic metabolic clearance of drugs. J Pharm Sci. 2012;101(11):4308–4326. doi: 10.1002/jps.23288. [DOI] [PubMed] [Google Scholar]
- 41.Paine SW, Menochet K, Denton R, McGinnity DF, Riley RJ. Prediction of human renal clearance from preclinical species for a diverse set of drugs that exhibit both active secretion and net reabsorption. Drug Metab Dispos. 2011;39(6):1008–1013. doi: 10.1124/dmd.110.037267. [DOI] [PubMed] [Google Scholar]
- 42.Mahmood I. Interspecies scaling of renally secreted drugs. Life Sci. 1998;63(26):2365–2371. doi: 10.1016/S0024-3205(98)00525-6. [DOI] [PubMed] [Google Scholar]
- 43.Mahmood I, Sahajwalla C. Interspecies scaling of biliary excreted drugs. J Pharm Sci. 2002;91(8):1908–1914. doi: 10.1002/jps.10174. [DOI] [PubMed] [Google Scholar]
- 44.Bi YA, Kazolias D, Duignan DB. Use of cryopreserved human hepatocytes in sandwich culture to measure hepatobiliary transport. Drug Metab Dispos. 2006;34(9):1658–1665. doi: 10.1124/dmd.105.009118. [DOI] [PubMed] [Google Scholar]
- 45.Abe K, Bridges AS, Brouwer KL. Use of sandwich-cultured human hepatocytes to predict biliary clearance of angiotensin II receptor blockers and HMG-CoA reductase inhibitors. Drug Metab Dispos. 2009;37(3):447–452. doi: 10.1124/dmd.108.023465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmitt W. General approach for the calculation of tissue to plasma partition coefficients. Toxicol In Vitro. 2008;22(2):457–67. [DOI] [PubMed]
- 47.Peyret T, Poulin P, Krishnan K. A unified algorithm for predicting partition coefficients for PBPK modeling of drugs and environmental chemicals. Toxicol Appl Pharmacol. 2010;249(3):197–207. doi: 10.1016/j.taap.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 48.Poulin P, Ekins S, Theil FP. A hybrid approach to advancing quantitative prediction of tissue distribution of basic drugs in human. Toxicol Appl Pharmacol. 2011;250(2):194–212. doi: 10.1016/j.taap.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 49.Poulin P, Theil FP. Development of a novel method for predicting human volume of distribution at steady-state of basic drugs and comparative assessment with existing methods. J Pharm Sci. 2009;98(12):4941–4961. doi: 10.1002/jps.21759. [DOI] [PubMed] [Google Scholar]
- 50.Jones RD, Jones HM, Rowland M, Gibson CR, Yates JW, Chien JY, et al. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 2: comparative assessment of prediction methods of human volume of distribution. J Pharm Sci. 2011 Mar 30. [DOI] [PubMed]
- 51.Graham H, Walker M, Jones O, Yates J, Galetin A, Aarons L. Comparison of in-vivo and in-silico methods used for prediction of tissue: plasma partition coefficients in rat. J Pharm Pharmacol. 2012;64(3):383–396. doi: 10.1111/j.2042-7158.2011.01429.x. [DOI] [PubMed] [Google Scholar]
- 52.Small H, Gardner I, Jones HM, Davis JD, Rowland M. Measurement of binding of basic drugs to acidic phospholipids using surface plasmon resonance and incorporation of the data into mechanistic tissue composition equations to predict steady-state volume of distribution. Drug Metab Dispos. 2011;39(10):1789–1793. doi: 10.1124/dmd.111.040253. [DOI] [PubMed] [Google Scholar]
- 53.An G, Morris ME. A physiologically based pharmacokinetic model of mitoxantrone in mice and scale-up to humans: a semi-mechanistic model incorporating DNA and protein binding. AAPS J. 2012;14(2):352–364. doi: 10.1208/s12248-012-9344-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu LX, Amidon GL. A compartmental absorption and transit model for estimating oral drug absorption. Int J Pharm. 1999;186(2):119–125. doi: 10.1016/S0378-5173(99)00147-7. [DOI] [PubMed] [Google Scholar]
- 55.Sugano K. Introduction to computational oral absorption simulation. Expert Opin Drug Metab Toxicol. 2009;5(3):259–293. doi: 10.1517/17425250902835506. [DOI] [PubMed] [Google Scholar]
- 56.Sugano K. Computational oral absorption simulation for low-solubility compounds. Chem Biodivers. 2009;6(11):2014–2029. doi: 10.1002/cbdv.200900101. [DOI] [PubMed] [Google Scholar]
- 57.Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001;50(Suppl 1):S41–S67. doi: 10.1016/S0169-409X(01)00179-X. [DOI] [PubMed] [Google Scholar]
- 58.Jamei M, Turner D, Yang J, Neuhoff S, Polak S, Rostami-Hodjegan A, et al. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009;11(2):225–237. doi: 10.1208/s12248-009-9099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Willmann S, Schmitt W, Keldenich J, Lippert J, Dressman JB. A physiological model for the estimation of the fraction dose absorbed in humans. J Med Chem. 2004;47(16):4022–4031. doi: 10.1021/jm030999b. [DOI] [PubMed] [Google Scholar]
- 60.Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(10):633–659. doi: 10.2165/11535960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 61.Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93(11):2645–2668. doi: 10.1002/jps.20178. [DOI] [PubMed] [Google Scholar]
- 62.Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–558. doi: 10.1038/clpt.2008.170. [DOI] [PubMed] [Google Scholar]
- 63.Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006;11(1–2):81–88. doi: 10.1016/S1359-6446(05)03638-X. [DOI] [PubMed] [Google Scholar]
- 64.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7(9):715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 65.Vugmeyster Y, DeFranco D, Szklut P, Wang Q, Xu X. Biodistribution of [125I]-labeled therapeutic proteins: application in protein drug development beyond oncology. J Pharm Sci. 2010;99(2):1028–1045. doi: 10.1002/jps.21855. [DOI] [PubMed] [Google Scholar]
- 66.Garg A, Balthasar JP. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn. 2007;34(5):687–709. doi: 10.1007/s10928-007-9065-1. [DOI] [PubMed] [Google Scholar]
- 67.Patlak CS, Goldstein DA, Hoffman JF. The flow of solute and solvent across a two-membrane system. J Theor Biol. 1963;5(3):426–442. doi: 10.1016/0022-5193(63)90088-2. [DOI] [PubMed] [Google Scholar]
- 68.Rippe B, Haraldsson B. Transport of macromolecules across microvascular walls: the two-pore theory. Physiol Rev. 1994;74(1):163–219. doi: 10.2466/pr0.1994.74.1.163. [DOI] [PubMed] [Google Scholar]
- 69.Ferl GZ, Wu AM, DiStefano JJ., 3rd A predictive model of therapeutic monoclonal antibody dynamics and regulation by the neonatal Fc receptor (FcRn) Ann Biomed Eng. 2005;33(11):1640–1652. doi: 10.1007/s10439-005-7410-3. [DOI] [PubMed] [Google Scholar]
- 70.Brambell FW, Hemmings WA, Morris IG. A theoretical model of gamma-globulin catabolism. Nature. 1964;203:1352–1354. doi: 10.1038/2031352a0. [DOI] [PubMed] [Google Scholar]
- 71.Poulin P, Jones RD, Jones HM, Gibson CR, Rowland M, Chien JY, et al. PHRMA CPCDC initiative on predictive models of human pharmacokinetics, part 5: prediction of plasma concentration-time profiles in human by using the physiologically-based pharmacokinetic modeling approach. J Pharm Sci. 2011. doi:10.1002/jps.22550. [DOI] [PubMed]
- 72.Bungay PJ, Tweedy S, Howe DC, Gibson KR, Jones HM, Mount NM. Pre-clinical and clinical pharmacokinetics of PF-02413873, a non-steroidal progesterone receptor antagonist. Drug Metab Dispos. 2011;39(8):1396–1405. doi: 10.1124/dmd.110.037234. [DOI] [PubMed] [Google Scholar]
- 73.Yamazaki S, Skaptason J, Romero D, Vekich S, Jones HM, Tan W, et al. Prediction of oral pharmacokinetics of cMet kinase inhibitors in humans: physiologically based pharmacokinetic model versus traditional one-compartment model. Drug Metab Dispos. 2011;39(3):383–393. doi: 10.1124/dmd.110.035857. [DOI] [PubMed] [Google Scholar]
- 74.Allan G, Davis J, Dickins M, Gardner I, Jenkins T, Jones H, et al. Pre-clinical pharmacokinetics of UK-453,061, a novel non-nucleoside reverse transcriptase inhibitor (NNRTI), and use of in silico physiologically based prediction tools to predict the oral pharmacokinetics of UK-453,061 in man. Xenobiotica. 2008;38(6):620–640. doi: 10.1080/00498250802069088. [DOI] [PubMed] [Google Scholar]
- 75.Edginton AN, Willmann S. Physiology-based simulations of a pathological condition: prediction of pharmacokinetics in patients with liver cirrhosis. Clin Pharmacokinet. 2008;47(11):743–752. doi: 10.2165/00003088-200847110-00005. [DOI] [PubMed] [Google Scholar]
- 76.Johnson TN, Boussery K, Rowland-Yeo K, Tucker GT, Rostami-Hodjegan A. A semi-mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin Pharmacokinet. 2010;49(3):189–206. doi: 10.2165/11318160-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 77.Zhao P, Vieira Mde L, Grillo JA, Song P, Wu TC, Zheng JH, et al. Evaluation of exposure change of nonrenally eliminated drugs in patients with chronic kidney disease using physiologically based pharmacokinetic modeling and simulation. J Clin Pharmacol. 2012;52(1 Suppl):91S–108S. doi: 10.1177/0091270011415528. [DOI] [PubMed] [Google Scholar]
- 78.Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. 2006;45(9):931–956. doi: 10.2165/00003088-200645090-00005. [DOI] [PubMed] [Google Scholar]
- 79.Bouzom F, Walther B. Pharmacokinetic predictions in children by using the physiologically based pharmacokinetic modelling. Fundam Clin Pharmacol. 2008;22(6):579–587. doi: 10.1111/j.1472-8206.2008.00648.x. [DOI] [PubMed] [Google Scholar]
- 80.Edginton AN, Schmitt W, Willmann S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin Pharmacokinet. 2006;45(10):1013–1034. doi: 10.2165/00003088-200645100-00005. [DOI] [PubMed] [Google Scholar]
- 81.Parrott N, Davies B, Hoffmann G, Koerner A, Lave T, Prinssen E, et al. Development of a physiologically based model for oseltamivir and simulation of pharmacokinetics in neonates and infants. Clin Pharmacokinet. 2011;50(9):613–623. doi: 10.2165/11592640-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 82.Bjorkman S. Prediction of drug disposition in infants and children by means of physiologically based pharmacokinetic (PBPK) modelling: theophylline and midazolam as model drugs. Br J Clin Pharmacol. 2005;59(6):691–704. doi: 10.1111/j.1365-2125.2004.02225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bjorkman S, Wada DR, Berling BM, Benoni G. Prediction of the disposition of midazolam in surgical patients by a physiologically based pharmacokinetic model. J Pharm Sci. 2001;90(9):1226–1241. doi: 10.1002/jps.1076. [DOI] [PubMed] [Google Scholar]
- 84.Plowchalk DR, Rowland Yeo K. Prediction of drug clearance in a smoking population: modeling the impact of variable cigarette consumption on the induction of CYP1A2. Eur J Clin Pharmacol. 2012;68(6):951–960. doi: 10.1007/s00228-011-1189-y. [DOI] [PubMed] [Google Scholar]
- 85.Ghobadi C, Johnson TN, Aarabi M, Almond LM, Allabi AC, Rowland-Yeo K, et al. Application of a systems approach to the bottom-up assessment of pharmacokinetics in obese patients: expected variations in clearance. Clin Pharmacokinet. 2011;50(12):809–822. doi: 10.2165/11594420-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 86.Rowland Yeo K, Aarabi M, Jamei M, Rostami-Hodjegan A. Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev Clin Pharmacol. 2011;4(2):261–274. doi: 10.1586/ecp.10.143. [DOI] [PubMed] [Google Scholar]
- 87.Grillo JA, Zhao P, Bullock J, Booth BP, Lu M, Robie-Suh K, et al. Utility of a physiologically-based pharmacokinetic (PBPK) modeling approach to quantitatively predict a complex drug-drug-disease interaction scenario for rivaroxaban during the drug review process: implications for clinical practice. Biopharm Drug Dispos. 2012;33(2):99–110. doi: 10.1002/bdd.1771. [DOI] [PubMed] [Google Scholar]
- 88.Jones HM, Dickins M, Youdim K, Gosset JR, Attkins NJ, Hay TL, et al. Application of PBPK modelling in drug discovery and development at Pfizer. Xenobiotica. 2012;42(1):94–106. doi: 10.3109/00498254.2011.627477. [DOI] [PubMed] [Google Scholar]
- 89.Lowe PJ, Tannenbaum S, Wu K, Lloyd P, Sims J. On setting the first dose in man: quantitating biotherapeutic drug-target binding through pharmacokinetic and pharmacodynamic models. Basic Clin Pharmacol Toxicol. 2010;106(3):195–209. doi: 10.1111/j.1742-7843.2009.00513.x. [DOI] [PubMed] [Google Scholar]
- 90.Stepensky D. Local versus systemic anti-tumour necrosis factor-alpha effects of adalimumab in rheumatoid arthritis: pharmacokinetic modelling analysis of interaction between a soluble target and a drug. Clin Pharmacokinet. 2012;51(7):443–455. doi: 10.2165/11599970-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 91.Pejovic M, Stankovic A, Mitrovic DR. Determination of the apparent synovial permeability in the knee joint of patients suffering from osteoarthritis and rheumatoid arthritis. Br J Rheumatol. 1995;34(6):520–524. doi: 10.1093/rheumatology/34.6.520. [DOI] [PubMed] [Google Scholar]
- 92.Narita T, Sasaki H, Hosoba M, Miura T, Yoshioka N, Morii T, et al. Parallel increase in urinary excretion rates of immunoglobulin G, ceruloplasmin, transferrin, and orosomucoid in normoalbuminuric type 2 diabetic patients. Diabetes Care. 2004;27(5):1176–1181. doi: 10.2337/diacare.27.5.1176. [DOI] [PubMed] [Google Scholar]
- 93.Comper WD, Haraldsson B, Deen WM. Resolved: normal glomeruli filter nephrotic levels of albumin. J Am Soc Nephrol. 2008;19(3):427–432. doi: 10.1681/ASN.2007090997. [DOI] [PubMed] [Google Scholar]
- 94.Kok DJ, Khan SR. Calcium oxalate nephrolithiasis, a free or fixed particle disease. Kidney Int. 1994;46(3):847–854. doi: 10.1038/ki.1994.341. [DOI] [PubMed] [Google Scholar]
- 95.Di L, Trapa P, Obach RS, Atkinson K, Bi YA, Wolford A, et al. A novel relay method for determining low-clearance values. Drug Metab Dispos. 2012;40(9):1860–1865. doi: 10.1124/dmd.112.046425. [DOI] [PubMed] [Google Scholar]
- 96.Bi YA, Kimoto E, Sevidal S, Jones HM, Barton HA, Kempshall S, et al. In vitro evaluation of hepatic transporter-mediated clinical drug-drug interactions: hepatocyte model optimization and retrospective investigation. Drug Metab Dispos. 2012;40(6):1085–1092. doi: 10.1124/dmd.111.043489. [DOI] [PubMed] [Google Scholar]
- 97.Jamei M, Dickinson GL, Rostami-Hodjegan A. A framework for assessing inter-individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: a tale of ‘bottom-up’ vs ‘top-down’ recognition of covariates. Drug Metab Pharmacokinet. 2009;24(1):53–75. doi: 10.2133/dmpk.24.53. [DOI] [PubMed] [Google Scholar]
- 98.Willmann S, Hohn K, Edginton A, Sevestre M, Solodenko J, Weiss W, et al. Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J Pharmacokinet Pharmacodyn. 2007;34(3):401–431. doi: 10.1007/s10928-007-9053-5. [DOI] [PubMed] [Google Scholar]
- 99.Hopkins JC, Leipold RJ. On the dangers of adjusting the parameters values of mechanism-based mathematical models. J Theor Biol. 1996;183(4):417–427. doi: 10.1006/jtbi.1996.0232. [DOI] [PubMed] [Google Scholar]
- 100.Heiskanen T, Kairemo K. Development of a PBPK model for monoclonal antibodies and simulation of human and mice PBPK of a radiolabelled monoclonal antibody. Curr Pharm Des. 2009;15(9):988–1007. doi: 10.2174/138161209787581968. [DOI] [PubMed] [Google Scholar]
- 101.Yeung YA, Leabman MK, Marvin JS, Qiu J, Adams CW, Lien S, et al. Engineering human IgG1 affinity to human neonatal Fc receptor: impact of affinity improvement on pharmacokinetics in primates. J Immunol. 2009;182(12):7663–7671. doi: 10.4049/jimmunol.0804182. [DOI] [PubMed] [Google Scholar]