

Abstract
We present a unified quantitative approach to predict the in vivo alteration in drug exposure caused by either cytochrome P450 (CYP) gene polymorphisms or CYP-mediated drug–drug interactions (DDI). An application to drugs metabolized by CYP2C19 is presented. The metrics used is the ratio of altered drug area under the curve (AUC) to the AUC in extensive metabolizers with no mutation or no interaction. Data from 42 pharmacokinetic studies performed in CYP2C19 genetic subgroups and 18 DDI studies were used to estimate model parameters and predicted AUC ratios by using Bayesian approach. Pharmacogenetic information was used to estimate a parameter of the model which was then used to predict DDI. The method adequately predicted the AUC ratios published in the literature, with mean errors of −0.15 and −0.62 and mean absolute errors of 0.62 and 1.05 for genotype and DDI data, respectively. The approach provides quantitative prediction of the effect of five genotype variants and 10 inhibitors on the exposure to 25 CYP2C19 substrates, including a number of unobserved cases. A quantitative approach for predicting the effect of gene polymorphisms and drug interactions on drug exposure has been successfully applied for CYP2C19 substrates. This study shows that pharmacogenetic information can be used to predict DDI. This may have important implications for the development of personalized medicine and drug development.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-012-9431-9) contains supplementary material, which is available to authorized users.
KEY WORDS: CYP2C19, drug interactions, personalized medicine, pharmacogenetics, quantitative prediction
INTRODUCTION
Cytochrome P450 (CYP) genetic polymorphisms and CYP-mediated drug interactions are important determinants of variability in drug exposure (1,2). For a drug that is a substrate of a polymorphic CYP, it is desirable to predict the magnitude of drug interactions caused by CYP inducers or inhibitors. Also, clinicians and pharmacologists may be interested in predicting the alteration in drug exposure in genotype/phenotype variants such as poor metabolizer (PM), intermediate metabolizer (IM), or ultrarapid metabolizer (UM) subjects with respect to the extensive metabolizer (EM) subjects.
Several approaches based on in vitro data have been proposed to predict the magnitude of drug–drug interactions (DDI), including mechanistic models (3) and physiologically based pharmacokinetic models (4). Those models are attractive but quite complex, and the quantification remains challenging. A much simpler in vivo modeling approach has been proposed by Ohno and colleagues for CYP3A4 inhibition and induction, with good predictive performance (5,6). Recently, our group has successfully applied this model to drugs metabolized by CYP2D6 (7).
In a recent Food and Drug Administration (FDA) draft guidance on clinical pharmacogenomics, it has been suggested that the alteration in CYP substrate drug exposure caused by either drug interactions or gene polymorphisms may be viewed as the same general question (8). For example, the increase in drug exposure observed in PM subjects with respect to EM subjects is similar to that caused by a strong inhibitor of the considered pathway in EM. Thus, pharmacokinetic studies performed in genetically identified subgroups may provide important information for the prediction of DDI.
In accordance with this view, we have recently derived a model to predict the change in drug exposure caused by CYP2D6 genetic polymorphisms based on the in vivo drug interaction model from Ohno et al., and such an approach showed good predictive performance (9). The objective of this study was to propose a general framework for the quantitative prediction of the effect of CYP genetic polymorphisms and CYP-mediated drug interactions on substrate drug exposure and to present an application for drugs metabolized by CYP2C19. The relative contribution of CYP2C19 to the clearance of marketed drugs is small, about 5% (2), but some widely prescribed drugs are metabolized by this pathway, such as clopidogrel, diazepam, or proton pump inhibitors. In addition, the clinical significance of CYP2C19 gene polymorphisms and DDI in drug therapy has been established (1,2,10).
METHODS
Background and Metrics
Our approach is based on frameworks proposed by Ohno (5) and ourselves (9) for drug interactions and gene polymorphisms, respectively. The general metrics used is the ratio of the altered substrate drug area under the curve (AUC*), where the alteration may be caused by either gene polymorphisms or drug interaction, to the reference AUC measured in patients with no mutation or no interaction. For CYP2C19 gene polymorphisms, the prediction of the AUC ratio is based on the following two-parameter equation, which has been described in detail elsewhere (9):
 |
1 |
where AUCXM is the substrate drug AUC in subjects with mutated CYP2C19 alleles, who may be either PM, IM, or UM, AUCEM is the reference drug AUC in wild-type, EM, CREM (contribution ratio) is the fraction of the apparent drug clearance due to CYP2C19 in EM, and FA (fractional activity) is the fraction of activity of CYP2C19 resulting from the combination of mutated alleles, with respect to the reference genotype (wild-type). The contribution ratio CR is basically an in vivo equivalent of the fraction metabolized (often denoted fm) by a certain CYP enzyme. The FA parameter characterizes the CYP2C19 genotype. The value of FA in homozygote wild-type EM individuals is 1, it is lower than 1 in IM and PM subjects, and greater than 1 in UM subjects.
For drug interactions by inhibition, the AUC ratio of the substrate drug is described as follows:
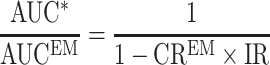 |
2 |
where AUCEM is the reference AUC of the drug when administered alone in EM, AUC* is the AUC of the drug when coadministered with a CYP2C19 inhibitor in EM individuals, CREM is the contribution ratio of the substrate drug (see above), and IR is the inhibition ratio, which is a measure of the inhibitor potency. The inhibition ratio ranges from 0 (the inhibitor has no effect on CYP2C19-mediated clearance of the victim drug) to 1 (the inhibitor reduces CYP2C19-mediated clearance of the victim drug to zero). For details on Eq. 2, see Ohno et al. (5) and Tod et al. (7). Of note, Eq. 2 only applies for DDI occurring in EM. It can be shown that the general equation for the quantitative prediction of drug interactions by inhibition in subjects with any genotype XM is as follows:
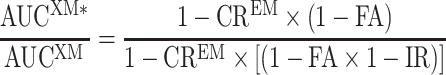 |
3 |
where AUCXM is the AUC of the substrate drug in subjects with genotype XM, AUCXM* is the AUC in subjects with genotype XM who are coadministered the inhibitor, CREM is the contribution ratio of the substrate drug in homozygote EM subjects, FA is the fraction of activity associated with genotype XM, and IR is the inhibition ratio of the inhibitor. The demonstration of Eq. 8 is provided as a Supplementary Material. In this application, we did not use this general equation but the simpler Eq. 2, as only drug interaction data from EM subjects were analyzed.
If the CR of a CYP2C19 substrate and the FA of a mutant genotype are known, one can predict the AUC increase in subjects with such a genotype using Eq. 1. Alternatively, Eq. 1 may be used to predict the value of one unknown parameter, CR or FA, provided that the other parameter is known, as well as the AUC increase. Similarly, Eq. 2 may be applied to predict either the AUC increase caused by a CYP2C19 inhibitor or the value of one unknown parameter, CR or IR, depending on available data.
As the contribution ratio CR is a common parameter in Eqs. 1 and 2, pharmacogenetic data may be used to estimate CRs of CYP2C19 substrates and then derive quantitative prediction of DDI, and vice versa. In this study, the CRs of various CYP2C19 substrates were first estimated from pharmacogenetic data using Eq. 1. Then, these CR estimates were used to predict the magnitude of DDI using Eq. 2. We chose this procedure because much more data were available from pharmacogenetic studies on CYP2C19 drugs than from DDI studies. A three-step approach was carried out for the analysis of both genotype and DDI data. First, initial values of the model parameters were estimated from a subset of published data, using Eqs. 1 and 2. Then, external validation of the AUC ratios predicted from those initial estimates was performed using a second set of published data. Finally, refined estimates of model parameters and AUC ratios were obtained by Bayesian orthogonal regression, using all the data from steps 1 and 2.
Genotype-Based Prediction of Drug Exposure
Step 1: Estimation of the Initial Values of CRs and FAs
The activity of CYP2C19 alleles has been classified as normal, null, decreased, or increased, based on in vitro and in vivo data (11). In the initial estimation, each allele was assumed to have its own contribution denoted as FAi and the fractional activity (FA) of any CYP2C19 genotype results from the allele combination as follows (9):
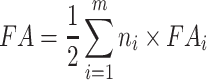 |
4 |
where m indicates the allele type (normal, null, decreased, or increased) and ni indicates the number of alleles of each category. Of note, Eq. 4 was used only for the initial estimation of the FAs. In the final estimation step, only the global activity of each allele combination FA was estimated, and so the assumptions of gene–dose effect and independence of allele activity did not stand in this final step.
The FAi of the reference allele, CYP2C19*1, was fixed at 1 (normal activity). While CYP2C19*2 and CYP2C19*3 alleles have been reported as inactive (11), we assumed very little activity, with FAi fixed at 0.01 for both alleles. For the gain-of-function CYP2C19*17 allele, FAi was estimated at 2.25 using Eq. 1 and data from a pharmacokinetic study performed with omeprazole in homozygote *17/*17 subjects (12).
For ease of reading, both inactive alleles *2 and *3 are denoted *2 throughout the article. As a result, six allele combinations along with their fractional activity were considered: *1*1 (FA = 1), *1*2 (FA = 0.5), *2*2 (FA = 0.01), *17/*17 (FA = 2.25), *1*17 (FA = 1.625), and *2*17 (FA = 1.13).
The contribution ratio CR of CYP2C19 substrates was calculated from AUC values measured in homozygous EM and homozygous PM, after simplification and rearrangement (see details in reference (9)), as follows:
 |
5 |
AUC values in EM and PM were retrieved from in vivo studies published up to December 2011. When several published AUC ratios were available in PM for a given substrate, data from multiple-dose studies were preferred over single-dose studies. When the various studies had similar design, the median AUC ratio reported in PM was selected to get a central tendency of the data. All data that had not been used during step 1 were considered in the external validation step.
Step 2: External Validation of Genotype-Based Predictions
The AUC ratios predicted by Eq. 1 with the initial estimates of CRs and FAs were compared to AUC ratios observed in a second set of published data. Predicted AUC ratios were plotted versus observed AUC ratios. Predicted values in the range 50–200% of the observed ratio were considered acceptable, in accordance with previous works (5,6). Bias and precision of predicted AUC ratios were calculated as the mean prediction error (i.e., predicted minus observed AUC ratio) and the mean absolute prediction error, respectively.
Step 3: Final Estimation of CRs, FAs, and AUC Ratios
A Bayesian approach was used to obtain refined estimates of CRs and FAs in the Winbugs software, version 1.4.3 (13). Equation 1, initial estimates of CRs and FAs, and all available data from steps 1 and 2 were imported into the Winbugs software to perform Bayesian orthogonal regression, as described in detail elsewhere (9).
Briefly, for each substrate i and genotype j, the predicted AUC ratio was coded in Winbugs as follows:
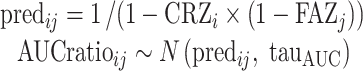 |
6 |
where predij and AUCratioij are the predicted and observed AUC ratios for each (CRi, FAj) pair, respectively, CRZi and FAZj are the refined (Bayesian posterior) estimates of the contribution ratio and fraction of activity, respectively, and tauAUC is the precision (i.e., reciprocal of the variance) of the AUC distribution. We assumed normal distribution for AUC ratios and logistic distribution for CRs and FAs. The mean of each distribution was set to the initial estimates found in step 1. We assumed that the precision of these distributions, denoted tau, followed a gamma distribution as follows: tauCR ∼ G(4, 1), tauFA ∼ G(4, 1), and tauAUC ∼ G(0.1, 2.5), for CR, FA, and AUC distributions, respectively. These gamma distributions were set so that the expected standard deviations of CRs and FAs on the logit scale and of AUC ratios were 0.5, 0.5, and 5, respectively.
In Bayesian statistics, the gamma distribution G(α, β) is commonly used as prior for the precision (i.e., the reciprocal of the variance) of a normal distribution (14). The mean of such a gamma distribution is α/β. In our application, the mean of the precisions denoted tau were 4 for CRs and FAs and 0.04 for AUC ratios. Therefore, the expected variances of the parameters are the reciprocal of those values, 0.25 and 25, and the expected standard deviations are 0.5 and 5 for CRs/FAs and AUC ratios, respectively.
The posterior distributions of the AUC ratios, CRs, and FAs were obtained by Monte Carlo Markov chain simulation in Winbugs. Convergence was assessed by checking the stability of the posterior distributions. Goodness-of-fit was assessed by visual examination of the residual scatter plots. The Bayesian posterior distributions were examined to detect multimodal distributions, which would have indicated a conflict between specified prior distribution and data. The means of the posterior distributions were used as point estimates of CRs, FAs, and AUC ratios. A 90% confidence interval was calculated for each result as the interval between the 5th and the 95th percentile of the posterior distribution of each variable.
Quantitative Prediction of Drug Interactions
Step 1: Estimation of the Initial Values of IRs
Point estimates of CRs obtained from the analysis of pharmacogenetic data described above, as well as AUC ratios from published drug interaction studies were used to estimate initial values of IRs, by rearranging Eq. 2 as shown below:
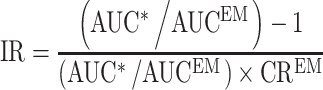 |
7 |
AUC* and AUCEM values for various (substrate, inhibitor) pairs were retrieved from studies published up to December 2011.
Step 2: External Validation of Drug Interaction Predictions
The AUC ratios predicted by Eq. 2 with the initial estimates of IRs and final estimates of CRs were compared to the AUC ratios observed in a second set of published data. Predictive performance of the model was assessed using the same criteria as those used in the analysis of pharmacogenetic data.
Step 3: Final Estimation of IRs and AUC Ratios
We used a Bayesian orthogonal regression procedure in Winbugs similar to that previously described, except that only IRs and AUC ratios were reestimated. CRs were fixed at their point estimates obtained from the previous Bayesian analysis of pharmacogenetic data (CRZi). Briefly, for each substrate i and inhibitor k:
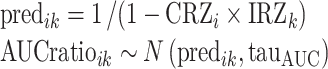 |
8 |
where predik and AUCratioik are the predicted and observed AUC ratios for each (CRi, IRk) pair, respectively, CRZi is the final point estimate of the contribution ratio, and IRZk is the refined (Bayesian posterior) estimate of the inhibition ratio. The precision tauAUC was set as stated above for genotype-based prediction (tauAUC ∼ G(0.1, 2.5)). We assumed logistic distribution of the IRs, with tauIR ∼ G(4, 1). Results were critically examined as explained above. Point estimates, as well as 90% confidence intervals, were calculated from the Bayesian posterior distribution of IRs and AUC ratios.
Prediction of AUC Ratios for Unpublished Substrate–Genotype and Substrate–Inhibitor Pairs
The final estimates of CRs, FAs, and IRs were used to calculate AUC ratios for all possible CYP2C19 substrate–genotype and substrate–inhibitor pairs, using Eqs. 1 and 2, respectively. This provided insights about the magnitude of the alteration in drug exposure for unpublished rare genotypes and drug interactions.
RESULTS
Genotype-Based Prediction
Initial Estimation
Ninety-nine AUC ratios from 42 published studies were available for 5 genotypes (*1*2, *2*2, *17*17, *1*17, and *2*17, with respect to the reference *1*1 genotype) and 25 CYP2C19 oral substrate drugs. Of note, these substrates included the R- and S-enantiomers of lansoprazole, the S-enantiomer of citalopram (escitalopram), and the active fraction of nelfinavir as separate entities, in addition to parent drugs, whose total was 21.
The initial values of FAs are given in the “Methods” section. The initial estimates of CRs for 25 CYP2C19 drug substrates are given in Table I, as well as literature references. The highest contribution of CYP2C19 to the oral drug clearance was observed for mephobarbital R (CR = 0.99), proguanil (CR = 0.93), lansoprazole S (CR = 0.87), omeprazole (CR = 0.85), and diazepam (CR = 0.84).
Table I.
Initial Estimate of the CYP2C19 CR for Various Substrate Drugs
| Drug | CR | Reference |
|---|---|---|
| Amitriptyline | 0.28 | (32) |
| Citalopram | 0.54 | (33) |
| Cilostazol | 0.30 | (34) |
| Clomipramine | 0.43 | (35) |
| Clopidogrel | 0.66 | (36) |
| Diazepam | 0.84 | (37) |
| Escitalopram | 0.46 | (38) |
| Fluoxetine | 0.66 | (39) |
| Gliclazide | 0.81 | (40) |
| Lansoprazole | 0.74 | (41) |
| Lansoprazole R | 0.75 | (17) |
| Lansoprazole S | 0.87 | (17) |
| Mephobarbital R | 0.99 | (42) |
| Moclobemide | 0.72 | (43) |
| Nelfinavir | 0.47 | (44) |
| Nelfinavir active moietya | 0.25 | (44) |
| Omeprazole | 0.85 | (45) |
| Pantoprazole | 0.80 | (21) |
| Praziquantel | 0.17 | (46) |
| Proguanil | 0.93 | (47) |
| Rabeprazole | 0.74 | (41) |
| Sertraline | 0.69 | (22) |
| Sibutramine | 0.71 | (48) |
| Trimipramine | 0.48 | (49) |
| Voriconazole | 0.67 | (50) |
CR contribution ratio
aThe active moiety of nelfinavir is nelfinavir + M8 active metabolite (nelfinavir hydroxy-t-butylamide)
External Validation
AUC ratios predicted using Eq. 1 and the initial estimates of FAs and CRs were compared with 75 AUC ratios reported in published studies. Published AUC ratios and corresponding references are provided in Table II. Figure 1 shows observed versus predicted AUC ratios. The mean error of prediction of the AUC ratio was −0.27 and the mean absolute error was 0.71. Only 5 out of 75 (6.7%) predicted AUC ratios were out of the 50–200% range of observed AUC ratios.
Table II.
Published AUC Ratios Used for the External Validation Step
| Drug | Genotype | FAa | Number of subjects | Observed AUC ratio | Reference |
|---|---|---|---|---|---|
| Amitriptyline | *1*2 | 0.5 | 18 | 1.29 | (32) |
| Citalopram | *2*2 | 0.01 | 5 | 1.23 | (51) |
| Citalopram | *1*2 | 0.5 | 7 | 1.44 | (33) |
| Cilostazol | *1*2 | 0.5 | 18 | 1.2 | (34) |
| Clomipramine | *1*2 | 0.5 | 25 | 1.41 | (35) |
| Clopidogrel | *1*2 | 0.5 | 8 | 1.67 | (36) |
| Diazepam | *1*2 | 0.5 | 6 | 2.47 | (37) |
| Escitalopram | *17*17 | 2.25 | 5 | 0.79 | (52) |
| Fluoxetine | *1*2 | 0.5 | 4 | 1.58 | (39) |
| Gliclazide | *2*2 | 0.01 | 3 | 3.42 | (53) |
| Gliclazide | *1*2 | 0.5 | 12 | 1.25 | (53) |
| Lansoprazole | *2*2 | 0.01 | 3 | 1.88 | (54) |
| Lansoprazole | *2*2 | 0.01 | 4 | 5.41 | (55) |
| Lansoprazole | *2*2 | 0.01 | 6 | 4.03 | (56) |
| Lansoprazole | *1*2 | 0.5 | 5 | 1.67 | (41) |
| Lansoprazole | *1*2 | 0.5 | 2 | 1.32 | (54) |
| Lansoprazole | *1*2 | 0.5 | 7 | 2.31 | (55) |
| Lansoprazole | *1*2 | 0.5 | 7 | 2.14 | (58) |
| Lansoprazole | *1*2 | 0.5 | 4 | 1.68 | (21) |
| Lansoprazole | *1*2 | 0.5 | 6 | 1.78 | (56) |
| Lansoprazole | *1*17 | 1.63 | 1 | 0.45 | (21) |
| Lansoprazole | *2*17 | 1.13 | 1 | 1.34 | (21) |
| Lansoprazole R | *1*2 | 0.5 | 6 | 1.46 | (17) |
| Lansoprazole S | *1*2 | 0.5 | 6 | 1.85 | (17) |
| Mephobarbital R | *1*2 | 0.5 | 10 | 4.4 | (42) |
| Nelfinavir | *1*2 | 0.5 | 22 | 1.49 | (44) |
| Nelfinavir active moietyb | *1*2 | 0.5 | 22 | 1.29 | (44) |
| Omeprazole | *2*2 | 0.01 | 6 | 7.42 | (58) |
| Omeprazole | *2*2 | 0.01 | 6 | 2.03 | (16) |
| Omeprazole | *2*2 | 0.01 | 6 | 9.03 | (59) |
| Omeprazole | *2*2 | 0.01 | 6 | 3.8 | (21) |
| Omeprazole | *2*2 | 0.01 | 6 | 7.54 | (56) |
| Omeprazole | *2*2 | 0.01 | 6 | 7.79 | (60) |
| Omeprazole | *2*2 | 0.01 | 8 | 5.68 | (61) |
| Omeprazole | *1*2 | 0.5 | 5 | 2.29 | (45) |
| Omeprazole | *1*2 | 0.5 | 6 | 1.72 | (58) |
| Omeprazole | *1*2 | 0.5 | 8 | 2.66 | (59) |
| Omeprazole | *1*2 | 0.5 | 2 | 3.67 | (21) |
| Omeprazole | *1*2 | 0.5 | 6 | 2.84 | (56) |
| Omeprazole | *1*2 | 0.5 | 6 | 2.85 | (60) |
| Omeprazole | *1*17 | 1.63 | 1 | 0.77 | (21) |
| Omeprazole | *2*17 | 1.13 | 1 | 1.83 | (21) |
| Pantoprazole | *1*2 | 0.5 | 7 | 2.16 | (62) |
| Pantoprazole | *1*2 | 0.5 | 2 | 4.2 | (21) |
| Pantoprazole | *1*17 | 1.63 | 1 | 0.79 | (21) |
| Pantoprazole | *2*17 | 1.13 | 1 | 1.41 | (21) |
| Proguanil | *1*2 | 0.5 | 5 | 2.99 | (47) |
| Rabeprazole | *2*2 | 0.01 | 6 | 3.13 | (63) |
| Rabeprazole | *2*2 | 0.01 | 4 | 5.26 | (45) |
| Rabeprazole | *2*2 | 0.01 | 4 | 4.34 | (64) |
| Rabeprazole | *2*2 | 0.01 | 6 | 3.73 | (56) |
| Rabeprazole | *2*2 | 0.01 | 5 | 2.68 | (65) |
| Rabeprazole | *1*2 | 0.5 | 5 | 1.71 | (41) |
| Rabeprazole | *1*2 | 0.5 | 8 | 1.38 | (63) |
| Rabeprazole | *1*2 | 0.5 | 5 | 3.02 | (45) |
| Rabeprazole | *1*2 | 0.5 | 6 | 2.05 | (64) |
| Rabeprazole | *1*2 | 0.5 | 6 | 1.57 | (56) |
| Rabeprazole | *1*2 | 0.5 | 8 | 1.1 | (65) |
| Sertraline | *1*2 | 0.5 | 22 | 1.4 | (22) |
| Sertraline | *17*17 | 2.25 | 5 | 0.99 | (22) |
| Sertraline | *1*17 | 1.63 | 40 | 1.1 | (22) |
| Sertraline | *2*17 | 1.13 | 7 | 1.9 | (22) |
| Sibutramine | *1*2 | 0.5 | 26 | 1.13 | (48) |
| Trimipramine | *1*2 | 0.5 | 7 | 2.04 | (49) |
| Voriconazole | *2*2 | 0.01 | 8 | 3.45 | (66) |
| Voriconazole | *2*2 | 0.01 | 5 | 2.78 | (23) |
| Voriconazole | *2*2 | 0.01 | 6 | 4.02 | (67) |
| Voriconazole | *2*2 | 0.01 | 4 | 2.90 | (68) |
| Voriconazole | *1*2 | 0.5 | 8 | 2.28 | (50) |
| Voriconazole | *1*2 | 0.5 | 11 | 1.56 | (23) |
| Voriconazole | *1*2 | 0.5 | 8 | 1.3 | (68) |
| Voriconazole | *1*2 | 0.5 | 6 | 2.88 | (67) |
| Voriconazole | *1*17 | 1.63 | 4 | 0.52 | (66) |
| Voriconazole | *1*17 | 1.63 | 8 | 0.82 | (23) |
| Voriconazole | *2*17 | 1.13 | 2 | 0.78 | (23) |
aInitial estimate of the fraction of CYP2C19 activity for each genotype, with respect to the reference genotype (*1*1)
bThe active moiety of nelfinavir is nelfinavir + M8 active metabolite (nelfinavir hydroxy-t-butylamide)
Fig. 1.
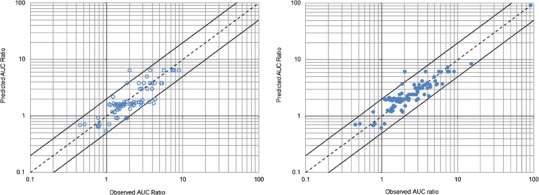
Observed area under the time–concentration curve (AUC) ratios versus genotype-based predictions from the external validation (left panel, open circles) and final estimation (right panel, filled circles) steps. The dashed line is the line of identity (y = x). The upper and lower solid lines represent y = 2x and y = 0.5x, respectively
Final Estimation of CRs and FAs
The final estimates of CRs and FAs with their confidence interval are presented in Tables III and IV, respectively. Table IV also indicates the frequency of CYP2C19 genotypes reported in the literature and associated phenotypes (57). The plot of observed AUC ratios versus predictions based on the final estimates of FAs and CRs is shown in Fig. 1 (n = 99). Compared with the initial estimation step, the predicted performance was improved, with mean error and mean absolute error of prediction of −0.15 and 0.62, respectively. Only one outlier remained at this step, the AUC ratio of omeprazole in *2*2 PM reported by Chen et al. (45), which was still overestimated.
Table III.
Final Estimate of CYP2C19 CR to the Apparent Oral Clearance of Various Substrate Drugs
| Drug | CRa | 90% CIb |
|---|---|---|
| Mephobarbital R | 0.99 | 0.99–1.0 |
| Proguanil | 0.89 | 0.87–0.90 |
| Lansoprazole S | 0.87 | 0.85–0.88 |
| Omeprazole | 0.84 | 0.82–0.86 |
| Diazepam | 0.84 | 0.81–0.86 |
| Pantoprazole | 0.80 | 0.76–0.83 |
| Gliclazide | 0.76 | 0.71–0.80 |
| Lansoprazole R | 0.74 | 0.68–0.79 |
| Lansoprazole | 0.73 | 0.67–0.78 |
| Rabeprazole | 0.72 | 0.65–0.77 |
| Moclobemide | 0.71 | 0.64–0.77 |
| Sibutramine | 0.69 | 0.61–0.75 |
| Voriconazole | 0.68 | 0.60–0.74 |
| Sertraline | 0.67 | 0.59–0.74 |
| Clopidogrel | 0.65 | 0.55–0.72 |
| Fluoxetine | 0.64 | 0.55–0.72 |
| Citalopram | 0.49 | 0.37–0.60 |
| Trimipramine | 0.49 | 0.36–0.60 |
| Nelfinavir | 0.46 | 0.33–0.58 |
| Clomipramine | 0.42 | 0.30–0.55 |
| Escitalopram | 0.45 | 0.33–0.57 |
| Cilostazol | 0.30 | 0.19–0.42 |
| Amitriptyline | 0.28 | 0.18–0.40 |
| Nelfinavir a.f. | 0.26 | 0.16–0.37 |
| Praziquantel | 0.18 | 0.10–0.27 |
CR contribution ratio, CI confidence interval
aCR was calculated as the mean of the Bayesian posterior distribution
bCI was calculated as the range between the 5th and the 95th percentiles of the Bayesian posterior distribution
Table IV.
Final Estimate of the FA of CYP2C19 for Various Genotypes with Respect to the Wild-Type (*1*1) Genotype
| Genotype | Frequencya (%) | Phenotypea | FAb | 90% CIc | |||
|---|---|---|---|---|---|---|---|
| Caucasian | Asian | African American | Hispanic | ||||
| *2*2 | 3.2 | 8.4 | 5.2 | 2 | PM | 0.005 | 0.002–0.008 |
| *1*2 | 16.8 | 44.4 | 18.8 | 17.2 | IM | 0.30 | 0.25–0.36 |
| *2*17 | 3.2 | 3.6 | 9.2 | 4.4 | Unknown | 0.80 | 0.50–1.19 |
| *1*17 | 22.8 | 5.6 | 20.4 | 20.4 | EM | 1.59 | 1.24–1.85 |
| *17*17 | 2.8 | 1.6 | 2.8 | 2.4 | UM | 2.03 | 1.28–2.62 |
FA fractional activity, CI confidence interval
aFrequencies and phenotypes as reported by Martis et al. (15). The frequencies of the reference *1*1 genotype reported in the study from Martis et al. were 49.2%, 36.4%, 38.4%, and 50.4% in Caucasians, Asians, African Americans, and Hispanics, respectively
bFA was calculated as the mean of the Bayesian posterior distribution
cCI was calculated as the range between the 5th and the 95th percentiles of the Bayesian posterior distribution
Quantitative Prediction of Drug Interactions
Initial Estimation of IRs
Fewer data were available for CYP2C19-mediated drug interactions. Twenty-two AUC ratios from 18 published studies were used in the analysis. The initial estimates of IRs for 10 CYP2C19 inhibitors are given in Table V, as well as literature references. Fluvoxamine (IR = 0.97), fluconazole (IR = 0.76), and voriconazole (IR = 0.66) were identified as the strongest CYP2C19 inhibitors. The proton pump inhibitors omeprazole and pantoprazole showed limited inhibition potency.
Table V.
Initial Estimate of the IR for Various CYP2C19 Inhibitors
| Inhibitor | Daily dose (mg) | IR | Reference |
|---|---|---|---|
| Clopidogrel | 75a | 0.26 | (16) |
| Fluconazole | 400/200b | 0.76 | (69) |
| Fluoxetine | 60 | 0.44 | (70) |
| Fluvoxamine | 50 | 0.97 | (60) |
| Moclobemide | 300 | 0.62 | (61) |
| Omeprazole | 40 | 0.43 | (71) |
| Pantoprazole | 80 | 0.25 | (72) |
| Ticlopidine | 300 | 0.52 | (73) |
| Voriconazole | 800/400b | 0.66 | (69) |
IR inhibition ratio
aA single dose of 300 mg was administered on the first day and then a 75-mg dose was administered once daily for three consecutive days
bThe daily dose was halved on the second day of the study. Doses are given as first dose/second dose
External Validation and Final Estimation
In the external validation step, model-based (Eq. 2) predictions of the AUC ratio were compared with 13 ratios reported in the literature. Values and references of published AUC ratios are shown in Table VI. The mean error and mean absolute error of predictions were −1.20 and 1.76, respectively. Figure 2 shows the observed versus predicted AUC ratios from both the external validation and final estimations steps of the analysis.
Table VI.
Published AUC Ratios Used for the External Validation Step
| Inhibitor | Substrate drug | Observed AUC ratio of substrate drug | Reference |
|---|---|---|---|
| Clopidogrel | Sibutramine | 2.27 | (74) |
| Fluconazole | Omeprazole | 6.29 | (18) |
| Fluvoxamine | Lansoprazole | 3.83 | (75) |
| Lansoprazole R | 8.99 | (17) | |
| Lansoprazole S | 13.97 | (17) | |
| Rabeprazole | 2.82 | (76) | |
| Diazepam | 2.80 | (77) | |
| Omeprazole | Moclobemide | 2.21 | (43) |
| Nelfinavir | 0.70 | (78) | |
| Diazepam | 1.64 | (79) | |
| Proguanil | 1.47 | (80) | |
| Clopidogrel | 1.51 | (72) | |
| Clopidogrel | 1.44 | (72) |
Fig. 2.
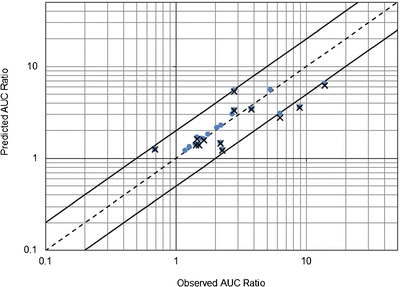
Observed area under the time–concentration curve (AUC) ratios versus model predictions for CYP2C19-mediated drug interactions. Plus signs predictions from the external validation step (n = 13), filled circles predictions from the final estimation step (n = 22). The dashed line is the line of identity (y = x). The upper and lower solid lines represent y = 2x and y = 0.5x, respectively
The final estimates of IRs for the 10 inhibitors are provided in Table VII, as well as their confidence intervals. As the inhibitor dose may influence the value of the inhibition ratio (7), the ranges of inhibitor doses used in published studies should be considered when interpreting the results. The AUC ratios predicted in the final analysis better correlated with the observed ratios (n = 22), as shown in Fig. 2, with mean error and mean absolute error of prediction of −0.62 and 1.05, respectively. After the final estimation step, three AUC ratios were slightly underestimated, for the R- and S-enantiomers of lansoprazole coadministered with fluvoxamine (25) and for omeprazole coadministered with fluconazole (64).
Table VII.
Final Estimate of the IR of Various CYP2C19 Inhibitors
| Inhibitor | Daily dose (mg)a | IRb | 90% CIc |
|---|---|---|---|
| Fluvoxamine | 50–150 | 0.98 | 0.95–0.99 |
| Fluconazole | 100–400 | 0.78 | 0.62–0.90 |
| Voriconazole | 400–800 | 0.64 | 0.43–0.82 |
| Moclobemide | 300 | 0.61 | 0.39–0.80 |
| Ticlopidine | 300 | 0.51 | 0.29–0.72 |
| Fluoxetine | 60 | 0.44 | 0.24–0.66 |
| Omeprazole | 40–80 | 0.43 | 0.24–0.64 |
| Clopidogrel | 75 | 0.28 | 0.13–0.48 |
| Pantoprazole | 80 | 0.26 | 0.12–0.45 |
IR inhibition ratio, CI confidence interval
aRange of the daily dose used in published drug interaction studies
bIR was calculated as the mean of the Bayesian posterior distribution
cCI was calculated as the range between the 5th and the 95th percentiles of the Bayesian posterior distribution
Prediction of AUC Ratios for All Substrate–Genotype and Substrate–Inhibitor Pairs
The Bayesian orthogonal regression analysis provided point estimates and confidence interval of the AUC ratios for all possible CYP2C19 substrate–genotype (n = 125) and substrate–inhibitor (n = 250) pairs, including unpublished cases. Figure 3 shows the predicted AUC ratios for 10 CYP2C19 substrate drugs in PM (*2*2) and UM (*17*17) subjects. The predicted AUC ratios of the same CYP2C19 substrates for all possible interactions with five CYP2C19 inhibitors are provided as Supplementary Material.
Fig. 3.
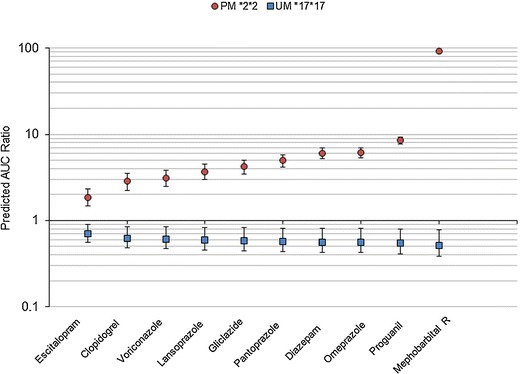
Model-based predictions of the AUC ratio for 10 CYP2C19 substrate drugs in poor metabolizer (PM) *2*2 and ultrarapid metabolizer (UM) *17*17 subjects. The squares and circles indicate the point estimate (mean of the Bayesian posterior distributions) of the AUC ratio in PM *2*2 and UM *17*17, respectively. The error bars indicate the 90% confidence interval of the AUC ratio
DISCUSSION
The variability in drug exposure caused by cytochrome gene polymorphisms and CYP-mediated drug interactions may have important clinical implications (10). For example, it has been shown that CYP2C19 gene polymorphisms influence the healing of gastroesophageal reflux disease and eradication rate of Helicobacter pylori infection treated by proton pump inhibitor-containing drug regimens (71). CYP2C19 alleles associated with both increased (CYP2C19*17) and decreased (CYP2C19*2) enzyme activity have been linked with cardiovascular outcomes in patients with coronary artery disease treated by clopidogrel (72).
In this study, a unified modeling approach for in vivo quantitative prediction of the effect of CYP gene polymorphisms and DDI has been presented and applied for CYP2C19 substrate drugs. This is not a fully new approach, as it has been first proposed by Ohno and colleagues for CYP3A4-mediated drug interactions (5) and then applied to CYP2D6 inhibition and CYP2D6 gene polymorphisms (7,9). However, in these previous works, DDI and genetic polymorphisms have been studied separately. Here, we used pharmacogenetic information to predict CYP2C19-mediated drug interactions. This was possible because the contribution ratio (CR), which is the substrate drug parameter in the model, is a common parameter in Eqs. 1 and 2. To our knowledge, such sequential approach for in vivo prediction of the effect of CYP gene polymorphisms and DDI has not been performed so far.
In the analysis of pharmacogenetic data, the model provided overall good predictive performance, as only one outlier remained in the final estimation of the AUC ratios. For *1*2 and *2*17 genotypes, the final estimates of FA (0.30 and 0.80, respectively) were quite different from the initial estimates (0.50 and 1.13, respectively). In the initial estimation step, we assumed gene–dose effect, as well as independent contribution of each allele FAi (see Eq. 4 in the “Methods” section) to estimate the FA values. However, in the final estimation step, only the total FA values of allele combinations were estimated and the individual contribution of each allele was not estimated. While a slightly increased CYP2C19 enzyme activity was initially expected in *2*17 subjects, the final estimate of FA (mean, 0.80; 90% CI, 0.50–1.19) suggests a slightly decreased activity instead. This finding is supported by the modest AUC increase in drug exposure reported in *2*17 subjects for omeprazole, pantoprazole, and lansoprazole (30) and sertraline (33). A limited decrease (AUC ratio, 0.78) in drug exposure in *2*17 subjects has been reported only for voriconazole so far (54). While more research in this specific subgroup is required, the magnitude of the change in drug exposure associated with the *2*17 genotype appears to be small anyway.
Fewer data were available to predict CYP2C19-mediated DDI. After the final estimation step, the predictive performance was acceptable, but three AUC ratios were underestimated, for omeprazole coadministered with fluconazole (predicted AUC ratio, 3.04; observed AUC ratio, 6.3 (64)) and for the R- and S-enantiomers of lansoprazole coadministered with fluvoxamine (predicted AUC ratios, 3.60 and 6.50; observed AUC ratios, 8.99 and 13.97, for R- and S-lansoprazole, respectively (25)). Interestingly, in the study from Miura and colleagues, the AUC of R- and S-lansoprazole were also measured in *2*2 and *2*3 PM subjects, and the observed AUC ratios (AUCPM/AUCEM) were 4.0 and 7.4 for R- and S-lansoprazole, respectively (25). So, fluvoxamine-induced AUC increase was significantly greater than that observed in homozygote PMs. This observation is not compatible with the theoretical basis of the method and indicates a conflict between pharmacogenetic and drug interaction data in this study.
The contribution ratios of substrate drugs and inhibition ratios of inhibitors may be compared with results from in vitro metabolism studies. Kita and colleagues studied the contribution of CYP2C19 to the in vitro metabolism of omeprazole and lansoprazole using antihuman CYP antibodies. They found percent inhibition of PPI hydroxylation by anti-CYP2C19 antibodies of 71.1–77.8% and 54.4–68.6% for omeprazole and lansoprazole, respectively (73). Relative metabolic contributions of CYP2C19 of 37% and 33% have been reported for S-citalopram (or escitalopram) and R-citalopram, respectively (74), while a 94–95% contribution has been reported for diazepam (75). Overall, the rank order of substrates in terms of relative contribution of CYP2C19 from these in vitro data (diazepam > omeprazole > lansoprazole > escitalopram) is in agreement with our results (see Table III). Quantitative discrepancies between in vitro estimates of CYP2C19 metabolic contribution and CRs are not surprising, as our approach uses the apparent oral drug clearance, not the intrinsic clearance. In addition, it has been shown that the various methods used for in vitro study of hepatic drug metabolism may provide significantly different results (75,76).
The in vitro [I]/Ki ratio, where [I] is the inhibitor concentration available to the enzyme and Ki is the inhibition constant, has been proposed as a predictor of in vivo magnitude of drug interactions (77), as the larger the [I]/Ki ratio, the greater the predicted AUC ratio and the inhibition potency. Using data from Obach et al., we calculated [I]/Ki ratios of 0.22, 1.37, 7.39, and 7.87 for moclobemide, ticlopidine, fluconazole, and fluvoxamine, respectively, using the free hepatic inlet maximal concentration estimated by the authors as the in vivo concentration available to the enzyme, [I] (78). Again, a fair agreement is observed with the inhibition potency provided by the IR parameter in our approach (ticlopidine < moclobemide < fluconazole < fluvoxamine; see Table VII), as our results show quite large overlapping in the 90% confidence interval of the IR for ticlopidine and moclobemide. Although available in vitro data are not sufficient to perform a systematic comparison, it appears that results from our in vivo modeling approach are in accordance with those from in vitro predictive approaches.
The paucity of data on drug interaction and in patients with rare genotypes, such as *2*17 subjects, is a clear limitation of the study. Also, this approach is based on a number of assumptions and has several limitations that have been discussed elsewhere in part (7,9). Briefly, the method is valid only for oral drugs with linear pharmacokinetics. For example, phenytoin, which is a substrate drug of both CYP2C9 and CYP2C19, was not included in the analysis because its pharmacokinetics is nonlinear (79). In addition, the model cannot deal with multiple, complex drug interaction patterns, and it can accommodate only one cytochrome pathway at a time. Some subjects may carry genetic mutations of several CYP metabolic pathways. For example, a haplotype with two CYP2C loss-of-function alleles (2C19*1–2C9*2–2C8*3) has been described in various ethnic groups, with a frequency ranging from 1.2% to 8.9% (57). This means that some individuals may be PM for both CYP2C9 and CYP2C8. For a drug that is a common CYP2C9 and CYP2C8 substrate, the application of our approach might lead to an overestimation of the CR of each individual CYP. In the future, it may be advantageous to refine the model in order to accommodate multiple CYP and mutations.
Despite the categorical nature of the CYP genotype, we used continuous distributions of the genotype parameter FA in our Bayesian modeling approach. Actually, the entire distribution of the FA parameter was described as a mixture of distributions, each genotype being characterized by its own distribution of the FA parameter. As illustrated by isoniazid clearance (80), mixture models are relevant methods to describe genetically determined characteristics that are observed at the phenotype level.
Finally, we only predict the magnitude of drug interactions in EM subjects. While Eq. 3 may be used for quantitative prediction of DDI in subjects with mutated genotype, little information exists on the drug interactions in genetic variants. Further work is required to assess the predictive ability of this general equation.
The translation of pharmacogenetic knowledge into personalized pharmacotherapy at the patients’ bedside has been quite laborious so far (10). This work may have important implication for routine patient care. The predicted AUC ratios may be used directly by clinicians for prior dose adjustment in routine when it seems clinically relevant to do so. Assuming that the exposure in EM is the target exposure to be achieved in an XM patient, one has to divide the usual maintenance dose by the predicted AUC ratio, AUCXM/AUCEM. This may be most useful for substrate–genotype or substrate–inhibitor pairs for which no clinical data have been reported so far. In such case, the prediction is based on all the available prior knowledge about this drug from other drug interactions and other genetic subgroups. However, one should be careful in the interpretation of the AUC ratio, taking into account the clinical behavior of the drug. For prodrugs, such as clopidogrel, that are converted to an active moiety by CYP2C19, the increase in drug exposure in PM subjects might lead to a decreased activity of the drug. It is noteworthy that this approach only provides an average prediction of the alteration in CYP2C19 substrate drug exposure, and so the clinical alteration may vary between individuals.
This work has shown that it is possible to use information from pharmacogenetic studies to predict drug interactions in vivo. While further research in other CYP is necessary to confirm this proof-of-concept of the FDA statement (8), this approach may have important implications for the optimization of the design of drug interaction and clinical pharmacogenetic studies in new drug development. For a new drug, once its contribution ratio has been estimated, the method may be used to predict the alteration in drug exposure for a number of drug interactions and in various genetic subgroups. This may be very useful to identify the most relevant situations that need to be confirmed by clinical studies.
CONCLUSION
A unified modeling approach for the quantitative prediction of the alteration in CYP substrate drug exposure caused by either genetic polymorphisms or drug interaction has been presented. Applied to drugs metabolized by CYP2C19, the approach provides quantitative predictions of the effect of five genotype variants and 10 inhibitors on the exposure to 25 oral substrate drugs. This work illustrates how pharmacokinetic information from genetic subgroups may be used to predict DDI and may have implications for both patients’ care and drug development.
ELECTRONIC SUPPLEMENTARY MATERIALS
Below is the link to the electronic supplementary material.
(DOC 39 kb)
(DOC 3563 kb)
ACKNOWLEDGMENTS
This study was not supported by any academic, company, or sponsor fund.
Conflict of Interest
The authors have no conflicts of interest that are relevant to the content of this study.
Footnotes
The Genophar II Working Group members are Charlotte Castellan, Bruno Charpiat, François Gueyffier, Behrouz Kassaï, and Patrice Nony.
REFERENCES
- 1.Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352(21):2211–2221. doi: 10.1056/NEJMra032424. [DOI] [PubMed] [Google Scholar]
- 2.Ingelman-Sundberg M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: the past, present and future. Trends Pharmacol Sci. 2004;25(4):193–200. doi: 10.1016/j.tips.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 3.Fahmi OA, Hurst S, Plowchalk D, Cook J, Guo F, Youdim K, et al. Comparison of different algorithms for predicting clinical drug–drug interactions, based on the use of CYP3A4 in vitro data: predictions of compounds as precipitants of interaction. Drug Metab Dispos. 2009;37(8):1658–1666. doi: 10.1124/dmd.108.026252. [DOI] [PubMed] [Google Scholar]
- 4.Rostami-Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov. 2007;6(2):140–148. doi: 10.1038/nrd2173. [DOI] [PubMed] [Google Scholar]
- 5.Ohno Y, Hisaka A, Suzuki H. General framework for the quantitative prediction of CYP3A4-mediated oral drug interactions based on the AUC increase by coadministration of standard drugs. Clin Pharmacokinet. 2007;46(8):681–696. doi: 10.2165/00003088-200746080-00005. [DOI] [PubMed] [Google Scholar]
- 6.Ohno Y, Hisaka A, Ueno M, Suzuki H. General framework for the prediction of oral drug interactions caused by CYP3A4 induction from in vivo information. Clin Pharmacokinet. 2008;47(10):669–680. doi: 10.2165/00003088-200847100-00004. [DOI] [PubMed] [Google Scholar]
- 7.Tod M, Goutelle S, Clavel-Grabit F, Nicolas G, Charpiat B. Quantitative prediction of cytochrome P450 (CYP) 2D6-mediated drug interactions. Clin Pharmacokinet. 2011;50(8):519–530. doi: 10.2165/11592620-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 8.US Food and Drug Administration. Guidance for industry. Clinical pharmacogenomics: premarketing evaluation in early phase clinical studies. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM243702.pdf (2011). Accessed 28 June 2012.
- 9.Tod M, Goutelle S, Gagnieu MC. Genotype-based quantitative prediction of drug exposure for drugs metabolized by CYP2D6. Clin Pharmacol Ther. 2011;90(4):582–587. doi: 10.1038/clpt.2011.147. [DOI] [PubMed] [Google Scholar]
- 10.Tomalik-Scharte D, Lazar A, Fuhr U, Kirchheiner J. The clinical role of genetic polymorphisms in drug-metabolizing enzymes. Pharmacogenomics J. 2008;8(1):4–15. doi: 10.1038/sj.tpj.6500462. [DOI] [PubMed] [Google Scholar]
- 11.The Human Cytochrome P450 (CYP) Allele Nomenclature Database. http://www.cypalleles.ki.se/. Accessed 28 June 2012.
- 12.Baldwin RM, Ohlsson S, Pedersen RS, Mwinyi J, Ingelman-Sundberg M, Eliasson E, et al. Increased omeprazole metabolism in carriers of the CYP2C19*17 allele; a pharmacokinetic study in healthy volunteers. Br J Clin Pharmacol. 2008;65(5):767–774. doi: 10.1111/j.1365-2125.2008.03104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spiegelhalter D, Thomas A, Best N, Lunn D. Winbugs 1.4.3 user manual. Cambridge: MRC Biostatistics Unit IoPH. 2007.
- 14.Congdon P. Bayesian statistical modelling. Chichester: Wiley; 2001. [Google Scholar]
- 15.Steimer W, Zopf K, von Amelunxen S, Pfeiffer H, Bachofer J, Popp J, et al. Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers. Clin Chem. 2004;50(9):1623–1633. doi: 10.1373/clinchem.2003.030825. [DOI] [PubMed] [Google Scholar]
- 16.Fudio S, Borobia AM, Pinana E, Ramirez E, Tabares B, Guerra P, et al. Evaluation of the influence of sex and CYP2C19 and CYP2D6 polymorphisms in the disposition of citalopram. Eur J Pharmacol. 2010;626(2–3):200–204. doi: 10.1016/j.ejphar.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 17.Yoo HD, Park SA, Cho HY, Lee YB. Influence of CYP3A and CYP2C19 genetic polymorphisms on the pharmacokinetics of cilostazol in healthy subjects. Clin Pharmacol Ther. 2009;86(3):281–284. doi: 10.1038/clpt.2009.90. [DOI] [PubMed] [Google Scholar]
- 18.Yokono A, Morita S, Someya T, Hirokane G, Okawa M, Shimoda K. The effect of CYP2C19 and CYP2D6 genotypes on the metabolism of clomipramine in Japanese psychiatric patients. J Clin Psychopharmacol. 2001;21(6):549–555. doi: 10.1097/00004714-200112000-00002. [DOI] [PubMed] [Google Scholar]
- 19.Kim KA, Park PW, Hong SJ, Park JY. The effect of CYP2C19 polymorphism on the pharmacokinetics and pharmacodynamics of clopidogrel: a possible mechanism for clopidogrel resistance. Clin Pharmacol Ther. 2008;84(2):236–242. doi: 10.1038/clpt.2008.20. [DOI] [PubMed] [Google Scholar]
- 20.Qin XP, Xie HG, Wang W, He N, Huang SL, Xu ZH, et al. Effect of the gene dosage of CgammaP2C19 on diazepam metabolism in Chinese subjects. Clin Pharmacol Ther. 1999;66(6):642–646. doi: 10.1016/S0009-9236(99)90075-9. [DOI] [PubMed] [Google Scholar]
- 21.Noehr-Jensen L, Zwisler ST, Larsen F, Sindrup SH, Damkier P, Nielsen F, et al. Impact of CYP2C19 phenotypes on escitalopram metabolism and an evaluation of pupillometry as a serotonergic biomarker. Eur J Clin Pharmacol. 2009;65(9):887–894. doi: 10.1007/s00228-009-0657-0. [DOI] [PubMed] [Google Scholar]
- 22.Liu ZQ, Cheng ZN, Huang SL, Chen XP, Ou-Yang DS, Jiang CH, et al. Effect of the CYP2C19 oxidation polymorphism on fluoxetine metabolism in Chinese healthy subjects. Br J Clin Pharmacol. 2001;52(1):96–99. doi: 10.1046/j.0306-5251.2001.01402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shao H, Ren XM, Liu NF, Chen GM, Li WL, Zhai ZH, et al. Influence of CYP2C9 and CYP2C19 genetic polymorphisms on pharmacokinetics and pharmacodynamics of gliclazide in healthy Chinese Han volunteers. J Clin Pharm Ther. 2010;35(3):351–360. doi: 10.1111/j.1365-2710.2009.01134.x. [DOI] [PubMed] [Google Scholar]
- 24.Ieiri I, Kishimoto Y, Okochi H, Momiyama K, Morita T, Kitano M, et al. Comparison of the kinetic disposition of and serum gastrin change by lansoprazole versus rabeprazole during an 8-day dosing scheme in relation to CYP2C19 polymorphism. Eur J Clin Pharmacol. 2001;57(6–7):485–492. doi: 10.1007/s002280100342. [DOI] [PubMed] [Google Scholar]
- 25.Miura M, Tada H, Yasui-Furukori N, Uno T, Sugawara K, Tateishi T, et al. Enantioselective disposition of lansoprazole in relation to CYP2C19 genotypes in the presence of fluvoxamine. Br J Clin Pharmacol. 2005;60(1):61–68. doi: 10.1111/j.1365-2125.2005.02381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi K, Morita J, Chiba K, Wanibuchi A, Kimura M, Irie S, et al. Pharmacogenetic roles of CYP2C19 and CYP2B6 in the metabolism of R- and S-mephobarbital in humans. Pharmacogenetics. 2004;14(8):549–556. doi: 10.1097/01.fpc.0000114764.78957.22. [DOI] [PubMed] [Google Scholar]
- 27.Yu KS, Yim DS, Cho JY, Park SS, Park JY, Lee KH, et al. Effect of omeprazole on the pharmacokinetics of moclobemide according to the genetic polymorphism of CYP2C19. Clin Pharmacol Ther. 2001;69(4):266–273. doi: 10.1067/mcp.2001.114231. [DOI] [PubMed] [Google Scholar]
- 28.Damle BD, Uderman H, Biswas P, Crownover P, Lin C, Glue P. Influence of CYP2C19 polymorphism on the pharmacokinetics of nelfinavir and its active metabolite. Br J Clin Pharmacol. 2009;68(5):682–689. doi: 10.1111/j.1365-2125.2009.03499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirai N, Furuta T, Moriyama Y, Okochi H, Kobayashi K, Takashima M, et al. Effects of CYP2C19 genotypic differences in the metabolism of omeprazole and rabeprazole on intragastric pH. Aliment Pharmacol Ther. 2001;15(12):1929–1937. doi: 10.1046/j.1365-2036.2001.01108.x. [DOI] [PubMed] [Google Scholar]
- 30.Hunfeld NG, Mathot RA, Touw DJ, van Schaik RH, Mulder PG, Franck PF, et al. Effect of CYP2C19*2 and *17 mutations on pharmacodynamics and kinetics of proton pump inhibitors in Caucasians. Br J Clin Pharmacol. 2008;65(5):752–760. doi: 10.1111/j.1365-2125.2007.03094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li XQ, Bjorkman A, Andersson TB, Gustafsson LL, Masimirembwa CM. Identification of human cytochrome P(450)s that metabolise anti-parasitic drugs and predictions of in vivo drug hepatic clearance from in vitro data. Eur J Clin Pharmacol. 2003;59(5–6):429–442. doi: 10.1007/s00228-003-0636-9. [DOI] [PubMed] [Google Scholar]
- 32.Coller JK, Somogyi AA, Bochner F. Association between CYP2C19 genotype and proguanil oxidative polymorphism. Br J Clin Pharmacol. 1997;43(6):659–660. doi: 10.1046/j.1365-2125.1997.00596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rudberg I, Hermann M, Refsum H, Molden E. Serum concentrations of sertraline and N-desmethyl sertraline in relation to CYP2C19 genotype in psychiatric patients. Eur J Clin Pharmacol. 2008;64(12):1181–1188. doi: 10.1007/s00228-008-0533-3. [DOI] [PubMed] [Google Scholar]
- 34.Kim KA, Song WK, Park JY. Association of CYP2B6, CYP3A5, and CYP2C19 genetic polymorphisms with sibutramine pharmacokinetics in healthy Korean subjects. Clin Pharmacol Ther. 2009;86(5):511–518. doi: 10.1038/clpt.2009.145. [DOI] [PubMed] [Google Scholar]
- 35.Kirchheiner J, Muller G, Meineke I, Wernecke KD, Roots I, Brockmoller J. Effects of polymorphisms in CYP2D6, CYP2C9, and CYP2C19 on trimipramine pharmacokinetics. J Clin Psychopharmacol. 2003;23(5):459–466. doi: 10.1097/01.jcp.0000088909.24613.92. [DOI] [PubMed] [Google Scholar]
- 36.Scholz I, Oberwittler H, Riedel KD, Burhenne J, Weiss J, Haefeli WE, et al. Pharmacokinetics, metabolism and bioavailability of the triazole antifungal agent voriconazole in relation to CYP2C19 genotype. Br J Clin Pharmacol. 2009;68(6):906–915. doi: 10.1111/j.1365-2125.2009.03534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu BN, Chen GL, He N, Ouyang DS, Chen XP, Liu ZQ, et al. Pharmacokinetics of citalopram in relation to genetic polymorphism of CYP2C19. Drug Metab Dispos. 2003;31(10):1255–1259. doi: 10.1124/dmd.31.10.1255. [DOI] [PubMed] [Google Scholar]
- 38.Ohlsson Rosenborg S, Mwinyi J, Andersson M, Baldwin RM, Pedersen RS, Sim SC, et al. Kinetics of omeprazole and escitalopram in relation to the CYP2C19*17 allele in healthy subjects. Eur J Clin Pharmacol. 2008;64(12):1175–1179. doi: 10.1007/s00228-008-0529-z. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Si D, Chen X, Lin N, Guo Y, Zhou H, et al. Influence of CYP2C9 and CYP2C19 genetic polymorphisms on pharmacokinetics of gliclazide MR in Chinese subjects. Br J Clin Pharmacol. 2007;64(1):67–74. doi: 10.1111/j.1365-2125.2007.02846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katsuki H, Nakamura C, Arimori K, Fujiyama S, Nakano M. Genetic polymorphism of CYP2C19 and lansoprazole pharmacokinetics in Japanese subjects. Eur J Clin Pharmacol. 1997;52(5):391–396. doi: 10.1007/s002280050307. [DOI] [PubMed] [Google Scholar]
- 41.Itagaki F, Homma M, Yuzawa K, Nishimura M, Naito S, Ueda N, et al. Effect of lansoprazole and rabeprazole on tacrolimus pharmacokinetics in healthy volunteers with CYP2C19 mutations. J Pharm Pharmacol. 2004;56(8):1055–1059. doi: 10.1211/0022357043914. [DOI] [PubMed] [Google Scholar]
- 42.Qiao HL, Hu YR, Tian X, Jia LJ, Gao N, Zhang LR, et al. Pharmacokinetics of three proton pump inhibitors in Chinese subjects in relation to the CYP2C19 genotype. Eur J Clin Pharmacol. 2006;62(2):107–112. doi: 10.1007/s00228-005-0063-1. [DOI] [PubMed] [Google Scholar]
- 43.Zalloum I, Hakooz N, Arafat T. Genetic polymorphism of CYP2C19 in a Jordanian population: influence of allele frequencies of CYP2C19*1 and CYP2C19*2 on the pharmacokinetic profile of lansoprazole. Mol Biol Rep. 2012;39(4):4195–4200. doi: 10.1007/s11033-011-1204-5. [DOI] [PubMed] [Google Scholar]
- 44.Sakai T, Aoyama N, Kita T, Sakaeda T, Nishiguchi K, Nishitora Y, et al. CYP2C19 genotype and pharmacokinetics of three proton pump inhibitors in healthy subjects. Pharm Res. 2001;18(6):721–727. doi: 10.1023/A:1011035007591. [DOI] [PubMed] [Google Scholar]
- 45.Chen BL, Chen Y, Tu JH, Li YL, Zhang W, Li Q, et al. Clopidogrel inhibits CYP2C19-dependent hydroxylation of omeprazole related to CYP2C19 genetic polymorphisms. J Clin Pharmacol. 2009;49(5):574–581. doi: 10.1177/0091270009333016. [DOI] [PubMed] [Google Scholar]
- 46.Uno T, Niioka T, Hayakari M, Yasui-Furukori N, Sugawara K, Tateishi T. Absolute bioavailability and metabolism of omeprazole in relation to CYP2C19 genotypes following single intravenous and oral administrations. Eur J Clin Pharmacol. 2007;63(2):143–149. doi: 10.1007/s00228-006-0251-7. [DOI] [PubMed] [Google Scholar]
- 47.Yasui-Furukori N, Takahata T, Nakagami T, Yoshiya G, Inoue Y, Kaneko S, et al. Different inhibitory effect of fluvoxamine on omeprazole metabolism between CYP2C19 genotypes. Br J Clin Pharmacol. 2004;57(4):487–494. doi: 10.1111/j.1365-2125.2003.02047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho JY, Yu KS, Jang IJ, Yang BH, Shin SG, Yim DS. Omeprazole hydroxylation is inhibited by a single dose of moclobemide in homozygotic EM genotype for CYP2C19. Br J Clin Pharmacol. 2002;53(4):393–397. doi: 10.1046/j.1365-2125.2002.01366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hunfeld NG, Touw DJ, Mathot RA, Mulder PG, Van Schaik RH, Kuipers EJ, et al. A comparison of the acid-inhibitory effects of esomeprazole and pantoprazole in relation to pharmacokinetics and CYP2C19 polymorphism. Aliment Pharmacol Ther. 2010;31(1):150–159. doi: 10.1111/j.1365-2036.2009.04150.x. [DOI] [PubMed] [Google Scholar]
- 50.Niioka T, Uno T, Yasui-Furukori N, Shimizu M, Sugawara K, Tateishi T. Identification of the time-point which gives a plasma rabeprazole concentration that adequately reflects the area under the concentration–time curve. Eur J Clin Pharmacol. 2006;62(10):855–861. doi: 10.1007/s00228-006-0184-1. [DOI] [PubMed] [Google Scholar]
- 51.Horai Y, Kimura M, Furuie H, Matsuguma K, Irie S, Koga Y, et al. Pharmacodynamic effects and kinetic disposition of rabeprazole in relation to CYP2C19 genotypes. Aliment Pharmacol Ther. 2001;15(6):793–803. doi: 10.1046/j.1365-2036.2001.00980.x. [DOI] [PubMed] [Google Scholar]
- 52.Shimizu M, Uno T, Yasui-Furukori N, Sugawara K, Tateishi T. Effects of clarithromycin and verapamil on rabeprazole pharmacokinetics between CYP2C19 genotypes. Eur J Clin Pharmacol. 2006;62(8):597–603. doi: 10.1007/s00228-006-0152-9. [DOI] [PubMed] [Google Scholar]
- 53.Wang G, Lei HP, Li Z, Tan ZR, Guo D, Fan L, et al. The CYP2C19 ultra-rapid metabolizer genotype influences the pharmacokinetics of voriconazole in healthy male volunteers. Eur J Clin Pharmacol. 2009;65(3):281–285. doi: 10.1007/s00228-008-0574-7. [DOI] [PubMed] [Google Scholar]
- 54.Weiss J, Ten Hoevel MM, Burhenne J, Walter-Sack I, Hoffmann MM, Rengelshausen J, et al. CYP2C19 genotype is a major factor contributing to the highly variable pharmacokinetics of voriconazole. J Clin Pharmacol. 2009;49(2):196–204. doi: 10.1177/0091270008327537. [DOI] [PubMed] [Google Scholar]
- 55.Shi HY, Yan J, Zhu WH, Yang GP, Tan ZR, Wu WH, et al. Effects of erythromycin on voriconazole pharmacokinetics and association with CYP2C19 polymorphism. Eur J Clin Pharmacol. 2010;66(11):1131–1136. doi: 10.1007/s00228-010-0869-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mikus G, Schowel V, Drzewinska M, Rengelshausen J, Ding R, Riedel KD, et al. Potent cytochrome P450 2C19 genotype-related interaction between voriconazole and the cytochrome P450 3A4 inhibitor ritonavir. Clin Pharmacol Ther. 2006;80(2):126–135. doi: 10.1016/j.clpt.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 57.Martis S, Peter I, Hulot JS, Kornreich R, Desnick RJ, Scott SA. Multi-ethnic distribution of clinically relevant CYP2C genotypes and haplotypes. Pharmacogenomics J. 2012. doi:10.1038/tpj.2012.10. [DOI] [PMC free article] [PubMed]
- 58.Saari TI, Laine K, Bertilsson L, Neuvonen PJ, Olkkola KT. Voriconazole and fluconazole increase the exposure to oral diazepam. Eur J Clin Pharmacol. 2007;63(10):941–949. doi: 10.1007/s00228-007-0350-0. [DOI] [PubMed] [Google Scholar]
- 59.Lemberger L, Rowe H, Bosomworth JC, Tenbarge JB, Bergstrom RF. The effect of fluoxetine on the pharmacokinetics and psychomotor responses of diazepam. Clin Pharmacol Ther. 1988;43(4):412–419. doi: 10.1038/clpt.1988.52. [DOI] [PubMed] [Google Scholar]
- 60.Wood N, Tan K, Purkins L, Layton G, Hamlin J, Kleinermans D, et al. Effect of omeprazole on the steady-state pharmacokinetics of voriconazole. Br J Clin Pharmacol. 2003;56(Suppl 1):56–61. doi: 10.1046/j.1365-2125.2003.02000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Angiolillo DJ, Gibson CM, Cheng S, Ollier C, Nicolas O, Bergougnan L, et al. Differential effects of omeprazole and pantoprazole on the pharmacodynamics and pharmacokinetics of clopidogrel in healthy subjects: randomized, placebo-controlled, crossover comparison studies. Clin Pharmacol Ther. 2011;89(1):65–74. doi: 10.1038/clpt.2010.219. [DOI] [PubMed] [Google Scholar]
- 62.Tateishi T, Kumai T, Watanabe M, Nakura H, Tanaka M, Kobayashi S. Ticlopidine decreases the in vivo activity of CYP2C19 as measured by omeprazole metabolism. Br J Clin Pharmacol. 1999;47(4):454–457. doi: 10.1046/j.1365-2125.1999.00914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bae JW, Jang CG, Lee SY. Effects of clopidogrel on the pharmacokinetics of sibutramine and its active metabolites. J Clin Pharmacol. 2011;51(12):1704–1711. doi: 10.1177/0091270010388651. [DOI] [PubMed] [Google Scholar]
- 64.Kang BC, Yang CQ, Cho HK, Suh OK, Shin WG. Influence of fluconazole on the pharmacokinetics of omeprazole in healthy volunteers. Biopharm Drug Dispos. 2002;23(2):77–81. doi: 10.1002/bdd.291. [DOI] [PubMed] [Google Scholar]
- 65.Yasui-Furukori N, Saito M, Uno T, Takahata T, Sugawara K, Tateishi T. Effects of fluvoxamine on lansoprazole pharmacokinetics in relation to CYP2C19 genotypes. J Clin Pharmacol. 2004;44(11):1223–1229. doi: 10.1177/0091270004269015. [DOI] [PubMed] [Google Scholar]
- 66.Uno T, Shimizu M, Yasui-Furukori N, Sugawara K, Tateishi T. Different effects of fluvoxamine on rabeprazole pharmacokinetics in relation to CYP2C19 genotype status. Br J Clin Pharmacol. 2006;61(3):309–314. doi: 10.1111/j.1365-2125.2005.02556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perucca E, Gatti G, Cipolla G, Spina E, Barel S, Soback S, et al. Inhibition of diazepam metabolism by fluvoxamine: a pharmacokinetic study in normal volunteers. Clin Pharmacol Ther. 1994;56(5):471–476. doi: 10.1038/clpt.1994.167. [DOI] [PubMed] [Google Scholar]
- 68.Fang AF, Damle BD, LaBadie RR, Crownover PH, Hewlett D, Jr, Glue PW. Significant decrease in nelfinavir systemic exposure after omeprazole coadministration in healthy subjects. Pharmacotherapy. 2008;28(1):42–50. doi: 10.1592/phco.28.1.42. [DOI] [PubMed] [Google Scholar]
- 69.Caraco Y, Tateishi T, Wood AJ. Interethnic difference in omeprazole’s inhibition of diazepam metabolism. Clin Pharmacol Ther. 1995;58(1):62–72. doi: 10.1016/0009-9236(95)90073-X. [DOI] [PubMed] [Google Scholar]
- 70.Funck-Brentano C, Becquemont L, Lenevu A, Roux A, Jaillon P, Beaune P. Inhibition by omeprazole of proguanil metabolism: mechanism of the interaction in vitro and prediction of in vivo results from the in vitro experiments. J Pharmacol Exp Ther. 1997;280(2):730–738. [PubMed] [Google Scholar]
- 71.Furuta T, Sugimoto M, Shirai N, Ishizaki T. CYP2C19 pharmacogenomics associated with therapy of Helicobacter pylori infection and gastro-esophageal reflux diseases with a proton pump inhibitor. Pharmacogenomics. 2007;8(9):1199–1210. doi: 10.2217/14622416.8.9.1199. [DOI] [PubMed] [Google Scholar]
- 72.Zabalza M, Subirana I, Sala J, Lluis-Ganella C, Lucas G, Tomas M, et al. Meta-analyses of the association between cytochrome CYP2C19 loss- and gain-of-function polymorphisms and cardiovascular outcomes in patients with coronary artery disease treated with clopidogrel. Heart. 2012;98(2):100–108. doi: 10.1136/hrt.2011.227652. [DOI] [PubMed] [Google Scholar]
- 73.Kita T, Sakaeda T, Baba T, Aoyama N, Kakumoto M, Kurimoto Y, et al. Different contribution of CYP2C19 in the in vitro metabolism of three proton pump inhibitors. Biol Pharm Bull. 2003;26(3):386–390. doi: 10.1248/bpb.26.386. [DOI] [PubMed] [Google Scholar]
- 74.von Moltke LL, Greenblatt DJ, Giancarlo GM, Granda BW, Harmatz JS, Shader RI. Escitalopram (S-citalopram) and its metabolites in vitro: cytochromes mediating biotransformation, inhibitory effects, and comparison to R-citalopram. Drug Metab Dispos. 2001;29(8):1102–1109. [PubMed] [Google Scholar]
- 75.Soars MG, Gelboin HV, Krausz KW, Riley RJ. A comparison of relative abundance, activity factor and inhibitory monoclonal antibody approaches in the characterization of human CYP enzymology. Br J Clin Pharmacol. 2003;55(2):175–181. doi: 10.1046/j.1365-2125.2003.01721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Venkatakrishnan K, von Moltke LL, Court MH, Harmatz JS, Crespi CL, Greenblatt DJ. Comparison between cytochrome P450 (CYP) content and relative activity approaches to scaling from cDNA-expressed CYPs to human liver microsomes: ratios of accessory proteins as sources of discrepancies between the approaches. Drug Metab Dispos. 2000;28(12):1493–1504. [PubMed] [Google Scholar]
- 77.Ito K, Brown HS, Houston JB. Database analyses for the prediction of in vivo drug–drug interactions from in vitro data. Br J Clin Pharmacol. 2004;57(4):473–486. doi: 10.1111/j.1365-2125.2003.02041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Obach RS, Walsky RL, Venkatakrishnan K, Gaman EA, Houston JB, Tremaine LM. The utility of in vitro cytochrome P450 inhibition data in the prediction of drug–drug interactions. J Pharmacol Exp Ther. 2006;316(1):336–348. doi: 10.1124/jpet.105.093229. [DOI] [PubMed] [Google Scholar]
- 79.Hung CC, Lin CJ, Chen CC, Chang CJ, Liou HH. Dosage recommendation of phenytoin for patients with epilepsy with different CYP2C9/CYP2C19 polymorphisms. Ther Drug Monit. 2004;26(5):534–540. doi: 10.1097/00007691-200410000-00012. [DOI] [PubMed] [Google Scholar]
- 80.Wilkins JJ, Langdon G, McIlleron H, Pillai G, Smith PJ, Simonsson US. Variability in the population pharmacokinetics of isoniazid in South African tuberculosis patients. Br J Clin Pharmacol. 2011;72(1):51–62. doi: 10.1111/j.1365-2125.2011.03940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 39 kb)
(DOC 3563 kb)