

Abstract
Drug polymer-based amorphous solid dispersions (ASD) are widely used in the pharmaceutical industry to improve bioavailability for poorly water-soluble compounds. Spray-drying is the most common process involved in the manufacturing of ASD material. However, spray-drying involves a high investment of material quantity and time. Lower investment manufacturing processes such as fast evaporation and freeze-drying (lyophilization) have been developed to manufacture ASD at the bench level. The general belief is that the overall performance of ASD material is thermodynamically driven and should be independent of the manufacturing process. However, no formal comparison has been made to assess the in vivo performance of material generated by different processes. This study compares the in vitro and in vivo properties of ASD material generated by fast evaporation, lyophilization, and spray-drying methods using griseofulvin as a model compound and hydroxypropyl methylcellulose acetate succinate as the polymer matrix. Our data suggest that despite minor differences in the formulation release properties and stability of the ASD materials, the overall exposure is comparable between the three manufacturing processes under the conditions examined. These results suggest that fast evaporation and lyophilization may be suitable to generate ASD material for oral evaluation. However, caution should be exercised since the general applicability of the present findings will need to be further evaluated.
KEY WORDS: amorphous, bioavailability, griseofulvin, in vitro, in vivo, solid dispersion
INTRODUCTION
Oral bioavailability is often influenced by factors such as the intestinal permeability, physicochemical properties, and metabolism. Among the physicochemical properties of poorly water-soluble drugs, solubility is considered the most critical factors affecting oral bioavailability. In the past decade, poor solubility has been a growing cause of poor oral bioavailability in the early development stage (1–10). Therefore, there has been a continuous effort in industry to improve the solubility of drug candidates. Despite these efforts, it is often difficult to incorporate solubility into a drug candidate while retaining adequate potency (2,11).
Formulation approaches have been used in both clinical and pre-clinical studies and offer an alternate means to improve oral bioavailability by improving both solubility and dissolution rate. Formulation approaches used to improve the oral bioavailability of poorly soluble compounds include inclusion complexation, nanoparticles, pro-drugs, co-solvents, micelles/emulsions, salts, co-crystals, and amorphous solids. Of these approaches, amorphous solids have recently drawn a high level of interest (1,2,12), and investigations on utilizing amorphous solids to improve oral bioavailability of poorly soluble drugs have been reported (1,2,12–18). Unlike crystalline material, an amorphous solid is characterized as having a molecular arrangement that lacks long range order. Consequently, the entropy (∆S) and free energy (∆G) of an amorphous solid is higher than that of its crystalline counterpart. It is this energy difference that often leads to significantly higher solubility and faster dissolution for a compound’s amorphous form compared to its crystalline form. However, the higher free energy of the amorphous form requires stabilization in order to prevent recrystallization (1,12). The current strategy to stabilize an amorphous solid is to create a solid dispersion amorphous drug polymer system (ASD). Various polymeric matrices have been reported to stabilize amorphous solids in solid dispersion drug polymer systems such as povidone, crospovidone, poloxamer, hydroxypropyl methylcellulose acetate succinate (HPMCAS), hydroxypropyl methylcellulose phthalate, hydroxypropyl methylcellulose, and hydroxypropyl-beta-cyclodextrin, polymethacrylates (1,2,9,12–19).
Various manufacturing processes have been developed for making solid dispersion amorphous drug polymer systems (ASD). These include fast evaporation, freeze-drying, spray-drying, and hot melt extrusion. Currently, it is unknown whether the in vivo performance of amorphous drug polymer systems made using these various processes is comparable. The spray-drying process is the most common process for making amorphous solid dispersions (SDD), and its use has been widely reported (1,2,9,12,14). However, the spray-drying process is both time-consuming and requires a large quantity of bulk drug making it very difficult to implement in the discovery setting where large numbers of candidates are made in small quantities (1). In order to overcome the throughput issues with the spray-drying process, newer processes such as solvent casting (fast evaporation) and freeze-drying (lyophilization) have been developed for manufacturing ASDs at smaller scales. These two simple processes can be executed on the milligram scale with good efficiency (1,2,20–26) and have been widely adopted in drug discovery setting (1,2,24,25).
Maximum stabilization of amorphous solid in a ASD requires intimate mixing of the amorphous solid with the polymer used for stabilization at the molecular level. With less optimal macroscopic mixing, inhibition of compound crystallization may not occur (26). Currently, it is not known whether ASD materials generated by different manufacturing processes are equivalent. To date, there has been no direct comparison of the in vivo performance of these various ASD manufacturing processes. Therefore, the scope of this work was to evaluate the performance of ASD materials generated by different processes both in vitro and in vivo. Our evaluation uses griseofulvin as a model compound and HPMCAS as a representative polymer matrix as its use is well reported (1,27–29). Three common processes used to make ASDs, namely quick evaporation, lyophilization, and spray dry, were compared. Information such as powder X-ray (PXRD) pattern, DSC/TGA, supersaturation solubility, dissolution, and solid-state NMP were collected from in vitro studies using ASD material made by all three processes. Finally, the in vivo performance of ASD made by all three processes was tested in a rat pharmacokinetic study.
MATERIALS AND METHODS
Materials and Instrumentation System
High-performance liquid chromatography (HPLC) grade acetonitrile was obtained from Burdick & Jackson (Muskegon, MI). The HPLC system used was an Agilent HP 1100 HPLC equipped with a diode array and a variable wavelength UV detector, and a quaternary solvent delivery system (Palo Alto, CA). Several analytical columns were tested, and an Alltech Alltima C8 (5 μm, 4.6 × 150 mm) was selected and used for analysis. The water purification system used was a Millipore Milli-Q system. All chemicals used for system validation were either synthesized internally or obtained from Aldrich (St. Louis, MO) and were used without further purification. Hydroxypropyl methylcellulose acetate succinate grade M (HPMCAS-M) was purchased from Shin-Etsu Chemical Co., Ltd. (Tokyo, Japan), lot # 6113225. For the fast evaporation solid dispersion (Evap ASD), an EZ-2 Plus centrifuge vacuum dry system from SP scientific (Stone Ridge, NY) was used for drying with the maximum temperature set at 80°C. A typical vacuum of 6–8 mbar or lower is often achieved during the drying. For the spray-dry solid dispersion (SDD), a Buchi B290 (Flawil, Switzerland) spray dryer coupled with high-performance cyclone and small-volume sample collector was used. For the freeze-dry solid dispersion (Lyo ASD), a Virtis AdVantage 2.0 BenchTop Freeze Dryer from SP Scientific (Gardiner NY) was used. The PXRD pattern was recorded at room temperature with a Rigaku (Texas, USA) MiniFlex II Desktop X-ray Powder Diffractometer. Radiation of Cu Kα at 30 KV −15 mA was used with 2θ increment rate of 3°/min. The scans run over a range of 2–40° 2θ with a step size of 0.02° and a step time of 2 s. The powder samples were placed on a flat Silicon Zero Background sample holder. The particle size distributions of the materials were measured by using a Microtrac (PA, USA) instrument in acidified aqueous. The average of triplicate measurements of each sample was used for the final particle size distribution. The particle size distribution was calculated based on the general purpose (normal sensitivity) analysis model and the following refractive indices: particle RI, 1.58; absorption, 1.0; and dispersant RI, 1.38. For pulverization and milling, a Retsch MM301 ball mill system (Haan, Germany) was used. Typically, sample was milled with a frequency of 20 1/s and duration of 5 min.
Fast Evaporation Solid Dispersion (Evap ASD) of Griseofulvin (20% Drug Load)
The Evap ASD was prepared following a procedure reported previously (1). Generally, solid molecular dispersions are reported as a percentage drug load (by weight) in HPMCAS-MF. For example, a 20% drug load consists of one part (by weight) compound and four parts (by weight) HPMCAS-MF. Griseofulvin (1.0 g) and HPMCAS (4.0 g) were dissolved as a 5 wt% solution in acetone. The solvent was quickly evaporated using the vacuum dry system and operating conditions described above. After drying, the sample was further dried under house vacuum at ambient temperature overnight. The dried Evap ASD was then pulverized by using a bench top micro-mill. The composition of the Evap ASD was measured by HPLC, solid state was checked by PXRD, and particle size was determined by the Microtrac.
Spray-Dry Solid Dispersion of Griseofulvin (20% Drug Load)
SDD preparation followed the procedure reported previously (21). Griseofulvin (1.0 g) and HPMCAS (4.0 g) were dissolved as a 5 wt% solution in acetone. Solutions were spray dried on a Buchi B290 (Flawil, Switzerland) spray dryer using a high-performance cyclone and small-volume sample collector. After spray drying, sample was further dried under house vacuum at ambient temperature overnight. The composition of the SDD was measured by HPLC, solid state was checked by PXRD, and particle size was determined by the Microtrack.
Lyophilized Solid Dispersion (Lyo ASD) of Griseofulvin (20% Drug Load)
Lyo ASD preparation followed the procedure listed as follows. Griseofulvin (1.0 g) and HPMCAS (4.0 g) were dissolved as an 8 wt% solution in DMSO. This mixture was lyophilized using a SP Scientific VirTis AdVantage 2.0 Benchtop Freeze Dryer. A 70-h procedure was used to remove DMSO from the compound. Prior to this process, preliminary freezing was accomplished by charging liquid nitrogen into the mixture then transferred into the freeze dryer. The initial freezing was done under vacuum at −70°C for 1.5 h at 500 mTorr pressure. This ensured that the entire solution stays frozen before primary drying was started. Primary drying was done to remove the bulk solvent via sublimation. From −70°C, the temperature was raised to −35°C, and the pressure was lowered to 100 mTorr for 1 h. The lyophilization pressure needs to be lower than the vapor pressure of DMSO in order to drive sublimation during this step. After drying at −35°C for 1 h, the temperature was raised to 5°C and dried for an additional 28 h at the same pressure. Primary drying ended with the last step at 15°C which is held for 16 h. Secondary drying removed any adsorbed solvent via desorption. The lyophilization pressure was lowered to 50 mTorr, and the temperature was raised to 35°C for 16 h. Secondary drying continued with the temperature lowering to 30°C and pressure lowering to 10 mTorr for 6 h. The final step of the lyophilization cycle has the temperature lowered to 25°C and the pressure raised back to 2,500 mTorr for 1 h. This 70-h recipe results in very low residual solvents in the sample (<1%). After finishing the lyophilization procedure, the sample was further dried under house vacuum at ambient temperature overnight. The dried Lyo ASD was then pulverized by using a bench top micro-mill. The composition of the Lyo ASD was measured by HPLC, solid state was checked by PXRD, and particle size was determined by the Microtrack.
DSC/TGA Analysis and Glass Transition (Tg) Determination
Differential scanning calorimetry was performed on a TA Instruments Q-1000 modulated DSC. A modulated differential scanning calorimeter (TA Instruments Q1000) was used to measure the melting point, glass transition temperature, and heat capacity of both crystalline and the 20% amorphous solid dispersions of griseofulvin with HPMCAS. Samples were initially cooled to 0°C for 5 min and were heated to 250°C at 2°C/min with modulation of ±1.0°C every 60 s. High purity indium was used to calibrate for the heat flow and heat capacity of the instrument. The thermal gravimetric analysis (TGA) experiments were carried out on a Thermal Analysis (New Castle, DE) TGA Q500 system which scans from 25°C to 250°C at a ramp rate of 10°C/min and modulation of ±0.5°C.
Supersaturation Study
For the supersaturation studies, a syringe/filter method was used. The test solution was held in a syringe from which samples are expelled through a filter at predetermined time points. In general, individually weighed griseofulvin ASD materials (40 mg) were placed in an empty disposable 20-mL syringe and 10 mL of pH 6.5 phosphate buffer with 0.01% Tween 80 was drawn into each syringe with extra air (5 mL). A 0.45-μm polyvinylidine difluoride syringe filter was then attached, and the syringe is rotated (100 rpm) on a wheel held in at RT. Analytical samples (100 uL) were expelled at pre-set time points; 50 uL of the samples was diluted 1:1 with DMSO, and the concentration of griseofulvin was determined by HPLC. The HPLC multiple solvent pump system was used for gradient elution. A total of two mobile phases were used to prepare the gradient. Solvent A contained acetonitrile with 0.1% TFA (v/v), and Solvent B contained Milli-Q water with 0.1% TFA (v/v). Flow rate was set at 1.5 ml/min for fast elution. At T = 0 min, the mobile phases (95% A and 5% B) were mixed by the HPLC pump and held for 0.5 min (isocratic elution). From T = 0.51 to T = 4.0 min, a linear gradient from 5% B to 100% B was applied and allowed to hold at 100% for 1 min (from 4.01 to 5.0 min). At T = 5.01 min, the system was set back to the initial condition, and the flow rate was set to 2.0 mL/min and allowed to equilibrate for 1 min to prepare for the next injection. A total of five wavelengths (220, 240, 254, 280, and 330 nm) were used for data collection for best monitoring. An external standard was injected, and 280 nm was selected to quantitatively study the concentration of griseofulvin in ASD materials and formulations.
Intrinsic Dissolution Study
For the intrinsic dissolution test, a paddle over stationary disk system (modified from a Vankel system) was used. Weighed material (50 mg) was added to a stainless steel die (8 mm) with plungers and then compressed into a flat surface by using a Carver Press (5,000 lbs for 10 s). The open end of the die was placed into a 500-mL beaker facing up. An exact amount of dissolution media (200 mL) was then added into the beaker (submerging the die) with an overhead paddle stirring at 50 rpm. Dissolution media used in our study is 50 mM pH 7.4 sodium phosphate buffer with the addition of 1.0% (w/v) of Tween 80 to ensure sink conditions respected to crystalline form were applicable during the study. Analytical samples (0.5 mL) were taken at 0, 1, 2, 3, 4, 5, 10, and 15 min, and replenished with fresh dissolution media each sampling. Samples were analyzed by HPLC with the method described above. The surface area was assumed constant during this short period of time. Duplicate samples were made and checked by PXRD to ensure the lack of polymorphic changes during the experiment. Dissolution rate was calculated based on the total amount of drug dissolved at each time point using the following equation:
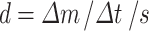 |
where
- d
the initial dissolution rate under this setting (micrograms per square millimeter per minute)
- ∆m
amount of drug dissolved in the media between t1 and t2
- ∆t
t1 − t2
- s
surface areas of the sample disk (πr2 = 50.2 mm2)
Stability Study
For the materials generated by each individual process, physical stability was evaluated by transferring approximately 200 mg of ASD material to a stability oven (controlled at 40°C and 75% RH) for a period of 2 weeks for stability evaluation. PXRD was used to evaluate ASD materials post-storage for crystalline content.
Solid-State NMR Spectroscopy
Solid-state nuclear magnetic resonance (NMR) experiments employed a Bruker Avance III spectrometer operating at 1H and 13C frequencies of 500.13 of 125.77 MHz, respectively. A Bruker double resonance narrow bore solids probe (Bruker BioSpin, Billerica, MA), doubly tuned for 1H and 13C and equipped with a 4-mm spinning module, was used for data collection. Samples were loaded into 4-mm ZrO2 rotors with Kel-F drive caps (Wilmad-LabGlass, Vineland, NJ). All experiments employed a 1H 90° pulse width of 2.9 ms, ramped cross polarization (CP), and SPINAL64 1H decoupling at a field of 86 kHz. A 5-pi version of Total Sideband Suppression was implemented for all experiments with 13C 180° pulses of 7.2 ms. The magic angle spinning rate was 8,000 ± 3 Hz, and the sample temperature was 298–300°K. Spectra of solid dispersions were acquired with a 4-s pulse delay, 2 ms CP contact time, and 1,944 scans. Crystalline griseofulvin data were acquired using a 2-s pulse delay, 8 ms CP contact time, and 486 scans. The dipolar dephased spectrum of crystalline griseofulvin was acquired using a 60-ms interruption in decoupling prior to acquisition. The melt-quenched amorphous griseofulvin spectrum was acquired with a 2-s pulse delay, 2 ms CP contact time, and 486 scans. A 13C-detected saturation recovery pulse sequence was utilized to measure 1H T1 relaxation times. The sequence used 16 1H 90° pulses for 1H saturation, and 16 recovery delays varied from 0.05 to 20 s with 64 scans for each slice. A 13C-detected CP pulse sequence with an additional 1H spin-locking period prior to CP was used to measure 1H T1r relaxation times. Sixteen 1H spin-locking times which varied from 0.05 to 50 ms were used to characterize T1r, with 256 or 512 scans collected for each slice. To calculate relaxation times of individual components in solid dispersions, griseofulvin was integrated from ∼34 to 47 ppm, while HPMCAS was integrated from ∼167 to 177 ppm. All relaxation data were fitted using KaleidaGraph 4.0 (Synergy Software, Reading, PA). Carbon peak assignments are based on predicted chemical shifts (ACD/Labs, Toronto, Canada) and dipolar dephasing data.
In Vivo Plasma Sample Analysis (LC/MS/MS)
For plasma sample analysis, LC/MS/MS system was used. Griseofulvin stocks were prepared at 0.1 mg/mL in methanol as working solutions. Intermediate mixed standard solutions at the desired concentrations for the calibration curve were made by serial dilution with methanol using working solutions. The internal standard spiking solution containing d8-dicyclohexyl urea was prepared at 1,000 ng/mL in 1:1 methanol/water. Calibration standards were prepared by spiking appropriate amounts of the intermediate mixed standard solutions into blank rat plasma and ranged from 1.02 to 6,670 ng/mL. Ten microliters of internal standard spiking solution (1,000 ng/ml) was added to each plasma sample and the calibration standards. The samples and calibration standards were extracted by protein precipitation using 150 μL acetonitrile. The samples were then vortexed for 5 min and centrifuged at 3,700 rpm for 15 min at 25°C. Post-centrifugation, 30 μL of supernatant was transferred to a shallow well injection plate which contained 120 μL of water. For the liquid chromatography–tandem mass spectrometry (LC-MS/MS) system, a Nexera UPLC injection system (Shimadzu, Kyoto, Japan) and a Shimadzu SIL-30AD solvent delivery system coupled with a Sciex API5500 QTrap turboionspray were used. The turboionspray source was operated under multiple reactions monitoring (MRM) mode for the quantitation of the compounds. The mass spectrometer was operated at unit mass resolution for both Q1 and Q3 quadrupoles. The chromatography separation was achieved on a Phenomenex Kinetex C18 (50 × 2.1 mm, 2.7 μm) column with gradient elution using mobile phase A of 0.1% formic acid in water and mobile phase B of 0.1% formic acid in acetonitrile. The LC flow rate was 0.7 mL/min, and the sample injection volume was 10 μL. The column temperature was set at 30°C. The MRM (parent/product) transition and lower limit of quantitation for griseofulvin is 353.0/165.0 and 3.05 ng/mL, respectively.
In Vivo Oral Pharmacokinetic Study in Rats
The purpose of this study was to examine the effect of ASD manufacturing process on the oral pharmacokinetics of grisoefulvin in male Sprague–Dawley rats (Charles River Laboratories, Hollister, CA). The study was conducted in accordance with the institutional guidelines for humane treatment of animals and was approved by the IACUC of Genentech. At study initiation, rats used weighed from 200 to 350 g and were 7 to 9 weeks of age. All animals were fasted overnight before dosing. Four groups of rats (n = 4 per group) received a single 75-mg/kg oral dose of griseofulvin as suspension prepared in corn oil (1 mL/kg) followed by 1 mL of 0.5% methylcellulose and 0.2% Tween 80 in water as chaser. A total of four groups were dosed. Group 1 was dosed with the griseofulvin bulk drug and HPMCAS physical mixture (match 20% drug load), Group 2 was dosed with the griseofulvin ASD made by quick evaporation, Group 3 was dosed with the griseofulvin ASD made by lyophilization, and Group 4 was dosed with the griseofulvin SDD made by spray drying. Blood samples (approximately 200 μL per sample) were collected from the carotid artery at predose, 2, 5, 15, and 30 min post-dose and 1, 2, 4, 8, and 24 h post-dose. All samples were collected into tubes containing potassium ethylenediaminetetraacetic acid as an anticoagulant. Blood samples were centrifuged within 30 min of collection, and plasma was harvested. Plasma samples were stored at ∼70°C until analysis for concentrations by a LC-MS/MS assay method. Compartmental methods were used to estimate pharmacokinetic parameters, and a two-tailed independent T test was used for statistical analysis for comparison.
RESULTS AND DISCUSSION
Thermal, PXRD, and DSC Analysis
The manufacturing of griseofulvin ASD material using rapid evaporation (Evap ASD), lyophilization (Lyo ASD), and spray-dry (SDD) processes was successful based on in vitro evaluations that were performed. The PXRD data shown in Fig. 1 suggested that all processes gave amorphous materials. HPLC potency analysis indicated that potencies of all ASD materials were within desired range (20.0 ± 0.5%). A result of the thermal analysis shown in Table I suggested all three ASD materials have single and similar Tg (glass transition temperature) and some minor differences in heat capacity (Cp). All the results above suggested that the materials generated by three different process were comparable macroscopically (30,31).
Fig. 1.
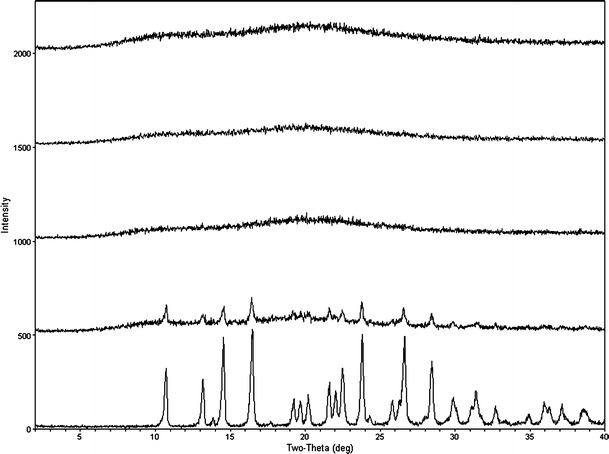
PXRD of griseofulvin (top to bottom) SDD, Evap ASD, Lyo ASD, physical mixture, API (SDD, Evap ASD, Lyo ASD, and physical mixture are made with HPMCAS with 20% drug load)
Table I.
Thermal Results of ASD Material (Griseofulvin HPMCAS 20/80) Generated Using Different Manufacturing Processes (Duplicate Measurements)
| Manufacturing process | T g (°C; two measurements) | C p (J/g °C; two Measurements) | ∆G (J/g; two measurements) |
|---|---|---|---|
| Rapid evaporation (Evap ASD) | 94.0/91.4 | 0.370/0.390 | −418/−434 |
| Lyophilization (Lyo ASD) | 91.6/88.3 | 0.376/0.418 | −475/−468 |
| Spray-dried (SDD) | 95.9/92.1 | 0.375/0.416 | −405/−428 |
Free energy differences between solid dispersions made through different processes were estimated by adopting a literature method (32,33). At a given temperature T, free energy difference between two solid dispersions ASD1 and ASD2 can be expressed as:
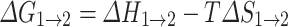 |
1 |
where ΔH1→2 and ΔS1→2 are the enthalpy and entropy differences between the two ASDs, respectively.
ΔH1→2 and ΔS1→2 at a given temperature T < Tg can be estimated through the measurement and integration of the isobaric heat capacity Cp within a temperature range between 37°C (310°K) to 150°C (423°K). In this study Cp was measured via the modulated DSC method outlined in the “Materials and Methods” section, and was integrated following the trapezoidal rule to yield ΔH1→2 and ΔS1→2 (32,33). The selection of 150°C as the upper temperature used in the ∆H estimation is based on the fact that the Tg for all ASD materials is around 90°C. Therefore, 50°C above the Tg should be sufficient enough to ensure all ASD are fully melted, and thus, the relative G (i.e., Evap ASD and SDD) should become zero. The estimated ∆G of each ASD material from duplicate measurements is presented in Table I. A comparison of ∆G at 37°C (Tref) should provide a rank order of the physical stability for the ASD materials tested. Our estimates of ∆G show the highest ∆G for the ASD material manufactured by lyophilization (Lyo) which is consistent with the PXRD physical stability data generated in accelerated conditions (i.e., 40°C/75% relative humidity). When PXRD was performed on samples that were placed in stability chamber under accelerated conditions, a crystalline signal was observed by day 3 for the Lyo ASD material but not the Evap ASD and the SDD material. Based upon the PXRD data, the Evap ASD and SDD stay amorphous for a much longer period (up to 28 days) under accelerated conditions.
Solid-State NMR, Supersaturation, and Dissolution Characterization
NMR spectroscopy was used to study molecular mobility as NMR relaxation rates are governed by the mobility of the sample (34–36) (Table II). Spin–lattice relaxation times (T1) are indicative of mobility on the Larmor timescale, or relatively rapid molecular motions in the tens to hundreds of MHz regime. 1H T1 measurements provide a picture of global mobility of the sample as 1H spin diffusion results in a uniform relaxation time across a given domain in a sample . If a two-component sample is not intimately mixed, spin diffusion becomes inefficient between the two domains, and unique 1H T1 times will be observed for each domain. The domain size limit of mixing homogeneity can be estimated using the equation relating length scale (L) to the time over which spin diffusion (D) takes place, in this case the relaxation time (T1) which is shown below in Eq. 2:
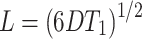 |
2 |
Table II.
Solid-State NMR 1H Relaxation Data for Griseofulvin, HPMCAS, and Solid Dispersions
| Sample | Griseofulvin 1H T 1 (s) | HPMCAS 1H T 1 (s) | Griseofulvin 1H T 1ρ (ms) | HPMCAS 1H T 1ρ (ms) |
|---|---|---|---|---|
| Crystalline griseofulvin | 1.05 ± 0.02 | – | 309 ± 43 | – |
| Amorphous griseofulvin | 1.08 ± 0.03 | – | 82.5 ± 2.6 | – |
| HPMCAS | – | 2.45 ± 0.10 | – | 8.81 ± 0.20 |
| 20:80 Griseofulvin/HPMCAS (spray-dried) | 1.61 ± 0.16 | 1.46 ± 0.06 | 15.0 ± 0.7 | 14.7 ± 0.4 |
| 20:80 Griseofulvin/HPMCAS (rapid evaporation) | 1.77 ± 0.15 | 1.81 ± 0.07 | 14.7 ± 0.7 | 13.6 ± 0.5 |
| 20:80 Griseofulvin/HPMCAS (lyophilization) | 2.66 ± 0.13 | 3.11 ± 0.14 | 13.6 ± 0.4 | 12.6 ± 0.3 |
Carbon-detected 1H T1 measurements afforded the resolution to measure the relaxation times of both griseofulvin and HPMCAS in these solid dispersions. The 13C solid-state NMR spectrum of crystalline griseofulvin is shown in Fig. 2, with carbon atom assignments provided for reference. Figure 3a–c illustrated the 13C solid-state NMR reference spectra for crystalline griseofulvin, neat amorphous griseofulvin, and HPMCAS, while Fig. 3d–f shows the spectra of the solid dispersions prepared via three different manufacturing processes. No crystalline griseofulvin (sharp, narrow peaks) is observed in any of the solid dispersion samples. Remaining DMSO [peak at 40 ppm (DMSO methyls)] from the preparation process was evident for the Lyo ASD sample (Fig. 3f). This is a known common problem for the Lyo process (1). Relaxation measurements were investigated in order to further discern differences among the three solid dispersions. Both the Evap ASD and SDD show uniform 1H T1 values, across both griseofulvin and HPMCAS components (Table II). Using the accepted spin diffusion rate for rigid polymer systems (0.8 nm2/ms), the T1 data indicate that the amorphous solid and the polymer are homogeneously mixed down to an estimated domain size of less than 93 nm in the Evap ASD and 85 nm in the SDD. Interestingly, the Lyo ASD showed longer 1H T1 times compared to the other two dispersions and of the two pure components, indicating reduced overall mobility. However, the griseofulvin T1 (2.66 s) and HPMCAS T1 (3.11 s) are statistically different to each other to infer possible phase separation (Table II). The longer absolute T1 values in the lyophilized sample may be an indication of reduced free volume in the Lyo ASD material due to a DMSO impurity in the same as shown by the peak at ∼40 ppm (Fig. 3f). DMSO has adequate rigidity to cross polarize in the NMR experiment. Thus, DMSO is a component of the solid dispersion that can accept hydrogen bonds from griseofulvin and HPMCAS, possibly reducing free volume and mobility of the entire dispersion.
Fig. 2.
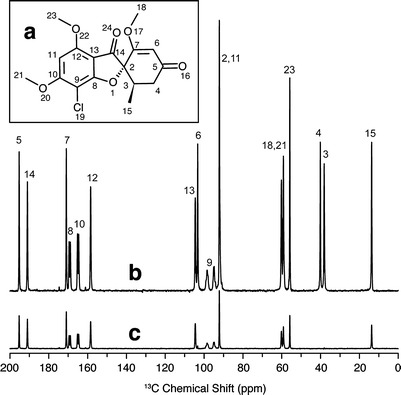
a Molecular structure of griseofulvin with numbering used in peak assignments; b 13C solid-state NMR spectrum of crystalline griseofulvin with peak assignments; c 13C solid-state NMR spectrum of crystalline griseofulvin with dipolar dephasing applied to remove CH and CH2 carbon resonances
Fig. 3.
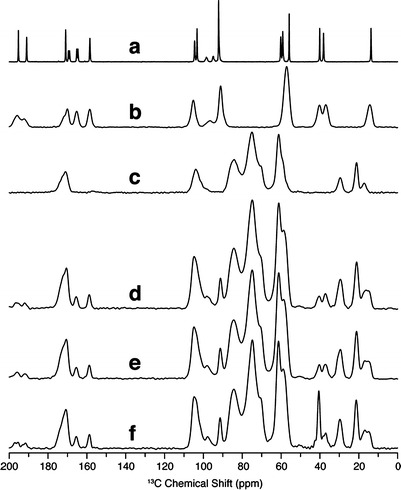
13C solid-state NMR spectra of a crystalline griseofulvin, b melt-quenched amorphous griseofulvin, c hydroxypropylmethylcellulose acetate succinate (HPMCAS), d spray-dried dispersion containing 20% griseofulvin and 80% HPMCAS, e centrifugal evaporated dispersion containing 20% griseofulvin and 80% HPMCAS, f lyophilized dispersion containing 20% griseofulvin and 80% HPMCAS
Spin–lattice relaxation time in the rotating frame (T1ρ) probes molecular motions on the order of tens of kilohertz, and is a good measurement of phase uniformity at smaller domain sizes. The SDD showed the best homogeneity as reflected in 1H T1 ρ, and this dispersion can be assumed to be homogeneous down to a domain size of <8 nm. While the T1 ρ values for each component in the Evap ASD and Lyo ASD samples are similar to each other, the differences indicate that it is likely there is some degree of heterogeneity in the lower nanometer length scale. Based on the 1H T1 ρ data, the predicted physical stability for each dispersion would be SDD > Evap ASD > Lyo ASD. This is consistent with the stability data generated using PXRD which suggested that the SDD and Evap ASD material was more stable than the Lyo ASD material.
The observed difference in stability between ASD material generated using various manufacturing processes is not surprising since each process has differences in both preparation time and the degree of solvent removal (1). Compared to Evap or Lyo, solvent removal is the most rapid for the SDD manufacturing process due to a very large surface area (small droplets) created during the process. Thus, SDD achieves the best homogeneity of the drug polymer mixtures. Differences in homogeneity of ASD material can have an impact on solubility and/or dissolution rate which may affect in vivo release characteristics. In the supersaturation study, the SDD material showed the quickest “onset” (5 min) to peak concentration when compared with Evap and Lyo (15 min; Fig. 4). However, the intrinsic dissolution rates suggested all three ASD materials were approximately equivalent. For the physical mixture, the dissolution rate is measured as 0.08 ± 0.04 μg/mm2/min. For Evap ASD, Lyo ASD, and SDD, the dissolution rate is measured as 0.30 ± 0.06, 0.30 ± 0.08, and 0.31 ± 0.08 μg/mm2/min, respectively. We speculate that the different onset times observed in the supersaturation may be explained by the different particle sizes of the material generated from each manufacturing process (Table III). Furthermore, the difference of supersaturation between SDD, Evap ASD, and Lyo ASD was not significant, suggesting in this case the homogeneity of the material has little influence on the acute solubility (Fig. 4). Since all three ASD materials converted back to crystalline in aqueous media at approximately the same rate (evident by solubility decreasing), no data suggest that in this case, homogeneity has significant influence on maintaining supersaturation upon dissolving. Overall, the long range homogeneity seems be a better predictor for stability upon storage which is a critical factor for decision making.
Fig. 4.
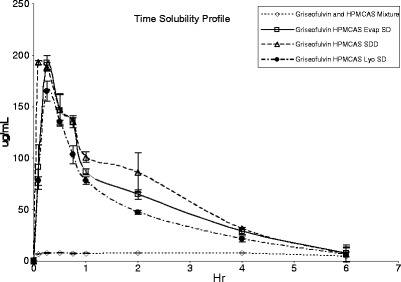
Plot of solubility vs time for supersaturation study (average of duplicate)
Table III.
Particle Size Analysis of Griseofulvin (Average ± SD)
| Test materials | D10 (μm) | D50 (μm) | D90 (μm) |
|---|---|---|---|
| Crystalline bulk drug griseofulvin | 3.6 ± 0.1 | 10.7 ± 0.1 | 19.0 ± 0.1 |
| Rapid evaporation (Evap ASD) | 21.0 ± 0.1 | 61.8 ± 0.3 | 154.9 ± 3.9 |
| Lyophilization (Lyo ASD) | 12.2 ± 0.0 | 33.1 ± 0.1 | 65.7 ± 0.7 |
| Spray-dried (SDD) | 3.7 ± 0.1 | 6.0 ± 0.3 | 9.5 ± 0.5 |
Rat Pharmacokinetic Study
A rat pharmacokinetic study was performed to evaluate the in vivo performance of the ASD materials generated by different processes. The in vivo pharmacokinetic data obtained by dosing griseofulvin in rats are summarized in Table IV, and the plasma concentration time profiles are shown in Fig. 5. Griseofulvin is known to be highly crystalline, and the results of our supersaturation study effect demonstrated that the supersaturation effect was sustained for only a very short period of time (Fig. 4). Because of this concern, a corn oil suspension (small dose volume) with aqueous chaser was used for the oral delivery of the ASD material to minimize crystallization before dosing (37). Overall, the in vivo results are in line with information generated from the in vitro studies. Rats dosed with griseofulvin ASD material showed significantly higher (p < 0.05) exposures (Cmax and AUC) when compared to rats dosed with physical mixture (Table IV). This finding is in good agreement with literature reports that the absorption of griseofulvin crystalline drug is a dissolution rate/solubility-controlled process (38–43). More importantly, overall exposure as assessed by AUC was not significantly different (p < 0.05) for ASD material generated by SDD, Evap ASD, and Lyo ASD manufacturing processes.
Table IV.
Pharmacokinetics of Griseofulvin (Griseofulvin HPMCAS 20/80) in Rats (n = 4) for Various Amorphous Solid Dispersions
| PK parameters/formulation | T max ± SD (h) | C max ± SD (uM) | AUC ± SD (uM × h) | CL/F ± SD (L/h/kg) |
|---|---|---|---|---|
| Physical mixture | 4.0 ± 0.0 | 3.2 ± 1.3 | 13.9 ± 5.2 | 17.1 ± 7.2 |
| Rapid evaporation (Evap ASD) | 4.0 ± 2.8 | 9.9 ± 4.9 | 50.4 ± 21.2 | 5.0 ± 2.6 |
| Lyophilization (Lyo ASD) | 1.2 ± 1.9 | 9.0 ± 3.1 | 31.9 ± 9.7 | 7.1 ± 2.0 |
| Spray-dried (SDD) | 0.2 ± 0.1 | 22.8 ± 8.1 | 46.2 ± 19.7 | 5.2 ± 2.1 |
Fig. 5.
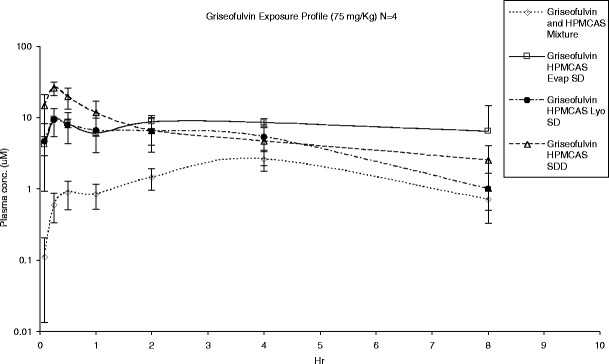
In vivo exposure of griseofulvin crystalline HPMCAS mixture and amorphous solid dispersion material generated using different manufacturing processes
Of note, no statistical difference (p < 0.05) was observed between both Cmax of the Evap ASD and Lyo ASD material. The ASD material generated by the SDD process resulted in a higher Cmax and shorter Tmax compared to the other ASD materials (p < 0.05). We speculate that this difference in Cmax and Tmax is related to a faster in vivo dissolution rate resulting from smaller particle size of the SDD material (Table III). This was also consistent with our observations from the supersaturation study (Fig. 4). We performed a pharmacokinetic analysis using a two-compartment model (44) in order to characterize the change in the absorption rate constant between Evap ASD and SDD material. The fitted absorption rate constant (ka) for SDD was approximately ten times higher than the Evap ASD (5 versus 0.4 h−1) which is consistent with a more rapid dissolution due to a smaller particle size observed for the SDD material. Despite this subtle difference in in vivo release by the SDD material, the overall exposure (i.e., AUC) was still comparable for ASD material generated using all three processes. Therefore, we conclude that both the Evap and Lyo manufacturing processes are suitable to generated ASD material for in vivo evaluation. However, the general applicability of our findings will need to be further validated, and other factors including particle size will need to be controlled. Studies of the in vivo performance of different ASD materials in higher animal species such as the dog would also be warranted.
CONCLUSIONS
Polymer-based amorphous solid dispersions have drawn much interest of late due to the increasing number of poorly soluble compounds in both the preclinical and clinical areas. Despite the high interest level, the implementation of amorphous solid dispersions at the drug discovery stage has been hampered mainly due to high material and time demands of the commonly used spray-drying process used to generate ASD material. More recently, several small-scale bench processes have been developed to make ASD materials without large investment. To date, there have been no direct comparative in vitro and in vivo evaluations of ASD material generated by the various manufacturing processes. Our study described the use of griseofulvin as model compound and compared the ASD (HPMCAS) materials generated by different processes. Base on the results of our study, we conclude that all three processes are very robust in terms of generating ASD material to evaluate the effect of ASD formulations for improving oral bioavailability of griseofulvin. Newer processes such as Evap and Lyo are reliable and suitable to generate ASD material for griseofulvin without large investments in time and compound quantity. The study results also suggest that some minor differences may lead to erroneous conclusions of physical stability for long-term storage. Thus, a careful evaluation of each technology should be conducted before selecting a process of producing solid dispersions.
ACKNOWLEDGMENTS
The authors would like to give special thanks to Dr. George Zografi for his valuable inputs to the manuscript.
REFERENCES
- 1.Chiang PC, Chou KJ, Cui Y, Sambrone A, Chan C, Hart R. Evaluation of spray-dried drug load and polymer by using a 96-well plate vacuum dry system for amorphous solid dispersion drug delivery. AAPS Pharm Sci Technol. 2012;13:713–22. doi: 10.1208/s12249-012-9795-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friesen DT, Shanker R, Crew M, Smithey DT, Curatolo WJ, Nightingale JAS. Hydroxypropyl methylcellulose acetate succinate-based spray-dried dispersions: an overview. Mol Pharm. 2008;5:1003–19. doi: 10.1021/mp8000793. [DOI] [PubMed] [Google Scholar]
- 3.Chiang PC, Thompson DC, Ghosh S, Heitmeier MR. A formulation-enabled pre-clinical efficacy assessment of a farnesoid X receptor (FXR) agonist, GW4064, in hamsters and cynomolgous monkeys. J Pharm Sci. 2011;100:4722–33. doi: 10.1002/jps.22664. [DOI] [PubMed] [Google Scholar]
- 4.Lipinski CA. Poor aqueous solubility—an industry wide problem in drug discovery. Am Pharm. 2002;5:82–5. [Google Scholar]
- 5.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2002;44:235–49. doi: 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 6.Lipinski, CA. 2003. Physicochemical properties and the discovery of orally active drugs: technical and people issues. Molecular informatics: confronting complexity. Proceedings of the Beilstein-Institut Workshop (Frankfurt, Germany)
- 7.Gardner CR, Walsh CT, Almarsson O. Drugs as materials: valuing physical form in drug discovery. Nat Rev Drug Discov. 2004;3:926–34. doi: 10.1038/nrd1550. [DOI] [PubMed] [Google Scholar]
- 8.Schroter C. Prioritizing molecules based on physicochemical characteristics. Am Pharm. 2006;9:60–7. doi: 10.1002/jps.3080090114. [DOI] [Google Scholar]
- 9.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50:47–60. doi: 10.1016/S0939-6411(00)00076-X. [DOI] [PubMed] [Google Scholar]
- 10.Anon. New drug development. GAO Report to Congress. GAO-07-49; 2006 November.
- 11.Ruben AJ, Kiso Y, Freire E. Overcoming roadblocks in lead optimization: a thermodynamic perspective. Chem Biol Drug Des. 2006;67:2–4. doi: 10.1111/j.1747-0285.2005.00314.x. [DOI] [PubMed] [Google Scholar]
- 12.Gao P. Amorphous pharmaceutical solids: characterization, stabilization, and development of marketable formulations of poorly soluble drugs with improved oral absorption. Mol Pharm. 2008;5:903–4. doi: 10.1021/mp800203k. [DOI] [PubMed] [Google Scholar]
- 13.Padden BE, Miller JM, Robbins T, Zocharski PD, Prasad L, Spence JK, et al. Amorphous solid dispersions as enabling formulation for discovery and early development. Am Pharm Rev. 2011;14:66–73. [Google Scholar]
- 14.Newman A, Knipp G, Zografi G. Commentary: assessing performance of amorphous solid dispersions. J Pharm Sci. 2012;101:1355–77. doi: 10.1002/jps.23031. [DOI] [PubMed] [Google Scholar]
- 15.Ivaniscevic I. Physical stability studies of miscible amorphous solid dispersions. J Pharm Sci. 2010;99:4005–12. doi: 10.1002/jps.22247. [DOI] [PubMed] [Google Scholar]
- 16.Hageman MJ, Miyake PJ, Stefanski KJ, He X, Rohrs BR, Mackin LA, et al. Solid state form of celecoxib having enhanced bioavailability. WO0141536. 2001.
- 17.Gupta P, Kakumanu VK, Bansal AK. Stability and solubility of celecoxib-PVP amorphous dispersions: a molecular perspective. Pharm Res. 2004;21(10):1762–9. doi: 10.1023/B:PHAM.0000045226.42859.b8. [DOI] [PubMed] [Google Scholar]
- 18.DiNunzio JC, Miller DA, Yang W, McGinity JW, Williams RO., III Amorphous compositions using concentration enhancing polymers for improved bioavailability of itraconazole. Mol Pharm. 2008;5:968–80. doi: 10.1021/mp800042d. [DOI] [PubMed] [Google Scholar]
- 19.Lipinski C, Lombardo F, Dominy B, Feeney P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 20.Hancock B, Parks M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm Res. 2000;17:397–404. doi: 10.1023/A:1007516718048. [DOI] [PubMed] [Google Scholar]
- 21.Imaizumi H. Stability and several physical properties of amorphous and crystalline forms of indomethacin. Chem Pharm Bull (Tokyo) 1980;29:983–7. doi: 10.1248/cpb.28.2565. [DOI] [PubMed] [Google Scholar]
- 22.Kaushal AM, Gupta P, Bansal AK. Amorphous drug delivery systems: molecular aspects, design and performance. Crit Rev Ther Drug Carrier Syst. 2004;21:133–93. doi: 10.1615/CritRevTherDrugCarrierSyst.v21.i3.10. [DOI] [PubMed] [Google Scholar]
- 23.Patel RC, Masnoon S, Patel MM, Patel NM. Formulation strategies for improving drug solubility using solid dispersions. Pharm Rev. 2009;7(6) ISSN: 1918–5561.
- 24.Mansky P, Dai W, Li S, Pollock-Dove C, Daehne K, Dong L, et al. Screening method to identify preclinical liquid and semi-solid formulations for low solubility compounds: miniaturization and automation of solvent casting and dissolution testing. J Pharm Sci. 2007;96:1548–63. doi: 10.1002/jps.20799. [DOI] [PubMed] [Google Scholar]
- 25.Shanbhag A, Rabel S, Nauka E, Casadevall G, Shivanand P, Eichenbaum G, et al. Method for screening of solid dispersion formulations of low-solubility compounds—miniaturization and automation of solvent casting and dissolution testing. Int J Pharm. 2008;351:209–18. doi: 10.1016/j.ijpharm.2007.09.042. [DOI] [PubMed] [Google Scholar]
- 26.Rumondor ACF, Ivanisevic I, Bates S, Alonzo DE, Taylor LS. Evaluation of drug-polymer miscibility in amorphous solid dispersion systems. Pharm Res. 2009;26:2523–34. doi: 10.1007/s11095-009-9970-7. [DOI] [PubMed] [Google Scholar]
- 27.Curatolo W, Nightingale JA, Herbig SM. Utility of hydroxypropylmethylcellulose acetate succinate (HPMCAS) for initiation and maintenance of drug supersaturation in the GI milieu. Pharm Res. 2009;26:1419–31. doi: 10.1007/s11095-009-9852-z. [DOI] [PubMed] [Google Scholar]
- 28.Al-Obaidi H, Brocchini S, Buckton G. Anomalous properties of spray dried solid dispersions. J Pharm Sci. 2009;98:4724–37. doi: 10.1002/jps.21782. [DOI] [PubMed] [Google Scholar]
- 29.Al-Obaidi H, Buckton G. Evaluation of griseofulvin binary and ternary solid dispersions with HPMCAS. AAPS PharmSciTech. 2009;10:1172–7. doi: 10.1208/s12249-009-9319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qian F, Zhu JHQ, Haddadin R, Gawel J, Garmise R, Hussain M. Is a distinctive single Tg a reliable indicator for the homogeneity of amorphous solid dispersion? Int J Pharm. 2010;395:232–5. doi: 10.1016/j.ijpharm.2010.05.033. [DOI] [PubMed] [Google Scholar]
- 31.Newman A, Enger D, Bates S, Ivanisevic I, Kelly R, Zografi G. Characterization of amorphous API:polymer mixtures using x-ray powder diffraction. J Pharm Sci. 2008;97:4840–56. doi: 10.1002/jps.21352. [DOI] [PubMed] [Google Scholar]
- 32.Murdande SB, Pikal MJ, Shanker RM, Bogner RH. Solubility advantage of amorphous pharmaceuticals: I. A thermodynamic analysis. J Pharm Sci. 2010;99:1254–64. doi: 10.1002/jps.21903. [DOI] [PubMed] [Google Scholar]
- 33.Murdande SB, Pikal MJ, Shanker RM, Bogner RH. Solubility advantage of amorphous pharmaceuticals: II. Application of quantitative thermodynamic relationships for prediction of solubility enhancement in structurally diverse insoluble pharmaceuticals. Pharm Res. 2010;27:2704–14. doi: 10.1007/s11095-010-0269-5. [DOI] [PubMed] [Google Scholar]
- 34.Ivanisevic I, Bates S, Chen P. Novel methods for the assessment of miscibility of amorphous drug-polymer dispersions. J Pharm Sci. 2009;98:3373–86. doi: 10.1002/jps.21717. [DOI] [PubMed] [Google Scholar]
- 35.Pham TN, Watson SA, Edwards AJ, Chavda M, Clawson JS, Strohmeier M, et al. Analysis of amorphous solid dispersions using 2D solid-state NMR and 1H T1 relaxation measurements. Mol Pharm. 2010;7:1667–91.A. doi: 10.1021/mp100205g. [DOI] [PubMed] [Google Scholar]
- 36.Newman A, Munson E. Characterizing miscibility in amorphous solid dispersions. Am Pharm Rev. 2012;15:92–8. [Google Scholar]
- 37.Chiang PC, South SA, Daniels JS, Anderson DR, Wene SP, Albin LA, et al. Aqueous vs. non-aqueous salt delivery strategies to enhance oral bioavailability of a mitogen activated protein kinase activated protein kinase (MK-2) inhibitor in rats. J Pharm Sci. 2009;98:248–56. doi: 10.1002/jps.21425. [DOI] [PubMed] [Google Scholar]
- 38.Wong SM, Kellaway IW, Murdan S. Enhancement of the dissolution rate and oral absorption of a poorly water soluble drug by formation of surfactant-containing microparticles. Int J Pharm. 2006;317:61–8. doi: 10.1016/j.ijpharm.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 39.Bates TR, Sequeira JA. Bioavailability of micronized griseofulvin from corn oil-in-water emulsion, aqueous suspension and commercial tablet dosage forms in humans. J Pharm Sci. 1975;64:793–7. doi: 10.1002/jps.2600640513. [DOI] [PubMed] [Google Scholar]
- 40.Chiou WL, Riegelman S. Preparation and dissolution characteristics of several fast-release solid dispersions of griseofulvin. J Pharm Sci. 1969;58:1505–10. doi: 10.1002/jps.2600581218. [DOI] [PubMed] [Google Scholar]
- 41.Chiou WL, Riegelman S. Oral absorption of griseofulvin in dogs: increased absorption via solid dispersion in dogs: increased absorption via solid dispersion in polyethylene glycol 6000. J Pharm Sci. 1970;59:937–42. doi: 10.1002/jps.2600590703. [DOI] [PubMed] [Google Scholar]
- 42.Tur KM, Ching HS, Baie S. Use of bioadhesive polymer to improve the bioavailability of griseofulvin. Int J Pharm. 1997;148:63–71. doi: 10.1016/S0378-5173(96)04827-2. [DOI] [Google Scholar]
- 43.Chiou WL, Riegelman S. Absorption characteristics of solid dispersed and micronized griseofulvin in man. J Pharm Sci. 1971;60:1376–80. doi: 10.1002/jps.2600600919. [DOI] [PubMed] [Google Scholar]
- 44.Gilbaldi M, Perrier D. Pharmacokinetics. 2. New York: Marcel Dekker; 1982. [Google Scholar]