

Abstract
The purposes of this study were to assess the efficiency of different nifedipine amorphous solid dispersions (ASDs) in achieving and maintaining supersaturation and to investigate the solubility–permeability interplay when increasing the apparent solubility via ASD formulations. Spray-dried ASDs of nifedipine in three different hydrophilic polymers, hydroxypropyl methylcellulose acetate succinate (HPMC-AS), copovidone, and polyvinylpyrrolidone (PVP), were prepared and characterized by powder X-ray diffraction and differential scanning calorimetry. The ability of these formulations to achieve and maintain supersaturation over 24 h was assessed. Then, nifedipine’s apparent intestinal permeability was investigated as a function of increasing supersaturation in the parallel artificial membrane permeability assay model and in the single-pass rat intestinal perfusion model. The efficiency of the different ASDs to achieve and maintain supersaturation of nifedipine was found to be highly polymer dependent; while a dispersion in HPMC-AS enabled supersaturation 20× that of the crystalline aqueous solubility, a dispersion in copovidone enabled 10×, and PVP allowed supersaturation of only 5× that of the crystalline solubility. Nifedipine flux across the intestine from supersaturated solutions was increased, and the apparent intestinal permeability was constant, irrespective of the degree of supersaturation or the polymer being used. In conclusion, while with other solubility-enabling approaches (e.g., surfactants, cyclodextrins, cosolvents), it is not enough to increase the apparent solubility, but to strike the optimal solubility–permeability balance, which limits the chances for successful drug delivery, the amorphous form emerges as a more advantageous strategy, in which higher apparent solubility (i.e., supersaturation) will be readily translated into higher drug flux and overall absorption.
Key words: amorphous solid dispersions, intestinal permeability, lipophilic drugs, oral absorption, solubility–permeability interplay
INTRODUCTION
It is frequently reported that due to modern drug discovery techniques (e.g., combinatorial chemistry, high-throughput screening) the percentage of drug candidates that are limited by poor aqueous solubility is increasing (1–3). Many solubility-enabling technologies are available for the pharmaceutical scientist to consider, including lipids, cosolvents, surfactants, nanoparticles, cyclodextrins, amorphous solid dispersions, and others (2,4–6). While significant increase of the apparent aqueous solubility may certainly be achieved by these solubility-enabling techniques, their effect on the drug flux through the intestinal membrane and the overall absorption is rather tricky; we have recently shown that a trade-off between apparent solubility increase and permeability decrease exists when using various solubility-enabling systems (7–12). This solubility–permeability trade-off may jeopardize the overall drug exposure and diminish the value of the solubility-enabling technologies.
The amorphous state has long been recognized as a way to increase the free energy and the apparent aqueous solubility of poorly soluble pharmaceuticals (13–18). Amorphous solid dispersion (ASD) technologies have emerged to allow stabilization of the amorphous state both in the dosage form and during supersaturation in the intestinal milieu, and delivery of some of the most important drugs of the twenty-first century has been made possible through amorphous solid dispersion technologies.
The aim of this study was to elucidate the performance of the amorphous form as an oral drug delivery practice for lipophilic molecules. We investigated the effects on both the solubility and the permeability aspects, both in vitro and in vivo, to allow a complete picture of the strengths/flaws of this technique. Then, a comparison of the ASD to other solubility-enabling techniques was made, to illustrate the uniqueness of the amorphous form practice.
MATERIALS AND METHODS
Materials
Nifedipine, MES buffer, KCl, NaCl, acetonitrile, and water (MS grade) were purchased from Sigma Chemical Co. (St. Louis, MO). Polyvinylpyrrolidone (PVP) (Kollidon® 30) and copovidone (Kollidon® VA 64) were obtained from BASF (Ludwigshafen, Germany). Hydroxypropyl methylcellulose acetate succinate (HPMC-AS) LF grade was obtained from Shin-Etsu Chemical Co. (Tokyo, Japan).
Preparation of Amorphous Solid Dispersion by Spray Drying
ASD powders of 10% nifedipine in PVP, copovidone, and HPMC-AS were prepared by spray drying. Drug and polymer were dissolved in acetone at a solids load of 5% (w/v), followed by spray drying using a Buchi mini-spray dryer B-290 (Flawil, Switzerland) at an inlet and outlet temperature of 80°C and 45°C, respectively, and feed solution infusion rate of approximately 5 mL/min. The resulting spray-dried particles were at a size range of ∼10 μm.
Powder X-Ray Diffraction
Powder X-ray diffraction (PXRD) was carried out on a theta/theta diffractometer (model Ultima™ II D/Max-2000-PC with model SA-HF3 3 kW X-ray generator, controlled by DMax2200PC series software, Rigaku Corp., Tokyo, Japan). X-rays were generated using a Cu anode with a generator power of 50 kV and 40 mA. Approximately 20 mg of each sample was pressed flat onto custom zero background silicon disks (Gem Dugout, State College, PA). Samples were scanned from 5–40° 2θ at 2° 2θ/min. Jade software (version 7.0, Materials Data, Inc., Livermore, CA) was used for data processing.
Modulated Differential Scanning Calorimetry
Modulated differential scanning calorimetry (mDSC) was carried out on a TA Instruments (New Castle, DE) Q100 DSC. About 7 mg of sample was weighed into an aluminum pan and covered with a pierced aluminum lid. Samples were then heated from 25°C to 190°C at 3°C/min under N2 purge at 50 mL/min. The temperature was modulated with amplitude of ±1°C and period of 60 s.
Determination of Supersaturation vs. Time
Solution stability of supersaturated solutions prepared from the nifedipine amorphous solid dispersions was carried out as previously described with minor adaptations (13,14,17). Briefly, supersaturated solutions of nifedipine were obtained by dissolving an appropriate amount of the 10% (w/w) nifedipine ASD powder into 10 mM MES buffer pH 6.5 to achieve supersaturated solutions of various times the equilibrium solubility of crystalline nifedipine (8 μg/mL). The resulting supersaturated solutions were then allowed to stand at 37°C with no agitation. The supersaturated solutions were sampled periodically and were analyzed for nifedipine content by ultra performance liquid chromatography (UPLC). As a control study to demonstrate true supersaturation in these experiments, the equilibrium solubility of crystalline nifedipine was measured as a function of the different polymer concentrations in the range of weight percent polymer (0.036–0.144% polymer for 5–20× supersaturation) used in the supersaturation studies and was found to be unchanged by the presence of the polymer.
Parallel Artificial Membrane Permeability Assay
Permeability studies through artificial membrane were carried out using the hexadecane-based parallel artificial membrane permeability assay (PAMPA), as described previously (9,19,20). Various levels of supersaturated nifedipine solutions (0.5 to 20 times equilibrium solubility) were prepared from the ASDs with pH 6.5 10 mM MES buffer. Millipore (Danvers, MA) 96-well MultiScreen-Permeability filter plates with 0.3 cm2 polycarbonate 0.45 mm filter support were used. The filter supports were impregnated with 15 μL of a 5% (v/v) hexadecane in hexane solution and were then allowed to dry for 1 h for complete evaporation of the hexane. The donor wells were then loaded with 200 μL of the supersaturated nifedipine solutions, and each receiver well was loaded with 300 μL of blank buffer. Four wells were loaded at each supersaturation degree to enable collection at different time points. Each experiment was repeated four times (n = 4). The donor plate was then placed upon the 96-well receiver plate, and the resulting PAMPA sandwich was incubated at room temperature (25°C).
Samples were collected every 30 min over 2 h, and the nifedipine concentration was determined by UPLC. Permeability coefficient (Papp) values were calculated from the linear plot of drug accumulated in the receiver side versus time, according to the equation Papp = (dQ/dt)/(A·C0), where dQ/dt is the steady-state appearance rate of nifedipine on the receiver side, C0 the initial concentration of nifedipine in the donor side, and A is the membrane surface area (0.048 cm2).
Single-Pass Intestinal Perfusion Studies in Rats
All animal experiments protocols were approved by the Ben-Gurion University of the Negev Animal Use and Care Committee (Protocol IL-60-11-2010). Animals were housed and handled according to the Ben-Gurion University of the Negev Unit for Laboratory Animal Medicine Guidelines. Male Wistar rats (Harlan, Israel) weighing 220–240 g were used for all studies. Rats were fasted overnight (12 h) prior to each experiment, with free access to water. Animals were randomly assigned to the different experimental groups.
Single-pass in situ jejunal perfusion studies were carried out as previously reported (21–23). Rats were anesthetized (1 mL/kg ketamine–xylazine 9%:1%) and placed on a 37°C surface (Harvard Apparatus Inc., Holliston, MA). A proximal jejunal segment of ∼10 cm was carefully exposed and cannulated on two ends with silicone tubing (Watson-Marlow Ltd, Wilmington, MA). Care was taken to avoid disturbance of the circulatory system, and the exposed segment was kept moist with 37°C normal saline solution. Various degrees of supersaturated solutions (0.5–20 times equilibrium solubility) were prepared from the nifedipine ASDs, in 10 mM MES buffer, pH 6.5, 135 mM NaCl, 5 mM KCl. All perfusate solutions were incubated in a 37°C water bath and were pumped through the intestinal segment (Watson-Marlow 205S, Wilmington, MA). The isolated segment was first rinsed with blank buffer (0.5 mL/min) to clean out any residual debris, and the test solutions were then perfused through the intestinal segment at a flow rate of 0.2 mL/min. To ensure steady-state conditions, the perfusion buffer was first perfused for 1 h, followed by an additional 1 h, during which samples were withdrawn at 10-min intervals. Samples were immediately assayed for nifedipine content. The length of each perfused segment was measured at the end of the experiment.
The effective permeability (Peff) through the rat jejunum in the single-pass intestinal perfusion studies was determined by the following equation:
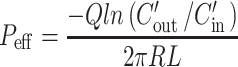 |
where Q is the perfusion flow rate (0.2 mL/min), C′out/C′in is the ratio of the outlet and inlet concentration of nifedipine that has been adjusted for water flux using phenol red as previously described (24–26), R is the radius of the intestine (set to 0.2 cm), and L is the length of the perfused intestinal segment.
Ultra Performance Liquid Chromatography
Drug concentrations were determined with a Waters (Milford, MA) Acquity UPLC H-Class system equipped with PDA detector and Empower software. Nifedipine was assayed using a Waters (Milford, MA) Acquity UPLC BEH C18 1.7 μm 2.1 × 100-mm column. The mobile phase consisted of 20:80 going to 80:20 (v/v) water/acetonitrile (0.1% TFA) over 5 min, at a flow rate of 0.5 mL/min.
Statistical Analysis
Supersaturation experiments were performed in triplicates, and all other experiments were n = 4. Values are expressed as mean ± standard deviation (SD). To determine statistically significant differences among the experimental groups, the nonparametric Kruskal–Wallis test was used for multiple comparisons and the two-tailed nonparametric Mann–Whitney U test for two-group comparison where appropriate. p < 0.05 was termed significant.
RESULTS AND DISCUSSION
The characterization of the crystalline and the ASD powders of nifedipine by PXRD and DSC are presented in Figs. 1 and 2, respectively. The PXRD pattern of the ASD was completely devoid of any diffraction peaks as compared to the crystalline pattern, indicating successful formation of the amorphous solid dispersion in all three polymers (Fig. 1). As additional evidence of amorphicity, the mDSC thermograms of the ASD powders showed a glass transition temperature (approximately 157°C, 104°C, and 100°C, for PVP, copovidone, and HPMC-AS, respectively), whereas the melting endotherm of crystalline nifedipine at ∼174°C was absent (Fig. 2).
Fig. 1.
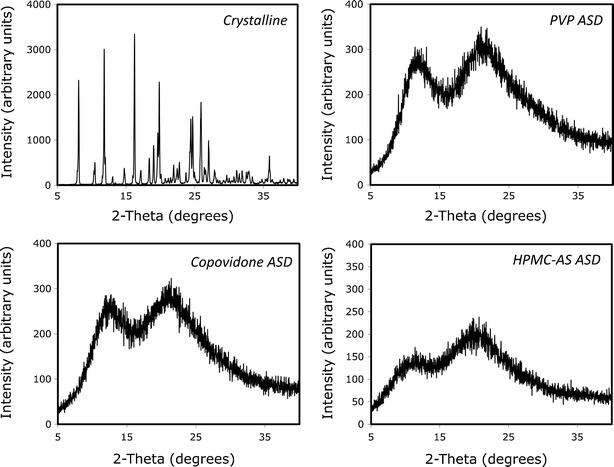
PXRD pattern of nifedipine crystalline powder and amorphous solid dispersions in PVP, copovidone, and HPMC-AS
Fig. 2.
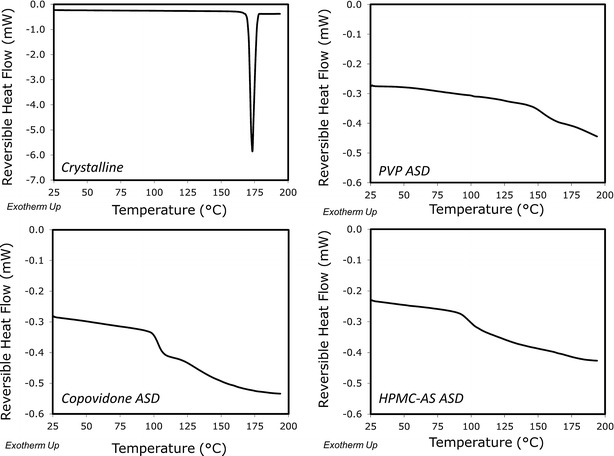
DSC thermogram of nifedipine crystalline powder and amorphous solid dispersions in PVP, copovidone, and HPMC-AS
The stability of supersaturated solutions of nifedipine obtained by dissolving the different ASD powders in MES buffer at concentrations up to 40× (320 μg/mL) the equilibrium solubility of crystalline nifedipine (8 μg/mL) is presented in Fig. 3. It can be seen that the ASD in HPMC-AS was able to maintain nifedipine supersaturated solutions of up to 160 μg/mL (20× the equilibrium solubility of crystalline nifedipine) for at least 5 h, providing sufficient time to carry out permeability studies. The ASD in copovidone allowed up to 80 μg/mL solutions (10×) to maintain supersaturation for sufficient time, and the PVP ASD maintained supersaturation of no more than 5× (40 μg/mL) the crystalline drug’s aqueous solubility. These results highlight the first advantage of using the amorphous form; even a supersaturated solution at 40× (320 μg/mL) was transiently stable for a couple of hours by the HPMC-AS-based ASD, demonstrating the extraordinary increase in apparent aqueous solubility afforded by the ASD, which has led to their widespread popularity and use. Furthermore, this study demonstrates a successful small-scale ASD screening; the primary objective in ASD screening is to identify the composition that enables in vivo exposure of a poorly water soluble compound and one that is stable, both chemically and physically. For this, a wide range of polymer systems can be screened. Polymers are critical components in ASDs because they act as carriers for the drug and inhibit crystallization in both the dosage form and in vivo. By remaining in an amorphous state during dissolution, the drug can achieve supersaturation and potentially greater absorption, when solubility is the limiting factor. We have clearly shown that a performance rank order of HPMC-AS > copovidone > PVP is evident for the polymer to be used in nifedipine ASDs (Fig. 3).
Fig. 3.
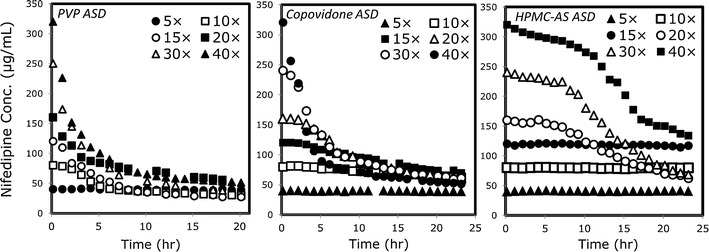
Supersaturation vs. time for nifedipine amorphous solid dispersions in PVP, copovidone, and HPMC-AS, dissolved at 5–40× the equilibrium solubility of crystalline nifedipine (8 μg/mL)
Based on the supersaturation behavior studies, we chose the concentrations to be used in the subsequent permeability studies. Figure 4 shows nifedipine flux (upper panel) and permeability (lower panel) in the PAMPA model, from undersaturated (0.5×), saturated (1×), and supersaturated solutions of nifedipine at concentrations up to 20× the equilibrium solubility of crystalline nifedipine, prepared from the different ASD powders. It can be seen that, irrespective of the polymer being used in the ASD, nifedipine’s flux increases markedly with increasing apparent solubility (i.e., supersaturation). As a result, the apparent permeability of nifedipine remained relatively constant with increasing apparent solubility in the PAMPA model. Similar findings were revealed in the in vivo rat intestinal perfusion studies (Fig. 5). The intestinal permeability of nifedipine remained unchanged with increasing apparent solubility, indicating higher drug flux as the supersaturation level increases, regardless of which ASD powder was used, and at all levels of supersaturation. Figure 6 compares the apparent flux of nifedipine as a function of apparent solubility increase for cases in which apparent solubility is obtained via supersaturation of an amorphous form vs. cases in which apparent solubility is obtained via solubilization (i.e., increase in apparent equilibrium solubility via addition of a solubilizer), including surfactant micellization, complexation with cyclodextrin, and solubilization by cosolvent. It can be seen that overall flux increased markedly with increasing apparent solubility via ASD as compared to the other approaches, in which increased solubility comes at the expense of decreased permeability with no increase in overall flux.
Fig. 4.
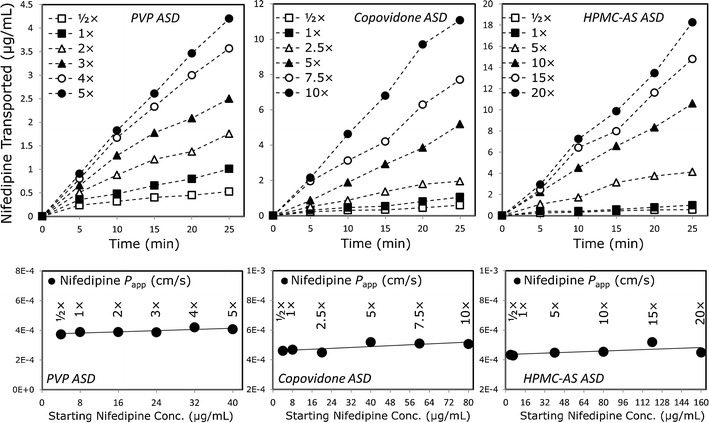
Flux (upper panel) and permeability (lower panel) of nifedipine as a function of increasing nifedipine concentration (i.e., supersaturation) in the PAMPA model. Supersaturated solutions were obtained via the nifedipine amorphous solid dispersion in PVP, copovidone, and HPMC-AS. n = 4 in each experimental group
Fig. 5.
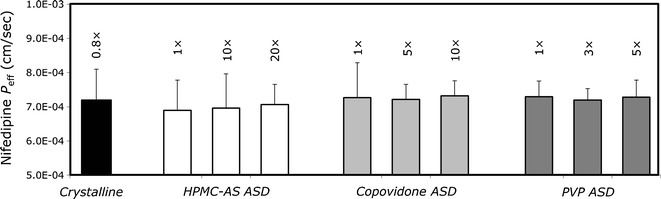
P eff (in centimeters per second) of nifedipine as a function of increasing apparent concentration (i.e., supersaturation) in the rat intestinal perfusion model. Supersaturated solutions were obtained via the nifedipine amorphous solid dispersion in PVP, copovidone, and HPMC-AS. Data are presented as average ± SD; n = 4 in each experimental group
Fig. 6.
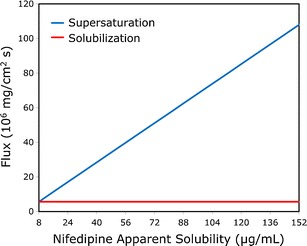
Theoretical flux (in milligrams per square centimeter per second) of nifedipine as a function of increasing nifedipine concentration for cases in which increased apparent solubility is obtained via supersaturation of the amorphous form vs. via solubilization methods, including surfactant micellization, complexation with cyclodextrin, and solubilization by cosolvent. The theoretical lines were constructed using the experimental rat intestinal permeability data (Fig. 5) and the equations modeling the solubility–permeability interplay described in detail in our previous publications (7–12,16)
This finding represents a novel insight in the field of oral delivery of lipophilic compounds; in a recent series of publications, we have investigated the interplay between apparent aqueous solubility increase and intestinal membrane permeability decrease, revealing the trade-off between these two parameters when using solubility-enabling technologies (8). We have shown this trade-off to be a universal phenomenon, rather than system dependent; at first, we showed and modeled the solubility–permeability trade-off when using cyclodextrin- (9,12) and surfactant-based (11) drug delivery systems, which could be well explained through the decreased free fraction of the drug inherent to these solubilization methods. Then, we revealed and modeled this trade-off in a more surprising setting, when using co-solvent based drug delivery systems, that allowed us to isolate the increased apparent solubility from free fraction considerations (7,10). We have shown that when increased apparent solubility is achieved in the intestinal milieu via addition of solubilizer (e.g., cyclodextrins, surfactants, cosolvents), the apparent partition coefficient between the intestinal membrane and the aqueous milieu is reduced. Since permeability is equal to the drug’s diffusion coefficient through the membrane times the membrane/aqueous partition coefficient divided by the membrane thickness, the decreased membrane/aqueous partitioning reduces the driving force for membrane partitioning and permeability, which explains the mechanism behind the solubility–permeability trade-off. In all of these cases, the increased apparent solubility afforded by the solubilization method failed to result in an increase in the drug flux, permeability was reduced, and hence, the success of the drug delivery system is questionable. In the current study, we reveal that using the amorphous form represents a way to overcome this trade-off; since supersaturation is a kinetic/non-equilibrium increase in apparent solubility, it will not affect the apparent membrane/aqueous lumen partition coefficient. Hence, the apparent membrane permeability is unaffected by supersaturation (Fig. 5), while flux (the product of apparent solubility × apparent permeability) increases dramatically (Figs. 4 and 6). In this way, the solubility–permeability interplay is circumvented and no longer exists in the case of supersaturation via the amorphous form, and solubility–permeability may be treated as independent parameters.
Since the solubility and the permeability have been identified as the two key parameters governing oral drug absorption (27–30), the importance and applicability of the findings presented in this paper emerge to be crucial in oral delivery of lipophilic drugs; while with other solubility-enabling approaches, it is not enough to increase the apparent solubility, but to strike the optimal solubility–permeability balance, which significantly limits the chances for successful drug delivery, the amorphous form emerges as a more advantageous strategy, in which higher apparent solubility (i.e., supersaturation) will be readily translated into higher drug flux and overall absorption.
CONCLUSIONS
In conclusion, we demonstrate the twofold advantage of the amorphous form as an oral drug delivery practice for lipophilic compounds: (1) the extraordinary increase in apparent aqueous solubility and (2) the concomitant increase in the drug’s flux through the intestinal membrane. These findings may allow the pharmaceutical scientist to be more strategic and successful when dealing with oral drug delivery of lipophilic compounds.
Acknowledgments
This work is a part of Avital Beig’s Ph.D. dissertation. The work was supported by a research grant from Abbott Laboratories.
References
- 1.Dahan A, Hoffman A. Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. J Control Release. 2008;129(1):1–10. doi: 10.1016/j.jconrel.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 2.Gao P, Shi Y. Characterization of supersaturatable formulations for improved absorption of poorly soluble drugs. AAPS J. 2012. doi:10.1208/s12248-012-9389-7. [DOI] [PMC free article] [PubMed]
- 3.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 4.Brouwers J, Brewster ME, Augustijns P. Supersaturating drug delivery systems: the answer to solubility-limited oral bioavailability? J Pharm Sci. 2009;98(8):2549–2572. doi: 10.1002/jps.21650. [DOI] [PubMed] [Google Scholar]
- 5.Dahan A, Miller J, Amidon G. Prediction of solubility and permeability class membership: provisional BCS classification of the world’s top oral drugs. AAPS J. 2009;11(4):740–746. doi: 10.1208/s12248-009-9144-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao Y, Carr RA, Spence JK, Wang WW, Turner TM, Lipari JM, et al. A pH-dilution method for estimation of biorelevant drug solubility along the gastrointestinal tract: application to physiologically based pharmacokinetic modeling. Mol Pharm. 2010;7(5):1516–1526. doi: 10.1021/mp100157s. [DOI] [PubMed] [Google Scholar]
- 7.Beig A, Miller JM, Dahan A. Accounting for the solubility-permeability interplay in oral formulation development for poor water solubility drugs: the effect of PEG-400 on carbamazepine absorption. Eur J Pharm Biopharm. 2012;81:386–391. doi: 10.1016/j.ejpb.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 8.Dahan A, Miller J. The solubility-permeability interplay and its implications in formulation design and development for poorly soluble drugs. AAPS J. 2012;14(2):244–251. doi: 10.1208/s12248-012-9337-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dahan A, Miller JM, Hoffman A, Amidon GE, Amidon GL. The solubility-permeability interplay in using cyclodextrins as pharmaceutical solubilizers: mechanistic modeling and application to progesterone. J Pharm Sci. 2010;99(6):2739–2749. doi: 10.1002/jps.22033. [DOI] [PubMed] [Google Scholar]
- 10.Miller JM, Beig A, Carr RA, Webster GK, Dahan A. The solubility-permeability interplay when using cosolvents for solubilization: revising the way we use solubility-enabling formulations. Mol Pharm. 2012;9(3):581–590. doi: 10.1021/mp200460u. [DOI] [PubMed] [Google Scholar]
- 11.Miller JM, Beig A, Krieg BJ, Carr RA, Borchardt TB, Amidon GE, et al. The solubility-permeability interplay: mechanistic modeling and predictive application of the impact of micellar solubilization on intestinal permeation. Mol Pharm. 2011;8(5):1848–1856. doi: 10.1021/mp200181v. [DOI] [PubMed] [Google Scholar]
- 12.Miller JM, Dahan A. Predicting the solubility-permeability interplay when using cyclodextrins in solubility-enabling formulations: model validation. Int J Pharm. 2012;430(1–2):388–391. doi: 10.1016/j.ijpharm.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 13.Alonzo D, Zhang G, Zhou D, Gao Y, Taylor L. Understanding the behavior of amorphous pharmaceutical systems during dissolution. Pharm Res. 2010;27(4):608–618. doi: 10.1007/s11095-009-0021-1. [DOI] [PubMed] [Google Scholar]
- 14.Alonzo DE, Gao Y, Zhou D, Mo H, Zhang GGZ, Taylor LS. Dissolution and precipitation behavior of amorphous solid dispersions. J Pharm Sci. 2011;100(8):3316–3331. doi: 10.1002/jps.22579. [DOI] [PubMed] [Google Scholar]
- 15.Frank KJ, Rosenblatt KM, Westedt U, Hölig P, Rosenberg J, Mägerlein M, et al. Amorphous solid dispersion enhances permeation of poorly soluble ABT-102: true supersaturation vs. apparent solubility enhancement. Int J Pharm. 2012;437(1–2):288–293. doi: 10.1016/j.ijpharm.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Gardner CR, Walsh CT, Almarsson O. Drugs as materials: valuing physical form in drug discovery. Nat Rev Drug Discov. 2004;3(11):926–934. doi: 10.1038/nrd1550. [DOI] [PubMed] [Google Scholar]
- 17.Miller JM, Beig A, Carr RA, Spence JK, Dahan A. A win-win solution in oral delivery of lipophilic drugs: supersaturation via amorphous solid dispersions increases apparent solubility without sacrifice of intestinal membrane permeability. Mol Pharm. 2012;9(7):2009–2016. doi: 10.1021/mp300104s. [DOI] [PubMed] [Google Scholar]
- 18.Newman A, Knipp G, Zografi G. Assessing the performance of amorphous solid dispersions. J Pharm Sci. 2012;101(4):1355–1377. doi: 10.1002/jps.23031. [DOI] [PubMed] [Google Scholar]
- 19.Wohnsland F, Faller B. High-throughput permeability pH profile and high-throughput alkane/water log P with artificial membranes. J Med Chem. 2001;44(6):923–930. doi: 10.1021/jm001020e. [DOI] [PubMed] [Google Scholar]
- 20.Miller JM, Dahan A, Gupta D, Varghese S, Amidon GL. Quasi-equilibrium analysis of the ion-pair mediated membrane transport of low-permeability drugs. J Control Release. 2009;137(1):31–37. doi: 10.1016/j.jconrel.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 21.Dahan A, Amidon G. Grapefruit juice and its constituents augment colchicine intestinal absorption: potential hazardous interaction and the role of P-glycoprotein. Pharm Res. 2009;26(4):883–892. doi: 10.1007/s11095-008-9789-7. [DOI] [PubMed] [Google Scholar]
- 22.Dahan A, Amidon GL. Small intestinal efflux mediated by MRP2 and BCRP shifts sulfasalazine intestinal permeability from high to low, enabling its colonic targeting. Am J Physiol Gastrointest Liver Physiol. 2009;297(2):G371–G377. doi: 10.1152/ajpgi.00102.2009. [DOI] [PubMed] [Google Scholar]
- 23.Dahan A, Sabit H, Amidon GL. Multiple efflux pumps are involved in the transepithelial transport of colchicine: combined effect of P-gp and MRP2 leads to decreased intestinal absorption throughout the entire small intestine. Drug Metab Dispos. 2009;37(10):2028–2036. doi: 10.1124/dmd.109.028282. [DOI] [PubMed] [Google Scholar]
- 24.Dahan A, Amidon GL. Segmental dependent transport of low permeability compounds along the small intestine due to P-glycoprotein: the role of efflux transport in the oral absorption of BCS class III drugs. Mol Pharm. 2009;6(1):19–28. doi: 10.1021/mp800088f. [DOI] [PubMed] [Google Scholar]
- 25.Dahan A, Sabit H, Amidon G. The H2 receptor antagonist nizatidine is a P-glycoprotein substrate: characterization of its intestinal epithelial cell efflux transport. AAPS J. 2009;11(2):205–213. doi: 10.1208/s12248-009-9092-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dahan A, West BT, Amidon GL. Segmental-dependent membrane permeability along the intestine following oral drug administration: evaluation of a triple single-pass intestinal perfusion (TSPIP) approach in the rat. Eur J Pharm Sci. 2009;36(2–3):320–329. doi: 10.1016/j.ejps.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 27.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 28.Dahan A, Lennernäs H, Amidon GL. The fraction dose absorbed, in humans, and high jejunal human permeability relationship. Mol Pharm. 2012;9(6):1847–1851. doi: 10.1021/mp300140h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dahan A, Miller JM, Hilfinger JM, Yamashita S, Yu LX, Lennernäs H, et al. High-permeability criterion for BCS classification: segmental/pH dependent permeability considerations. Mol Pharm. 2010;7(5):1827–1834. doi: 10.1021/mp100175a. [DOI] [PubMed] [Google Scholar]
- 30.Lennernäs H. Intestinal permeability and its relevance for absorption and elimination. Xenobiotica. 2007;37(10–11):1015–1051. doi: 10.1080/00498250701704819. [DOI] [PubMed] [Google Scholar]