

Abstract
Macrophages have a critical function in the recognition and engulfment of dead cells. In some settings, macrophages also actively signal programmed cell death. Here we show that during developmentally scheduled vascular regression, resident macrophages are an obligatory participant in a signaling switch that favors death over survival. This switch occurs when the signaling ligand angiopoietin 2 has the dual effect of suppressing survival signaling in vascular endothelial cells (VECs) and stimulating Wnt ligand production by macrophages. In response to the Wnt ligand, VECs enter the cell cycle and in the absence of survival signals, die from G1 phase of the cell cycle. We propose that this mechanism represents an adaptation to ensure that the macrophage and its disposal capability are on hand when cell death occurs.
Keywords: Macrophage, Angiopoietin, Wnt, Programmed cell death, Vascular regression, Cell cycle
INTRODUCTION
Macrophages have a critical function in tissue homeostasis because they can rapidly recognize and engulf dead cells (Savill et al., 2002). Rapid identification and engulfment of apoptotic cells by macrophages prevents the release of pro-inflammatory intracellular components that when chronically released can be deleterious. Indeed, recognition of apoptotic cells by macrophages results in the release of anti-inflammatory agents including TGFβ (Savill et al., 2002; Lucas et al., 2003). In many circumstances where macrophages encounter apoptotic cells, the macrophage is believed to have a after-the-fact role in disposal and immune modulation.
There is emerging evidence to suggest that in some cases, macrophages play an active role in inducing programmed cell death. This was first appreciated after macrophage ablation experiments resulted in the persistence of viable cells in ocular vessel networks that failed to undergo scheduled regression (Lang and Bishop, 1993). The proposal that macrophages can actively signal cell death has received support from a number of systems including C. elegans, where caspase (CED-3) partial function combined with recognition pathway mutants demonstrate that recognition is a backup stimulus for apoptosis (Hoeppner et al., 2001; Reddien et al., 2001). Although there is currently no mechanistic parallel apparent, discovery of macrophage-induced (Lang and Bishop, 1993; Diez-Roux and Lang, 1997) and phagocyte-enhanced (Hoeppner et al., 2001; Reddien et al., 2001) apoptosis in mice and worms may suggest an activity functionally conserved in metazoans.
Signaling mechanisms accounting for macrophage-induced cell death are now being described. In the developing chick retina, it has been demonstrated that macrophage-related microglia are a source of nerve growth factor (NGF) that causes neurons to die (Frade et al., 1996), and in mice, microglia promote programmed cell death of Purkinje cells through production of superoxide ions (Marin-Teva et al., 2004; Mallat et al., 2005). In the mouse eye the canonical Wnt pathway (Logan and Nusse, 2004) is crucial for scheduled vascular regression. In this system, resident macrophages produce Wnt7b and through close contact with vascular endothelial cells (VECs) signal cell death (Lobov et al., 2005).
The angiopoietin signaling system plays a key role in the regulation of angiogenesis, vascular homeostasis and vascular regression (Yancopoulos et al., 2000). One angiopoietin receptor is the conventional tyrosine kinase receptor Tie2 (Dumont et al., 1994). Expression of Tie2 is largely restricted to endothelial cells but can also be found in haematopoietic cells and a subset of tumor-associated monocytes and eosinophils (Iwama et al., 1993; Dumont et al., 1994; Feistritzer et al., 2004; De Palma et al., 2005). More recent work has indicated that integrins can serve as alternative receptors for the angiopoietins (Carlson et al., 2001) and that integrins and Tie2 can form a complex (Cascone et al., 2005).
Angiopoietin 1 (Ang1; also known as Angpt1 – Mouse Genome Informatics) and angiopoietin 2 (Ang2; also known as Angpt2) are the two best characterized Tie2 ligands. Ang1 (Suri et al., 1996) is an agonist and elicits many responses including cell-survival through the PI3-kinase-Akt signaling pathway (Datta et al., 1999; Peters et al., 2004). Under some (Gale et al., 2002; Lobov et al., 2002) but not all (Kim et al., 2000b; Gale et al., 2002) circumstances, Ang2 (Maisonpierre et al., 1997) is a signaling antagonist and inhibits the action of Ang1. As an antagonist of the PI 3-kinase-Akt signaling pathway, Ang2 activity is synonymous with survival signal withdrawal. Ang2 promotes vessel destabilization (Maisonpierre et al., 1997) and, in the presence of vascular endothelial cell growth factor (VEGF), is an important angiogenic stimulus (Yancopoulos et al., 2000; Lobov et al., 2002). Under conditions where VEGF signaling is inhibited or absent, the action of Ang2 in destabilizing vessels can lead to cell death and capillary regression (Yancopoulos et al., 2000; Lobov et al., 2002).
The hyaloid vessels of the eye serve as a model system to further our understanding of the mechanisms of vascular regression (Lang and Bishop, 1993; Diez-Roux and Lang, 1997; Diez-Roux et al., 1999; Ito and Yoshioka, 1999; Meeson et al., 1999; Ylikarppa et al., 2003; Ohlmann et al., 2004; Lobov et al., 2005). In the current study we sought to explain the mechanisms by which hyaloid VECs undergo apoptosis and examined the possibility of cooperation between the Wnt and angiopoietin pathways. We show that during scheduled vascular regression, macrophages are an obligatory participant in a signaling switch that favors death over survival. This switch occurs when Ang2 has the dual effect of suppressing PI 3-kinase-Akt survival signaling in VECs and stimulating Wnt7b production in macrophages. In response to Wnt7b, VECs enter the cell cycle and die in the G1 phase of the cell cycle due to Ang2-mediated withdrawal of survival signals. We propose that close coupling of macrophage function to the cell death program is an adaptation to ensure that a ‘professional’ phagocyte is on hand when cell death occurs.
MATERIALS AND METHODS
Mouse genotyping
Genotyping of Lrp5lacZ (Kato et al., 2002), Wnt7blacZ (Lobov et al., 2005), Wnt7bd1 (Lobov et al., 2005), Ang2lacZ (Gale et al., 2002), VE-cadherin:tTa (Sun et al., 2005), TET:myrAkt (Sun et al., 2005) and TOPGAL (DasGupta and Fuchs, 1999) mice were performed as previously described.
Dissections, immunostaining and imaging
Hyaloid vessel preparations were generated as previously described (Lobov et al., 2005) with the exception that dissections were performed without gelatin. X-Gal staining was done according to established protocols (Shu et al., 2002). Indirect immunofluorescent staining and BrdU labeling were performed as previously described (Diez-Roux et al., 1999). Primary antibodies were as follows: anti-BrdU (1:100, Dako), anti-β-catenin (1:50, BD Transduction). Secondary antibodies labeled with Alexa fluorochromes (Molecular Probes) were used at a 1:500 dilution. TUNEL labeling of apoptotic cells was performed using the In Situ Cell Death Detection Kit (Roche Applied Science).
Cell culture experiments
Microvascular endothelial cells (MVECs; Cambrex) from skin were grown to 60–70% confluency, serum starved overnight and stimulated with Ang1 (500 ng/ml) and/or Wnt3a (250 ng/ml) in combination with the Akt inhibitor SH6 (Kozikowski et al., 2003; Castillo et al., 2004) (10 μM) or the PI 3-kinase inhibitor wortmannin (250 nM) for 4 hours or 12 hours and labeled for β-catenin.
Intravitreal injection
Intravitreal injection was performed with glass needles pulled on a model P87 flaming-pipette puller and beveled with a K. T. Brown Type model BV-10 diamond beveller (Sutter Instrument Company, Novato, CA). Materials injected included 120 nl of 10 μM SH6 (Alexis Biochemicals) and 120 nl of 50 μM Ang1 or Ang2 (R&D Systems).
Laser capture microdissection
Macrophages were captured from whole-mount hyaloid vessels of P3 CD1 mice, injected with Ang2 intravitreally (see above) using the Veritas Microdissection System. Control eyes were sham injected with PBS. For each experiment, 5–7 ng/ml of RNA was used.
RT-PCR
RNA from dissected hyaloid vessel preparations was purified using the Qiagen MicroEasy RNA Isolation Kit. Subsequent RT-PCR was performed using the OneStep RT-PCR Kit (Qiagen). The following primers (shown 5′ to 3′) were used:
Gapdh: forward (F), ACTCCACTCACGGCAAATTC; reverse (R), CACATTGGGGGTAGGAACAC.
Ang1: F1, AGGCTTGGTTTCTCGTCAGA; R1, TCTGCACAGTCTCGAAATGG.
Wnt7b: F, AAGAACTCCGAGTAGGGAGTCG; R, TGCGT TGTACTTCTCCTTGAGC.
Wnt7b: 2nd round F, CCGAGTAGGGAGTCGAGAGG; R, CACACCGTGACACTTACATTCC.
Statistical analysis
Vessel number was quantified using established methods (Ito and Yoshioka, 1999). At least four hyaloid vessel preparations were quantified for each experiment. All data are presented with standard error bars. The Student’s t-test and one-way ANOVA with Tukey’s test were used to assess statistical significance.
RESULTS
Wnt and angiopoietin pathways cooperate to mediate hyaloid vessel regression
We have previously demonstrated that Wnt signaling is important in hyaloid vessel regression (Lobov et al., 2005). However, mechanistically it has been difficult to reconcile how Wnt signaling could culminate in death of VECs. This prompted us to investigate other pathways that might be required. The Tie2 antagonist Ang2 can induce vessel regression in the absence of VEGF, and the Tie2 agonist Ang1 is linked with survival of endothelial cells (Yancopoulos et al., 2000; Lobov et al., 2002). Furthermore, like Wn7bd1 mutant mice (Lobov et al., 2005) Ang2 mutants have persistent hyaloid vessels (Gale et al., 2002) suggesting that the Wnt and angiopoietin pathways function in concert to regulate regression. To investigate the nature of this cooperation we assessed genetic interaction between mutant loci representing each pathway [Wnt7bd1 (Lobov et al., 2005) and Ang2lacZ (Gale et al., 2002)]. By 8 days after birth (P8) in wildtype mice (Fig. 1A,B) hyaloid vessel regression is all but complete. By contrast, Wnt7bd1/d1 (Fig. 1C,D) and Ang2lacZ/lacZ (Fig. 1E,F) mutants show persistent vessels. Unlike Wnt7bd1/d1 mutant vessels, those from Ang2lacZ/lacZ mice show an unusual hypercellularity (Fig. 1H). Compound heterozygotes show a persistence phenotype (Fig. 1J) that is more severe than that of either single heterozygote (Fig. 2A) and similar in degree to that of the Wnt7bd1 homozygote (Fig. 2A). Very similar responses are observed when interactions between the Wnt pathway co-receptor gene Lrp5lacZ and Ang2lacZ are assessed (data not shown). These data are consistent with a cooperative action between the Wnt and angiopoietin pathways in regulating vessel regression.
Fig. 1. Genetic Interaction between the Wnt and the angiopoietin pathways.
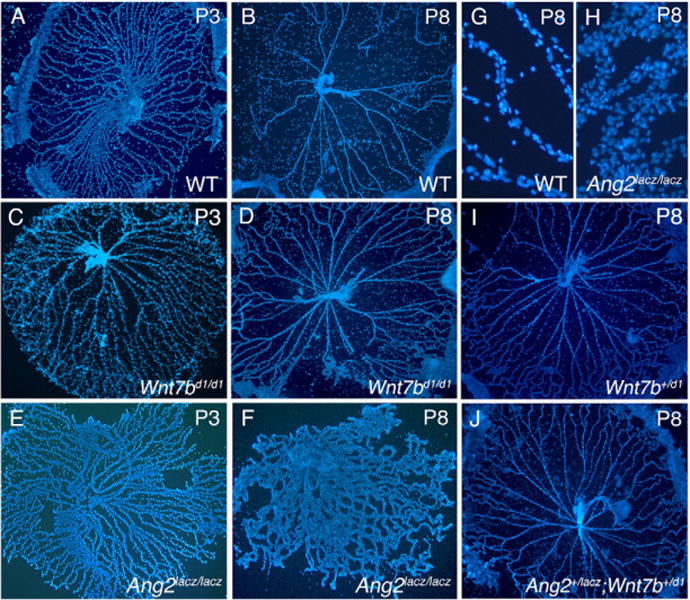
(A–J) Hyaloid vessels from WT (A,B,G), Wnt7bd1/d1 (C,D), Ang2lacZ/lacZ (E,F,H), Wnt7b+/d1 (I) and Ang2+/lacZ; Wnt7b+/d1 (J) animals of the indicated ages stained with Hoechst 33258. (H) Hypercellularity of Ang2lacZ/lacZ hyaloid capillaries compared with WT (G) at P8. Magnifications: 50 × in A–F,I,J; 400 × in G,H.
Fig. 2. The Wnt and angiopoietin pathways are finely balanced in regulating cell death, proliferation and hyaloid regression.
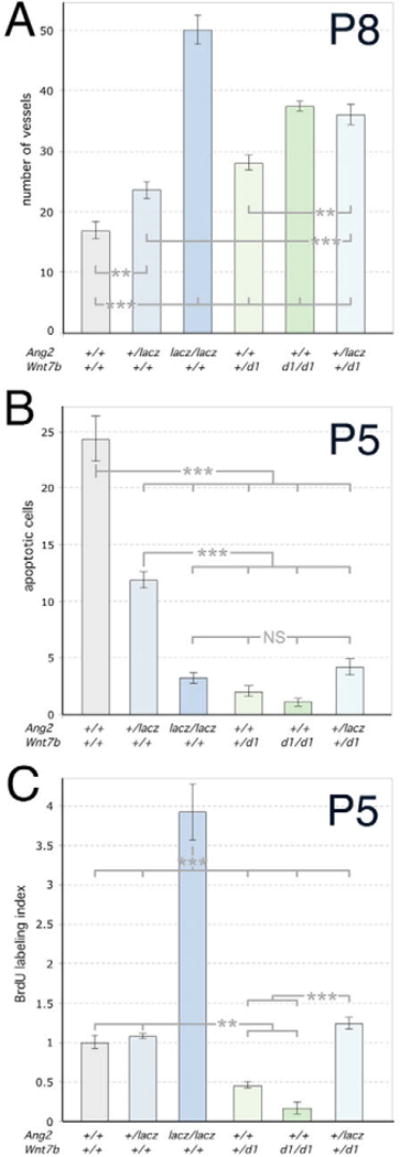
(A–C) Numbers of hyaloid vessels at P8 (A), apoptotic cells at P5 (B) and BrdU-labeled cells at P5 (C) in animals of the indicated genotypes. Significance was assessed using one-way ANOVA. All genotypes were compared to WT and were significantly different as indicated by the asterisks. Brackets indicate the groups that were compared using oneway ANOVA. Significance levels: **P ≤ s0.01, > 0.001; ***P ≤ s0.001, > 0.
Proliferation and death are exquisitely sensitive to Wnt-Angiopoietin pathway interaction
Evidence for cooperative action of the two pathways also emerges when proliferation and cell death are quantified (Fig. 2B,C). Ang2lacZ heterozygotes and homozygotes have progressively reduced levels of cell death as determined by TUNEL analysis (Fig. 2B). There is a similar but more pronounced response in Wnt7bd1 mutants (Fig. 2B). Double heterozygotes show a level of cell death significantly less than Ang2lacZ heterozygotes (Fig. 2B) suggesting that Wnt7b modulates the Ang2 pro-apoptotic function. An examination of BrdU labeling levels is also revealing (Fig. 2C). The Ang2lacZ heterozygote is essentially unchanged whereas the Ang2lacZ homozygote has a dramatically increased BrdU labeling index (Fig. 2C), consistent with the distinct hyperplastic phenotype of the hyaloid vessels (Fig. 1H). Wnt7bd1 mutants show the opposite response, with the heterozygote having less than half the wild-type level of labeling (Fig. 2C). Combining Ang2 heterozygosity with Wnt7b+/d1 completely rescues the proliferation defect in the Wnt mutant. We conclude that for proliferation and cell death, Wnt7b and Ang2 mutations show a genetic interaction and that the pathways are finely balanced in regulating cellular responses.
The Akt component of Tie2 signaling is critical for cell survival
Wnt7b is expressed by hyaloid-associated macrophages (Lobov et al., 2005). X-Gal staining of hyaloid vessels from Ang2+/lacZ mice revealed that Ang2 is expressed in capillary cells (Fig. 3A). The X-Gal staining pattern (Fig. 3B, red arrowheads) was characteristic of pericyte processes. Resident macrophages (Fig. 3B, red dashed rings) did not stain. Small-diameter capillaries that typically have minimal pericyte investment also had limited staining (Fig. 3B, red bracket). Immunofluorescent staining of hyaloid vessels from wild-type mice with desmin, a marker for pericytes, also showed a similar pattern of staining as the X-Gal-positive cells (Fig. 3C, white arrowheads). X-Gal staining of hyaloid vessels from Tie2-lacZ mice (Schlaeger et al., 1997) showed a contrasting pattern of labeling similar to that of VECs (Fig. 3D). This suggested that the source of Ang2 required for hyaloid vessel regression was the pericyte.
Fig. 3. Angiopoietin pathway responses downstream of Akt regulate hyaloid regression.
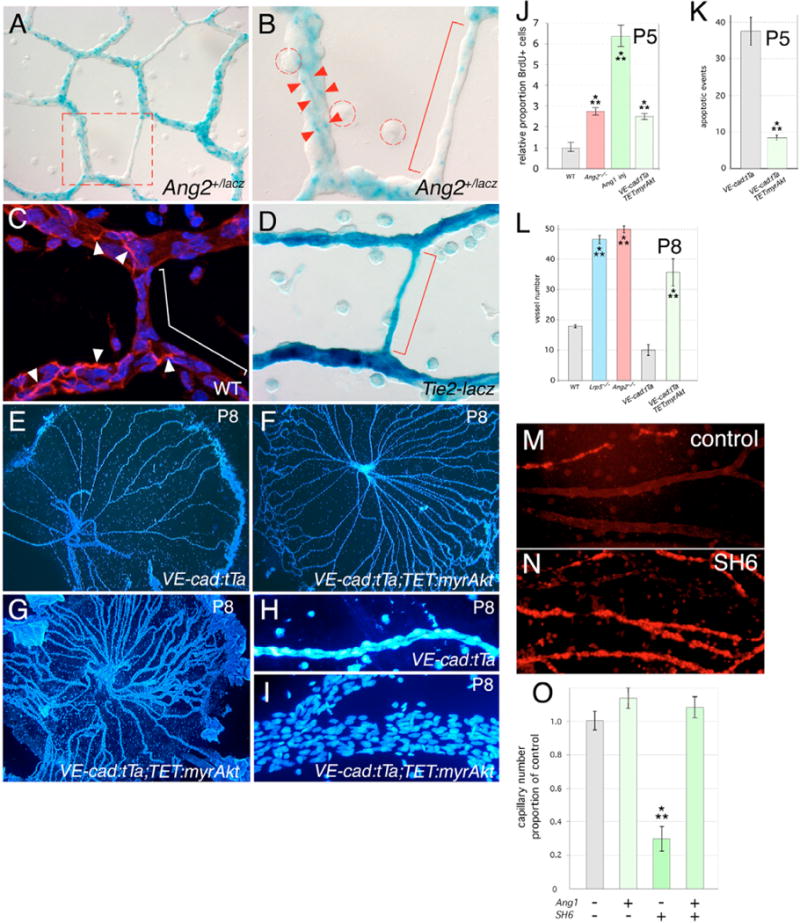
(A,B) X-Gal staining of P5 hyaloid vessels from Ang2+/lacZ mice. Dashed red box in A indicates region shown at higher magnification in B. Red arrowheads indicate X-Gal-labeled cell processes, the red bracket indicates a small capillary with limited X-Gal staining. Macrophages are indicated by the dashed red circles. (C) Pericytes from P5 hyaloid vessels of wild-type mice labeled with desmin (red). Nuclei are stained with Hoechst 33258 (blue). White arrowheads indicate cell processes of the pericytes, the white bracket indicates areas with no desmin staining. (D) X-Gal staining of P5 hyaloid vessels from a Tie2-lacZ mouse in which lacZ is expressed in vascular endothelial cells (VECs). The red bracket indicates a small capillary with uniform X-Gal staining. (E–I) Hoechst 33258-labeled hyaloid vessels from control (E,H) and VE-cadherin:tTa; TET:myrAkt (F,G,I) transgenic animals. (J) Proportion of BrdU-positive cells in P5 hyaloid vessels of WT, Ang2lacZ/lacZ, WT injected with Ang1 and VE-cadherin:tTa;TET:myrAkt mice. (K) Number of apoptotic events at P5 in the hyaloid vessels from mice of the indicated genotypes. (L) Hyaloid vessel number at P8 in mice of the indicated genotypes. (M,N) TUNEL labeling of hyaloid vessel preparations with no treatment (M) or after intravitreal injection of SH6 (N). (O) Quantification of healthy hyaloid vessel number as a proportion of wild type after injection of the indicated reagents at P4 and assessment at P5. Magnifications: 50 × (E–G), 200 × (A), 400 × (M,N), 630 × (B,C,D,H,I). Significance levels: **P ≤ s0.01, > 0.001; ***P ≤ s0.001, > 0.
The angiopoietin pathway incorporates the serine-threonine kinase Akt (Kim et al., 2000a). Akt is known to have multiple effects, including suppression of cell death (Datta et al., 1999) and in some settings, stabilization of β-catenin (Moore and Lemischka, 2006). Since Ang2 is an antagonist, loss-of-function Ang2 should be equivalent to gain-of-function Akt. To determine whether this was the case, we used the TET transactivation system (Corbel and Rossi, 2002) to express a dominant active form of Akt in VECs. When tetracycline is withdrawn, bitransgenic VE-cadherin:tTa;TET:myrAkt mice express myrAkt in VECs (Sun et al., 2005). Withdrawal of tetracycline at the day of birth followed by BrdU labeling at P5 showed that Akt gain-of-function mice have an increased labeling index (Fig. 3J, light green bar) that is comparable to that obtained with the intra-vitreal injection of Ang1 (an alternative strategy for pathway activation; Fig. 3J, green bar) or when the Ang2 gene is mutated (Fig. 3J, red bar). These data indicate that activation of the angiopoietin pathway stimulates proliferation, that proliferation responses occur downstream of Akt, and confirms that Ang1 and Ang2 behave as agonist and antagonist, respectively.
Akt gain-of-function phenocopies other aspects of the Ang2lacZ/lacZhyaloid persistence phenotype (Fig. 3E–L). In VE-cadherin:tTa;TET:myrAkt mice, levels of apoptosis are low at P5 (Fig. 3K), vessel number is greater at P8 (Fig. 3L) and although there is some variability (Fig. 3F,G) many vessels have the hypercellularity (Fig. 3I) characteristic of the Ang2 mutant (Fig. 1H). Limited stability of Ang1 after intra-vitreal injection precluded assessment of vessel number at P8. To perform Akt loss-of-function experiments, we took advantage of the Akt inhibitor SH6 (Kozikowski et al., 2003; Castillo et al., 2004) and compared the consequences of injecting Ang1, SH6 or both, into the vitreous of wild-type mice at P4. Hyaloid vessels were dissected at P5 and TUNEL analysis performed (Fig. 3M,N) so that we could quantify the total remaining healthy capillaries (Fig. 3O). SH6 injection resulted in a dramatic increase in cell death and precocious hyaloid regression (Fig. 3M–O). Importantly, the specificity of the SH6 cell death response was demonstrated by the rescue of precocious regression when Ang1 was co-injected (Fig. 3O). The ability of Ang1 to reverse SH6-induced cell death may indicate that the inhibitor is in equilibrium with Akt and can be competed out if sufficient activated Akt is provided by Ang1 stimulation. Combined, these data indicate that Akt is a key mediator of both proliferation and survival responses in VECs of the hyaloid system.
Stabilization of β-catenin via Akt has been documented in other systems (Fukumoto et al., 2001; Sharma et al., 2002) and this was one possible explanation for the cooperative action of Wnt and angiopoietin pathways during hyaloid regression. We examined this possibility in a culture system using β-catenin labeling of microvascular endothelial cells (MVECs) under various treatment conditions. MVECs were serum-starved overnight to remove the influence of other agents, and then incubated for 12 hours (Fig. 4A–F) with serum-free medium as a control (Fig. 4A), with recombinant Wnt3a (Fig. 4B) or with Ang1 (Fig. 4C). Whereas there were changes in the strength of β-catenin labeling in other regions of the cells, Ang1, like Wnt3a, was found to stimulate translocation of β-catenin to the nucleus. Prevention of nuclear β-catenin localization, when wortmannin (a PI 3-kinase inhibitor; Fig. 4D) or SH6 (an Akt inhibitor; Fig. 4E) were added in combination with Ang1, provided evidence that β-catenin stabilization was downstream of PI 3-kinase and Akt. Quantification also showed that at the concentrations of recombinant ligands chosen, Ang1 was at least as potent as Wnt3a in stimulating this response (Fig. 4F). Furthermore, the inability of wortmannin or SH6 to prevent β-catenin nuclear localization in response to Wnt3a emphasizes that the Wnt and angiopoietin pathway are alternative β-catenin stabilization options (Fig. 4F). We also performed experiments in which MVECs were incubated with Ang1, Wnt3a or both for 4 hours (Fig. 4F, right panel). Since the combined factors gave a synergistic response, this also suggested the Wnt and angiopoietin pathways cooperate to stabilize β-catenin.
Fig. 4. The Tie2 and canonical Wnt pathways are integrated through Akt.

(A–E) Immunofluorescence detection of β-catenin in primary MVECs either untreated (A) or treated with Wnt3a (B), Ang1 (C), Ang1 + wortmannin (D) or Ang1 + SH6 (E). Upper and lower parts of each panel show β-catenin labeling alone and merged nuclear and β-catenin labeling, respectively. (F) Proportion of MVECS with nuclear β-catenin detectable by immunofluorescence under the conditions indicated. For this quantification, the significance levels are: **P < 0.005, ****P < 0.00005.
To determine whether β-catenin might be stabilized via the angiopoietin pathway in vivo, we performed β-catenin immunolabeling on hyaloid preparations from various mouse mutants. β-catenin associated with junctional complexes in wild-type hyaloid vessels is abundant (Fig. 5A). By contrast, the nuclear β-catenin indicative of normal Wnt pathway signaling is difficult to detect. To improve our ability to detect nuclear β-catenin, we developed a labeling protocol in which we performed two rounds of primary antibody incubation and detected each round with color-differentiated secondary antibodies (Fig. 5A–L). This may work more effectively than a single round of primary antibody because the first round saturates abundant junctional β-catenin epitopes. Thus, as expected, β-catenin levels in wild-type (Fig. 5A–C) and Lrp5 mutant (Fig. 5D–F) hyaloid vessels were high at junctions but nuclei were not obviously positive. By contrast, in both the Ang2 homozygote (Fig. 5G–I) and the myrAkt gain-of-function mice (Fig. 5J–L) nuclear β-catenin signal was robust. The response was somewhat patchy in the Akt gain-of-function mice, perhaps due to non-uniform gene expression induced by tetracycline withdrawal. Junctional β-catenin was downregulated with Akt gain of function (Fig. 5K,L). These observations indicate that in VECs, Akt activates β-catenin.
Fig. 5. The angiopoietin pathway activates Wnt target genes via Akt andβ-catenin.
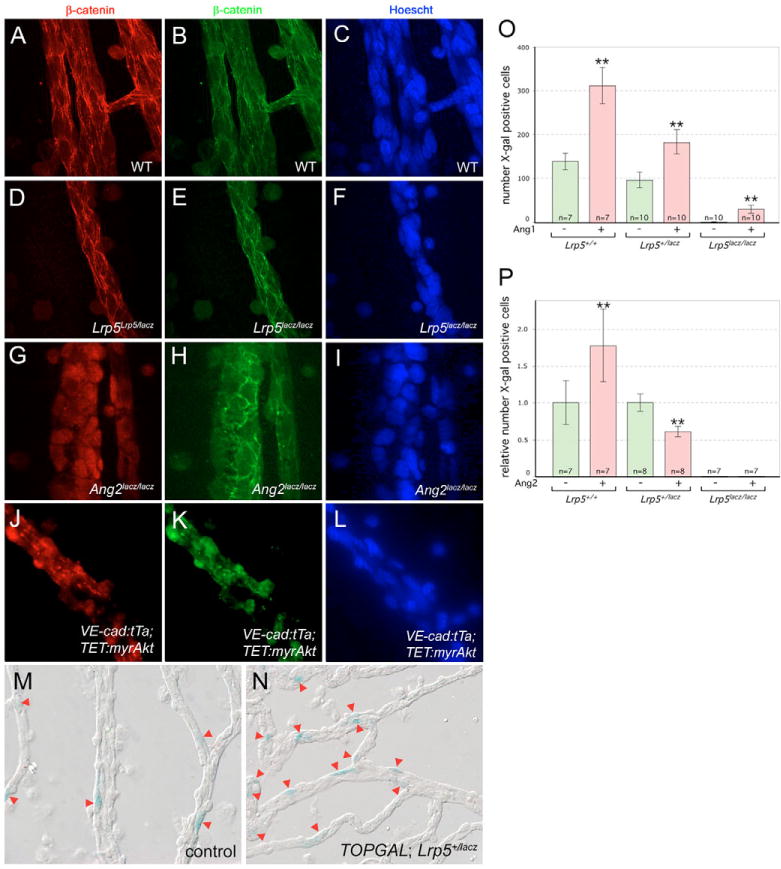
(A–L) Immunofluorescent staining for β-catenin (green and red with two rounds of labeling – see text) and nuclei (blue) in hyaloid preparations in mice of the indicated genotypes. (M,N) X-Gal staining of hyaloid vessels (red arrowheads) from TOPGAL; Lrp5+/lacZ animals that were (M) mock injected intra-vitreally or (N) injected with Ang1 intra-vitreally. (O,P) Quantification of X-Gal-stained cell numbers (as in M and N) from hyaloid vessels of the indicated genotypes injected with Ang1 (O) or Ang2 (P). Significance levels: **P ≤ s0.01, > 0.001.
As an alternative way to assess angiopoietin-regulated Wnt pathway responses, we quantified TOPGAL reporter expression in the hyaloid vessels when Ang1 or 2 was injected into the eye. Previously we validated the TOPGAL transgene – a lacZ open reading frame regulated by Lef/TCF binding sites – as a Wnt pathway reporter for VECs of the hyaloid capillaries (Lobov et al., 2005). We have also demonstrated that lacZ expression and X-Gal staining in Lrp5lacZ/lacZ mice is very weak in all VECs, and therefore can be easily distinguished from the strong, sporadic VEC staining of the TOPGAL transgene (Lobov et al., 2005). Intra-vitreal injection of recombinant Ang1 increased the number of TOPGAL-expressing cells regardless of the Lrp5 status of the experimental mouse (Fig. 5M–O). This indicates that the angiopoietin pathway can activate a model Wnt pathway target gene and is consistent with Akt-mediated stabilization of β-catenin.
Injection of Ang2 into TOPGAL mice gave differing responses depending on Lrp5 status. In Lrp5 homozygous mutants, TOPGAL-positive cell number was close to zero and, as might be anticipated from an antagonist, injection of Ang2 did not elicit a change (Fig. 5P). In Lrp5 heterozygous mutants also, as expected, Ang2 reduced the number of TOPGAL-expressing cells (Fig. 5P). Surprisingly, in wild-type mice, Ang2 injection reproducibly elicited the opposite response and increased the number of TOPGAL-expressing cells (Fig. 5P). In seeking an explanation for this, we considered the possibility that besides regulating angiopoietin pathway signaling, Ang2 might have an additional activity regulating macrophage production of Wnt7b. Using the Wnt7blacZ allele, we assessed gene expression on the Ang2lacZ heterozygous and homozygous backgrounds and found that when Ang2 activity was absent, Wnt7b expression in macrophages was lost (Fig. 6A,B). Since it could be argued that macrophages in the Ang2lacZ null might be poorly differentiated and therefore incapable of expressing Wnt7b, we also performed a gain-of-function experiment. Ang2 was injected intra-vitreally in CD1 animals, hyaloid macrophages were isolated by laser capture microdissection and Wnt7b RT-PCR performed (Fig. 6C). Though Wnt7b expression in the control eye showed some variation, from being undetectable to being weakly present, in every experiment Ang2 injection resulted in upregulation of Wnt7b. These data show that directly or indirectly, Ang2 is required for Wnt7b expression in macrophages. This was a straightforward explanation for the increased number of cells expressing TOPGAL with injection of Ang2. Combined with the above analysis, this suggests that Ang2 has two functions, the pro-apoptotic suppression of Akt and the pro-cycle activation of macrophage Wnt7b. Expression of Ang1 in the hyaloid vessel complex (Fig. 6D) suggested that Ang1 and Ang2 had the opportunity to function as agonist and antagonist.
Fig. 6. Angiopoietin signaling regulates Wnt7b expression and cell cycle stage-dependent cell death.
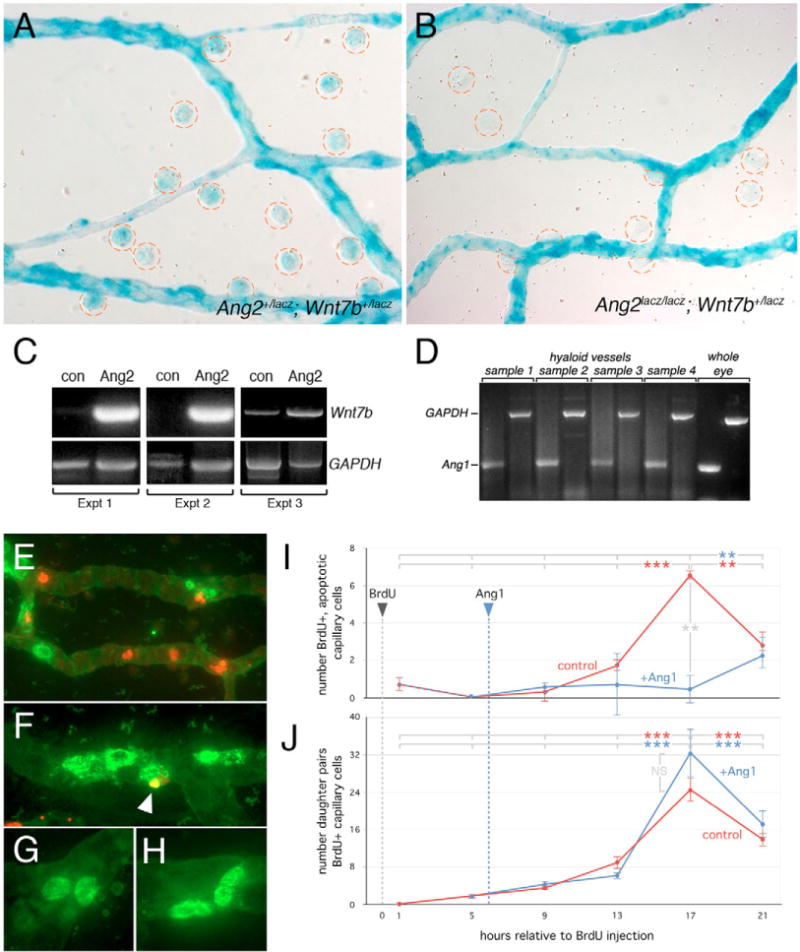
(A,B) X-Gal staining of hyaloid vessels from Ang2lacZ/+; Wnt7blacZ/+ (A) and Ang2lacZ/lacZ; Wnt7blacZ/+ (B) animals. Macrophages are circled in red. (C)Wnt7b and Gapdh RT-PCR of laser captured macrophages from hyaloid vessels of P3 wild-type animals, injected intra-vitreally with Ang2 (Ang2) or sham injected (con). Data from three independent experiments are presented. (D)Ang1 and Gapdh RT-PCR from wild-type P5 hyaloid vessel mRNA. Whole eye RNA was used as a control. (E–H) Combined BrdU (green) and TUNEL (red) labeling of hyaloid vessels showing BrdU-positive VECs that are apoptotic (F, indicated by white arrowhead) and pairs of BrdU-positive daughter nuclei (G,H). (I,J) BrdU labeled cell fate mapping: red lines show the number of BrdU-TUNEL double labeled nuclei (I) and BrdU-positive daughter pairs (J) in left eye hyaloid vessels assessed over a 21 hour time-course after BrdU incorporation. The blue lines in I and J show similar quantification in the right, contralateral eye after Ang1 injection at t = 6 hours. Significance levels: **P ≤ 0.01, > 0.001; ***P ≤ 0.001, > 0.
Cell cycle entry is required for hyaloid VEC apoptosis
Cell death is sometimes coupled to cell cycle progression (Diez-Roux et al., 1999; Lowe et al., 2004). This strategy of cell death regulation could explain why Wnt7b – a promoter of cell cycle entry in hyaloid VECs (Lobov et al., 2005) – is required for cell death. To address the relationship between cell cycle entry and cell death directly, we used BrdU labeling to track the fate of a synchronous cohort of hyaloid VECs through the cell cycle. BrdU was injected at t = 0 hours and hyaloid vessel preparations fixed at t = 1 hour, then at 4-hour intervals subsequently. We scored the number of cells that had both TUNEL and BrdU labeling (Fig. 6E,F). This revealed that BrdU-labeled, apoptotic cells rose to a peak, 17 hours after the BrdU injection (Fig. 6I, red line). The existence of this peak indicates that cell death is dependent on cell cycle stage. By scoring the occurrence of paired, BrdU-labeled nuclei that are the products of cell division (Fig. 6G,H), we were also able to define when, on average, the cohort of BrdU-labeled cells left M phase and entered G1 phase. The peak of G1 phase daughter cells corresponded with the peak of BrdU-positive, apoptotic cells at 17 hours after BrdU (Fig. 6J, red line). These data (confirmed in two independent experiments) indicate that cell cycle progression and cell death are coupled, and that cells die from G1 phase of the cell cycle.
To examine the role of the angiopoietin pathway in regulating cell cycle stage-dependent cell death we injected Ang1 intra-vitreally into the right eye of each animal 6 hours after the BrdU injection. This resulted in the absence of a cell death peak at 17 hours (Fig. 6I, blue line) but did not significantly influence the appearance of daughter pairs (Fig. 6J, blue line). These data indicate that Ang1 suppresses cell death and that the effects of this are observed in the G1 phase of the cell cycle. Combined with previous work (Lobov et al., 2005) these experiments show that the Wnt and angiopoietin pathways are integrated through the cell cycle – Wnt7b promotes cell cycle entry and Ang2 promotes cell death from G1 phase.
DISCUSSION
In the current study we sought to explain how the Wnt and the angiopoietin pathways cooperate to regulate scheduled vascular regression. We have shown that there are multiple levels of pathway integration that constitute an Ang2-mediated cell death switch (Fig. 7).
Fig. 7. Model of the angiopoietin 2-dependent cell death switch.
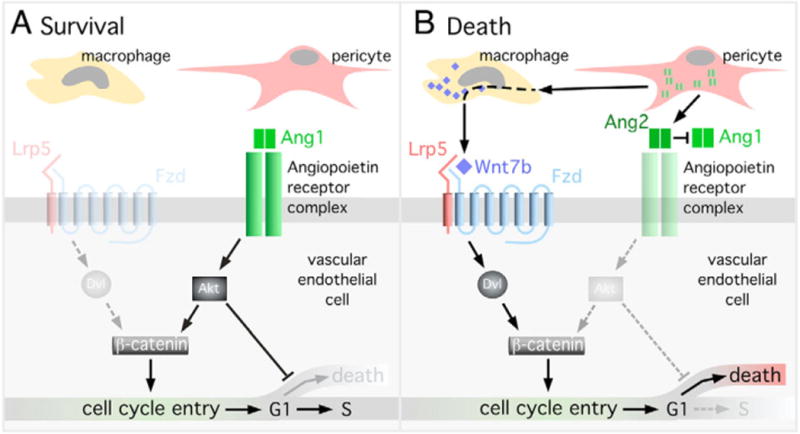
(A) Hyaloid VECs will survive when Ang1 activates Akt, suppresses cell death and promotes proliferation through β-catenin stabilization. (B) Hyaloid VECs will die when Ang2 promotes cell death via suppression of Akt and cell cycle entry by upregulating Wnt7b in macrophages. In the absence of survival signals the cycling hyaloid VECs will undergo apoptosis in the G1 phase of the cell cycle.
Mechanism of the cell death switch
Current data indicate that cell cycle entry is a crucial element of the switch because it is a precondition for cell death, an observation consistent with coupling between cell cycle progression and cell death in other systems (Lowe et al., 2004). In the hyaloid vessels, Wnt pathway activation is a stimulus for cell cycle entry and essential for cell death (Lobov et al., 2005). An explanation for the Wnt pathway requirement emerges from the demonstration that hyaloid VECs die from G1 phase of the cell cycle when Ang2 suppresses survival signaling (Fig. 6).
Much remains unknown about the mechanism of cell cycle-cell death coupling. It has been observed that in general, cells are more sensitive to death stimuli when they are actively cycling. One mechanism for coupling proliferation and death appears to be the ability of growth promoting proteins such as Myc [a Wnt pathway target (He et al., 1998)] to sensitize cells to pro-apoptotic stimuli (including survival signal withdrawal) through enhanced cytochrome C release and caspase 9 activation (Evan and Vousden, 2001). Furthermore, in many settings where proliferating cells die, they do so at cell cycle checkpoints. The p53 genotoxic damage control checkpoint (Sherr and Roberts, 1995) and the so-called Restriction Point (Pardee, 1989; Planas-Silva and Weinberg, 1997) at which survival factors are critical (Zhu et al., 1996; Assoian, 1997; Planas-Silva and Weinberg, 1997) can result in apoptosis in G1 phase of the cell cycle.
A hyaloid VEC will survive (death switch in the off position, Fig. 7A) when Ang1 is the predominant influence. This activates Akt and suppresses cell death, presumably through the well-described inhibition of the pro-apoptotic mediators FoxO and Bad (Datta et al., 1999). Our data also indicate that activation of the angiopoietin pathway is accompanied by proliferation. In this system, proliferation is at least partly a consequence of Akt-mediated stabilization and nuclear translocation of β-catenin (Fig. 7A).
When the death switch is in the on position (Fig. 7B) Ang2 has two functions. The first function is to suppress Akt activity and promote cell death (Datta et al., 1999). However, in suppressing Akt, Ang2 also eliminates the cell cycle entry stimulus (via β-catenin) that is a prerequisite for cell death. Thus, the second function of Ang2, which is upregulation of macrophage Wnt7b expression, appears to be an adaptation to provide an alternative pathway to stimulate cell cycle entry and coupled cell death. How Ang2 upregulates Wnt7b expression in macrophages is currently unclear. This could be an indirect consequence of VECs producing a signaling molecule in response to Ang2 ligation of Tie2. Alternatively, macrophages might respond directly to the presence of Ang2 through the integrins that represent alternative receptors for the angiopoietins (Carlson et al., 2001; Camenisch et al., 2002; Cascone et al., 2005).
Macrophage function in homeostasis and disease
In his phagocytosis theory, Nobelist Illya Metchinikoff first formulated the idea that macrophages could ‘police’ the tissues as a way of reconciling inherent chaos (Tauber, 2003). Indeed, the ability of macrophages to identify, engulf and dispose of foreign matter and apoptotic cells gives them a crucial role in tissue homeostasis. Even a modest reduction in this function can result in the chronic inflammation that promotes diseases such as cancer and diabetes (Wellen and Hotamisligil, 2005; Condeelis and Pollard, 2006). Being widely distributed and highly mobile, macrophages are perfectly suited to this homeostatic function.
In a few documented circumstances, macrophages take on the additional role of actively signaling apoptosis in a target cell. A number of macrophage products have been implicated: nerve growth factor (NGF) where macrophage-related microglia cause neurons to die in the chick (Frade et al., 1996) and superoxide ions where microglia signal apoptosis of Purkinje cells in mouse brain slice cultures (Marin-Teva et al., 2004; Mallat et al., 2005). An additional example – where the dead cell recognition machinery is a back-up pathway for caspase pathway activation in C. elegans – is important because it implies that phagocyte-induced cell death is a highly conserved activity that will probably be found throughout the metazoans (Hoeppner et al., 2001; Reddien et al., 2001).
In the current work we describe a cell death signaling mechanism that involves three cell types, the pericyte, the macrophage and the vascular endothelial cell. Ang2, which is probably produced by pericytes, is a critical initiator of a switch in the pattern of signaling that results in VEC cell cycle entry and cell death (Fig. 7). In this system, the macrophage is the critical source of Wnt7b (Lobov et al., 2005) that is required for VEC cell cycle entry. In turn, Wnt7b expression in macrophages requires Ang2. This arrangement of regulatory interactions means that participation of the macrophage is obligatory. If the macrophage is absent, there would be no source of Wnt7b to stimulate cell cycle entry, and no cell death. Ang2-dependent Wnt7b expression may represent the optimal way to ensure that when cell death occurs the macrophage and its dead cell recognition and disposal capabilities are present. We anticipate that this or related mechanisms may be generally applicable.
Macrophages have been implicated in a variety of diseases, often where angiogenesis is a prominent component of the disease process. One example is the vision-compromising choroidal neovascularization that occurs in the wet form of age-related macular degeneration (AMD) (Ferrara and Kerbel, 2005). In this setting, according to depletion experiments (Espinosa-Heidmann et al., 2003; Sakurai et al., 2003) macrophages enhance the formation of new blood vessels. Another example is the activity of tumor-associated macrophages (TAMs) in enhancing tumor angiogenesis and tumor progression (Condeelis and Pollard, 2006). In a mouse breast cancer model that closely mimics the human disease (Lin et al., 2003), macrophage deficiency leads to tumors that are poorly vascularized and in a response that is probably related, essentially non-metastatic (Lin et al., 2006).
In diseases like AMD and cancer where macrophages play a prominent role, it will be interesting to determine whether macrophage production of Wnt ligands is a factor contributing to disease progression. Based on the current work, macrophage Wnts are a stimulus for VEC proliferation and in the absence of a second, modulating signal (Ang2 in the hyaloid system, Fig. 7) could stimulate angiogenesis and promote disease progression. Further work will be needed to address these questions.
Acknowledgments
We thank Mr Paul Speeg for excellent technical assistance. We are indebted to Dr Tom Sato for providing the Ang2lacZ mouse line prior to publication. This work was supported by the following: NIH RO1 HL071049 (L.B.), NIH RO1s EY10559, EY15766, EY16241 and EY17848 and by funds from the Abrahamson Pediatric Eye Institute Endowment at Children’s Hospital Medical Center of Cincinnati (R.A.L.).
References
- Assoian RK. Anchorage-dependent cell cycle progression. J Cell Biol. 1997;136:1–4. doi: 10.1083/jcb.136.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camenisch G, Pisabarro MT, Sherman D, Kowalski J, Nagel M, Hass P, Xie MH, Gurney A, Bodary S, Liang XH. ANGPTL3 stimulates endothelial cell adhesion and migration via integrin alpha vbeta 3 and induces blood vessel formation in vivo. J Biol Chem. 2002;277:17281–17290. doi: 10.1074/jbc.M109768200. [DOI] [PubMed] [Google Scholar]
- Carlson TR, Feng Y, Maisonpierre PC, Mrksich M, Morla AO. Direct cell adhesion to the angiopoietins mediated by integrins. J Biol Chem. 2001;276:26516–26525. doi: 10.1074/jbc.M100282200. [DOI] [PubMed] [Google Scholar]
- Cascone I, Napione L, Maniero F, Serini G, Bussolino F. Stable interaction between alpha5beta1 integrin and Tie2 tyrosine kinase receptor regulates endothelial cell response to Ang-1. J Cell Biol. 2005;170:993–1004. doi: 10.1083/jcb.200507082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo SS, Brognard J, Petukhov PA, Zhang C, Tsurutani J, Granville CA, Li M, Jung M, West KA, Gills JG. Preferential inhibition of Akt and killing of Akt-dependent cancer cells by rationally designed phosphatidylinositol ether lipid analogues. Cancer Res. 2004;64:2782–2792. doi: 10.1158/0008-5472.can-03-1530. [DOI] [PubMed] [Google Scholar]
- Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Corbel SY, Rossi FM. Latest developments and in vivo use of the Tet system: ex vivo and in vivo delivery of tetracycline-regulated genes. Curr Opin Biotechnol. 2002;13:448–452. doi: 10.1016/s0958-1669(02)00361-0. [DOI] [PubMed] [Google Scholar]
- DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–4568. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, Naldini L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–226. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Diez-Roux G, Lang RA. Macrophages induce apoptosis in normal cells in vivo. Development. 1997;124:3633–3638. doi: 10.1242/dev.124.18.3633. [DOI] [PubMed] [Google Scholar]
- Diez-Roux G, Argilla M, Makarenkova H, Ko K, Lang RA. Macrophages kill capillary cells in G1 phase of the cell cycle during programmed vascular regression. Development. 1999;126:2141–2147. doi: 10.1242/dev.126.10.2141. [DOI] [PubMed] [Google Scholar]
- Dumont DJ, Gradwohl G, Fong GH, Puri MC, Gertsenstein M, Auerbach A, Breitman ML. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev. 1994;8:1897–1909. doi: 10.1101/gad.8.16.1897. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Suner IJ, Hernandez EP, Monroy D, Csaky KG, Cousins SW. Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3586–3592. doi: 10.1167/iovs.03-0038. [DOI] [PubMed] [Google Scholar]
- Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- Feistritzer C, Mosheimer BA, Sturn DH, Bijuklic K, Patsch JR, Wiedermann CJ. Expression and function of the angiopoietin receptor Tie-2 in human eosinophils. J Allergy Clin Immunol. 2004;114:1077–1084. doi: 10.1016/j.jaci.2004.06.045. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- Frade JM, Rodriguez-Tebar A, Barde YA. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature. 1996;383:166–168. doi: 10.1038/383166a0. [DOI] [PubMed] [Google Scholar]
- Fukumoto S, Hsieh CM, Maemura K, Layne MD, Yet SF, Lee KH, Matsui T, Rosenzweig A, Taylor WG, Rubin JS. Akt participation in the Wnt signaling pathway through Dishevelled. J Biol Chem. 2001;276:17479–17483. doi: 10.1074/jbc.C000880200. [DOI] [PubMed] [Google Scholar]
- Gale NW, Thurston G, Hackett SF, Renard R, Wang Q, McClain J, Martin C, Witte L, Witte MH, Jackson D. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter is rescued by Angiopoietin-1. Dev Cell. 2002;3:411–423. doi: 10.1016/s1534-5807(02)00217-4. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Hoeppner DJ, Hengartner MO, Schnabel R. Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans. Nature. 2001;412:202–206. doi: 10.1038/35084103. [DOI] [PubMed] [Google Scholar]
- Ito M, Yoshioka M. Regression of the hyaloid vessels and pupillary membrane of the mouse. Anat Embryol. 1999;200:403–411. doi: 10.1007/s004290050289. [DOI] [PubMed] [Google Scholar]
- Iwama A, Hamaguchi I, Hashiyama M, Murayama Y, Yasunaga K, Suda T. Molecular cloning and characterization of mouse TIE and TEK receptor tyrosine kinase genes and their expression in hematopoietic stem cells. Biochem Biophys Res Commun. 1993;195:301–309. doi: 10.1006/bbrc.1993.2045. [DOI] [PubMed] [Google Scholar]
- Kato M, Patel MS, Levasseur R, Lobov I, Chang BH, Glass DA, 2nd, Hartmann C, Li L, Hwang TH, Brayton CF, et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157:303–314. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Kim HG, So JN, Kim JH, Kwak HJ, Koh GY. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3′-Kinase/Akt signal transduction pathway. Circ Res. 2000a;86:24–29. doi: 10.1161/01.res.86.1.24. [DOI] [PubMed] [Google Scholar]
- Kim I, Kim JH, Moon SO, Kwak HJ, Kim NG, Koh GY. Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Oncogene. 2000b;19:4549–4552. doi: 10.1038/sj.onc.1203800. [DOI] [PubMed] [Google Scholar]
- Kozikowski AP, Sun H, Brognard J, Dennis PA. Novel PI analogues selectively block activation of the pro-survival serine/threonine kinase Akt. J Am Chem Soc. 2003;125:1144–1145. doi: 10.1021/ja0285159. [DOI] [PubMed] [Google Scholar]
- Lang RA, Bishop MJ. Macrophages are required for cell death and tissue remodeling in the developing mouse eye. Cell. 1993;74:453–462. doi: 10.1016/0092-8674(93)80047-i. [DOI] [PubMed] [Google Scholar]
- Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, Pollard JW. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66:11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- Lobov IB, Brooks PC, Lang RA. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc Natl Acad Sci USA. 2002;99:11205–11210. doi: 10.1073/pnas.172161899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobov IB, Rao S, Carroll TJ, Vallance JE, Ito M, Ondr JK, Kurup S, Glass DA, Patel MS, Shu W. WNT7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature. 2005;437:417–421. doi: 10.1038/nature03928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- Lucas M, Stuart LM, Savill J, Lacy-Hulbert A. Apoptotic cells and innate immune stimuli combine to regulate macrophage cytokine secretion. J Immunol. 2003;171:2610–2615. doi: 10.4049/jimmunol.171.5.2610. [DOI] [PubMed] [Google Scholar]
- Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- Mallat M, Marin-Teva JL, Cheret C. Phagocytosis in the developing CNS: more than clearing the corpses. Curr Opin Neurobiol. 2005;15:101–107. doi: 10.1016/j.conb.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41:535–547. doi: 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- Meeson AP, Argilla M, Ko K, Witte L, Lang RA. VEGF deprivation-induced apoptosis is a component of programmed capillary regression. Development. 1999;126:1407–1415. doi: 10.1242/dev.126.7.1407. [DOI] [PubMed] [Google Scholar]
- Moore KA, Lemischka IR. Stem cells and their niches. Science. 2006;311:1880–1885. doi: 10.1126/science.1110542. [DOI] [PubMed] [Google Scholar]
- Ohlmann AV, Adamek E, Ohlmann A, Lutjen-Drecoll E. Norrie gene product is necessary for regression of hyaloid vessels. Invest Ophthalmol Vis Sci. 2004;45:2384–2390. doi: 10.1167/iovs.03-1214. [DOI] [PubMed] [Google Scholar]
- Pardee AB. G1 events and regulation of cell proliferation. Science. 1989;246:603–608. doi: 10.1126/science.2683075. [DOI] [PubMed] [Google Scholar]
- Peters KG, Kontos CD, Lin PC, Wong AL, Rao P, Huang L, Dewhirst MW, Sankar S. Functional significance of Tie2 signaling in the adult vasculature. Recent Prog Horm Res. 2004;59:51–71. doi: 10.1210/rp.59.1.51. [DOI] [PubMed] [Google Scholar]
- Planas-Silva MD, Weinberg RA. The restriction point and control of cell proliferation. Curr Opin Cell Biol. 1997;9:768–772. doi: 10.1016/s0955-0674(97)80076-2. [DOI] [PubMed] [Google Scholar]
- Reddien PW, Cameron S, Horvitz HR. Phagocytosis promotes programmed cell death in C. elegans. Nature. 2001;412:198–202. doi: 10.1038/35084096. [DOI] [PubMed] [Google Scholar]
- Sakurai E, Anand A, Ambati BK, van Rooijen N, Ambati J. Macrophage depletion inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3578–3585. doi: 10.1167/iovs.03-0097. [DOI] [PubMed] [Google Scholar]
- Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- Schlaeger TM, Bartunkova S, Lawitts JA, Teichmann G, Risau W, Deutsch U, Sato TN. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc Natl Acad Sci USA. 1997;94:3058–3063. doi: 10.1073/pnas.94.7.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Chuang WW, Sun Z. Phosphatidylinositol 3-kinase/Akt stimulates androgen pathway through GSK3beta inhibition and nuclear beta-catenin accumulation. J Biol Chem. 2002;277:30935–30941. doi: 10.1074/jbc.M201919200. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- Shu W, Jiang YQ, Lu MM, Morrisey EE. Wnt7b regulates mesenchymal proliferation and vascular development in the lung. Development. 2002;129:4831–4842. doi: 10.1242/dev.129.20.4831. [DOI] [PubMed] [Google Scholar]
- Sun JF, Phung T, Shiojima I, Felske T, Upalakalin JN, Feng D, Kornaga T, Dor T, Dvorak AM, Walsh K. Microvascular patterning is controlled by fine-tuning the Akt signal. Proc Natl Acad Sci USA. 2005;102:128–133. doi: 10.1073/pnas.0403198102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- Tauber AI. Metchnikoff and the phagocytosis theory. Nat Rev Mol Cell Biol. 2003;4:897–901. doi: 10.1038/nrm1244. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- Ylikarppa R, Eklund L, Sormunen R, Kontiola AI, Utriainen A, Maatta M, Fukai N, Olsen BR, Pihlajaniemi T. Lack of type XVIII collagen results in anterior ocular defects. FASEB J. 2003;17:2257–2259. doi: 10.1096/fj.02-1001fje. [DOI] [PubMed] [Google Scholar]
- Zhu X, Ohtsubo M, Bohmer RM, Roberts JM, Assoian RK. Adhesion-dependent cell cycle progression linked to the expression of cyclin D1, activation of cyclin E-cdk2, and phosphorylation of the retinoblastoma protein. J Cell Biol. 1996;133:391–403. doi: 10.1083/jcb.133.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]