

Summary
Free fatty acids (FFA) cause apoptosis of pancreatic β-cells and might contribute to β-cell loss in type 2 diabetes via the induction of endoplasmic reticulum (ER) stress. We studied here the molecular mechanisms implicated in FFA-induced ER stress initiation and apoptosis in INS-1E cells, FACS-purified primary β-cells and human islets exposed to oleate and/or palmitate. Treatment with saturated and/or unsaturated FFA led to differential ER stress signaling. Palmitate induced more apoptosis and markedly activated the IRE1, PERK and ATF6 pathways, owing to a sustained depletion of ER Ca2+ stores, whereas the unsaturated FFA oleate led to milder PERK and IRE1 activation and comparable ATF6 signaling. Non-metabolizable methyl-FFA analogs induced neither ER stress nor β-cell apoptosis. The FFA-induced ER stress response was not modified by high glucose concentrations, suggesting that ER stress in primary β-cells is primarily lipotoxic, and not glucolipotoxic. Palmitate, but not oleate, activated JNK. JNK inhibitors reduced palmitate-mediated AP-1 activation and apoptosis. Blocking the transcription factor CHOP delayed palmitate-induced β-cell apoptosis. In conclusion, saturated FFA induce ER stress via ER Ca2+ depletion. The IRE1 and resulting JNK activation contribute to β-cell apoptosis. PERK activation by palmitate also contributes to β-cell apoptosis via CHOP.
Keywords: Pancreatic β-cell, Islet, Endoplasmic reticulum stress, Fatty acid, Oleate, Palmitate, Lipotoxicity, Apoptosis, Type 2 diabetes
Introduction
Insulin deficiency is present early in the pathogenesis of type 2 diabetes (T2D) and worsens over time (Cnop et al., 2007b; Festa et al., 2006; Larsson and Ahren, 2000; Matthews et al., 1998; Weyer et al., 1999; Xiang et al., 2006). Insufficient insulin secretion is probably due to both functional pancreatic-β-cell defects and loss of β-cell mass (Butler et al., 2003; Clark et al., 1988; Klöppel et al., 1985; Sakuraba et al., 2002; Yoon et al., 2003) by apoptosis (Butler et al., 2003; Del Guerra et al., 2005; Marchetti et al., 2004).
The worldwide prevalence of T2D is rapidly increasing owing to major modifications in dietary habits and decreased physical activity. Western diets rich in saturated (animal) fats cause quantitative and qualitative changes in plasma lipid levels, leading to obesity and insulin resistance, and thereby increasing the demand on β-cells. Concomitantly, β-cells are exposed to increased levels of free fatty acids (FFA) (Porte, Jr and Kahn, 2001) of dietary origin or released by adipose tissues. Chronic exposure to high FFA concentrations causes β-cell apoptosis (Cnop et al., 2001; Cnop et al., 2005; Maedler et al., 2001) and might contribute to the increased β-cell apoptosis rates in T2D (Butler et al., 2003). In some studies, the combination of high levels of both glucose and FFA increased β-cell toxicity, and this was termed glucolipotoxicity (El-Assaad et al., 2003).
The main role of pancreatic β-cells is the synthesis and release of insulin in response to glucose and other nutrients. Proinsulin mRNA represents 20% of the total β-cell mRNA expression (Van Lommel et al., 2006) and glucose-regulated insulin translation can account for up to 50% of total protein synthesis in β-cells (Schuit et al., 1988). For the endoplasmic reticulum (ER) to cope with the nutrient-induced changes in protein synthesis, β-cells deploy homeostatic mechanisms to balance the newly synthesized protein load against the ER protein-folding capacity (Eizirik et al., 2008). This cellular response is known as the unfolded protein response (UPR) or ER stress response (Ron and Walter, 2007; Zhang and Kaufman, 2006). When the response fails to preserve and/or restore ER function, it can culminate in cell death (Eizirik et al., 2008; Marciniak and Ron, 2006).
ER stress leads to integrated transcriptional and translational responses (schematized in supplementary material Fig. S1) through activation of the ER membrane proteins IRE1 (inositol requiring ER-to-nucleus signal kinase 1; also known as ERN1), ATF6 (activating transcription factor 6) and PERK (PKR-like ER kinase; also known as EIF2AK3). These ER stress transducers remain inactive when bound to the ER chaperone BiP [immunoglobulin heavy-chain binding protein; also known as glucose-regulated protein (GRP)78 and HSPA5], and when BiP dissociates from their luminal side to assist in protein folding, the transducers are activated, signaling ER-chaperone depletion. Active IRE1 alternatively splices XBP1 (X-box binding protein 1) mRNA. Spliced XBP1 (XBP1s) induces genes involved in ER expansion, in protein folding and in the degradation of misfolded proteins (Lee et al., 2005; Lee et al., 2003; Marciniak and Ron, 2006). Active ATF6 translocates to the Golgi and is cleaved from the membrane by site-1 protease (S1P; also known as MBTPS1) and S2P (also known as MBTPS2) (Haze et al., 1999). ATF6 induces transcription of ER chaperones, such as BiP. PERK phosphorylates the eukaryotic translation initiation factor (eIF)2α, thereby inhibiting global protein synthesis and decreasing the functional demand on the ER. In parallel, translation of selected proteins, such as ATF4, is facilitated and expression of downstream C/EBP homologous protein [CHOP, also known as GADD153 and DDIT3 (Fornace, Jr et al., 1989)] is induced (Marciniak and Ron, 2006). The integrated result of the ER stress response is attenuation of protein translation paralleled by upregulation of ER chaperones and ER folding capacity, and degradation of irreversibly misfolded proteins. When this response fails to solve the ER stress, apoptosis is triggered. How ER stress leads to apoptosis remains unclear, but it might involve activation of the transcription factors ATF3 and CHOP, Jun N-terminal kinase (JNK), the Akt inhibitor TRB3, caspases 4 and 12, and the mitochondrial apoptosis pathway; the latter process might be activated through the downregulation of Bcl-2, activation of Bax/Bak, Bim, p53, PUMA and NOXA (Eizirik et al., 2008).
Evidence for pancreatic β-cell ER stress in diabetes includes the increased expression of ER stress markers in mouse islets from diabetic db/db mice (Laybutt et al., 2007; Yusta et al., 2006). Three recent reports provide histological (Huang et al., 2007; Laybutt et al., 2007) and electron microscopic (Marchetti et al., 2007) evidence of ER stress in islets of T2D patients. Gene-profiling studies did not detect marked differences in the expression of genes related to the ER stress response in laser capture micro-dissected human islets from T2D and non-diabetic individuals (Marchetti et al., 2007), perhaps indicating a state of chronic compensation (Lin et al., 2007; Rutkowski et al., 2006). Culture of these islets at high glucose levels, however, induced a more marked increase in BiP and XBP1 as compared with islets from non-diabetic donors (Marchetti et al., 2007). Taken together, these data suggest that ER stress is present in human β-cells in diabetes. Because high glucose levels can mildly activate ER stress signaling (Elouil et al., 2007), the issue remains whether it merely activates the β-cell stress response after diabetes onset, or whether other nutrient mediators trigger an ER stress response in β-cells and thereby contribute to the pathogenesis of T2D.
ER stress has been proposed to link high-fat-diet-induced obesity with insulin resistance in rodents (Ozcan et al., 2004). We have suggested that this cellular stress response represents a molecular mechanism common to the two main defects of T2D, namely pancreatic β-cell failure and insulin resistance, based on our observation that ER stress is triggered by FFA in a β-cell line (Kharroubi et al., 2004). Several studies have since extended these observations and demonstrated that palmitate, and to a lesser extent oleate, activates an ER stress response in the INS-1(E) and MIN6 β-cell lines (Cnop et al., 2007a; Karaskov et al., 2006; Laybutt et al., 2007), but whether this occurs in primary β-cells is unknown. Furthermore, the mechanism through which FFA activate ER stress and eventually trigger apoptosis has not been determined.
In the present study, we examined: (1) whether FFA induce an ER stress response in INS-1E cells, primary FACS-purified rat β-cells and human islets; (2) whether glucose-potentiation of lipotoxic ER stress contributes to glucolipotoxicity; (3) the mechanisms by which FFA trigger an ER stress response in β-cells; and (4) which mediators in the ER stress response activate the β-cell apoptotic pathway. This study demonstrates that FFA induce ER stress signaling in primary rodent and human β-cells, and sheds light on the mechanisms involved in triggering FFA-induced ER stress and the subsequent execution of β-cell apoptosis.
Results
Glucolipotoxicity in pancreatic β-cells
Oleate and palmitate induced apoptosis in INS-1E cells at low (5.6 mM), medium (11 mM) and high (28 mM) glucose concentrations (Fig. 1A). The saturated FFA palmitate caused more apoptosis than the unsaturated oleate over 12–72 hours (supplementary material Fig. S2A), whereas the combination of the two FFA was not toxic, confirming previous observations (Cnop et al., 2001; Maedler et al., 2001). Oleate and palmitate activated caspase-12 in INS-1E cells (2±0% caspase-12-positive cells for oleate, 11±2% for palmitate, compared with 1±0% in the control after 14 hours; n=5; P<0.05 for oleate or palmitate vs control). The synthetic ER stressor thapsigargin was used as a positive control (thapsigargin: 15±0% caspase-12-positive cells; n=5; P<0.05 vs control) and necrosis-inducing H2O2 as a negative control (50 μM; 1±0%; n=3). Glucose dose-dependently potentiated oleate- and palmitate-induced toxicity in INS-1E cells, doubling the apoptosis rates at 28 mM glucose (Fig. 1A).
Fig. 1.

Glucolipotoxicity in rat β-cells. INS-1E cells (A) and rat primary β-cells (B) were cultured for 72 hours in the presence or absence of oleate (0.5 mM, gray bars), palmitate (0.5 mM, black bars) or an equimolar combination of oleate and palmitate (0.25 mM each, hatched bars) at glucose concentrations (G, shown in mM) as indicated. The results are the means ± s.e.m. of 7–15 independent experiments. **P<0.01, ***P<0.001 vs respective glucose control (i.e. not exposed to FFA and cultured at the same glucose concentration); l, P<0.05 vs low glucose (5.6 or 6.1 mM); m, P<0.05 vs medium glucose (10 or 11 mM).
In FACS-purified primary rat β-cells, FFA also caused apoptosis, with palmitate being more toxic than oleate (Fig. 1B and supplementary material Fig. S2B). The death of primary β-cells was preceded by activation of caspase-12 (1.3±0.1% caspase-12-positive cells for oleate, 5.9±0.7% for palmitate, compared with 0.8±0.1% in the control after 24 hours; n=3; P<0.05 for oleate or palmitate vs control) and caspases 3 and 7 (oleate: 2.1±0.2-fold increase compared with control after 48 hours; palmitate: 3.0±0.8-fold increase, n=3). The glucose effect was less marked in rat primary β-cells, with glucose only modestly increasing palmitate-and oleate-induced apoptosis after 3 (Fig. 1B) and 6 (supplementary material Fig. S2C) days. In contrast to INS-1E cells, and in keeping with previous observations (Hoorens et al., 1996), apoptosis of primary β-cells tended to increase following long-term culture at 6.1 mM glucose (supplementary material Fig. S2C). Cell exposure to an equimolar concentration of oleate+palmitate did not increase apoptosis.
Palmitate was also toxic to human islets (Fig. 2), but this was not potentiated by glucose. Neither oleate nor high glucose levels induced human islet cell death after 3–6 days (Fig. 2).
Fig. 2.

Glucolipotoxicity in human islets. Human islets were cultured for 72 hours or 6 days in the presence or absence of oleate (gray bars) or palmitate (black bars) at a glucose concentration of 6.1 or 28 mM, as indicated. The results are the means ± s.e.m. of four to seven independent experiments. *P<0.05, **P<0.01 vs respective control.
FFA and glucose activate ER stress pathways in β-cells
In a subsequent series of experiments, we examined the effects of FFA and glucose on the three main UPR branches, under the control of PERK, ATF6 and IRE1 (supplementary material Fig. S1).
The PERK pathway
Palmitate, but not oleate or the combination of oleate and palmitate, induced PERK and eIF2α phosphorylation in INS-1E cells (Fig. 3) after 6–24 hours, an effect that was augmented at 28 mM glucose (supplementary material Fig. S3). Downstream in the PERK-eIF2α pathway, we observed induction of ATF3 mRNA and protein (Fig. 3), and this was clearly potentiated by glucose (supplementary material Fig. S4), as previously reported (Hartman et al., 2004). The transcription factor CHOP, which was shown to be an ATF4 target gene in β-cells (Pirot et al., 2007), was induced by palmitate by a factor of four after 6 hours at 5.6 and 11 mM glucose (Fig. 3), and it doubled at 28 mM glucose (supplementary material Fig. S5). We have previously shown that palmitate induces CHOP at the protein level (Cnop et al., 2007a; Pirot et al., 2007). Palmitate-induced CHOP mRNA expression peaked to eightfold at 12 hours and decreased at later time points. A concentration of 28 mM glucose did not modify CHOP expression at 24–48 hours (supplementary material Fig. S5). By contrast, oleate or oleate plus palmitate induced CHOP expression progressively to twofold higher than in controls (Fig. 3) and in these conditions high glucose decreased CHOP expression (supplementary material Fig. S5).
Fig. 3.

FFA activate PERK signaling. INS-1E cells were cultured in the presence or absence of oleate (O, gray diamonds), palmitate (P, black squares) or oleate plus palmitate (OP, white circles) at 11 mM glucose. Western blots using phospho-specific PERK (P-PERK), phospho-specific eIF2α (P-eIF2α) and anti-ATF3 antibodies after 6–12 hours exposure to FFA are shown. Total eIF2α or β-actin was used as control for protein loading. One representative experiment for three to six similar experiments is shown, as well as mean optical density measurements of the western blots following 6-, 12- and 24- hour FFA exposure; n=2–6. ATF3 and CHOP mRNA expression following a 6- to 48-hour FFA exposure was analyzed by real-time PCR, normalized for the expression level of the housekeeping gene GAPDH and expressed as fold induction of the control. The results represent means ± s.e.m. of 6–11 independent experiments. *P<0.05, **P<0.01 vs control.
The ATF6 pathway
As a downstream target of ATF6 activation, we examined BiP mRNA expression. Oleate and palmitate, as well as their combination, led to a 2- to 2.5-fold induction of the ER chaperone BiP after 24 hours (Fig. 4). At 28 mM glucose, BiP expression levels were up to threefold lower as compared with 5.6 or 11 mM (supplementary material Fig. S6).
Fig. 4.

FFA activate IRE1 and ATF6 signaling. INS-1E cells were cultured in the presence or absence of oleate (O, gray diamonds), palmitate (P, black squares) or oleate plus palmitate (OP, white circles) at a glucose concentration of 11 mM. The mRNA expression of BiP, XBP1s and XBP1t following a 6- to 48-hour FFA exposure was analyzed by real-time PCR, normalized for the expression level of the housekeeping gene GAPDH and expressed as fold induction. The results represent means ± s.e.m. of 6–11 independent experiments. For the measurement of UPR luciferase reporter activity, INS-1E cells were co-transfected with the UPR reporter, which is responsive to ATF6 and XBP1s, and with the internal control pRL-CMV, encoding Renilla luciferase. After overnight transfection, the cells were exposed for 24 hours to FFA. The firefly results were normalized for Renilla luciferase activity and are means ± s.e.m. of ten independent experiments. *P<0.05, **P<0.01, ***P<0.001 vs control (C).
ATF6 activation also induces XBP1 transcription (Eizirik et al., 2008). Native XBP1 mRNA (XBP1t) was similarly upregulated by all FFA tested (Fig. 4) and its expression decreased in the presence of 28 mM glucose (supplementary material Fig. S7). To further assess the activation of ATF6 and XBP1, we used a UPR luciferase reporter construct, containing ATF6- and XBP1-binding sites. All FFA tested activated the reporter (Fig. 4) and again a decrease in reporter activity was observed with increasing glucose concentrations (supplementary material Fig. S8).
The IRE1 pathway
Activation of IRE1 leads to alternative splicing and activation of the transcription factor XBP1. Compared with controls, XBP1 splicing was earlier and more marked in the presence of palmitate, intermediate with the combination of oleate and palmitate, and modest with oleate alone (Fig. 4). The palmitate-mediated activation of the IRE1 and PERK branches tapered over time, with levels comparable to the other FFA conditions at 48 hours (Fig. 4). With increasing glucose concentrations, a decrease in XBP1s message was observed (supplementary material Fig. S9), as was seen for CHOP (supplementary material Fig. S5) and BiP (supplementary material Fig. S6).
FFA-induced ER stress signaling in rat primary β-cells and human islets
In line with the pattern of ER stress signaling in INS-1E cells, palmitate induced a marked activation of the PERK pathway in FACS-purified rat primary β-cells after 24 hours (Fig. 5A). Thus, ATF3 was eightfold upregulated, compared with a twofold induction by oleate, but glucose did not have a potentiating effect. Palmitate also increased CHOP expression by twofold (Fig. 5A). IRE1 was activated by palmitate, as shown by XBP1 splicing, and there was a trend for BiP induction (Fig. 5A). Addition of oleate to palmitate prevented induction of the ER stress markers, even leading to a decrease in XBP1s (Fig. 5A). There was a clear ER stress response in human islets exposed to palmitate for 48 hours (Fig. 5B). ATF3 and CHOP were markedly upregulated, again in a glucose-concentration-independent manner. ER stress markers were not induced by 28 mM glucose alone, in line with previous findings (Marchetti et al., 2007). Palmitate, but not oleate, caused XBP1 splicing and ATF6-dependent BiP induction (Fig. 5B).
Fig. 5.

FFA activate ER stress signaling in rat primary β-cells and human islets. (A) FACS-purified rat β-cells were cultured for 24 hours in the presence of oleate (gray bars), palmitate (black bars) or oleate plus palmitate (hatched bars) at glucose concentrations of 10 and 28 mM. (B) Human islets were cultured for 48 hours in the absence (white bars) or presence of oleate (gray bars) or palmitate (black bars) at glucose concentrations of 6.1 and 28 mM. ATF3, CHOP, XBP1s and BiP mRNA expression was analyzed by real-time PCR, normalized for the expression level of the housekeeping genes GAPDH (for rat β-cells) or β-actin (for human islets) and expressed as fold induction of the control. The results represent means ± s.e.m. of three (human) or four to seven (rat) independent experiments. *P<0.05, **P<0.01, ***P<0.001 vs control (white bars).
Mechanisms contributing to FFA- and glucose-induced ER stress and β-cell apoptosis
We next focused on the mechanisms contributing to FFA-induced ER stress and β-cell apoptosis. For this purpose, we selected an intermediate glucose concentration (11 mM), at which the levels of apoptosis and ER stress induced by FFA in INS-1E cells are comparable with those in rat primary β-cells.
FFA metabolism is required for ER stress and apoptosis induction
The non-metabolizable FFA methylpalmitate is taken up by cells but it is not converted to acyl-CoA for subsequent esterification in the ER or β-oxidation in mitochondria (Parker et al., 2003). The methyl fatty acid was not toxic to INS-1E cells (Fig. 6), whereas palmitate induced apoptosis, as demonstrated before (see Fig. 1A). Whereas palmitate upregulated the diverse ER stress markers tested, methylpalmitate induced no or a negligible increase in these markers (Fig. 6). Similar observations were made with methyloleate (data not shown).
Fig. 6.
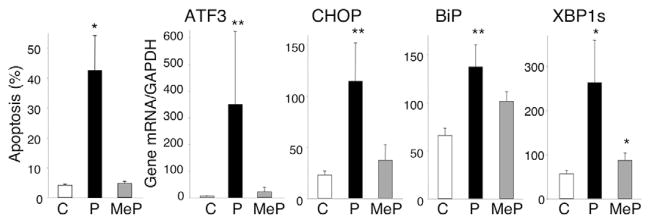
Non-metabolized non-toxic methyl FFA do not elicit ER stress. INS-1E cells were exposed to control (C), palmitate (0.5 mM, P) or methyl-palmitate (0.5 mM, MeP) at 11 mM glucose. After 72 hours, the percentage of apoptotic cells was determined; n=4–5 (left). mRNA expression of ER stress markers was analyzed after 24 hours by real-time PCR and normalized for the housekeeping gene GAPDH; n=4. *P<0.05, **P<0.01 vs control.
FFA deplete ER Ca2+ stores
The ER stress response can be activated in conditions of ER Ca2+ depletion (Paschen, 2003) and we next examined whether this contributes to the triggering of ER stress by FFA. In Fura-2-loaded cells, acute stimulation with thapsigargin blocks the SERCA (sarcoendoplasmic-reticulum pump Ca2+-ATPase; also known as ATP2A2) pump and the ensuing peak in cytoplasmic fluorescence provides an indirect measure of ER Ca2+ stores. This method was applied to INS-1E cells following a 24 hour FFA exposure. As a positive control, we treated cells for 24 hours with thapsigargin. Following the latter treatment, acute exposure to thapsigargin did not induce a cytoplasmic Ca2+ peak (data not shown). Oleate and, to a greater extent, palmitate depleted ER Ca2+ (Fig. 7A), reducing the area under the Ca2+ curve by 25 and 40%, respectively (P<0.05). Because this assay only reflects the ER Ca2+ stores, we investigated whether reduced Ca2+ content is due to reduced ER Ca2+ uptake and measured this using an ER-targeted aequorin (Fig. 7B). Palmitate caused an early (3 hours) impairment in ER Ca2+ uptake, which was maintained up to 24 hours. The combination of FFA resulted in a mild decrease in ER Ca2+ uptake after 6–12 hours and oleate decreased ER Ca2+ storage after 24 hours (P<0.05). SERCA mRNA expression was not inhibited in INS-1E cells exposed to FFA between 6 and 48 hours (data not shown), suggesting that palmitate has direct effects on SERCA pump activity.
Fig. 7.

FFA deplete ER calcium stores in β-cells. (A) INS-1E cells were cultured for 24 hours in the presence of oleate (O, red line), palmitate (P, green line) or control (C, black line) at a glucose concentration of 11 mM. The intracellular Ca2+ concentration was measured at baseline and following acute thapsigargin stimulation (arrow). The traces shown are the means of 5–6 independent experiments (278–368 cells/experiment). (B) Following infection with an ER-targeted aequorin-encoding adenovirus, INS-1E cells were cultured for 3–24 hours in the presence of oleate (O, red line), palmitate (P, green line), an equimolar mixture of oleate and palmitate (OP, blue line) or control (C, black line) at a glucose concentration of 11 mM. Prior to the measurements, cells were depleted of Ca2+ and the aequorin was reconstituted with coelenterazine n in Ca2+-free KRBB. External CaCl2 (1.5 mM) was then reintroduced at t=20 seconds to allow the reestablishment of ER Ca2+ levels. The results are the means ± s.e.m. of four independent experiments. Bold diamond symbols represent Ca2+ values significantly different from control condition (P<0.05).
Role of ATF3 and CHOP in FFA-induced β-cell apoptosis
Because of the prominent induction of ATF3 and CHOP by palmitate in clonal β-cells, and in rat and human primary β-cells (Figs 3 and 5), and their role in ER-stress-induced cell death (Hartman et al., 2004; Oyadomari et al., 2001), we examined whether these transcription factors play a role in palmitate-induced β-cell apoptosis. Mouse islet cells from ATF3−/− mice were not protected from apoptotic death following a 48 hour (data not shown) or 72 hour palmitate exposure (supplementary material Fig. S10A). Because congenital ATF3 deficiency in these mice might activate compensatory signaling mechanisms, we also used RNA interference to examine whether ATF3 knockdown protects β-cells from ER stress. Using two different ATF3 small interfering RNAs (siRNAs), we obtained a 70–80% inhibition of ATF3 induction by the SERCA blocker cyclopiazonic acid (CPA; used as a positive control for ER stress) or palmitate in INS-1E cells (supplementary material Fig. S10B,C) or primary β-cells (supplementary material Fig. S10D). This efficient knockdown did not, however, protect against CPA- or palmitate-induced apoptosis (supplementary material Fig. S10C,D). On the contrary, apoptosis rates increased in the ATF3 knockdown cells, suggesting that ATF3 induction might play a neutral (mouse islets) or even an anti-apoptotic (INS-1E and rat β-cells) role in the context of palmitate-induced β-cell death. Further work is required to clarify these unexpected findings.
We next examined whether CHOP knockdown protects β-cells from ER stress. CHOP siRNA (from Ambion) transfection resulted in efficient CHOP inhibition in INS-1E cells exposed to CPA (Fig. 8A) or palmitate (Fig. 8B), respectively reducing CHOP protein expression levels by 75% and 60%, compared with non-transfected or negative (inactive)-siRNA-transfected cells. This resulted in a 50% reduction of CPA-induced apoptosis after 6 hours (Fig. 8C), and a lesser protection after 24 hours (trend to a 20% reduction, Fig. 8C). CHOP-depleted cells were also partially protected against palmitate-induced apoptosis after 14 hours (40% less apoptosis, Fig. 8D), but not after 24 hours, suggesting that CHOP inhibition delays but does not prevent palmitate-induced apoptosis (Fig. 8D). The knockdown and anti-apoptotic data obtained with another CHOP siRNA (Huang et al., 2007) were similar (data not shown).
Fig. 8.

CHOP knockdown delays CPA- and FFA-mediated β-cell apoptosis. INS-1E cells were transfected with siRNA against CHOP mRNA (si, gray bars), negative (inactive) siRNA (N, black bars) or empty liposomes (C, white bars) and exposed to CPA (positive control), 0.5 mM palmitate or vehicle (control) for 6–24 hours. (A,B) One representative CHOP western blot after 24-hour exposure and optical densitometry of CHOP protein expression from four independent experiments are shown. Results are expressed as CHOP protein normalized to β-actin protein (used as a loading control) and are means ± s.e.m. (C,D) Percentage of cells undergoing apoptosis after exposure to CPA for 6–24 hours (C) or palmitate for 14–24 hours (D). Results are means ± s.e.m. of four to seven independent experiments. *P<0.05, **P<0.01, ***P<0.001 vs control; #P<0.05 vs non-transfected cells.
Role of JNK in FFA-induced β-cell apoptosis
Both chemical and small-peptide inhibitory approaches were used to study the contribution of JNK in FFA-induced β-cell apoptosis. Oleate did not result in JNK activation, as measured using an AP-1 reporter construct, and JNK inhibitors did not alter oleate-induced apoptosis (data not shown). Both SP600125 and L-TAT-JNKi inhibited palmitate-induced JNK activation by 30–40% in INS-1E cells (Fig. 9A). This inhibition reduced palmitate-mediated apoptosis by around 20% (Fig. 9B).
Fig. 9.

Involvement of JNK in FFA-induced β-cell apoptosis. (A) INS-1E cells were co-transfected with the AP-1 reporter and the internal control pRL-CMV, encoding Renilla luciferase. After transfection and overnight recovery, the cells were exposed for 24 hours to control or palmitate (0.5 mM) at 11 mM glucose in the presence or absence of the JNK inhibitors SP600125 (25 μM, gray bars) or L-TAT-JNKi (1 μM, black bars), and assayed for firefly and Renilla luciferase activities. The results were normalized for Renilla luciferase activity and are means ± s.e.m. of four to seven independent experiments. (B) After 72 hours, the percentage of apoptotic cells was determined. n=8–14. *P<0.001 vs control; #P<0.01 vs similar condition without JNK inhibitor.
Glucose can sensitize INS-1E cells to ER-stress-mediated apoptosis
Misfolded proteins might trigger an ER stress response when insufficient chaperones are available in the ER to promote protein-chaperone interaction. We have previously shown that interferon-γ sensitizes β-cells to subsequent ER stress by decreasing ER-chaperone expression (Pirot et al., 2006). To evaluate whether a similar mechanism mediates the glucose potentiation of lipotoxicity in INS-1E cells, we examined whether a pretreatment with high concentrations of glucose sensitizes cells to apoptosis induced by the synthetic ER stressor CPA. CPA induced apoptosis in INS-1E cells in a dose-dependent manner (supplementary material Fig. S11A). At 28 mM glucose, the percentage of apoptotic INS-1E cells tripled as compared with apoptosis at 11 mM glucose. In line with the minor glucose-FFA potentiating effect in primary β-cells, 28 mM glucose only slightly increased CPA-induced β-cell apoptosis (supplementary material Fig. S11B), suggesting that glucose sensitization to ER stress is important in INS-1E cells but is not a major contributor to apoptosis in primary β-cells.
Discussion
We and others have previously shown that FFA induce ER stress and apoptosis in β-cell lines (Cnop et al., 2007a; Karaskov et al., 2006; Kharroubi et al., 2004; Laybutt et al., 2007), but the mechanisms involved have not been elucidated. The present detailed time-course study in INS-1E cells demonstrates that saturated and unsaturated FFA lead to differential ER stress signaling. The PERK pathway is markedly activated by palmitate, and much less so by oleate. The IRE1 pathway is activated by palmitate, but less by the combination of FFA or oleate alone. By contrast, all FFA, including the non-toxic combination of oleate and palmitate, induce to a similar extent BiP and XBP1t, two markers for ATF6 activation. We presently extended these observations to FACS-purified rat primary β-cells and human islets. The FFA-mediated ER stress response and apoptosis in primary β-cells were more marked in the presence of palmitate. Little or no increase was seen when combining FFA with high glucose concentrations in these cells, suggesting that ER stress in rat and human primary β-cells is primarily lipotoxic and not glucolipotoxic.
Quantitative and qualitative changes in dietary fatty-acid composition are rapidly reflected in circulating and cellular FFA. It is conceivable that the increased saturated fat intake typical of Western diets causes an in vivo ER stress response in pancreatic β-cells similar to the response observed in vitro in palmitate-treated β-cells. This might contribute to β-cell dysfunction and β-cell loss by apoptosis in individuals with a genetic predisposition for T2D (Kashyap et al., 2003). Recent reports using post-mortem material provide histological evidence of ER stress in islets from T2D patients, including increased ATF3 (Hartman et al., 2004), CHOP, BiP and DNAJC3 expression (Laybutt et al., 2007), nuclear localization of CHOP (Huang et al., 2007), and expansion of β-cell ER surface (Marchetti et al., 2007). ER stress signaling thus seems to be present in human β-cells in T2D and might be involved in the pathogenesis of the disease. ER stress in T2D can be triggered by islet amyloid polypeptide (Casas et al., 2007; Huang et al., 2007) and our present data identify FFA as a potential nutrient trigger of β-cell ER stress. High-fat feeding and obesity also cause ER stress in mouse liver and fat tissues (Nakatani et al., 2005; Ozcan et al., 2004), and this impairs insulin signaling in a JNK-dependent fashion. These effects are probably mediated by saturated fats (Wang et al., 2006; Wei et al., 2006). ER stress could thus be one of the molecular mechanisms underlying both insulin resistance and β-cell failure in T2D.
The non-metabolizable methyl fatty acids are non-toxic and induce little or no ER stress (present data). Thus, FFA need to be metabolically active to trigger ER stress and apoptosis. Inhibition of ceramide formation from palmitate has been shown to reduce FFA toxicity (Lupi et al., 2002). FFA serve as an important source of energy through their mitochondrial β-oxidation (Berne, 1975; Malaisse et al., 1983). Excess FFA are esterified into triglycerides in the ER, and FFA flux into triglycerides augments substantially with increased glucose availability (Briaud et al., 2001; Moffitt et al., 2005). It is unlikely that triglyceride synthesis per se activates the ER stress response, because oleate and the FFA combination lead to more triglyceride formation than does palmitate (Cnop et al., 2001; Moffitt et al., 2005) but is associated with a lesser ER stress response (present data). The lack of glucose potentiation of lipotoxic signaling also argues against a causal role of triglyceride synthesis in ER stress triggering. Palmitate and oleate cause, respectively, spindle- and droplet-shaped triglyceride accumulation, and both forms are present following exposure to a mixture of FFA (Cnop et al., 2001; Moffitt et al., 2005). It has been suggested that the physicochemical properties of tripalmitin cause palmitate toxicity (Moffitt et al., 2005), but the presence of this triglyceride in oleate+palmitate-exposed β-cells suggests that tripalmitin accumulation per se is not sufficient to trigger ER stress and apoptosis. High glucose levels greatly increase the demand on the β-cell ER in terms of synthesis of proteins, mostly proinsulin (Schuit et al., 1988). High glucose concentration has been shown to induce BiP expression in cultured rat islets (Elouil et al., 2007) and IRE1 activation in mouse islets (Lipson et al., 2006; Lipson et al., 2008). In INS-1E cells, 28 mM glucose decreased BiP expression and XBP1 splicing, and this was accompanied by increased FFA toxicity. The downregulation of such β-cell defense mechanisms has been shown to sensitize cells to ER stress (Pirot et al., 2006), and a clear sensitization to CPA was observed in INS-1E cells pretreated with high glucose concentrations (supplementary material Fig. S11). Our present data show that high glucose levels do not, however, significantly modulate lipotoxic ER stress in primary β-cells or human islets, suggesting that ER stress in these cells is mainly a consequence of FFA exposure and does not depend on the augmented functional demand posed by high levels of glucose.
Using two complementary methods, we demonstrate here that palmitate decreases ER Ca2+ stores in INS-1E cells. In a previous study, Karaskov et al. did not observe ER Ca2+ depletion using fura-2 measurements in INS-1 cells exposed to FFA for 6 hours (Karaskov et al., 2006). A possible explanation for the discrepancy is that those experiments were carried out in the absence of extracellular Ca2+ and in the presence of 10 mM glucose (Karaskov et al., 2006), compared with 1 mM Ca2+ and 1.1 mM glucose in our study. The absence of extracellular Ca2+ might deplete β-cell ER Ca2+ stores and intermediate glucose concentrations might reduce the effect of Ca2+-releasing agents (Herchuelz and Malaisse, 1980), decreasing the sensitivity of the method. ER Ca2+ depletion activates the ER stress response in the case of thapsigargin and CPA, which inhibit the SERCA pump. Overexpression of the Na/Ca exchanger, a major Ca2+-extruding mechanism in β-cells, also depletes ER Ca2+ stores with subsequent ER stress and caspase-12 activation in a rat β-cell line (Diaz-Horta et al., 2002). Cytokines also trigger ER stress in β-cells via nitric-oxide-mediated inhibition of SERCA (Cardozo et al., 2005). Interestingly, ER Ca2+ depletion by FFA was apparently not mediated through transcriptional inhibition of SERCA, as is the case for cytokines. Whether changes in SERCA protein level, or catalytic activity, are involved remain to be clarified. We also considered the possibility that ER Ca2+ depletion might enhance Ca2+ entry via store-operated (Smyth et al., 2006) or voltage-gated Ca2+ channels (Miura et al., 1997), thus raising cytosolic, and possibly mitochondrial, free Ca2+ levels (Rutter et al., 2006), to provide a cue for apoptosis (Choi et al., 2007). However, arguing against such a mechanism, both palmitate and oleate in fact tended to decrease resting cytosolic Ca2+ (Fig. 7). Disruption of ER Ca2+ homeostasis has previously been shown to hamper protein folding (Kuznetsov et al., 1992; Lodish and Kong, 1990) and trigger an ER stress response (Paschen, 2003). Accumulation of ganglioside sialoglycolipids in neurons (Tessitore et al., 2004) or cholesterol in macrophages (Feng et al., 2003) similarly induces ER stress by depleting ER Ca2+. Palmitate was previously shown to deplete ER Ca2+ in Chinese hamster ovary cells (Borradaile et al., 2006). The activation of the different branches of the ER stress response might depend on the magnitude and duration of ER Ca2+ depletion, both of which were greater with palmitate than with oleate or the FFA combination. Changes in ER lipid composition might also affect ER function by altering a putative lipid-droplet-mediated dislocation of misfolded proteins from the ER (Ploegh, 2007).
The ER stress response aims to restore ER homeostasis by attenuating protein translation, upregulating ER chaperones (thereby increasing the ER folding capacity) and degrading misfolded proteins (Eizirik et al., 2008). In cases in which the response fails to solve the ER stress, the apoptosis program is initiated. The induction of different branches of the ER stress response is signal and cell dependent (Eizirik et al., 2008), and the time course of the induction of a particular branch might determine cell fate (Lin et al., 2007). In human embryonic kidney cells, rapid (8 hours) attenuation of IRE1 signaling seems to be pro-apoptotic, because artificial maintenance of IRE1 activation delayed apoptosis (Lin et al., 2007). We observed a clear IRE1 activation by palmitate that persisted for 24 hours. Oleate caused minor, and the FFA combination caused intermediate, XBP1 splicing. IRE1 can recruit tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2), and activate JNK and downstream pro-apoptotic signaling (Urano et al., 2000). The IRE1-TRAF2 complex can also lead to NF-κB activation (Kaneko et al., 2003), which is known to be proapoptotic in β-cells (Cnop et al., 2005). The IRE1-TRAF2 association is also required for the activation of pro-caspase-12, which might contribute to execution of ER-stress-triggered apoptosis in rodent β-cells (Nakagawa et al., 2000; Yoneda et al., 2001). We have previously shown that FFA do not activate NF-κB in pancreatic β-cells (Cnop et al., 2005; Kharroubi et al., 2004). A clear activation of JNK and caspase-12 was, however, observed with palmitate (present data). The IRE1 activation by palmitate might at least in part contribute to β-cell apoptosis, because JNK inhibitors partially protected β-cells from palmitate.
Activation of the PERK pathway might be pro- or anti-apoptotic, depending on the source and extent of ER stress. The activation of PERK signaling by palmitate peaks at 12 hours and subsequently diminishes. This late decrease might be due to GADD34 induction, downstream of ATF4 and CHOP, and consequent protein-phosphatase-1-mediated dephosphorylation of eIF2α (Cnop et al., 2007a; Novoa et al., 2001). This negative-feedback mechanism alleviates repression of protein translation in the course of the ER stress response. Little PERK pathway activation is seen with oleate or the FFA mixture. Prolonged activation of the PERK branch, especially in the presence of FFA, leads to β-cell death (Cnop et al., 2007a), whereas mice with deletions of different components of the PERK pathway have progressive β-cell loss and diabetes (Harding et al., 2001; Scheuner et al., 2001; Zhang et al., 2002; Zhang et al., 2006). The PERK pathway induces apoptosis via ATF4 overexpression and consequent CHOP (Oyadomari and Mori, 2004; Zinszner et al., 1998) and ATF3 (Jiang et al., 2004; Ma et al., 2002) induction. Because of the marked ATF3 and CHOP induction by FFA, we examined their role in apoptosis. Whereas ATF3-deficient β-cells are partially protected against cytokine- and nitric-oxide-induced apoptosis (Hartman et al., 2004), no protection (or even aggravation of apoptosis) was observed against FFA using RNA interference or ATF3-knockout mice (present data). CHOP, by contrast, contributes to the initiation of palmitate-induced β-cell apoptosis. Several studies point to a pro-apoptotic effect of CHOP downstream of irremediable ER stress (Benavides et al., 2005; Oyadomari and Mori, 2004; Tajiri et al., 2006), but this effect might depend on the parallel expression of other components of the ER stress response (Williams and Lipkin, 2006). Most ER-stress-related pro-apoptotic signals ultimately lead to caspase activation (Rao et al., 2004). In addition to caspase-12, which has been proposed as a specific mediator of ER-stress-induced apoptosis in rodent cells, we observed activation of the effector caspases 3 and 7.
Toxic as well as non-toxic FFA activated ATF6 expression, leading to progressive increases in BiP and XBP1t that were maintained up to 48 hours. Hence, ATF6 signaling does not contribute to apoptosis; instead, it might be β-cell protective, because BiP overexpression protected clonal β-cells against palmitate (Laybutt et al., 2007).
In conclusion, saturated FFA deplete ER Ca2+ and induce a lipotoxic ER stress response in clonal β-cells, rat primary β-cells and human islets. JNK and CHOP in the IRE1 and PERK pathways contribute to the subsequent execution of β-cell apoptosis, whereas ATF6 activation might be anti-apoptotic. Intervention strategies in the former pathways should be developed to prevent lipotoxic β-cell loss, whereas promotion of ATF6 signaling might boost β-cell defense mechanisms in T2D.
Materials and Methods
Culture of primary FACS-purified rat β-cells, mouse islet cells and INS-1E cells
Male Wistar rats (Charles River Laboratories Belgium, Brussels, Belgium) were housed and used according to the guidelines of the Belgian Regulations for Animal Care. Rat islets were isolated by collagenase digestion followed by hand picking under a stereomicroscope. For β-cell isolation, islets were dispersed and β-cells purified by autofluorescence-activated cell sorting (FACS; FACStar, Becton Dickinson and Co., Sunnyvale, CA) (Pipeleers et al., 1985; Rasschaert et al., 2005). The preparations used in the present experiments contained 89±1% β-cells (n=19). Purified β-cells were pre-cultured overnight in Ham’s F-10 medium with 10 mM glucose, 2 mM glutamine, 50 μM 3-isobutyl-1-methylxanthine, 5% heat-inactivated fetal bovine serum (FBS), 0.5% charcoal-absorbed bovine serum albumin (BSA, Albumin Fraktion V; Boehringer, Indianapolis, IN, USA), 50 U/ml penicillin and 50 μg/ml streptomycin (Ling et al., 1994). During FFA exposure, cells were cultured in the same medium with 1% BSA and no serum, at a glucose concentration of 6.1, 10 or 28 mM.
Mouse islets were isolated from C57BL/6N (wt, Charles River) and ATF3-deficient mice (Hartman et al., 2004), as previously described (Pavlovic et al., 1999). The hand-picked islets were then dispersed with 5 mg/ml dispase (Roche) and dispersed cells were cultured in medium as used for primary rat β-cells.
The rat insulin-producing INS-1E cell line (a kind gift from C. Wollheim, Centre Medical Universitaire, Geneva, Switzerland) was cultured in RPMI 1640 (with GlutaMAX-I) containing 5% FBS, 10 mM HEPES, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin and 50 μM 2-mercaptoethanol (Asfari et al., 1992). The INS-1E cells (passages 55–75) were plated and cultured for 48 hours prior to their FFA exposure in RPMI 1640 supplemented with 1% BSA, 1% FCS, 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM 2-mercaptoethanol and a glucose concentration of 5.6, 11 or 28 mM. Oleate and palmitate (sodium salt, Sigma Aldrich, St Louis, MO, USA) were dissolved in 90% ethanol, heated to 60°C, and used in a 1:100 dilution (at a final concentration of 0.5 mM) (Cnop et al., 2001; Cnop et al., 2002; Kharroubi et al., 2004). The molar ratio of FFA:BSA in the culture medium was 3.4, corresponding to unbound oleate and palmitate concentrations of 47 and 27 nM, respectively, as previously calculated (Cnop et al., 2001). The FFA mixture contains 0.25 mM of each. The control condition contained a similar dilution of ethanol. Methyloleate and methylpalmitate (Sigma) were dissolved in 90% ethanol and used at 0.5 mM. The SERCA blockers CPA and thapsigargin (both from Sigma) were dissolved in DMSO and used at concentrations of, respectively, 25 and 1 μM (Cardozo et al., 2005), unless otherwise indicated. The control condition contained a similar dilution of DMSO. The chemical JNK inhibitor SP600125 (Merck-Calbiochem) was dissolved in DMSO, and the peptide JNK inhibitor L-TAT-JNKi (a kind gift of C. Bonny and M. Mathieu; XigenPharma, Lausanne, Switzerland) was dissolved in culture medium at concentrations previously used (Bonny et al., 2001; Ozcan et al., 2004).
Human islets
Islets were isolated from seven organ donors (age 62±6 years; body mass index 23.9±1.3 kg/m2) in Pisa, Italy, with the approval of the local Ethics Committee. Causes of death were trauma (n=3) and cardiovascular events (n=4). Islets were isolated by enzymatic digestion and density-gradient purification, as previously described (Lupi et al., 2002), placed in M199 culture medium containing 5.5 mM glucose and cultured in a CO2 incubator. Their functional status was determined using glucose-stimulated insulin release and was 2.8±0.4 (expressed as stimulation index). The human islets were shipped to Brussels for study within 1–5 days of isolation. After overnight recovery in Ham’s F-10 medium containing 6.1 mM glucose, 2 mM GlutaMAX, 50 μM 3-isobutyl-1-methylxanthine, 1% BSA, 50 U/ml penicillin, 50 μg/ml streptomycin and 10% FCS, islets were exposed to FFA in the same medium without FCS for 2–6 days. In these experiments, 6.1 mM glucose was selected as the standard glucose concentration, based on previous experiments indicating better preservation of human islet function at this glucose level (Eizirik et al., 1992). The percentage of β-cells, assessed in three dispersed-islet preparations following staining with mouse monoclonal anti-insulin antibody (1:1000, Sigma) and donkey anti-mouse IgG rhodamine (1:200, Jackson ImmunoResearch Europe, Soham, Cambridgeshire, UK), was 60±6%.
Assessment of β-cell viability
The percentage of viable, apoptotic and necrotic cells was determined following incubation for 15 minutes with the DNA-binding dyes propidium iodide (PI, 5 μg/ml, Sigma) and Hoechst 33342 (HO, 5 μg/ml, Sigma) (Hoorens et al., 1996). The cells were examined by inverted fluorescence microscopy (excitation at 365 nm for HO and at 546 nm for PI). Viable cells were identified by their intact nuclei with blue fluorescence (HO), necrotic cells by their intact nuclei with red fluorescence (PI) and apoptotic cells by their fragmented nuclei, exhibiting either a blue (HO; early apoptosis) or red (PI; late apoptosis) fluorescence (Hoorens et al., 1996). A minimum of 500 cells was counted in each experimental condition. Viability was evaluated by two independent observers, one of them being unaware of sample identity. The agreement between findings obtained by the two observers was >90%. Data are expressed as percent apoptosis or as apoptotic index (Cnop et al., 2001) when evaluating anti-apoptotic compounds. For the semiquantitative evaluation of the viability of human islets, the percentage of dead cells was estimated following HO-PI staining by two to four observers.
Western blot experiments
Western blot analysis of protein expression and phosphorylation in FFA-treated INS-1E cells was performed as previously described (Cnop et al., 2007a), using antibodies against phospho-eIF2α (1:1000), phospho-PERK (1:1000), β-actin (1:2000, all from Cell Signaling, Beverly, MA, USA), eIF2α (1:500) or ATF3 (1:200, the latter two from Santa Cruz Biotechnology, Santa Cruz, CA) as primary antibodies, and a horseradish-peroxidase-labeled donkey anti-rabbit antibody (1:3000 dilution, Santa Cruz Biotechnology) as secondary antibody. The protein-specific signals were detected using chemiluminescence Supersignal (Pierce, Rockford, IL) and quantified using Aida1D analysis software (Fujifilm).
Analysis by real-time PCR of mRNA expression of ER stress markers
Poly(A)+ RNA was isolated from INS-1E cells, rat primary β-cells and human islets, and reverse transcribed (Chen et al., 2001). The real-time PCR amplification reaction was done in a volume of 20 μl containing 2.5–5.0 mM MgCl2, 0.5 μM forward and reverse primers, 10 μl SYBR Green PCR master mix (Qiagen, Hilden, Germany), and 2 μl cDNA. Standards for each gene were prepared using appropriate primers in a conventional PCR reaction and quantification of the PCR product was done fluorometrically using SYBR Green. The samples were assayed on a LightCycler instrument (Roche Diagnostics, Mannheim, Germany) and their concentration was calculated as copies per μl using the standard curve (Overbergh et al., 1999). The expression level of the gene of interest was normalized to the highest value and corrected for the expression of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH for rat cells) or β-actin (for human islets). The rat ATF3 primers were F 5′-CTCCTGGGTCACTGGTGTTT-3′ and R 5′-AGTGCACAGGAAGCCAGTTT-3′ (yielding a PCR fragment of 568 bp) for standard PCR, and F 5′-GCTGGAGTCAGTCACCATCA-3′ and R 5′-ACACTTGGCAGCAGCAATTT-3′ (PCR fragment of 127 bp) for real-time PCR. The human ATF3 primers were F 5′-ATCTCCTTCACCGTGGCTAC-3′ and R 5′-AGGACCTGCCATCATACTGC-3′ (PCR fragment of 357 bp) for standard PCR, and F 5′-GCTGTC ACCA - CGTGCAGTAT-3′ and R 5′-TTTGTGTTAACGCTGGGAGA-3′ (PCR fragment of 92 bp) for real time PCR. The other primers have been reported elsewhere (Cnop et al., 2007a; Kharroubi et al., 2004; Marchetti et al., 2007; Pirot et al., 2006; Welsh et al., 2005).
Measurements of ER Ca2+ concentration
Measurement of the cytoplasmic Ca2+ concentration [Ca2+]i was performed using fura-2 acetoxymethyl ester as previously described (Cardozo et al., 2005; Diaz-Horta et al., 2002). Where indicated, 1 μM thapsigargin was added to induce ER Ca2+ release.
For direct ER Ca2+ measurements, INS-1E cells were seeded onto glass coverslips and, at 50% confluency, infected with an adenovirus encoding an ER-lumen-targeted aequorin, a Ca2+ sensitive bioluminescent protein (Montero et al., 1995), at a multiplicity of infection of 30 for 24 hours. Cells were treated with FFA and depleted of Ca2+ by incubation with ionomycin (10 μM), monensin (10 μM) and CPA (10 μM) in modified Krebs-Ringer bicarbonate buffer (KRBB, 140 mM NaCl, 3.5 mM KCl, 0.5 mM NaH2PO4, 0.5 mM MgSO4, 3 mM glucose, 10 mM HEPES and 2 mM NaHCO3, pH 7.4) supplemented with 1 mM EGTA, for 5 minutes at 4°C. Aequorin was reconstituted with its cofactor coelenterazine n (5 μM) for 2 hours at 4°C in KRBB with 1 mM EGTA. After this, cells were perifused with KRBB with 1.5 mM Ca2+ at a flow rate of 2 ml/minute in a thermostatted chamber (37°C) in a photomultiplier tube (ThornEMI). At the end of all experiments, cells were lysed in a hypotonic Ca2+-rich solution (100 μM digitonin and 10 mM Ca2+ in H2O) to discharge the remaining aequorin pool for calibration of the aequorin signal (Mitchell et al., 2001).
Luciferase reporter studies
To evaluate the functionality of the FFA-induced transcriptional activation of the IRE1 and ATF6 pathways, we used a UPRE luciferase reporter construct kindly provided by Ron Prywes (Columbia University, New York, NY). This element functions as a binding site for both XBP1 (Yoshida et al., 2001) and ATF6 (Wang et al., 2000). To assess JNK activation, we used a luciferase reporter kindly provided by P. Johnson (University of Southampton, Southampton General Hospital, Southampton, UK) and Z. Dong (University of Minnesota, Austin, MN) containing four AP1 response elements (Dong et al., 1994). INS-1E cells were co-transfected with the luciferase reporter and pRL-CMV (Promega, Madison, WI) as internal control using Lipofectamine 2000 (Invitrogen, Baesley, Scotland) (Darville and Eizirik, 1998). Luciferase activities were assayed with the dual-luciferase-reporter assay system (Promega) as previously described (Darville and Eizirik, 1998; Kutlu et al., 2003).
RNA interference
siRNA against CHOP was purchased from Ambion (Austin, TX, USA) or generated according to Huang et al. (Huang et al., 2007). Negative controls of 21-nucleotide duplex RNA with no known sequence homology were purchased from Ambion or generated (Huang et al., 2007). Because the negative siRNAs gave similar results, apoptosis data were pooled. siRNAs targeting two different regions of the ATF3 mRNA were generated using BLOCK-iT RNAi Designer Invitrogen software (#1 5′-AUCUCCAGAGGUCUGUUGUUGAUGG-3′ and #2 5′-UGGAGAGUGUGAAUGCCGAACUGAA-3′). A scrambled sequence of siRNA #1 (5′-AUCG - ACUGACGGAUCUUUGUUGUGG-3′) was generated as a negative control (Invitrogen, Paisley, GB). Transfection of siRNA was done using the lipid carrier Dharmafect (Dharmacon, Chicago, IL). Lipid-RNA complexes were formed in Optimem1 in a proportion of 0.8 and 1.25 μl of Dharmafect to 150 nM of siRNA for INS-1E and primary β-cells, respectively, at room temperature for 20 minutes. The complex was added to cells in antibiotic-free medium at a final concentration of 30 nM siRNA for overnight transfection. The transfection efficiency was >90% as measured using an FITC-conjugated siRNA (siGLO, Dharmacon). Afterwards, cells were cultured for a 24-hour recovery period and subsequently exposed to ER stressors for the indicated time.
Caspase activation
Caspase-3 and caspase-7 activation was measured with the Caspase-Glo 3/7 assay (Promega, Madison, WI, USA) following the manufacturer’s instructions. Briefly, FACS-purified rat β-cells (5000–8000 cells/condition) were cultured in white-walled 96-well plates. At the end of the treatment, 100 μl of Caspase-Glo 3/7 Reagent was added to the wells containing the β-cells in 100 μl culture medium. The plates were placed on a shaker (300–500 rpm) for 30 seconds and incubated at room temperature for 2 hours. Luminescence was measured in a plate-reading luminometer.
Caspase-12 activation was measured with the CaspGlow Caspase-12 staining kit (Biovision, Mountain View, CA) according to the supplier’s instructions. Briefly, cells were incubated with FITC-ATAD-FMK for 1 hour at 37°C, thrice washed, incubated for 15 minutes with HO and examined by inverted fluorescence microscopy. The number of caspase-12-positive cells was counted and expressed as a percentage of HO-positive cells.
Statistical analysis
Data are presented as means ± s.e.m. Comparisons were performed by two-sided paired or ratio t-test or by ANOVA followed by paired t-test with the Bonferroni correction for multiple comparisons. A P value <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Linda Hendershot for data discussions, Zeynep Dogusan for advice on the western blotting, Ilham Kharroubi for help with the initial viability experiments, and Marie-Anne Neef, Gilbert Vandenbroeck, Maryse Urbain, Jeanine Schoonheydt, Nasira El Amrite and Anissa Iabkriman for expert technical assistance. This work was supported by the European Union [Integrated Project EuroDia LSHM-CT-2006-518153 and STREP Savebeta contract 036903 in the Framework Programme 6 (FP6) of the European Community]; the Belgian Program on Interuniversity Poles of Attraction initiated by the Belgian State (IUAP P6/40); a Belgian Lipid Club research fellowship; by grants from the Octave Dupont Foundation (Royal Academy of Science of Belgium), the Fonds National de la Recherche Scientifique (FNRS), Fonds de la Recherche Scientifique Médicale (FRSM) and Actions de Recherche Concertées de la Communauté Française, Belgium (M.C. and D.L.E.). A.K.C. is a Research Associate of the FNRS, Belgium. G.A.R. was supported by Wellcome Trust Programme Grants 067081/Z/02/Z, 081958/Z/07/Z, MRC (UK) Research Grant G0401641, NIH RO1 Project Grant RO1 DK071962-01, and an Imperial College Research Studentship (for E.B.).
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/121/14/2308/DC1
References
- Asfari M, Janjic D, Meda P, Li G, Halban PA, Wollheim CB. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology. 1992;130:167–178. doi: 10.1210/endo.130.1.1370150. [DOI] [PubMed] [Google Scholar]
- Benavides A, Pastor D, Santos P, Tranque P, Calvo S. CHOP plays a pivotal role in the astrocyte death induced by oxygen and glucose deprivation. Glia. 2005;52:261–275. doi: 10.1002/glia.20242. [DOI] [PubMed] [Google Scholar]
- Berne C. The metabolism of lipids in mouse pancreatic islets. The oxidation of fatty acids and ketone bodies. Biochem J. 1975;152:661–666. doi: 10.1042/bj1520661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF. Cell-permeable peptide inhibitors of JNK: novel blockers of β-cell death. Diabetes. 2001;50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res. 2006;47:2726–2737. doi: 10.1194/jlr.M600299-JLR200. [DOI] [PubMed] [Google Scholar]
- Briaud I, Harmon JS, Kelpe CL, Segu VB, Poitout V. Lipotoxicity of the pancreatic β-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes. 2001;50:315–321. doi: 10.2337/diabetes.50.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- Cardozo AK, Ortis F, Storling J, Feng YM, Rasschaert J, Tonnesen M, Van Eylen F, Mandrup-Poulsen T, Herchuelz A, Eizirik DL. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes. 2005;54:452–461. doi: 10.2337/diabetes.54.2.452. [DOI] [PubMed] [Google Scholar]
- Casas S, Gomis R, Gribble FM, Altirriba J, Knuutila S, Novials A. Impairment of the ubiquitin-proteasome pathway is a downstream endoplasmic reticulum stress response induced by extracellular human islet amyloid polypeptide and contributes to pancreatic β-cell apoptosis. Diabetes. 2007;56:2284–2294. doi: 10.2337/db07-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MC, Proost P, Gysemans C, Mathieu C, Eizirik DL. Monocyte chemoattractant protein-1 is expressed in pancreatic islets from prediabetic NOD mice and in interleukin-1β-exposed human and rat islet cells. Diabetologia. 2001;44:325–332. doi: 10.1007/s001250051622. [DOI] [PubMed] [Google Scholar]
- Choi SE, Kim HE, Shin HC, Jang HJ, Lee KW, Kim Y, Kang SS, Chun J, Kang Y. Involvement of Ca2+-mediated apoptotic signals in palmitate-induced MIN6N8a β cell death. Mol Cell Endocrinol. 2007;272:50–62. doi: 10.1016/j.mce.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, Cooper GJ, Holman RR, Turner RC. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988;9:151–159. [PubMed] [Google Scholar]
- Cnop M, Hannaert JC, Hoorens A, Eizirik DL, Pipeleers DG. Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes. 2001;50:1771–1777. doi: 10.2337/diabetes.50.8.1771. [DOI] [PubMed] [Google Scholar]
- Cnop M, Hannaert JC, Pipeleers DG. Troglitazone does not protect rat pancreatic β cells against free fatty acid-induced cytotoxicity. Biochem Pharmacol. 2002;63:1281–1285. doi: 10.1016/s0006-2952(02)00860-2. [DOI] [PubMed] [Google Scholar]
- Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(Suppl 2):S97–S107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- Cnop M, Ladriere L, Hekerman P, Ortis F, Cardozo AK, Dogusan Z, Flamez D, Boyce M, Yuan J, Eizirik DL. Selective inhibition of eukaryotic translation initiation factor 2α dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic β-cell dysfunction and apoptosis. J Biol Chem. 2007a;282:3989–3997. doi: 10.1074/jbc.M607627200. [DOI] [PubMed] [Google Scholar]
- Cnop M, Vidal J, Hull RL, Utzschneider KM, Carr DB, Schraw T, Scherer PE, Boyko EJ, Fujimoto WY, Kahn SE. Progressive loss of β-cell function leads to worsening glucose tolerance in first-degree relatives of subjects with type 2 diabetes. Diabetes Care. 2007b;30:677–682. doi: 10.2337/dc06-1834. [DOI] [PubMed] [Google Scholar]
- Darville MI, Eizirik DL. Regulation by cytokines of the inducible nitric oxide synthase promoter in insulin-producing cells. Diabetologia. 1998;41:1101–1108. doi: 10.1007/s001250051036. [DOI] [PubMed] [Google Scholar]
- Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, Torri S, Pollera M, Boggi U, Mosca F, et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes. 2005;54:727–735. doi: 10.2337/diabetes.54.3.727. [DOI] [PubMed] [Google Scholar]
- Diaz-Horta O, Kamagate A, Herchuelz A, Van Eylen F. Na/Ca exchanger overexpression induces endoplasmic reticulum-related apoptosis and caspase-12 activation in insulin-releasing BRIN-BD11 cells. Diabetes. 2002;51:1815–1824. doi: 10.2337/diabetes.51.6.1815. [DOI] [PubMed] [Google Scholar]
- Dong Z, Birrer MJ, Watts RG, Matrisian LM, Colburn NH. Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells. Proc Natl Acad Sci USA. 1994;91:609–613. doi: 10.1073/pnas.91.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizirik DL, Korbutt GS, Hellerstrom C. Prolonged exposure of human pancreatic islets to high glucose concentrations in vitro impairs the β-cell function. J Clin Invest. 1992;90:1263–1268. doi: 10.1172/JCI115989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29:42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- El-Assaad W, Buteau J, Peyot ML, Nolan C, Roduit R, Hardy S, Joly E, Dbaibo G, Rosenberg L, Prentki M. Saturated fatty acids synergize with elevated glucose to cause pancreatic β-cell death. Endocrinology. 2003;144:4154–4163. doi: 10.1210/en.2003-0410. [DOI] [PubMed] [Google Scholar]
- Elouil H, Bensellam M, Guiot Y, Vander Mierde D, Pascal SM, Schuit FC, Jonas JC. Acute nutrient regulation of the unfolded protein response and integrated stress response in cultured rat pancreatic islets. Diabetologia. 2007;50:1442–1452. doi: 10.1007/s00125-007-0674-4. [DOI] [PubMed] [Google Scholar]
- Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- Festa A, Williams K, D’Agostino R, Jr, Wagenknecht LE, Haffner SM. The natural course of β-cell function in nondiabetic and diabetic individuals: the Insulin Resistance Atherosclerosis Study. Diabetes. 2006;55:1114–1120. doi: 10.2337/diabetes.55.04.06.db05-1100. [DOI] [PubMed] [Google Scholar]
- Fornace AJ, Jr, Nebert DW, Hollander MC, Luethy JD, Papathanasiou M, Fargnoli J, Holbrook NJ. Mammalian genes coordinately regulated by growth arrest signals and DNA-damaging agents. Mol Cell Biol. 1989;9:4196–4203. doi: 10.1128/mcb.9.10.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk–/– mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Hartman MG, Lu D, Kim ML, Kociba GJ, Shukri T, Buteau J, Wang X, Frankel WL, Guttridge D, Prentki M, et al. Role for activating transcription factor 3 in stress-induced β-cell apoptosis. Mol Cell Biol. 2004;24:5721–5732. doi: 10.1128/MCB.24.13.5721-5732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herchuelz A, Malaisse WJ. Regulation of calcium fluxes in rat pancreatic islets: dissimilar effects of glucose and of sodium ion accumulation. J Physiol. 1980;302:263–280. doi: 10.1113/jphysiol.1980.sp013241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoorens A, Van de Casteele M, Kloppel G, Pipeleers D. Glucose promotes survival of rat pancreatic β cells by activating synthesis of proteins which suppress a constitutive apoptotic program. J Clin Invest. 1996;98:1568–1574. doi: 10.1172/JCI118950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CJ, Lin CY, Haataja L, Gurlo T, Butler AE, Rizza RA, Butler PC. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated β-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes. 2007;56:2016–2027. doi: 10.2337/db07-0197. [DOI] [PubMed] [Google Scholar]
- Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol. 2004;24:1365–1377. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Niinuma Y, Nomura Y. Activation signal of nuclear factor-κB in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biol Pharm Bull. 2003;26:931–935. doi: 10.1248/bpb.26.931. [DOI] [PubMed] [Google Scholar]
- Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic β-cell apoptosis. Endocrinology. 2006;147:3398–3407. doi: 10.1210/en.2005-1494. [DOI] [PubMed] [Google Scholar]
- Kashyap S, Belfort R, Gastaldelli A, Pratipanawatr T, Berria R, Pratipanawatr W, Bajaj M, Mandarino L, DeFronzo R, Cusi K. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes. 2003;52:2461–2474. doi: 10.2337/diabetes.52.10.2461. [DOI] [PubMed] [Google Scholar]
- Kharroubi I, Ladriere L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Free fatty acids and cytokines induce pancreatic β-cell apoptosis by different mechanisms: role of nuclear factor-κB and endoplasmic reticulum stress. Endocrinology. 2004;145:5087–5096. doi: 10.1210/en.2004-0478. [DOI] [PubMed] [Google Scholar]
- Klöppel G, Löhr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res. 1985;4:110–125. doi: 10.1159/000156969. [DOI] [PubMed] [Google Scholar]
- Kutlu B, Darville MI, Cardozo AK, Eizirik DL. Molecular regulation of monocyte chemoattractant protein-1 expression in pancreatic β-cells. Diabetes. 2003;52:348–355. doi: 10.2337/diabetes.52.2.348. [DOI] [PubMed] [Google Scholar]
- Kuznetsov G, Brostrom MA, Brostrom CO. Demonstration of a calcium requirement for secretory protein processing and export. Differential effects of calcium and dithiothreitol. J Biol Chem. 1992;267:3932–3939. [PubMed] [Google Scholar]
- Larsson H, Ahren B. Glucose intolerance is predicted by low insulin secretion and high glucagon secretion: outcome of a prospective study in postmenopausal Caucasian women. Diabetologia. 2000;43:194–202. doi: 10.1007/s001250050029. [DOI] [PubMed] [Google Scholar]
- Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, Biden TJ. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–763. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005;24:4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Z, Hannaert JC, Pipeleers D. Effect of nutrients, hormones and serum on survival of rat islet beta cells in culture. Diabetologia. 1994;37:15–21. doi: 10.1007/BF00428772. [DOI] [PubMed] [Google Scholar]
- Lipson KL, Fonseca SG, Ishigaki S, Nguyen LX, Foss E, Bortell R, Rossini AA, Urano F. Regulation of insulin biosynthesis in pancreatic β cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006;4:245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Lipson KL, Ghosh R, Urano F. The role of IRE1α in the degradation of insulin mRNA in pancreatic β-cells. PLoS ONE. 2008;3:e1648. doi: 10.1371/journal.pone.0001648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish HF, Kong N. Perturbation of cellular calcium blocks exit of secretory proteins from the rough endoplasmic reticulum. J Biol Chem. 1990;265:10893–10899. [PubMed] [Google Scholar]
- Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, Patane G, Boggi U, Piro S, Anello M, et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that β-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes. 2002;51:1437–1442. doi: 10.2337/diabetes.51.5.1437. [DOI] [PubMed] [Google Scholar]
- Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY. Distinct effects of saturated and monounsaturated fatty acids on β-cell turnover and function. Diabetes. 2001;50:69–76. doi: 10.2337/diabetes.50.1.69. [DOI] [PubMed] [Google Scholar]
- Malaisse WJ, Best L, Kawazu S, Malaisse-Lagae F, Sener A. The stimulus-secretion coupling of glucose-induced insulin release: fuel metabolism in islets deprived of exogenous nutrient. Arch Biochem Biophys. 1983;224:102–110. doi: 10.1016/0003-9861(83)90193-5. [DOI] [PubMed] [Google Scholar]
- Marchetti P, Del Guerra S, Marselli L, Lupi R, Masini M, Pollera M, Bugliani M, Boggi U, Vistoli F, Mosca F, et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J Clin Endocrinol Metab. 2004;89:5535–5541. doi: 10.1210/jc.2004-0150. [DOI] [PubMed] [Google Scholar]
- Marchetti P, Bugliani M, Lupi R, Marselli L, Masini M, Boggi U, Filipponi F, Weir GC, Eizirik DL, Cnop M. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia. 2007;50:2486–2494. doi: 10.1007/s00125-007-0816-8. [DOI] [PubMed] [Google Scholar]
- Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- Matthews DR, Cull CA, Stratton IM, Holman RR, Turner RC. UKPDS 26, Sulphonylurea failure in non-insulin-dependent diabetic patients over six years. UK Prospective Diabetes Study (UKPDS) Group. Diabet Med. 1998;15:297–303. doi: 10.1002/(SICI)1096-9136(199804)15:4<297::AID-DIA572>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Mitchell KJ, Pinton P, Varadi A, Tacchetti C, Ainscow EK, Pozzan T, Rizzuto R, Rutter GA. Dense core secretory vesicles revealed as a dynamic Ca2+ store in neuroendocrine cells with a vesicle-associated membrane protein aequorin chimaera. J Cell Biol. 2001;155:41–51. doi: 10.1083/jcb.200103145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura Y, Henquin JC, Gilon P. Emptying of intracellular Ca2+ stores stimulates Ca2+ entry in mouse pancreatic β-cells by both direct and indirect mechanisms. J Physiol. 1997;503:387–398. doi: 10.1111/j.1469-7793.1997.387bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JH, Fielding BA, Evershed R, Berstan R, Currie JM, Clark A. Adverse physicochemical properties of tripalmitin in beta cells lead to morphological changes and lipotoxicity in vitro. Diabetologia. 2005;48:1819–1829. doi: 10.1007/s00125-005-1861-9. [DOI] [PubMed] [Google Scholar]
- Montero M, Brini M, Marsault R, Alvarez J, Sitia R, Pozzan T, Rizzuto R. Monitoring dynamic changes in free Ca2+ concentration in the endoplasmic reticulum of intact cells. EMBO J. 1995;14:5467–5475. doi: 10.1002/j.1460-2075.1995.tb00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, Ozawa K, Ogawa S, Hori M, Yamasaki Y, et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem. 2005;280:847–851. doi: 10.1074/jbc.M411860200. [DOI] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbergh L, Valckx D, Waer M, Mathieu C. Quantification of murine cytokine mRNAs using real time quantitative reverse transcriptase PCR. Cytokine. 1999;11:305–312. doi: 10.1006/cyto.1998.0426. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Takeda K, Takiguchi M, Gotoh T, Matsumoto M, Wada I, Akira S, Araki E, Mori M. Nitric oxide-induced apoptosis in pancreatic β cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci USA. 2001;98:10845–10850. doi: 10.1073/pnas.191207498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Parker SM, Moore PC, Johnson LM, Poitout V. Palmitate potentiation of glucose-induced insulin release: a study using 2-bromopalmitate. Metabolism. 2003;52:1367–1371. doi: 10.1016/s0026-0495(03)00279-8. [DOI] [PubMed] [Google Scholar]
- Paschen W. Endoplasmic reticulum: a primary target in various acute disorders and degenerative diseases of the brain. Cell Calcium. 2003;34:365–383. doi: 10.1016/s0143-4160(03)00139-8. [DOI] [PubMed] [Google Scholar]
- Pavlovic D, Chen MC, Gysemans CA, Mathieu C, Eizirik DL. The role of interferon regulatory factor-1 in cytokine-induced mRNA expression and cell death in murine pancreatic β-cells. Eur Cytokine Netw. 1999;10:403–412. [PubMed] [Google Scholar]
- Pipeleers DG, in’t Veld PA, Van de Winkel M, Maes E, Schuit FC, Gepts W. A new in vitro model for the study of pancreatic A and B cells. Endocrinology. 1985;117:806–816. doi: 10.1210/endo-117-3-806. [DOI] [PubMed] [Google Scholar]
- Pirot P, Eizirik DL, Cardozo AK. Interferon-γ potentiates endoplasmic reticulum stress-induced death by reducing pancreatic beta cell defence mechanisms. Diabetologia. 2006;49:1229–1236. doi: 10.1007/s00125-006-0214-7. [DOI] [PubMed] [Google Scholar]
- Pirot P, Ortis F, Cnop M, Ma Y, Hendershot LM, Eizirik DL, Cardozo AK. Transcriptional regulation of the endoplasmic reticulum stress gene chop in pancreatic insulin-producing cells. Diabetes. 2007;56:1069–1077. doi: 10.2337/db06-1253. [DOI] [PubMed] [Google Scholar]
- Ploegh HL. A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature. 2007;448:435–438. doi: 10.1038/nature06004. [DOI] [PubMed] [Google Scholar]
- Porte D, Jr, Kahn SE. β-Cell dysfunction and failure in type 2 diabetes: potential mechanisms. Diabetes. 2001;50(Suppl 1):S160–S163. doi: 10.2337/diabetes.50.2007.s160. [DOI] [PubMed] [Google Scholar]
- Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- Rasschaert J, Ladriere L, Urbain M, Dogusan Z, Katabua B, Sato S, Akira S, Gysemans C, Mathieu C, Eizirik DL. Toll-like receptor 3 and STAT-1 contribute to double-stranded RNA + interferon-γ-induced apoptosis in primary pancreatic β-cells. J Biol Chem. 2005;280:33984–33991. doi: 10.1074/jbc.M502213200. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter GA, Tsuboi T, Ravier MA. Ca2+ microdomains and the control of insulin secretion. Cell Calcium. 2006;40:539–551. doi: 10.1016/j.ceca.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. Reduced β-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia. 2002;45:85–96. doi: 10.1007/s125-002-8248-z. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Schuit FC, In’t Veld PA, Pipeleers DG. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc Natl Acad Sci USA. 1988;85:3865–3869. doi: 10.1073/pnas.85.11.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth JT, Dehaven WI, Jones BF, Mercer JC, Trebak M, Vazquez G, Putney JW., Jr Emerging perspectives in store-operated Ca2+ entry: roles of Orai, Stim and TRP. Biochim Biophys Acta. 2006;1763:1147–1160. doi: 10.1016/j.bbamcr.2006.08.050. [DOI] [PubMed] [Google Scholar]
- Tajiri S, Yano S, Morioka M, Kuratsu J, Mori M, Gotoh T. CHOP is involved in neuronal apoptosis induced by neurotrophic factor deprivation. FEBS Lett. 2006;580:3462–3468. doi: 10.1016/j.febslet.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Tessitore A, del P, Martin M, Sano R, Ma Y, Mann L, Ingrassia A, Laywell ED, Steindler DA, Hendershot LM, d’Azzo A. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol Cell. 2004;15:753–766. doi: 10.1016/j.molcel.2004.08.029. [DOI] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Van Lommel L, Janssens K, Quintens R, Tsukamoto K, Vander Mierde D, Lemaire K, Denef C, Jonas JC, Martens G, Pipeleers D, et al. Probe-independent and direct quantification of insulin mRNA and growth hormone mRNA in enriched cell preparations. Diabetes. 2006;55:3214–3220. doi: 10.2337/db06-0774. [DOI] [PubMed] [Google Scholar]
- Wang D, Wei Y, Pagliassotti MJ. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 2006;147:943–951. doi: 10.1210/en.2005-0570. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem. 2000;275:27013–27020. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275–E281. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- Welsh N, Cnop M, Kharroubi I, Bugliani M, Lupi R, Marchetti P, Eizirik DL. Is there a role for locally produced interleukin-1 in the deleterious effects of high glucose or the type 2 diabetes milieu to human pancreatic islets? Diabetes. 2005;54:3238–3244. doi: 10.2337/diabetes.54.11.3238. [DOI] [PubMed] [Google Scholar]
- Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest. 1999;104:787–794. doi: 10.1172/JCI7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BL, Lipkin WI. Endoplasmic reticulum stress and neurodegeneration in rats neonatally infected with borna disease virus. J Virol. 2006;80:8613–8626. doi: 10.1128/JVI.00836-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang AH, Wang C, Peters RK, Trigo E, Kjos SL, Buchanan TA. Coordinate changes in plasma glucose and pancreatic β-cell function in Latino women at high risk for type 2 diabetes. Diabetes. 2006;55:1074–1079. doi: 10.2337/diabetes.55.04.06.db05-1109. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem. 2001;276:13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, Yoo SJ, Kang MI, Cha BY, Lee KW, et al. Selective β-cell loss and α-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab. 2003;88:2300–2308. doi: 10.1210/jc.2002-020735. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Yusta B, Baggio LL, Estall JL, Koehler JA, Holland DP, Li H, Pipeleers D, Ling Z, Drucker DJ. GLP-1 receptor activation improves β cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006;4:391–406. doi: 10.1016/j.cmet.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. Protein folding in the endoplasmic reticulum and the unfolded protein response. Handb Exp Pharmacol. 2006;2006:69–91. doi: 10.1007/3-540-29717-0_3. [DOI] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2α kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic β cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 2006;4:491–497. doi: 10.1016/j.cmet.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.