

Abstract
Prostate cancer is the most common malignancy and the second leading cause of cancer-related deaths in men. One common treatment is androgen-deprivation therapy, which reduces symptoms in most patients. However, over time, patients develop tumors that are androgen-independent and ultimately fatal. The mechanisms that cause this transition remain largely unknown, and as a result, there are no effective treatments against androgen-independent prostate cancer. As a model platform, we used the LNCaP cell line and its androgen-independent derivative, LNCaP-SF. Utilizing stable isotope labeling with amino acids in cell culture coupled to mass spectrometry, we assessed the differential global protein expression of the two cell lines. Our proteomic analysis resulted in the quantification of 3355 proteins. Bioinformatic prioritization resulted in 42 up-regulated and 46 down-regulated proteins in LNCaP-SF cells relative to LNCaP cells. Our top candidate, HMGCS2, an enzyme involved in ketogenesis, was found to be 9-fold elevated in LNCaP-SF cells, based on peptide ratios. After analyzing the remaining enzymes of this pathway (ACAT1, BDH1, HMGCL, and OXCT1), we observed increased expression of these proteins in the LNCaP-SF cells, which was further verified using Western blotting. To determine whether these enzymes were up-regulated in clinical samples, we performed quantitative PCR and immunohistochemistry on human prostate cancer tissues, from which we observed significantly increased transcript and protein levels in high-grade cancer (Gleason grade ≥ 8). In addition, we observed significant elevation of these enzymes in the LuCaP 96AI castration-resistant xenograft. Further assessment of ACAT1 on human castration-resistant metastatic prostate cancer tissues revealed substantially elevated expression of ACAT1 in these specimens. Taken together, our results indicate that enzymes of the ketogenic pathway are up-regulated in high-grade prostate cancer and could serve as potential tissue biomarkers for the diagnosis or prognosis of high-grade disease.
Prostate cancer is the most commonly diagnosed cancer, and the second leading cause of cancer-related deaths among men in North America (1). In the early stages of cancer development, prostate cancer cells rely on androgens for their growth and survival. Androgen deprivation remains one of the most widely used therapies for metastatic and recurring prostate cancers (2). However, patients often regress to the more lethal androgen-independent prostate cancer (AIPC)1 or hormone-refractory prostate cancer, which is characterized by poor prognosis (3, 4). Effective therapeutic management is unavailable for AIPC. Thus, studying the molecular changes that occur during the progression to androgen independence remains of utmost importance in order to gain a better understanding of this process, as well as to generate biomarkers and targeted therapies.
The androgen receptor (AR) signaling cascade has been demonstrated to be an important pathway that is activated during androgen independence. Its activation has been documented to occur through AR gene amplifications, AR gene mutations, changes in co-regulators or steroidogenic enzymes, or alternative proteins via outlaw pathways (3–9). Although the AR pathway appears to be a key player in the development of androgen independence, it is important to note that numerous “bypass pathways,” the so-called AR-independent pathways, have also been suggested to be implicated in this process (10).
LNCaP, a hormone-dependent prostate cancer cell line, has often been used to model the progression of prostate cancer to androgen independence (11). Many groups have been able to generate androgen-independent sub lines of LNCaP by culturing them for extended periods in androgen-deprived media, a process that selects for clones that have gained the ability to grow in the absence of androgens (12–16). Many of these androgen-independent LNCaP sublines, such as LNCaP-SF, have features similar to those seen in clinical cases of hormone-refractory prostate cancer, including increased AR expression, and they represent excellent model systems for the investigation of AIPC progression (12–18).
Proteomics using mass spectrometry is a maturing analytical technique that has provided the opportunity to characterize the proteome of virtually any biological specimen. Such studies have been conducted to identify novel prostate cancer biomarkers, as well as key molecules implicated in cancer progression, using various cell lines and biological samples, specifically, tissues and fluids; however, markers with sufficient clinical relevance have yet to be introduced into practice (19–25). Prostate-specific antigen, the currently utilized biomarker for prostate cancer diagnosis, lacks specificity, as it becomes elevated in a variety of prostatic diseases, including benign prostate hyperplasia and prostatitis (26). The tissue specificity of prostate-specific antigen has also been questioned, as it has been found in the sera of women with breast cancer and in nipple aspirate fluid (27).
In this study, we utilized a high-throughput quantitative proteomics approach to delineate important modulators implicated in the development of AIPC, using the LNCaP/LNCaP-SF model system. Stable isotope labeling of amino acids in cell culture (SILAC) coupled to mass spectrometry was employed to comparatively quantify over 3300 proteins in LNCaP and LNCaP-SF cells. Several proteins of the ketogenesis pathway (HMGCS2, ACAT1, BDH1, HMGCL, and OXCT1) were up-regulated in LNCaP-SF cells. These ketogenic proteins were over-expressed in high-grade human prostate cancer samples, with ACAT1 displaying the greatest change in expression. In addition, ACAT1 expression was also elevated in castration-resistant metastatic lesions from prostate cancer patients. These results demonstrate that enzymes involved in the ketogenesis pathway could serve as potential biomarkers for high-grade prostate cancers, as well as provide new targets for therapeutic intervention.
MATERIALS AND METHODS
Cells and Reagents
The human prostate cancer LNCaP cell line and the androgen-independent subline LNCaP-SF cells were kindly provided by Dr. Atsushi Mizokami and maintained in DMEM (Wisent, St. Bruno, Quebec, Canada) supplemented with either 10% (v/v) FBS (HyClone) for LNCaP cells or 10% charcoal stripped FBS (HyClone) for LNCaP-SF cells at 37 °C with 5% CO2 in a humidified incubator.
SILAC
LNCaP and LNCaP-SF cells were seeded at low confluency (∼25%) in T75 flasks. SILAC media was prepared from customized DMEM lacking two essential amino acids: l-arginine and l-lysine (Athena ES, Baltimore, MD). Heavy amino acids, l-Arg6 (13C) and l-Lys8 (13C and 15N), were supplemented to the medium to generate the “heavy ” medium (Cambridge Isotope Laboratories, Andover, MA). For control or “light ” medium, l-arginine and l-lysine were supplemented into the medium (Sigma). Additionally, both heavy and light media were further supplemented with dialyzed FBS (Invitrogen). LNCaP cells were metabolically labeled with light medium, and LNCaP-SF cells were metabolically labeled with heavy SILAC conditioned medium in T25 flasks. Three independent biological replicates were used for both LNCaP and LNCaP-SF cells. A minimum of five doubling times was ensured, and the growth medium was changed every two to three days. Cells were grown for an additional 48 h and were then detached non-enzymatically, washed twice in PBS, and centrifuged at 1500 × g for 5 min. Cell pellets were kept at −80 °C until further processing.
Sample Preparation
Cell pellets were lysed using 250 μl of 0.1% RapiGest (Waters Inc., Milford, MA) in 25 mm ammonium bicarbonate and were subsequently sonicated three times for 30 s. The resulting cell lysates were then centrifuged for 15 min at 15,000 × g and 4 °C. Protein lysates from both LNCaP and LNCaP-SF cells were quantified using the BCA assay (Thermo Scientific) and mixed in a 1:1 ratio to obtain a total of 250 μg total protein in each replicate (125 μg protein from heavy-labeled and 125 μg from light-labeled condition). Proteins were then heat-denatured at 85 °C for 15 min, reduced with 10 mm DTT (Sigma-Aldrich, Madison, WI) for 10 min at 70 °C, alkylated with 20 mm iodoacetamide (Sigma-Aldrich) for 60 min with shaking in the dark, and trypsin-digested (Promega) at a ratio of 1:50 (trypsin:protein concentration) overnight at 37 °C. The resulting tryptic peptides were reconstituted in 200 μl of 0.26 m formic acid in 5% acetonitrile (mobile phase A) buffer.
Strong Cation Exchange on a High-performance Liquid Chromatography System
To reduce sample complexity, the samples were subjected to strong cation exchange using the Agilent 1100 high-performance liquid chromatography (HPLC) system. Tryptic peptides were initially diluted to 500 μl with strong cation exchange mobile phase A (0.26 m formic acid in 5% acetonitrile; pH 2–3) and loaded directly onto a 500 μl loop connected to a PolySULFOETHYL A™ column with a 2-μm pore size and a diameter of 5 μm (The Nest Group, Inc., Southborough, MA). Peptides were eluted using an elution buffer that contained all components of mobile phase A with the addition of 1 m ammonium formate, which was introduced at 10 min and further increased to 20% at 30 min and 100% at 45 min. A total of 14 fractions were collected for mass spectrometric analysis. The HPLC fractions were C18-extracted using ZipTipC18 micropipette tips (Millipore, Billerica, MA) and eluted in 5 μl of Buffer B (90% acetonitrile, 0.1% formic acid, 10% water, and 0.02% trifluoroacetic acid). An additional 80 μl of Buffer A (95% water, 0.1% formic acid, 5% acetonitrile, and 0.02% trifluoroacetic acid) was added to the samples.
Mass Spectrometry
40 μl of each fraction were injected into an autosampler on the EASY-nLC system (Proxeon Biosystems, Odense, Denmark). Firstly, the peptides were collected onto a 3-cm C18 column (inner diameter of 200 μm) and were then eluted onto a resolving 5-cm analytic C18 column (inner diameter of 75 μm) with an 8-μm tip (New Objective, Woburn, MA). This HPLC system was coupled online to an LTQ-Orbitrap XL (Thermo Fisher Scientific) mass spectrometer using a nano-electrospray ionization source (Proxeon Biosystems, West Palm Beach, FL) in data-dependent mode. The fractions were run on a 60-min gradient consisting of 3 min at 1%–14% Buffer B (100% acetonitrile, 0.1% formic acid), 44 min at 14%–40% Buffer B, 5 min at 40%–65% Buffer B, 3 min at 65%–100% Buffer B, and 5 min at 100% Buffer B. The eluted peptides were subjected to one full MS1 scan (450–1450 m/z) in the Orbitrap at 60,000 resolution, followed by six data-dependent MS2 scans in the linear ion trap. The following parameters were enabled: monoisotopic precursor selection, charge state screening, and dynamic exclusion. In addition, charge states of +1 and >4 and unassigned charge states were not subjected to MS2 fragmentation.
Protein Identification and Quantitation Using MaxQuant Software
The resulting mass spectra were analyzed using MaxQuant Software (version 1.1.1.25), which generates the extracted ion current-based quantitation for SILAC pairs. Raw MS files were loaded directly into MaxQuant, and identification and quantitation of individual peptides were conducted in protein groups. The MaxQuant searches were executed against the International Protein Index human protein database (version 3.62, 167,894 forward and reverse protein sequences) and decoy database. All entries were filtered using a false positive rate of 1%, and all false positives were removed. The following search parameters were used: one missed and/or nonspecific cleavage permitted, carbamidomethylation (57 m/z) on cysteine fixed modification, and oxidation (methionine) and acetal (N-terminus proteins) variable modifications. The mass tolerance for precursor ions was initially 20 ppm and then was adjusted to 6 ppm, followed by recalibration in MaxQuant. The mass tolerance for fragment ions was 0.5 Da. The quantification of proteins was based on the normalized heavy/light (H/L) ratios, as determined by MaxQuant. The MaxQuant results can be found in supplementary File S1. Complete protein and peptide lists, as well as the underlying RAW files, are available on the Peptide Atlas Database. Annotated spectra for single peptide identifications are provided in supplementary File S2.
Data Analysis
Protein groups from MaxQuant were exported to Excel files that displayed the results of three independent SILAC runs with their corresponding H/L ratios. The normalized average H/L ratio for each protein was the final quantitative value used to filter and select for candidates. To visualize and assess networks of over-expressed and under-expressed candidates, Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA) software was used. Using this software, pathway analysis was performed, obtaining information on canonical pathways and molecular networks that had been altered, as determined by Fisher's exact test.
Reverse-transcription and Quantitative PCR
Total RNA was isolated from LNCaP and LNCaP-SF cells using an RNeasy Kit (Qiagen Hilden, Germany). cDNA was generated from 1 μg of total RNA using the Superscript II cDNA synthesis kit (Invitrogen). Quantitative PCR was conducted using 1X SYBR reagent (Applied Biosystems, Foster City, CA), and transcript levels of HMGCS2, ACAT1, BDH1, HMGCL, OXCT1, DHRS2, AGR2, HSD11B2, ALDH6, MAOA, MAOB, TYMP, ARG2, and SQSTM1 were measured on a 7500 ABI system. All quantitative PCR data were normalized to tata-binding protein expression. Sequences of all primers used are shown in supplemental Table S1.
For clinical validation, the TissueScan Prostate Cancer cDNA Array II was used (Origene, Rockville, MD). Clinical information for these patients can be found in supplemental Table S2. Quantitative PCR was conducted on these samples using the same SYBR green reagent as mentioned above.
Protein Extraction
LNCaP and LNCaP-SF cell pellets were washed with PBS (pH 7.4) three times, and cells were lysed using 1% SDS solution. The samples were then sonicated three times on ice in 30-s intervals using a MISONIX immersion tip sonicator (Q SONICA LLC, CT), and centrifuged at 15000 × g for 15 °C minutes at 4 °C. Protein concentrations were quantified using a BCA protein assay kit (Thermo Scientific, Newtown, CT).
LuCaP96 and LuCaP96AI xenograft tissues, as described elsewhere (28), were frozen in liquid nitrogen and then ground into a fine powder using a mortar and pestle. The resulting powdered tissue was lysed using 1% SDS solution, and this was followed by sonication on ice for three 30-s intervals. The samples were then centrifuged at 14000 × g for 15 min at 4 °C, and the resulting supernatant was used for further analysis.
Western Blotting
The protein expression of ACAT1, HMGCS2, BDH1, OXCT1, and HMGCL was assessed using Western blot analysis. Roughly 30 μg of total protein from each sample was loaded onto an SDS-PAGE gel (4%–15%, Bio-Rad) and transferred onto PVDF membranes (Bio-Rad). Membranes were then incubated with 5% blocking solution (2.5 g skim milk powder in Tris buffer solution containing 0.1% Tween) overnight at 4 °C. Membranes were incubated with rabbit polyclonal antibody against ACAT1, BDH1, OXCT1, HMGCL, (Sigma) or HMGCS2 (Protein Tech, Chicago, IL) for 1 h at room temperature. The membranes were then washed six times (three 15-min washes followed by three 5-min washes) with Tris buffer solution containing 0.1% Tween. Membranes were then incubated with goat anti-rabbit secondary antibody conjugated to horseradish peroxidase (Jackson Laboratories, Bar Harbor, Maine) for 1 h at room temperature. After being washed with Tris buffer solution containing 0.1% Tween, proteins were detected using the ECL detection reagent (Siemens, Berlin, Germany). The expression of GAPDH (Cell Signaling Technology, M: Danvers, MA) or B-actin (Abcam, Cambridge, UK) was used as an internal standard.
β-hydroxybutyrate Detection Assay
LNCaP and LNCaP-SF cells were grown in T25 flasks in serum-free media for 48 h, and the resulting cell supernatants were collected and assayed for β-hydroxybutyrate levels using a β-hydroxybutyrate (Ketone Body) Colorimetric Assay Kit (Cayman Chemical, Ann Arbor, MI), according to the manufacturers protocol. Total β-hydroxybutyrate values were normalized to the total protein levels in the supernatants.
Immunohistochemistry
Prostate cancer tissue microarrays consisting of 8 normal and 40 cancer cores were purchased from US BioMax (Rockville, MD). The metastatic prostate cancer tissue microarray was developed and provided by the GU Cancer Research laboratories at the University of Washington (Seattle, WA). Human tissue microarrays of fixed paraffin-embedded metastatic tissues from 23 rapid autopsy patients who died of prostate cancer (consisting of three tissue microarray blocks with two replicate cores per metastatic site) were used for immunohistochemical analyses. All patients had castration-resistant prostate cancer at the time of autopsy, defined by the presence of a rising level of serum prostate-specific antigen following medical or surgical castration. Clinical information for these patients can be found in supplemental Table S3.
Tissue microarrays were deparaffinized in xylene and rehydrated using ethanol. Endogenous peroxidase was reduced using hydrogen peroxide for 10 min and washed with PBS. Antigen retrieval was then performed using citrate buffer in a microwave for 10 min. Slides were then blocked for 5 min in casein and incubated overnight with the following primary antibodies: ACAT1 (1:1000), BDH1 (1:2000), HMGCL (1:1200), HMGCS2 (1:600), and OXCT1 (1:1000). Rabbit IgG was used on a duplicate slide to serve as a negative control. Following 10 min of PBS washing, slides were placed in secondary antibody for 30 min using the BGX kit (Biogenex, Fremont, CA). After a 10-min wash in PBS, slides were developed with the addition of 3,3′-diaminobenzidine (DAB) for 5 min. Slides were then counterstained with hematoxylin, dehydrated, and coverslipped.
Statistical Analysis
All gene expression studies on cell lines and xenografts consisting of normalized expressions were compared using a two-tailed t test (GraphPad Prism Software). Gene expression studies on human prostate cancer and normal tissue were compared using a non-parametric Mann–Whitney test (GraphPad Prism Software). Finally, χ2 tests were used to compare different groups from the immunohistochemistry data. Differences were considered significant if the p value was less than 0.05. All data are expressed as means ± S.E. of the mean.
RESULTS
Quantitative Proteomic Profiling of LNCaP versus LNCaP-SF Cells
To identify potential modulators of AIPC, we performed global proteomic profiling of the LNCaP cell line and its androgen-independent counterpart, LNCaP-SF, using SILAC. This approach provided a robust and comprehensive method of detecting differential proteomic expression between both cell lines, as each was grown in distinct amino acid isotopic media (Fig. 1A). Using MaxQuant software, we were able to identify and quantify 3416 proteins with one peptide hit. Based on the H/L ratio for each protein, a numerical value representing the differential protein expression between LNCaP-SF cells (grown in heavy media) and LNCaP cells (grown in light media) was assigned. The majority of proteins (85%) fell within 1 standard deviation of the total average ratio (H/L ratio between 0.67 and 1.5) among the three replicates, indicating that there were no significant differences with respect to protein levels between the two cell lines (Fig. 1B). Among the three biological replicates, we observed a reproducibility of 80% to 90% with respect to the H/L ratio between independent experiments (supplemental Fig. S1). In addition, H/L ratios of various housekeeping proteins, including β-actin (H/L = 1.37), GAPDH (H/L = 0.85), HSP90 (H/L = 0.92), and α-tubulin (H/L = 1.06), all remained within 1 standard deviation of their average ratio (supplemental Table S4).
Fig. 1.

SILAC-based quantitative proteomic profiling of prostate cancer progression to androgen independence. A, flow chart depicting the steps involved in quantitative proteomic profiling of LNCaP and LNCaP-SF cells. LNCaP cells were grown in medium containing 12C-Arg/Lys, whereas LNCaP-SF were grown in 13C-15N-Arg/Lys medium, and both cell lines were allowed to grow for a minimum of five doubling times. Cells were lysed and total protein from LNCaP and LNCaP-SF cell lysates was mixed in a 1:1 ratio. Following trypsin digestion and fractionation with strong cation exchange chromatography, peptides were identified using MS/MS, and the resulting data were analyzed with MaxQuant Software. B, following protein identification using MaxQuant, over 3400 proteins were quantified, with the majority having a 1:1 ratio, indicating no change in protein expression between the two cell lines. C, 42 proteins were found to be up-regulated in LNCaP-SF cells using stringent criteria (greater than 2.5-fold H/L ratio in three experimental replicates). D, Ingenuity Pathway Analysis revealed alterations in central nodes such as p53, ESR1, SP1, TGFβ, and TNF signaling pathways.
In order to identify candidates that were either up- or down-regulated in LNCaP-SF cells relative to LNCaP cells, we used H/L values that were 2 standard deviations from the mean H/L ratio of all proteins identified, which corresponded to H/L ratios > 2.5 as over-expressed hits and H/L ratios < 0.4 as under-expressed. Each of the candidates had to meet these cut-offs in all three independent biological replicates in order to be further considered. After implementing these stringent criteria, 42 (Fig. 1C, supplemental Table S5) and 46 proteins (supplemental Table S6) were found to be up- and down-regulated, respectively, in LNCaP-SF cells relative to LNCaP cells.
Analysis of Candidate Proteins
After assessing the candidate lists, we identified proteins that were previously studied in the context of prostate cancer progression, including AGR2, ALDH1A3, S100P MAOA, AADACL1, and SOD2 among the over-expressed and IGFBP2, STMN1, DNMT1, ATAD2, KPNA2, and ADI1 among the under-expressed proteins (29–40). We next subjected our candidate list to pre-clustering pathway analysis using Ingenuity Pathway Analysis. This analysis revealed that the top molecular networks of the over-expressed candidates were cancer and cellular development (Fig. 1D). Specifically, “lipid metabolism” and “small molecular biochemistry” were the top molecular function clusters. The over-expressed candidates had central nodes in the NF-kB, p53, ESR1, TGFβ, TNF, and SP1 signaling cascades, all of which have been previously documented as being associated with prostate cancer progression (41–45). With regard to our under-expressed candidates, the top molecular networks were (i) DNA replication, recombination, and repair; cell cycle; cellular assembly; and organization, and (ii) cell death, cellular growth, and proliferation. Our top under-expressed candidate was SEMG1, with a 13-fold down-regulation in LNCaP-SF cells (H/L of 0.08), and its functionally related protein, SEMG2, was also under-expressed.
The top over-expressed candidate, HMGCS2, which had a 9-fold increase in LNCaP-SF cells, is an enzyme involved in ketogenesis, a metabolic pathway that provides lipid-derived energy during times of carbohydrate deprivation (46). We identified another protein in this pathway, OXCT1, with a more than 2-fold increase in LNCaP-SF cells, which led us to investigate the remainder of the proteins involved in this pathway. We were able to identify all five proteins within the ketogenic pathway (HMGCS2, HMGCL, BDH1, ACAT1, and OXCT1), and four of them—HMGCS2, OXCT1, BDH1, and ACAT1—exhibited increased protein expression in LNCaP-SF cells based on our initial SILAC experiments. The normalized H/L ratios for each protein were 9.24 for HMGCS2, 2.15 for OXCT1, 1.88 for ACAT1, 1.75 for BDH1, and 1.37 for HMGCL (Fig. 3A). These preliminary results suggest that the ketogenesis pathway may be up-regulated during the progression of prostate cancer to androgen-independence.
Fig. 3.

Analysis of gene and protein expression levels of enzymes involved in the ketogenic pathway (ACAT1, BDH1, OXCT1, HMGCL, and HMGCS2) in LNCaP and LNCaP-SF cells. A, enzymes of ketogenic pathway quantified in our analysis. B, C, gene and protein expression validation using real-time PCR and Western blotting confirmed the significant over-expression of HMGCS2, OXCT1, ACAT1, and BDH1 at the mRNA level (*p < 0.05, two-tailed t test) with all enzymes displaying elevated expression at the protein level. D, β-hydroxybutyrate secretion was found to be significantly elevated in the conditioned medium of LNCaP-SF cells (*p < 0.05, two-tailed t test).
In Vitro Validation of Candidates
The gene expressions of our top candidates were further validated in vitro using real-time PCR. Validation of the proposed over-expressed genes revealed that 7 out of the top 10 over-expressed candidates (HMGCS2, DHRS2, AGR2, HSD11B2, ALDH6, MAOA, and MAOB) had increased gene expression in LNCaP-SF cells (Fig. 2), whereas three candidates (TYMP, ARG2, and SQSTM1) did not show any significant changes in gene expression. Considering that we used RNA to verify our protein discovery results, it is important to note that several post-transcriptional factors might play a role in the differences observed between RNA and protein expression.
Fig. 2.
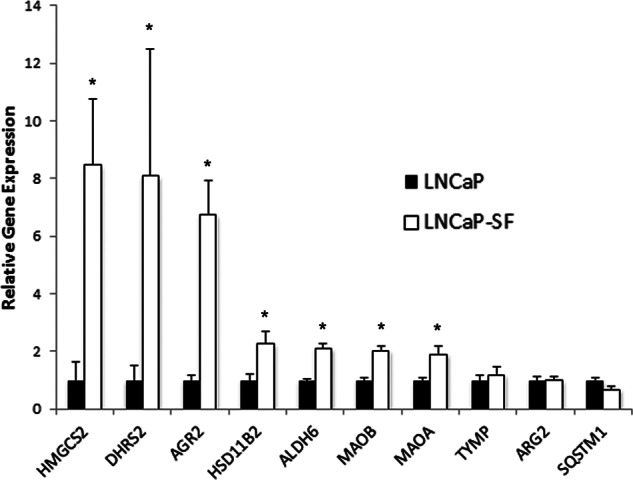
Validation of the top over-expressed candidates using real-time PCR in LNCaP and LNCaP-SF cells. Seven of the 10 candidates (HMGCS2, DHRS2, AGR2, HSD11B2, ALDH6, MAOA, and MAOB) showed up-regulation in transcript levels in LNCaP-SF cells, and three (TYMP, ARG2, and SQSTM1) displayed no significant changes in mRNA expression. *p < 0.05, two-tailed t test.
Additionally, we also performed quantitative PCR validation of the genes associated with the ketogenesis pathway. Four genes (HMGCS2, OXCT1, ACAT1, and BDH1) had increased expression in LNCaP-SF cells, whereas HMGCL displayed relatively similar expression levels in the two cell lines (Fig. 3B). Protein validation of the ketogenic pathway enzymes using Western blotting supported our preliminary data, demonstrating an up-regulation of our identified enzymes in LNCaP-SF cells, with HMGCS2, OXCT1, and ACAT1 having the greatest changes in protein expression (Fig. 3C). Finally, because this pathway is responsible for the generation of ketone bodies, we assessed the secretion levels of β-hydroxybutyrate, the most common ketone body, in these cell lines. We found a statistically significant 2-fold increase in β-hydroxybutyrate levels in the conditioned medium of LNCaP-SF cells relative to LNCaP cells (Fig. 3D). These preliminary in vitro results imply that the ketogenic pathway enzymes identified in our preliminary analysis, as well as the associated ketone bodies, exhibit elevated expression in LNCaP-SF cells.
Ketogenic Pathway Gene Transcript and Protein Levels Are Increased during High-grade Prostate Cancer
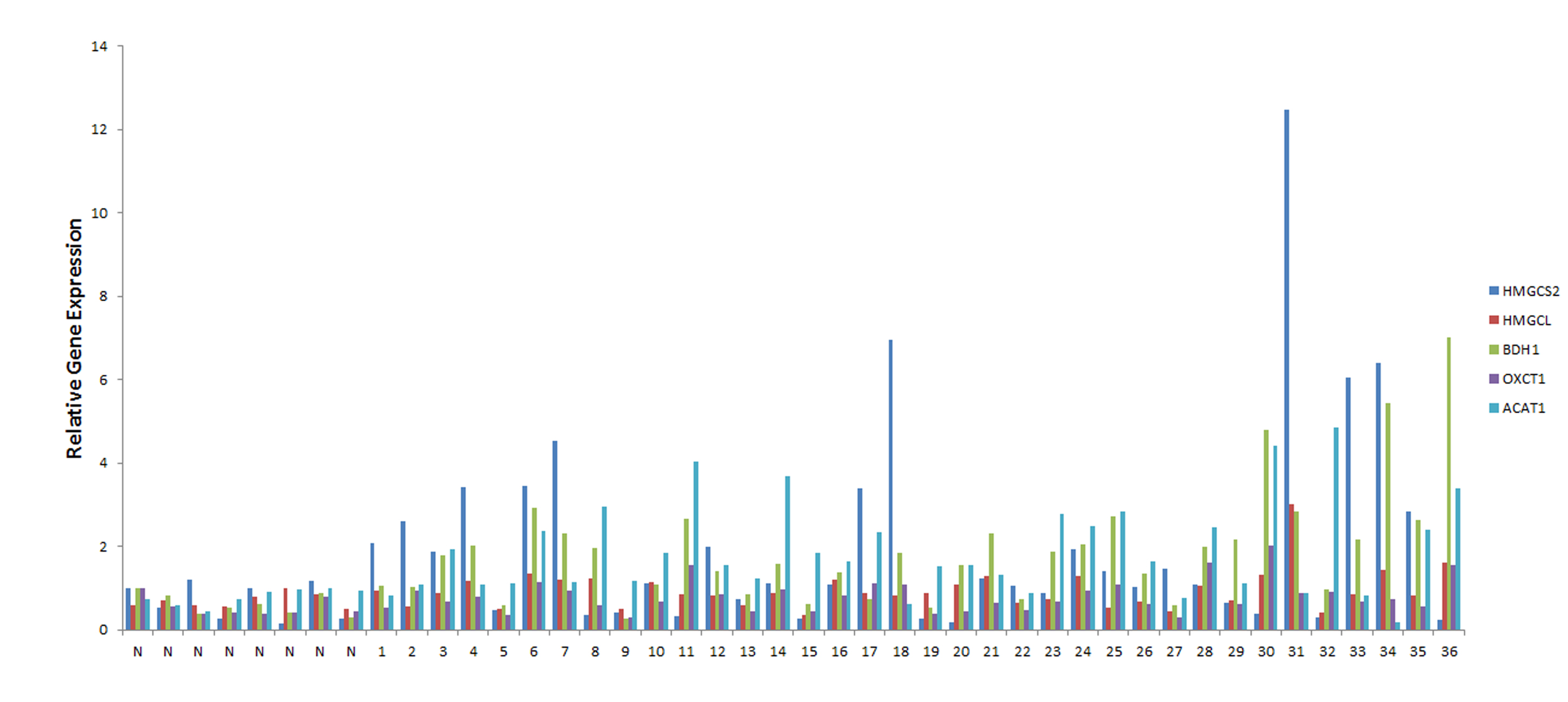
To determine whether the ketogenesis pathway genes play a role during prostate cancer progression, we measured their transcript levels in normal and human tumor tissue samples. Using real-time PCR, we found that HMGCS2 (p = 0.048), OXCT1 (p = 0.04), BDH1 (p = 0.0008), and ACAT1 (p = 0.009) have significantly increased gene expression in prostate cancer relative to normal tissue (Fig. 4). In particular, ACAT1 and BDH1 displayed the most prominent changes in gene expression, with 3-fold and 4-fold increases, respectively. HMGCL expression was slightly elevated in prostate cancer; however, this was not statistically significant (p = 0.075). Interestingly, of the 36 prostate cancer patients analyzed, 20 had at least a 2-fold increase in expression of at least two ketogenic enzymes relative to the 8 normal patients analyzed (supplemental Fig. S2). Among these 20 patients, 4 had increases in two ketogenic genes, 12 had increases in three genes, 3 had increases in four genes, and 1 patient had increases in all five ketogenic genes.
Fig. 4.
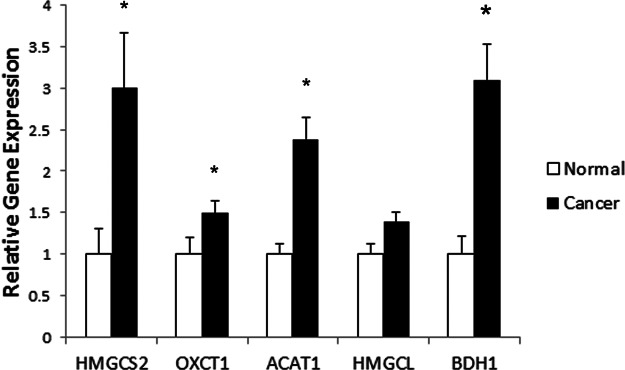
Gene expression profiling of ketogenic pathway enzymes in normal and prostate cancer human tissue. Based on a TissueScan Prostate Cancer cDNA Array II consisting of 8 normal and 36 prostate cancer specimens, the gene expression profiles of HMGCS2, OXCT1, BDH1, and ACAT1 all showed significantly elevated mRNA expression in cancer relative to normal specimens (*p < 0.05; **p < 0.01, Mann–Whitney test).
In addition, using immunohistochemistry, we assessed the protein expression levels of the ketogenesis enzymes in various normal and prostate cancer samples. We devised a scoring system to assess protein expression, whereby each core was scored with a 0, 1, 2, or 3, which correspond to no staining, low staining, moderate staining, and high staining, respectively. All ketogenic enzymes had preferentially higher staining patterns in high-grade prostate cancers (Gleason grade ≥ 8). The expression profiles of each enzyme followed an increasing pattern, going from normal prostate samples to low-grade prostate cancer (Gleason ≤ 7), to high-grade prostate cancer (Fig. 5 and supplemental Table S7). In ACAT1, normal cores had very little positive staining (13% stained with a score of 2 or greater), moderate levels of staining were observed in low-grade prostate cancer cores (46%), and almost all high-grade prostate cancer cores had intense staining (86%). For BDH1, there was moderate or high staining in 63% of normal cores, 77% of low-grade prostate cancer samples, and 87% of high-grade prostate cancer samples. HMGCL also had similar staining patterns, with moderate or high staining in 50% of normal cores, 70% of low-grade cancer cores, and 93% of high-grade prostate cancer cores. Both BDH1 and HMGCL proteins did not exhibit sharp changes in expression, as in the case of ACAT1. OXCT1 also displayed increased staining intensity in high-grade prostate cancers (66%) relative to normal (25%) and low-grade samples (27%). Finally, the top candidate from our initial proteomic experiments, HMGCS2, had similar staining intensities in both normal and low-grade cancers (50% in both), but it also displayed increased staining in high-grade prostate cancer samples (65%). These results indicate that enzymes involved in the ketogenic pathway appear to be over-expressed during the development of high-grade prostate cancers and therefore might play a role in the progression of prostate cancer to an advanced-stage disease.
Fig. 5.

Protein expression of ketogenic pathway enzymes in human prostate cancer specimens of varying grades. A, representative immunohistochemistry images of HMGCS2, HMGCL, BDH1, OXCT1, and ACAT1 expression in normal, low-grade (Gleason ≤ 7), and high-grade (Gleason ≥ 8) prostate cancer specimens under light microscopy (20×). B, immunohistochemical staining was quantified using a scoring scale of 0, 1, 2, and 3 corresponding to no staining, low staining, moderate staining, and high staining, respectively, as blindly determined by a pathologist.
The Ketogenic Pathway Is Activated in the LuCaP96AI AIPC Xenograft Model
To evaluate whether ketogenic enzymes play a role during the progression to AIPC, xenografts of LuCaP 96 and its androgen-independent counterpart, LuCaP 96AI, were utilized. The transcript and protein levels of the five ketogenic enzymes were assessed using quantitative PCR and Western blotting, respectively. It was found that HMGCS2 (p = 0.014), BDH1 (p = 0.003), ACAT1 (p = 0.024), and HMGCL (p < 0.001) all displayed statistically significant increases in expression in the LuCaP 96AI xenograft-derived cells (Fig. 6A). ACAT1 and BDH1 had the most prominent gene expression differences, exhibiting almost 3-fold increases in their transcript levels in the androgen-independent xenograft. Both HMGCS2 and HMGCL displayed roughly 2-fold increases in expression within LuCaP 96AI cells. Surprisingly, our results indicate that OXCT1 exhibited decreased expression in the LuCaP 96AI xenograft. Protein expression profiles corresponded well with our gene expression data, as ACAT1, BDH1, HMGCL, and HMGCS2 all displayed increased protein expression in LuCaP 96AI cells (Fig. 6B). Taken together, these results further support the involvement of altered ketogenic enzyme profiles in the development of AIPC.
Fig. 6.
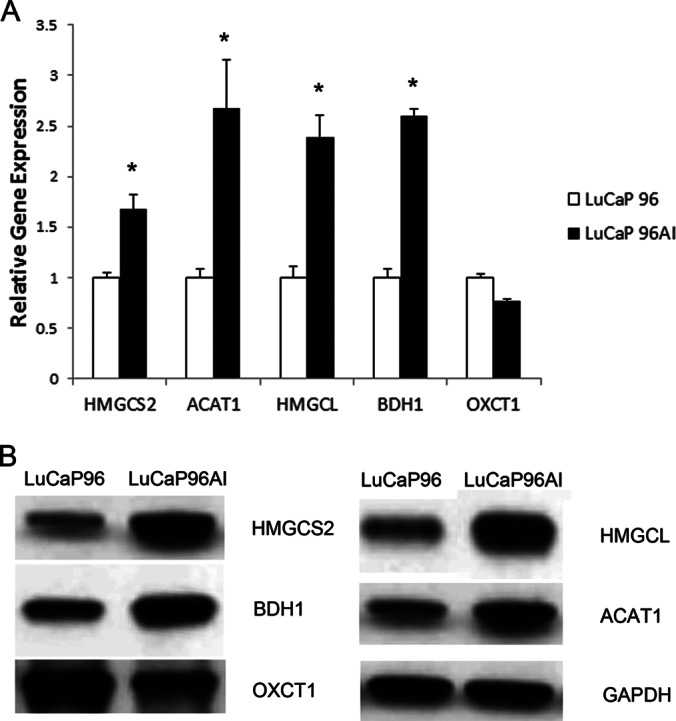
Expression of ketogenic pathway enzymes in in vivo LuCaP 96 xenograft and its androgen-independent xenograft LuCaP 96AI. A, gene expression profiling reveals statistically significant up-regulation of HMGCS2, ACAT1, HMGCL, and BDH1 (*p < 0.05, two-tailed t test) at the transcript level. B, protein expression profiles from Western blot analysis corresponded well with gene expression data; ACAT1, BDH1, HMGCL, and HMGCS2 all displayed increased protein expression in LuCaP 96AI cells.
ACAT1 Is Highly Expressed in Castration-resistant Metastatic Prostate Cancers
Based on all these observations, ACAT1 was identified as the most promising candidate, because its expression was found to be almost absent in normal samples and gradually increased in advanced-grade prostate cancers. Increased ACAT1 transcript and protein expression were also demonstrated in both prostate cancer samples and an androgen-independent xenograft model. Therefore, we aimed to examine ACAT1 expression in various castration-resistant metastatic prostate cancer samples, to identify whether it is dysregulated during hormone-refractory prostate cancer. Using our reported scoring system in a tissue microarray containing metastatic lesions to the bone, lymph nodes, lung, and liver, we demonstrated increased ACAT1 staining in all metastatic sites, with bone metastases presenting with highest expression (Fig. 7). Specifically, we observed 36.8% of lung and liver metastases containing no or low staining, 31.6% staining moderately, and 31.6% of cores having high staining. With respect to the lymph node metastasis cores, we found 32.1% with low staining, 46.4% with moderate staining, and 21.4% with high staining. Bone metastatic lesions had the most prominent ACAT1 expression patterns, as 13.5% of cores had low staining, 47.3% had moderate staining, and 39.2% had high staining. In contrast, in normal prostate cores, ACAT1 staining was very low—the majority of the samples (87.5%) had little or no staining, and a small proportion (12.5%) had moderate ACAT1 expression. Following statistical analysis, ACAT1 expression was found to be significantly up-regulated in lung and liver (p = 0.0329), lymph node (p = 0.0121), and bone (p = 0.0001) prostate cancer metastatic lesions relative to normal prostate samples (Table I). Based on these results, ACAT1 expression is up-regulated in castration-resistant prostate cancer metastases, with bone metastatic lesions having the most prominent expression patterns.
Fig. 7.

Expression of ACAT1 in castration-resistant metastatic prostate cancer specimens of the bone, lymph node, liver, and lungs. A, representative immunohistochemistry images of ACAT1 staining in normal tissue, liver metastasis, lung metastasis, lymph node metastasis, and bone metastasis of the prostate are shown. Images were taken under light microscopy (20×). B, immunohistochemical staining was quantified using a scoring scale of 0, 1, 2, and 3 corresponding to no staining, low staining, moderate staining, and high staining, respectively, as blindly determined by a pathologist.
Table I. ACAT1 expression in castration-resistant metastatic prostate cancer specimens.
| Tissue type | Number of positive samplesa | Staining percentage (%) | p value compared to normalb |
|---|---|---|---|
| Normal prostate | 1/8 | 12.5 | NA |
| Lung and liver metastasis | 12/19 | 63.2 | 0.0329 |
| Lymph node metastasis | 19/28 | 67.9 | 0.0121 |
| Bone metastasis | 64/74 | 86.5 | 0.0001 |
a Positive staining is defined as a score of 2 or higher, based on the pathologist's score assessment.
b p value was calculated using a χ2 test.
DISCUSSION
Prostate cancer is a curable disease upon early detection, as the tumor is most often localized to the prostate. However, once the cancer has spread outside of the prostate gland, effective treatments are limited, with radiation therapy, chemotherapy, and the gold standard of hormonal therapy usually failing in the long run. Although hormonal therapy is initially very effective in reducing tumor growth and volume, cancer cells eventually gain resistance and grow in the absence of circulating androgens. This is the stage at which the disease becomes fatal, as no other targeted therapies are available. Therefore, studies focusing on understanding the development and progression of AIPC are crucial in order to develop targeted therapeutic strategies.
LNCaP is a widely used cell line for the investigation of androgen-dependent prostate cancer and has been utilized by numerous groups to develop an androgen-independent clone (12–18). However, several limitations exist in using this model. For instance, LNCaP cells carry a mutation in the ligand-binding domain of the AR, making it more “promiscuous, ” whereby the AR can be activated by molecules other than dihydrotestosterone, including various anti-androgens. However, during prostate cancer progression, mutations within the AR gene—specifically, the ligand binding domain—become more common, so this mutation reflects one mode of gaining a survival advantage for cancer cells. Also, phenotypic characteristics of the various LNCaP androgen-independent sublines generated by others accurately represent common features usually presented in clinical cases of AIPC, including increased AR expression. Therefore, this model remains quite valuable for studying the progression of prostate cancer.
In this study, by performing powerful, high-throughput, global proteomic profiling of an in vitro LNCaP cell line model of AIPC progression, we identified over 3400 proteins and specified over 80 proteins as differentially regulated between LNCaP and LNCaP-SF. To our knowledge, this is the most comprehensive quantitative proteomics study to date aiming at understanding the mechanisms of prostate cancer progression. To internally validate our approach, we observed among our candidates (42 over-expressed and 46 under-expressed) a plethora of proteins previously studied or implicated in prostate cancer progression. Six of the up-regulated genes (AGR2, ALDH1A3, S100P MAOA, AADACL1, and SOD2) have been previously deemed to be involved in promoting prostate cancer progression (29–34). For example, AGR2, which exhibited almost a 3-fold increase in LNCaP-SF cells, has been previously shown to be involved in prostate cancer metastasis. Likewise, six of the under-expressed candidates (IGFBP2, STMN1, DNMT1, ATAD2, KPNA2, and ADI1) have also been previously implicated in prostate cancer progression (35–40). Interestingly, two of our top under-expressed candidates, SEMG1 and SEMG2, have been previously shown to be decreased in expression in two androgen-independent cell lines (PC3 and DU145), which corresponds well with our results (61).
Our top over-expressed candidate, HMGCS2, which was 9-fold elevated, is an enzyme involved in the ketogenic pathway. This significant dysregulation prompted us to investigate additional candidates of the ketogenesis pathway. Performing clinical validation on a variety of samples, we observed that these enzymes were over-expressed during prostate cancer, specifically in high-grade prostate cancer. The most interesting of these enzymes, ACAT1, was also found to be highly expressed in castration-resistant metastatic prostate cancer specimens.
In order for cancer cells to proliferate and survive, they must meet their high energy demand for carrying out integral cellular processes. Cell growth, proliferation, and migration require large amounts of energy in the form of ATP, and by using alternative energy-producing pathways, cancerous cells gain a survival advantage. The ketogenic pathway is such an alternative energy-producing pathway, primarily responsible for the production of ketone bodies from fatty acids via the breakdown of acetyl-CoA, a key molecule formed during fatty acid metabolism (47). Acetyl-CoA, under normal high-glucose conditions, is oxidized, resulting in the formation of the high-energy molecules NADH and FADH2 in the citric acid cycle. However, when levels of acetyl-CoA are higher than required for the citric acid cycle, it is used for the biosynthesis of ketone bodies through the aid of five cellular enzymes. From our initial proteomics data, we identified all five of these enzymes as up-regulated in LNCaP-SF cells, and we also found an increase in β-hydroxybutyrate, the most common ketone body, in the secretome of these cells. These observations suggest that the ketogenesis pathway might be an alternate energy-producing mechanism through which prostate cancer cells gain a survival advantage allowing them to become increasingly aggressive and gain androgen-independent properties. During androgen deprivation, prostate cancer cells lose a critical signaling cascade via AR activation, resulting in the decreased expression of AR-regulated genes. A recent study by Massie et al. identified key energy-producing networks, including glucose uptake and glycolysis, as regulated by AR signaling (48). A decrease in the activity of the glycolytic pathway places prostate cancer cells under stress to generate energy in a quick manner in order to carry out necessary cellular functions. One avenue through which such an effect can be achieved is to increase energy production through the breakdown of fatty acids via the β-oxidation pathway (49).
Fatty acid oxidation has been widely studied with respect to prostate cancer progression, specifically, as a means of providing an important source of bioenergy (49, 50). Various proteins involved in the metabolism of fatty acids have been determined to be altered during prostate cancer (49, 50). Fatty acid synthase (FASN) has been one of the most widely studied of these proteins, as it has been found to be over-expressed at both mRNA and protein levels in prostate cancer (51). Interestingly, the highest levels of fatty acid synthase are associated with androgen-independent bone metastatic lesions (51). Other frequently described fatty acid oxidation pathway protein alterations observed in prostate cancer include a loss of stearoyl-CoA desaturase expression and increases in d-functional protein and α-methylacyl-CoA racemase expression (52–54). α-methylacyl-CoA racemase expression in particular has been associated with increased prostate cancer risk (54, 55).
For the first time, we identified the ketogenic pathway as a novel bioenergetic pathway potentially involved in the progression of prostate cancer from low-grade to high-grade disease, followed by androgen independence. With the exception of ACAT1, the ketogenesis pathway has not been investigated in prostate cancer. Specifically, ACAT1 was shown to be involved in the androgen-mediated cholesterol metabolism in prostate cancer cell lines (56). In another recent study, ACAT1 protein expression was found to be elevated in LNCaP androgen-independent xenografts, further suggesting its importance during prostate cancer progression (57). Both studies focused on ACAT1 and its involvement with cholesterol metabolism, an important metabolic pathway for cholesterol biosynthesis. Cholesterol metabolism has also been found to be altered during the progression of prostate cancer to AIPC, as free cholesterol from increased biosynthesis or uptake is a potential resource for intratumoral de novo androgen synthesis. Thus, an alternative hypothesis rationalizing the observed over-expression of ketogenic enzymes during high-grade prostate cancer and AIPC is to provide more efficient production of cholesterol, which in turn can act as a precursor for the generation of androgens. In order to better understand the mechanism of action of these ketogenic enzymes in prostate cancer progression, further studies using RNA interference technology in prostate cancer cell lines and mouse models need to be conducted. In addition, to further support our results, expression studies on a larger number of samples, including more diverse clinical samples, such as hormone naïve and hormone-refractory specimens, are needed to provide additional insight into the importance of these enzymes in prostate cancer. ACAT1 is of particular interest, as it was found to be significantly elevated in prostate cancer metastatic lesions. ACAT1 inhibitors have been previously investigated for various other diseases (58, 59) and present an interesting avenue of therapeutic intervention to potentially treat high-grade and metastatic prostate cancers.
In recent years, the field of cancer bioenergetics has enjoyed a resurgence, specifically pertaining to alterations of metabolic pathways during carcinogenesis. Displaying what was initially described as the Warburg effect, tumor cells were observed to switch from oxidative phosphorylation to anaerobic glycolysis, even in the presence of oxygen (60). As a result of the identification of mutations in genes that encode for enzymes for specific metabolic pathways, it has become apparent that malignant cells will gain a survival advantage if they are able to produce greater amounts of energy with which to carry out important cellular tasks such as rapid cellular growth and proliferation.
Overall, in this study, through the use of quantitative proteomics and validation on clinical samples, we have demonstrated that (i) many proteins become altered during the progression of prostate cancer to androgen independence, (ii) the ketogenic pathway enzymes become over-expressed during high-grade prostate cancers, and (iii) ACAT1 becomes highly elevated in metastatic prostate cancer. Going forward, the complete understanding of the function of these ketogenic pathway proteins with respect to prostate cancer pathophysiology will depend on the development of relevant in vitro and in vivo models. However, our present findings provide sufficient evidence that the ketogenic-pathway-associated enzymes play an important role during prostate cancer pathogenesis and might be an interesting area for therapeutic intervention for a disease that, to date, lacks targeted treatments.
Supplementary Material
Acknowledgments
We acknowledge the support of the University Health Network (UHN) Pathology Research Program (PRP) for performing the immunohistochemistry experiments. We thank Apostolos Dimitromanolakis for assistance in analyzing the proteomics data and statistical analysis. We thank the patients and their families who were willing to participate in the Prostate Cancer Donor Program, for without them research of this nature would not be possible. We also acknowledge Lawrence True, Eva Corey, and Robert Vessella and the rapid autopsy teams in the Urology Department at the University of Washington.
Footnotes
* This material is the result of work supported by resources from the Pacific Northwest Prostate Cancer SPORE (P50CA97186), a PO1 NIH grant (PO1CA085859), and the Richard M. Lucas Foundation. Punit Saraon was supported by the Ontario Graduate Scholarship (OGS) and the Scace Graduate Prostate Cancer Fellowship.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- ACAT1
- acetyl-coenzyme A acetyltransferase 1
- AIPC
- androgen-independent prostate cancer
- AR
- androgen receptor
- BDH1
- D-beta-hydroxybutyrate dehydrogenase
- H/L
- heavy/light (ratio)
- HMGCL
- 3-hydroxymethyl-3-methylglutaryl-CoA lyase
- HMGCS2
- 3-hydroxy-3-methylglutaryl-CoA synthase 2
- HPLC
- high-performance liquid chromatography
- OXCT1
- succinyl-CoA:3-ketoacid-coenzyme A transferase 1
- SILAC
- stable isotope labeling of amino acids in cell culture.
REFERENCES
- 1. Gronberg H. (2003) Prostate cancer epidemiology. Lancet 361, 859–864 [DOI] [PubMed] [Google Scholar]
- 2. Javidan J., Deitch A. D., Shi X. B., de Vere White R. W. (2005) The androgen receptor and mechanisms for androgen independence in prostate cancer. Cancer Invest. 23, 520–528 [DOI] [PubMed] [Google Scholar]
- 3. Saraon P., Jarvi K., Diamandis E. P. (2011) Molecular alterations during progression of prostate cancer to androgen independence. Clin. Chem. 57, 1366–1375 [DOI] [PubMed] [Google Scholar]
- 4. Denmeade S. R., Isaacs J. T. (2002) A history of prostate cancer treatment. Nat. Rev. Cancer 2, 389–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crawford E. D., Petrylak D. (2010) Castration-resistant prostate cancer: descriptive yet pejorative? J. Clin. Oncol. 28, e408. [DOI] [PubMed] [Google Scholar]
- 6. Chodak G. W., Kranc D. M., Puy L. A., Takeda H., Johnson K., Chang C. (1992) Nuclear localization of androgen receptor in heterogeneous samples of normal, hyperplastic and neoplastic human prostate. J. Urol. 147, 798–803 [DOI] [PubMed] [Google Scholar]
- 7. Ruizeveld de Winter J. A., Trapman J., Vermey M., Mulder E., Zegers N. D., van der Kwast T. H. (1991) Androgen receptor expression in human tissues: an immunohistochemical study. J. Histochem. Cytochem. 39, 927–936 [DOI] [PubMed] [Google Scholar]
- 8. Sadi M. V., Walsh P. C., Barrack E. R. (1991) Immunohistochemical study of androgen receptors in metastatic prostate cancer. Comparison of receptor content and response to hormonal therapy. Cancer 67, 3057–3064 [DOI] [PubMed] [Google Scholar]
- 9. Debes J. D., Tindall D. J. (2004) Mechanisms of androgen-refractory prostate cancer. N. Engl. J. Med. 351, 1488–1490 [DOI] [PubMed] [Google Scholar]
- 10. Edwards J., Bartlett J. M. (2005) The androgen receptor and signal-transduction pathways in hormone-refractory prostate cancer. Part 2: androgen-receptor cofactors and bypass pathways. BJU Int. 95, 1327–1335 [DOI] [PubMed] [Google Scholar]
- 11. Wilding G., Chen M., Gelmann E. P. (1989) Aberrant response in vitro of hormone-responsive prostate cancer cells to antiandrogens. Prostate 14, 103–115 [DOI] [PubMed] [Google Scholar]
- 12. Iwasa Y., Mizokami A., Miwa S., Koshida K., Namiki M. (2007) Establishment and characterization of androgen-independent human prostate cancer cell lines, LN-REC4 and LNCaP-SF, from LNCaP. Int. J. Urol. 14, 233–239 [DOI] [PubMed] [Google Scholar]
- 13. Wu T. T., Sikes R. A., Cui Q., Thalmann G .N., Kao C., Murphy C. F., Yang H., Zhau H. E., Balian G., Chung L. W. (1998) Establishing human prostate cancer cell xenografts in bone: induction of osteoblastic reaction by prostate-specific antigen-producing tumors in athymic and SCID/bg mice using LNCaP and lineage-derived metastatic sublines. Int. J. Cancer 77, 887–894 [DOI] [PubMed] [Google Scholar]
- 14. Thalmann G. N., Sikes R. A., Wu T. T., Degeorges A., Chang S. M., Ozen M., Pathak S., Chung L. W. (2000) LNCaP progression model of human prostate cancer: androgen-independence and osseous metastasis. Prostate 44, 91–103 [DOI] [PubMed] [Google Scholar]
- 15. Igawa T., Lin F. F., Lee M. S., Karan D., Batra S. K., Lin M. F. (2002) Establishment and characterization of androgen-independent human prostate cancer LNCaP cell model. Prostate 50, 222–235 [DOI] [PubMed] [Google Scholar]
- 16. Zhou J. R., Yu L., Zerbini L. F., Libermann T. A., Blackburn G. L. (2004) Progression to androgen-independent LNCaP human prostate tumors: cellular and molecular alterations. Int. J. Cancer 110, 800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shi X. B., Ma A. H., Tepper C. G., Xia L., Gregg J. P., Gandour-Edwards R., Mack P. C., Kung H. J., deVere White R. W. (2004) Molecular alterations associated with LNCaP cell progression to androgen independence. Prostate 60, 257–271 [DOI] [PubMed] [Google Scholar]
- 18. Konig J. J., Teubel W., van Steenbrugge G. J., Romijn J. C., Hagemeijer A. (1999) Characterization of chromosome 8 aberrations in the prostate cancer cell line LNCaP-FGC and sublines. Urol. Res. 27, 3–8 [DOI] [PubMed] [Google Scholar]
- 19. Tyson D. R., Ornstein D. K. (2008) Proteomics of cancer of hormone-dependent tissues. Adv. Exp. Med. Biol. 630, 133–147 [DOI] [PubMed] [Google Scholar]
- 20. Zheng Y., Xu Y., Ye B., Lei J., Weinstein M. H., O'Leary M. P., Richie J. P., Mok S. C., Liu B. C. (2003) Prostate carcinoma tissue proteomics for biomarker discovery. Cancer 98, 2576–2582 [DOI] [PubMed] [Google Scholar]
- 21. Sardana G., Jung K., Stephan C., Diamandis E. P. (2008) Proteomic analysis of conditioned media from the PC3, LNCaP, and 22Rv1 prostate cancer cell lines: discovery and validation of candidate prostate cancer biomarkers. J. Proteome Res. 7, 3329–3338 [DOI] [PubMed] [Google Scholar]
- 22. Alaiya A. A., Al-Mohanna M., Aslam M., Shinwari Z., Al-Mansouri L., Al-Rodayan M., Al-Eid M., Ahmad I., Hanash K., Tulbah A., Bin Mahfooz A., Adra C. (2011) Proteomics-based signature for human benign prostate hyperplasia and prostate adenocarcinoma. Int. J. Oncol. 38, 1047–1057 [DOI] [PubMed] [Google Scholar]
- 23. Skvortsov S., Schäfer G., Stasyk T., Fuchsberger C., Bonn G. K., Bartsch G., Klocker H., Huber L. A. (2011) Proteomics profiling of microdissected low- and high-grade prostate tumors identifies Lamin A as a discriminatory biomarker. J. Proteome Res. 10, 259–268 [DOI] [PubMed] [Google Scholar]
- 24. Saraon P., Musrap N., Cretu D., Karagiannis G. S., Batruch I., Smith C., Drabovich A. P., Trudel D., van der Kwast T., Morrissey C., Jarvi K. A., Diamandis E. P. (2012) Proteomic profiling of androgen-independent prostate cancer cell lines reveals a role for protein S during the development of high grade and castrate-resistant prostate cancer. J. Biol. Chem. 287, 34019–34031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khan A. P., Poisson L. M., Bhat V. B., Fermin D., Zhao R., Kalyana-Sundaram S., Michailidis G., Nesvizhskii A. I., Omenn G. S., Chinnaiyan A. M., Sreekumar A. (2010) Quantitative proteomic profiling of prostate cancer reveals a role for miR-128 in prostate cancer. Mol. Cell. Proteomics 9, 298–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Diamandis E. P. (1998) Prostate-specific antigen: its usefulness in clinical medicine. Trends Endocrinol. Metab. 9, 310–316 [DOI] [PubMed] [Google Scholar]
- 27. Sauter E. R., Scott S., Hewett J., Kliethermes B., Ruhlen R. L., Basarakodu K., de la Torre R. (2008) Biomarkers associated with breast cancer are associated with obesity. Cancer Detect. Prev. 32, 149–155 [DOI] [PubMed] [Google Scholar]
- 28. Montgomery R. B., Mostaghel E. A., Vessella R., Hess D. L., Kalhorn T. F., Higano C. S., True L. D., Nelson P. S. (2008) Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 68, 4447–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maresh E. L., Mah V., Alavi M., Horvath S., Bagryanova L., Liebeskind E. S., Knutzen L. A., Zhou Y., Chia D., Liu A. Y., Goodglick L. (2010) Differential expression of anterior gradient gene AGR2 in prostate cancer. BMC Cancer 10, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Touma S. E., Perner S., Rubin M. A., Nanus D. M., Gudas L. J. (2009) Retinoid metabolism and ALDH1A2 (RALDH2) expression are altered in the transgenic adenocarcinoma mouse prostate model. Biochem. Pharmacol. 78, 1127–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Basu G. D., Azorsa D. O., Kiefer J. A., Rojas A. M., Tuzmen S., Barrett M. T., Trent J. M., Kallioniemi O., Mousses S. (2008) Functional evidence implicating S100P in prostate cancer progression. Int. J. Cancer 123, 330–339 [DOI] [PubMed] [Google Scholar]
- 32. Flamand V., Zhao H., Peehl D. M. (2010) Targeting monoamine oxidase A in advanced prostate cancer. J. Cancer Res. Clin. Oncol. 136, 1761–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chang J. W., Nomura D. K., Cravatt B. F. (2011) A potent and selective inhibitor of KIAA1363/AADACL1 that impairs prostate cancer pathogenesis. Chem. Biol. 18, 476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quirós I., Sáinz R. M., Hevia D., García-Suárez O., Astudillo A., Rivas M., Mayo J. C. (2009) Upregulation of manganese superoxide dismutase (SOD2) is a common pathway for neuroendocrine differentiation in prostate cancer cells. Int. J. Cancer 125, 1497–1504 [DOI] [PubMed] [Google Scholar]
- 35. Degraff D. J., Aguiar A. A., Sikes R. A. (2009) Disease evidence for IGFBP-2 as a key player in prostate cancer progression and development of osteosclerotic lesions. Am. J. Transl. Res. 1, 115–130 [PMC free article] [PubMed] [Google Scholar]
- 36. Chung M. K., Kim H. J., Lee Y. S., Han M. E., Yoon S., Baek S. Y., Kim B. S., Kim J. B., Oh S. O. (2010) Hedgehog signaling regulates proliferation of prostate cancer cells via stathmin1. Clin. Exp. Med. 10, 51–57 [DOI] [PubMed] [Google Scholar]
- 37. Chen M. F., Chen W. C., Chang Y. J., Wu C. F., Wu C. T. (2010) Role of DNA methyltransferase 1 in hormone-resistant prostate cancer. J. Mol. Med. 88, 953–962 [DOI] [PubMed] [Google Scholar]
- 38. Zou J. X., Guo L., Revenko A. S., Tepper C. G., Gemo A. T., Kung H. J., Chen H. W. (2009) Androgen-induced coactivator ANCCA mediates specific androgen receptor signaling in prostate cancer. Cancer Res. 69, 3339–3346 [DOI] [PubMed] [Google Scholar]
- 39. Mortezavi A., Hermanns T., Seifert H. H., et al. (2011) KPNA2 expression is an independent adverse predictor of biochemical recurrence after radical prostatectomy. Clin. Cancer Res. 17, 1111–1121 [DOI] [PubMed] [Google Scholar]
- 40. Oram S. W., Ai J., Pagani G. M., Hitchens M. R., Stern J. A., Eggener S., Pins M., Xiao W., Cai X., Haleem R., Jiang F., Pochapsky T. C., Hedstrom L., Wang Z. (2007) Expression and function of the human androgen-responsive gene ADI1 in prostate cancer. Neoplasia 9, 643–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suh J., Rabson A. B. (2004) NF-kappaB activation in human prostate cancer: important mediator or epiphenomenon? J. Cell. Biochem. 91, 100–117 [DOI] [PubMed] [Google Scholar]
- 42. Cohen M. B., Rokhlin O. W. (2009) Mechanisms of prostate cancer cell survival after inhibition of AR expression. J. Cell. Biochem. 10, 363–371 [DOI] [PubMed] [Google Scholar]
- 43. Balistreri C. R., Caruso C., Carruba G., Miceli V., Candore G. (2011) Genotyping of sex hormone-related pathways in benign and malignant human prostate tissues: data of a preliminary study. OMICS 15, 369–374 [DOI] [PubMed] [Google Scholar]
- 44. Wegiel B., Evans S., Hellsten R., Otterbein L. E., Bjartell A., Persson J. L. (2010) Molecular pathways in the progression of hormone-independent and metastatic prostate cancer. Curr. Cancer Drug Targets 10, 392–401 [DOI] [PubMed] [Google Scholar]
- 45. Sankpal U. T., Goodison S., Abdelrahim M., Basha R. (2011) Targeting sp1 transcription factors in prostate cancer therapy. J. Med. Chem. 7, 518–525 [DOI] [PubMed] [Google Scholar]
- 46. Puisac B., Ramos M., Arnedo M., Menao S., Gil-Rodriíguez M. C., Teresa-Rodrigo M. E., Pié A., de Karam J. C., Wesselink J. J., Giménez I., Ramos F. J., Casals N., Gómez-Puertas P., Hegardt F. G., Pié J. (2011) Characterization of splice variants of the genes encoding human mitochondrial HMG-CoA lyase and HMG-CoA synthase, the main enzymes of the ketogenesis pathway. Mol. Biol. Rep. 39, 4777–85 [DOI] [PubMed] [Google Scholar]
- 47. Nakamura M. T., Cheon Y., Li Y., Nara T. Y. (2004) Mechanisms of regulation of gene expression by fatty acids. J. Lipids 39, 1077–1083 [DOI] [PubMed] [Google Scholar]
- 48. Massie C. E., Lynch A., Ramos-Montoya A., Boren J., Stark R., Fazli L., Warren A., Scott H., Madhu B., Sharma N., Bon H., Zecchini V., Smith D. M., Denicola G. M., Mathews N., Osborne M., Hadfield J., Macarthur S., Adryan B., Lyons S. K., Brindle K. M., Griffiths J., Gleave M. E., Rennie P. S., Neal D. E., Mills I. G. (2011) The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 30, 2719–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu Y. (2006) Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 9, 230–234 [DOI] [PubMed] [Google Scholar]
- 50. Benedettini E., Nguyen P., Loda M. (2008) The pathogenesis of prostate cancer: from molecular to metabolic alterations. Diagn. Histopathol. 14, 195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rossi S., Graner E., Febbo P., Weinstein L., Bhattacharya N., Onody T., Bubley G., Balk S., Loda M. (2003) Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol. Cancer Res. 1, 707–715 [PubMed] [Google Scholar]
- 52. Moore S., Knudsen B., True L. D., Hawley S., Etzioni R., Wade C., Gifford D., Coleman I., Nelson P. S. (2005) Loss of stearoyl-CoA desaturase expression is a frequent event in prostate carcinoma. Int. J. Cancer 114, 563–571 [DOI] [PubMed] [Google Scholar]
- 53. Zha S., Ferdinandusse S., Hicks J. L., Denis S., Dunn T. A., Wanders R. J., Luo J., De Marzo A. M., Isaacs W. B. (2005) Peroxisomal branched chain fatty acid beta-oxidation pathway is upregulated in prostate cancer. Prostate 63, 316–323 [DOI] [PubMed] [Google Scholar]
- 54. Luo J., Zha S., Gage W. R., Dunn T. A., Hicks J. L., Bennett C. J., Ewing C. M., Platz E. A., Ferdinandusse S., Wanders R. J., Trent J. M., Isaacs W. B., De Marzo A. M. (2002) Alpha-methylacyl-CoA racemase: a new molecular marker for prostate cancer. Cancer Res. 62, 2220–2226 [PubMed] [Google Scholar]
- 55. Kumar-Sinha C., Shah R. B., Laxman B., Tomlins S. A., Harwood J., Schmitz W., Conzelmann E., Sanda M. G., Wei J. T., Rubin M. A., Chinnaiyan A. M. (2004) Elevated alpha-methylacyl-CoA racemase enzymatic activity in prostate cancer. Am. J. Path. 164, 787–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Locke J. A., Wasan K. M., Nelson C. C., Guns E. S., Leon C. G. (2008) Androgen-mediated cholesterol metabolism in LNCaP and PC-3 cell lines is regulated through two different isoforms of acyl-coenzyme A:cholesterol acyltransferase (ACAT). Prostate 68, 20–33 [DOI] [PubMed] [Google Scholar]
- 57. Leon C. G., Locke J. A., Adomat H. H., Etinger S. L., Twiddy A. L., Neumann R. D., Nelson C. C., Guns E. S., Wasan K. M. (2010) Alterations in cholesterol regulation contribute to the production of intratumoral androgens during progression to castration-resistant prostate cancer in a mouse xenograft model. Prostate 70, 390–400 [DOI] [PubMed] [Google Scholar]
- 58. Leon C., Hill J. S., Wasan K. M. (2005) Potential role of acyl-coenzyme A:cholesterol transferase (ACAT) inhibitors as hypolipidemic and antiatherosclerosis drugs. Pharm. Res. 22, 1578–1588 [DOI] [PubMed] [Google Scholar]
- 59. Yoshinaka Y., Shibata H., Kobayashi H., Kuriyama H., Shibuya K., Tanabe S., Watanabe T., Miyazaki A. (2010) A selective ACAT-1 inhibitor, K-604, stimulates collagen production in cultured smooth muscle cells and alters plaque phenotype in apolipoprotein E-knockout mice. Atherosclerosis 213, 85–91 [DOI] [PubMed] [Google Scholar]
- 60. McKnight S. L. (2010) On getting there from here. Science 330, 1338–1339 [DOI] [PubMed] [Google Scholar]
- 61. Canacci A. M., Izumi K., Zheng Y., Gordetsky J., Yao J. L., Miyamoto H. (2011) Expression of semenogelins I and II and its prognostic significance in human prostate cancer. Prostate 71, 1108–1114 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}