

Abstract
Nuclear DNA in eukaryotic cells is assembled into the hierarchical chromatin structure via a process that is dynamically affected by the combinatorial set of post-translational modifications (PTMs) of histones in a dynamic manner responsive to physiological and environmental changes. The precise quantification of these complex modifications is challenging. Here we present a robust MS-based quantitative proteomics method for studying histone PTMs using 15N metabolically labeled histones as the internal reference. Using this approach, we identified Tetrahymena trithorax related 1 (Txr1p) as a histone methyltransferase in Tetrahymena thermophila and characterized the relationships of the Txr1p and Ezl2p methyltransferases to histone H3 modification. We identified 32 PTMs in more than 60 tryptic peptides from histone H3 of the ciliate model organism Tetrahymena thermophila, and we quantified them (average coefficient of variation: 13%). We examined perturbations to histone modification patterns in two knockout strains of SET-domain-containing histone methyltransferases (HMT). Knockout of TXR1 led to progressively decreased mono-, di-, and tri-methylation of H3K27 and apparent reduced monomethylation of H3K36 in vivo. In contrast, EZL2 knockout resulted in dramatic reductions in both di- and tri-methylation of H3K27 in vivo, whereas the levels of monomethylation of H3K27 increased significantly. This buildup of monomethyl H3K27 is consistent with its role as a substrate for Ezl2p. These results were validated via immunoblotting using modification site-specific antibodies. Taken together, our studies define Txr1p as an H3K27 monomethylation-specific HMT that facilitates the buildup of H3K27 di- and trimethylation by the canonical H3K27-specific HMT, Ezl2p. Our studies also delineate some of the interdependences between various H3 modifications, as compensatory increases in monomethylation at H3K4, H3K23, and H3K56 were also observed for both TXR1 and ELZ2 mutants.
Histones, and especially their N-terminal tails, are subject to various covalent post-translational modifications (PTMs)1 including acetylation, methylation, phosphorylation, ubiquitination, and citrullination (1–3). A combinatorial set of PTMs on one or more histones, deposited by histone-modifying enzymes, effectively serves to modulate various DNA pathways, including gene expression and replication as postulated in the histone code hypothesis (4, 5). Prominent among the PTMs is the reversible epigenetic mark, lysine methylation, present in mono-, di-, and trimethylation states. Methyl groups are added to the ε-amine of the lysyl residue by histone methyltransferases (HMTs) and removed by histone demethylases (6–9). Different lysine methylation states are often associated with different—sometimes even opposite—biological functions. For example, the monomethylation of histone residues H3K9, H3K27, and H4K20 is linked to active transcription, whereas their trimethylation states are associated with transcriptional repression (6, 10). The functional distinction of the different methylation states is further underscored by the presence of divergent state-specific HMTs, such as SETDB1/SETDB2 (for H3K9Me1) and SUV39H1/SUV39H2 (for H3K9Me2 and H3K9Me3) (11), SETD8 (for H4K20Me1) (12), and SUV4–20 (for H4K20Me2 and H4K20Me3) (13). It is a major challenge in the field of epigenetics to unravel the mechanism regulating histone lysine methylation events, which are dynamically affected by many factors and implicated in various biological processes. The modifications of histone lysines are dynamic, and mono-, di-, and trimethylated residues are generally considered to be progressively methylated in vivo (14).
Most HMTs for lysine methylation contain a conserved catalytic domain, the SET (suppressor of variegation, enhancer of zeste, trithorax) domain (15). HMTs, in particular lysine methyltransferases, have been implicated in human diseases, including cancers (16). In the past decade, a large number of HMTs have been identified in a wide range of eukaryotic organisms, and they have been classified according to their sequence homology into subfamilies, whose members generally share the substrate specificity. Arabidopsis trithorax related 5 (ATXR5) and ATXR6, the founding members of a recently identified HMT subfamily, were first isolated as proliferating cell nuclear antigen interacting proteins in Arabidopsis thaliana (17). Both ATXR5 and ATXR6 feature a divergent SET domain (17), a plant homeodomain (PHD) finger that binds the modified histones (18–21), and a proliferating cell nuclear antigen interacting protein box that binds proliferating cell nuclear antigen (22). Homologues of ATXR5 and ATXR6 are found in plants but not in animals. The atxr5 atxr6 double mutant exhibits reduced H3K27Me1 levels (23), supporting the notion that ATXR5 and ATXR6 function as the H3K27 monomethylation-specific HMTs.
Tetrahymena trithorax related 1 (Txr1p) was recently identified as a putative HMT in the ciliate model organism Tetrahymena thermophila (encoded by the TXR1 gene) via homology to Arabidopsis ATXR5 and ATXR6. Txr1p carries two PHD domains (PHD1 and PHD2), one proliferating cell nuclear antigen interacting protein box (QKLIEDYF), and one C-terminal SET domain (Fig. 1), all of which are consistent with its being a bona fide member of the ATXR5/ATXR6 subfamily of HMTs. In Tetrahymena, there are also three homologues of the canonical H3K27-specific HMT enhancer of zeste, referred to as EZL1, EZL2, and EZL3, respectively (24, 25). Only EZL2 is expressed at significant levels (24, 25) and required for H3K27 di- and trimethylation in asexually dividing cells (see below).
Fig. 1.

Domain structure analysis of protein Txr1p. A, the proliferating cell nuclear antigen interacting protein box, the C-terminal SET domain, and two N-terminal PHD domains (PHD1 and PHD2) suggest that Txr1p is a putative HMT homologous to plant ATXR5/ATXR6. B, amino acid sequence alignment of SET domains in Txr1p and Arabidopsis homologues ATXR5 and ATXR6. Identical and similar residues are darkly and lightly shaded, respectively.
Mass spectrometry (MS) has played an important role in the study of histone PTMs for the following reasons: (1) MS is capable of simultaneously monitoring multiple PTMs; and (2) it can identify and quantify known and unknown PTMs in histones that cannot be easily determined via other analytical approaches such as micro-sequencing by Edman degradation or immunoblotting with modification site-specific antibodies (26–30). The quantification of histone PTMs can be achieved via a label-free strategy based on the relative intensities of extracted ion chromatograms of precursors (31) or, more accurately through stable isotope labeling techniques such as SILAC or iTRAQ (32–35). However, the analysis of histone PTMs via LC/MS is particularly challenging because of the enormous number of isoforms generated by the combination of various densely deposited PTMs (29). The problem is further exacerbated by the basicity of histones, which, after trypsin digestion, generate peptides too small or hydrophilic to be effectively retained on reversed-phase HPLC columns and analyzed via MS. Chemical derivatization using propionic anhydride was introduced in order to overcome some of these challenges (36, 37). Briefly, the propionylated histones are only cleaved after arginyl residues when digested with trypsin and thus generate nicely sized, more hydrophobic peptides that can be more readily analyzed via LC/MS. When samples are chemically labeled with light and heavy isotopes (d0/d10-propionic anhydride), the relative levels of individual modifications from the two samples can be quantified by means of MS (32). The deuterium isotope effect on chromatography and the variations in differential labeling from one peptide to another, however, place some limitations on the precise quantification of histone PTMs. If metabolic labeling is used for quantification, variations in propionylation can be minimized because different physiological samples are combined prior to reaction with propionic anhydride.
The major aims of this study were to determine the roles of the Txr1p and Ezl2p methyltransferases in shaping histone modification patterns, with the focus on H3. This was achieved through quantifying the levels of various histone modifications in TXR1 and EZL2 knockout cells, as well as the wild-type cells. For this purpose, we developed a robust MS-based quantitative proteomics method for the study of histone PTMs using 15N metabolically labeled histones as internal standards spiked into histone preparations as references. The general strategy and experimental design for this uniform labeling technique are illustrated in Fig. 2. Similar studies using isotope-labeled tissue or cells as a global internal standard have been carried out in mammals (38, 39). Overall, more than 60 unique H3 tryptic peptides were successfully quantified using this technique with small statistical variation (average coefficient of variation (CV): ∼13%). Similar to Super-SILAC, which combines a mixture of several stable-isotope-labeled cell lines to serve as internal standards for MS-based analysis (40), our method provides a cost-effective alternative for studying protein PTMs, especially in systems for which the SILAC medium is not available or cannot be easily formulated and for which iTRAQ-based quantitative proteomics techniques are not easily applicable.
Fig. 2.
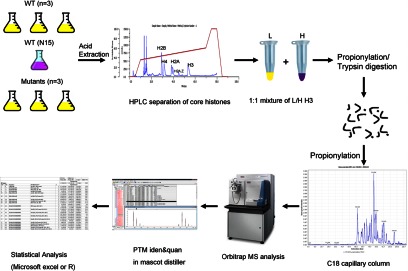
Experimental design. Three wild-type Tetrahymena cells and HMT mutants were normally grown in SPP medium, and one wild-type Tetrahymena cell was metabolically labeled with 15N stable isotopes as a global reference. Once bulk histones were acid extracted, individual core histones were separated via reversed-phase HPLC. The light form and the heavy form of H3 were equally mixed and chemically propionylated before and after trypsin digestion. The digests were resolved using a C18 capillary column before they were analyzed via mass spectrometer. The commercial software Mascot Distiller was used to quantitate PTM changes in wild-type and knockout cells. Finally, normalization and statistical analysis of histone PTMs were performed using Microsoft Excel or R.
EXPERIMENTAL PROCEDURES
Construction of TXR1 and EZL2 Knockout Strains
To generate the TXR1 and ELZ2 knockout constructs, the genomic regions flanking TXR1 or ELZ2 were PCR amplified from the wild-type Tetrahymena cells and fused with the neo4 cassette (41), which confers paromomycin resistance to Tetrahymena cells. The constructs were introduced into Tetrahymena cells via standard biolistic transformations (42). Transformants were selected for paromomycin resistance and complete replacement was finally confirmed via quantitative PCR.
Cell Culture and 15 N Metabolic Labeling of Tetrahymena Cellular Proteome
Tetrahymena thermophila wild-type strain CU428 (Tetrahymena Stock Center) and TXR1 and ELZ2 knockout strains were grown in 1× SPP medium (2% protease peptone, 0.2% dextrose, 0.1% yeast extract, 0.003% sequestrine) at 30 °C with gentle shaking. Logarithmic-phase cells (2 × 105/ml) were collected for subsequent experiments.
For 15N labeling of wild-type Tetrahymena cells, 15N-labeled Escherichia coli BL21 cells were grown in [15N] M9 minimal medium (30 mm Na2HPO4, 2 g/l KH2PO4, 0.5 g/l NaCl, 300 μm Na2SO4, 1 mm MgSO4, 0.3 mm CaCl2, 1 μg/ml biotin, 1 μg/ml thiamine, 10 g/l glucose, and 1 g/l (15NH4)2SO4 (Cambridge Isotope Laboratories, Andover, MA)) supplemented with 15N-substituted Bioexpress (Cambridge Isotope Laboratories). Briefly, E. coli BL21 cells were inoculated into a small LB starter culture and incubated at 37 °C with vigorous shaking until reaching the logarithmic phase (OD600: 0.5 to 1). 1.0 ml of the starter culture was inoculated into 500 ml of the [15N] M9 media and incubated overnight at 37 °C with vigorous shaking. Stationary-phase E. coli BL21 cells were collected via centrifugation, and the cell pellet was resuspended in 500 ml of 1× phosphate buffer (0.2 g/l KCl, 1.15 g/l Na2HPO4, 0.2 g/l KH2PO4). Added into this labeling medium were inoculated Tetrahymena cells from a logarithmic-phase small starter culture grown in 1× SPP medium. The culture was incubated at 30 °C with gentle shaking for 48 h, with 15N-labeled E. coli BL21 cells as the only nitrogen source. The labeled Tetrahymena cells were collected via centrifugation for subsequent nuclear preparations.
Nuclear Preparation, Histone Acid Extraction, and HPLC Purification
The procedure for isolating the macronuclei from Tetrahymena cells was adapted from a previously reported protocol (43). Briefly, Tetrahymena cells were resuspended in 200 ml of medium A (0.1 m sucrose, 2 mm MgCl2, 4% gum arabic, 10 mm Tris, 5 mm EDTA, 10 mm butyric acid, 1 mm iodoacetamide, 1 mm PMSF, adjusted to pH 6.5). Cells were disrupted via vigorous blending in the presence of 1-octanol (0.7 ml). Tetrahymena macronuclei were pelleted by means of differential centrifugation. They were then acid extracted with 1 ml of 0.4 N sulfuric acid, as reported elsewhere (44). The acid-extracted histones were precipitated by TCA (20% w/v). After being washed once with acidified actone (0.2% HCl) and once with acetone, the histone samples were air-dried and resuspended in 500 μl of water.
The histone samples were further purified on a C8 reversed-phase HPLC column (Vydac Part No. 208TP54, 250 mm × 4.6 mm) on a Rainin Rabbit HPLC with 5 ml/min pump heads, with the HPLC run conditions as reported elsewhere (45). Briefly, the HPLC column was equilibrated with 100% solvent A (5% acetonitrile in 0.1% TFA) for 5 min, and then 35% solvent B was added (90% acetonitrile in 0.1% TFA) and the column was equilibrated for another 5 min. A 60-min gradient to 65% solvent B was applied to elute core histones from the column. Finally, the column was washed with 100% solvent B for 10 min and then re-equilibrated with 100% solvent A. HPLC fractions were vacuum dried, resuspended in deionized water, and evaluated via 15% SDS-PAGE, and those containing individual histones were combined. Concentrations of the purified histones were determined via the Bradford method (Bio-Rad).
Chemical Derivatization, Protein Digestion, and Quantitative MS Analysis of Histone PTMs
For each biological replicate (n = 3), 5 μg of histone H3 from wild-type, ΔTXR1, or ΔEZL2 cells grown in 1× SPP medium was mixed with an equal amount of 15N-labeled H3 separated and purified from wild-type cells. The two-step chemical derivatization of histone H3 with propionic anhydride, adopted from the work of Garcia et al. (37), was performed before and after trypsin digestion to increase the hydrophobicity of tryptic peptides, as histones are very basic proteins with relatively short retention times on a reversed-phase column. Briefly, dried histone H3 samples were resuspended in 5 μl 100 mm ammonium bicarbonate. Samples were then treated with 20 μl propionylation reagent made with 3:1 (v/v) anhydrous methanol (Alfa Aesar, Ward Hill, MA):propionic anhydride (Sigma Aldrich), and this was immediately followed by the addition of ∼15 μl ammonium hydroxide (Sigma Aldrich) to raise the pH to 8.0. The reaction mixtures were incubated for 15 min at 50 °C and then concentrated to ∼5 μl in a SpeedVac concentrator. The propionylation reaction was performed twice to ensure the maximum conversion of primary amines to propionyl amides. Generally, more than 95% propionylation efficiency was achieved after two rounds of chemical derivatization. No significant evidence of Asn or Gln deamidation (a potential side reaction of propionylation) for H3 peptides was found through database searches. Propionylated samples were again brought up in 100 mm ammonium bicarbonate buffer and in-solution digested with sequencing-grade trypsin (Promega, Madison, WI) at a ratio of 1:20 (enzyme:substrate) at 37 °C for 6 h. The reactions were quenched by TFA (10% w/v). A second round of propionylation was performed as described above to convert the newly generated N termini to propionyl amides. Finally, the reaction mixture was vacuum dried and reconstituted in 0.1% formic acid. After filtering (Millipore Ultracel YM-10), the sample was stored in a −20° Celsius freezer until the MS analysis.
The histone-derived peptides were resolved with a C18 capillary column (3 μm, 300 Å, 150 mm × 100 μm; CVC Technologies, Fontana, CA) on an Eksigent HPLC with a non-ferrous solvent path. A linear gradient for peptide separation was used as follows: run 100% solvent A (HPLC-grade water with 0.1% formic acid) over 5 min; run a 0% to 40% gradient against solvent B (90% acetonitrile in 0.1% formic acid) over 90 min; and finally run 10 min 100% solvent B followed by 15 min re-equilibrium with 100% solvent A. The resolved peptides were then introduced into a Thermo Fisher Orbitrap XL mass spectrometer at a 200 nl/min flow rate connected to a nano-ESI source operated in positive ion mode. The mass spectrometer was operated in data dependent mode at a resolution of 30,000 on MS1, followed by eight collision-induced dissociation tandem MS with 30% normalized collision energy. All precursor ions were placed on a dynamic exclusion list for 120 s. Two polydimethylcyclosiloxane ions (m/z = 429.088735 and 445.12002, respectively) were selected for internal mass calibration.
Raw data were processed in Mascot distiller (version 2.4, Matrix Sciences, London, UK), and spectra were searched against the NCBI Tetrahymena database using the Mascot search engine (version 2.2.07, Matrix Sciences). A false discovery rate was estimated from the protein decoy database. The mass error tolerance was 10 ppm for the precursor ion and 0.8 Da for the fragment ions. N-terminal propionylation was set as a fixed modification, and variable modifications were as follows: acetylation (K), methylation (KR), propionylation (K), monomethylation and propionylation (K, +70 Da), and phosphorylation (STY). We allowed up to five missed cleavages for trypsin digestion to compensate for the expected missed cleavages due to propionylation and PTMs on lysyl residues. Peptides were analyzed and quantified using the 15N metabolic labeling method in the Mascot Distiller software. Peptide ratios were normalized against total histone H3, as the light and heavy forms were equally loaded based on the protein quantification (Bradford method) of histone samples and standards. In detail, all replicates were normalized based on the weighted average of the ratios of all H3 peptides according to their intensities. That is, normalization forced the average peptide ratio of light to heavy forms to be 1. Statistical analysis was done in Microsoft Excel or R. All spectra assigned with PTMs were manually validated based on the following criteria: (1) the most abundant ions should be assigned as b or y ions; (2) they should have more than three spectra observed; (3) generally, there were at least three consecutive peaks identified bracketing an assigned PTM residue; (4) the precursor has a mass error less than 6 ppm, and the fragment ion mass accuracy is less than 0.8 Da; and (5) rules such as the proline effect, the loss of water on ST, and the loss of ammonia on K,R,N,Q were also taken into consideration.
Immuno-blot Analysis of H3K27 Methylation and Acetylation
Wild-type, ΔTXR1, or ΔEZL2 histone H3 were resolved via 15% SDS-PAGE and transferred onto the Immobilon® P membrane (Millipore). Blots were then washed, blocked with nonfat dry milk, and incubated overnight with rabbit polyclonal antibodies to H3K27Me1 (1:5000, Millipore, catalog number 07–448), H3K27Me2 (1:5000, Millipore, catalog number 07–452), and H3K27Me3 (1:5000, Millipore, catalog number 07–449) and mouse monoclonal antibody to H3K27Ac (1:50000, Wako Chemicals, Richmond, VA catalog number 306–3484). Blots were washed and then incubated with appropriate peroxidase-labeled secondary antibodies. Finally, the signal was visualized and processed with the Bio-Rad imaging system after being developed using the ECL chemiluminescent reagent (GE Healthcare).
RESULTS AND DISCUSSION
We developed an MS-based quantitative proteomics method based on spiking uniformly 15N-labeled histones into normally 14N-labeled histone preparations to allow quantitative analysis of histone PTMs in Tetrahymena thermophila. In this method, wild-type Tetrahymena cells were metabolically labeled with 15N stable isotopes as described in “Experimental Procedures,” and the 15N-labeled histones were purified as described. In parallel, wild-type, ΔTXR1, and ΔEZL2 Tetrahymena cells were grown in the standard medium without 15N and the histones were similarly purified. Purified histone H3 from wild-type or knockout cells was spiked with 15N-labeled histone H3, which served as an internal reference in the experiment. This allowed a single preparation of 15N-labeled histones to be used across many biological experiments with normally labeled histones, providing a common standard for quantification and minimizing one potential source of variation. The peptide ratios could be precisely determined from the isotope distributions of the light and heavy forms of the tryptic peptides. As both forms were treated under the same experimental conditions and 15N-labeled H3 was equally spiked across all biological replicates prior to propionylation and trypsinization, variations of chemical reactions were minimized, leading to a more accurate determination of the ratios. Finally, the peptide ratio of wild-type versus ΔTXR1 or ΔEZL2 cell could be calculated from each ratio of light versus heavy peptide. The overall experimental procedure is outlined in Fig. 2.
There are several reasons that we used this uniform metabolic labeling strategy instead of the general metabolic labeling technique in which cellular proteomes from two different physiological states are directly compared with each other as they are grown in chemically identical media. SILAC medium is not commercially available and cannot be easily prepared for the growth of Tetrahymena cells. In addition, Tetrahymena cells grow poorly in chemically defined media. Because Tetrahymena can feed on bacteria, we achieved the metabolic incorporation of 15N stable isotopes from E. coli grown with 15N ammonium sulfate as the sole nitrogen source. However, the preparation of 15N-labeled E. coli and the routine use of those cells as the food source for binary experiments on Tetrahymena histones would be expensive and would contribute to variability in labeling. In addition, growing Tetrahymena cells on E. coli greatly slows their growth rates and can potentially change the histone modification patterns. In our method, both control (wild-type) and knockout cells were grown in a standard medium without bacteria, and peptides in each group were compared with the 15N labeled histones, which were equally spiked into the samples across all experiments. This provides an internal reference for histones from different groups to be compared with. It also removes the practical limitation in commercial SILAC on the number of different types of cells to be compared, which is a result of the fact that only a triplex experiment can be performed.
False Discovery Rate, Sequence Coverage, and Number of PTMs Identified and Quantified in Histone H3
Overall, the false discovery rate of peptide identifications evaluated from the decoy database is below 1% at the Mascot identity threshold and less than 3% at the homology threshold (see supplemental Table S1). The sequence coverage generated from trypsin digestion is 76% for histone H3 (Fig. 3), which includes the major H3 and two minor variants, H3.3 and H3.4 (46, 47). A total of 64 chemically unique peptides have been successfully identified and statistically quantified in histone H3, 22 of which are unique to the minor variant H3.3. A total of 32 PTMs were successfully identified and localized in the major H3, H3.3, and H3.4 forms, and 18 of these were statistically quantified. The PTMs identified in this study were K4Me1, K9Ac, K14Ac, K14Me1, K18Ac, K23Ac, K23Me1, K23Me2, K23Me3, K27Me1, K27Me2, K27Me3, K27Ac, K36Me1, K36Me2, R40Me2, K56Me1, R83Me1, and K79Me1 in the major H3 histone; K9Ac, K14Ac, K18Ac, K23Ac, K23Me2, K27Me1, K27Me2, K36Me1, K36Me2, K37Me3, and K56Ac in H3.3; and K36Ac and K36Me1 in H3.4 (Fig. 3). The peptides containing H3K4 were short and hydrophilic, and thus were poorly retained in the reversed-phase HPLC column and scarcely recovered via MS. This accounts for the discrepancy between the well-documented presence of high levels of H3K4 methylation (48) and the low representation in our MS analysis. Lists of all peptides and PTMs statistically quantified in the major H3 and H3.3 are shown in Table I and supplemental Table S2, respectively. All PTMs identified from wild-type and ΔTXR1 cells and their modification site information in all three H3 variants are listed in supplemental Table S3, and their corresponding tandem mass spectra are shown in supplemental Fig. S1. Complete information on all peptides identified is provided in supplemental Table S6 in Excel spreadsheet format.
Fig. 3.

Histone H3 variants in Tetrahymena thermophila. About 76% sequence coverage (bold letters) was achieved in all three histone H3 variants. Sequence variations in these variants are shown in rectangular frames. A total of 32 PTMs identified in three variants are labeled by different types of marks.
Table I. Quantification of all valid peptides and PTMs in histone H3 major form.
| WT/15N WT |
TXR1/15N WT |
p valuea | Final ratio |
Normalized |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Mean (S.D.) | Mean (S.D.) | TXR1/WT | TXR1/WT | ||||||
| H3 major | Protein-level ratio |
||||||||
| 1.26 (0.03) | 1.01 (0.19) | 0.13464 | 0.80 | 1.07 | |||||
| Start | End | Sequence | Modificationsb | z | Peptide ratio-level ratios | ||||
| 3 | 8 | TKQTAR | Me1(K4); Pr(N-term) | 2 | 0.74 (0.06) | 0.93 (0.04) | 0.01503 | 1.26 | 1.78 |
| 9 | 17 | KSTGAKAPR | Pr(N-term, K14) | 2 | 0.94 (0.04) | 0.68 (0.09) | 0.022982 | 0.73 | 1.02 |
| 9 | 17 | KSTGAKAPR | Pr(N-term, K9, K14) | 1 | 0.82 (0.04) | 0.53 (0.05) | 0.002279 | 0.65 | 0.91 |
| 9 | 17 | KSTGAKAPR | Ac(K14); Pr(N-term) | 2 | 1.40 (0.35) | 1.25 (0.11) | 0.538428 | 0.89 | 1.24 |
| 18 | 26 | KQLASKAAR | Ac(K18, K23); Pr(N-term) | 1 | 1.19 (0.08) | 1.17 (0.13) | 0.816231 | 0.98 | 1.36 |
| 2 | 1.15 (0.04) | 1.16 (0.12) | 0.872236 | 1.01 | 1.41 | ||||
| 18 | 26 | KQLASKAAR | Ac(K23); Pr(N-term, K18) | 1 | 0.98 (0.04) | 0.90 (0.11) | 0.322315 | 0.91 | 1.27 |
| 2 | 0.99 (0.03) | 0.96 (0.11) | 0.685553 | 0.97 | 1.35 | ||||
| 18 | 26 | KQLASKAAR | Ac(K18); Pr(N-term, K23) | 2 | 0.99 (0.03) | 0.96 (0.10) | 0.669132 | 0.97 | 1.35 |
| 18 | 26 | KQLASKAAR | Me1(K23); Pr(N-term, K18, K23) | 2 | 0.63 (0.17) | 1.13 (0.09) | 0.019933 | 1.79 | 2.52 |
| 18 | 26 | KQLASKAAR | Pr(N-term, K18, K23) | 1 | 1.18 (0.05) | 0.87 (0.10) | 0.016895 | 0.74 | 1.02 |
| 2 | 1.20 (0.05) | 0.96 (0.17) | 0.126337 | 0.80 | 1.12 | ||||
| 27 | 40 | KSAPATGGIKKPHR | Pr(N-term, K36, K37) | 2 | 2.16 (0.40) | 5.25 (1.03) | 0.023132 | 2.43 | 3.34 |
| 3 | 2.18 (0.48) | 5.20 (0.98) | 0.018246 | 2.39 | 3.29 | ||||
| 27 | 40 | KSAPATGGIKKPHR | Me1(K27); Pr(N-term, K36, K37) | 2 | 1.36 (0.05) | 0.25 (0.03) | 8.29 × 10−5 | 0.19 | 0.26 |
| 3 | 1.30 (0.04) | 0.27 (0.03) | 1.47 × 10−5 | 0.21 | 0.29 | ||||
| 27 | 40 | KSAPATGGIKKPHR | Me2(K27); Pr(N-term, K36, K37) | 2 | 0.98 (0.08) | 0.40 (0.05) | 0.000824 | 0.41 | 0.56 |
| 3 | 0.87 (0.03) | 0.34 (0.04) | 0.000115 | 0.40 | 0.55 | ||||
| 27 | 40 | KSAPATGGIKKPHR | Me3(K27); Pr(N-term, K36, K37) | 2 | 0.63 (0.03) | 0.44 (0.04) | 0.040226 | 0.70 | 0.92 |
| 3 | 0.61 (0.04) | 0.38 (0.05) | 0.005474 | 0.63 | 0.91 | ||||
| 27 | 40 | KSAPATGGIKKPHR | Me1(K27, K36); Pr(N-term, K36, K37) | 2 | 0.59 (0.22) | 0.05 (0.02) | 0.049941 | 0.09 | 0.13 |
| 3 | 0.74 (0.08) | 0.06 (0.00) | 0.004257 | 0.08 | 0.11 | ||||
| 27 | 40 | KSAPATGGIKKPHR | Ac(K27); Pr(N-term, K36, K37) | 2 | 0.99 (0.10) | 1.06 (0.09) | 0.37916 | 1.08 | 1.50 |
| 27 | 40 | KSAPATGGIKKPHR | Me1(K27); Pr(N-term, K27, K36, K37) | 2 | 1.35 (0.04) | 0.18 (0.02) | 4.17 × 10−5 | 0.13 | 0.18 |
| 27 | 40 | KSAPATGGIKKPHR | Pr(N-term, K27, K36, K37) | 2 | 2.19 (0.43) | 4.98 (0.92) | 0.020405 | 2.27 | 3.12 |
| 27 | 40 | KSAPATGGIKKPHR | Me1(K36); Pr(N-term, K27, K36, K37) | 2 | 1.30 (0.09) | 0.15 (0.01) | 0.001829 | 0.12 | 0.16 |
| 27 | 40 | KSAPATGGIKKPHR | Me1(K27, K36); Pr(N-term, K27, K36, K37) | 2 | 0.79 (0.10) | 0.06 (0.00) | 0.005714 | 0.08 | 0.11 |
| 27 | 40 | KSAPATGGIKKPHR | Me1(K36); Me2(K27); Pr(N-term, K36, K37) | 3 | 0.58 (0.08) | 0.37 (0.02) | 0.160328 | 0.64 | 1.02 |
| 28 | 40 | SAPATGGIKKPHR | Pr(N-term, K36, K37) | 2 | 4.92 (0.82) | 4.98 (2.56) | 0.978241 | 1.01 | 1.24 |
| 41 | 49 | FRPGTVALR | Pr(N-term) | 2 | 1.31 (0.15) | 1.01 (0.17) | 0.087001 | 0.77 | 1.07 |
| 53 | 63 | KYQKSTDLLIR | Pr(N-term, K56) | 2 | 1.24 (0.12) | 1.00 (0.15) | 0.093877 | 0.80 | 1.11 |
| 53 | 63 | KYQKSTDLLIR | Me1(K56); Pr(N-term, K56) | 2 | 0.66 (0.14) | 1.17 (0.04) | 0.019107 | 1.77 | 2.49 |
| 54 | 63 | YQKSTDLLIR | Pr(N-term) | 1 | 1.13 (0.01) | 0.87 (0.01) | 0.394434 | 0.77 | 0.90 |
| 2 | 1.38 (0.21) | 1.01 (0.40) | 0.001585 | 0.73 | 1.23 | ||||
| 70 | 83 | LVRDIAHEFKAELR | Pr(N-term, K79) | 2 | 1.06 (0.19) | 1.02 (0.16) | 0.825064 | 0.96 | 1.34 |
| 73 | 83 | DIAHEFKAELR | Pr(N-term, K79) | 1 | 1.26 (0.24) | 1.04 (0.24) | 0.569269 | 0.83 | 1.26 |
| 2 | 1.13 (0.24) | 1.03 (0.15) | 0.461435 | 0.91 | 1.06 | ||||
| 73 | 83 | DIAHEFKAELR | Me1(R83); Pr(N-term, K79) | 2 | 1.16 (0.03) | 1.06 (0.25) | 0.675039 | 0.92 | 1.17 |
| 116 | 128 | RVTIMTKDMQLAR | Pr(N-term, K122) | 2 | 0.99 (0.30) | 0.99 (0.12) | 0.746262 | 1.00 | 1.30 |
| 3 | 1.00 (0.35) | 0.92 (0.12) | 0.982936 | 0.92 | 1.39 | ||||
| 117 | 128 | VTIMTKDMQLAR | Pr(N-term, K122) | 1 | 1.05 (0.25) | 1.10 (0.19) | 0.808235 | 1.05 | 1.45 |
| 2 | 1.07 (0.24) | 1.06 (0.17) | 0.959774 | 0.99 | 1.37 | ||||
a Species with significant p values (p < 0.05) are boxed. The p value is calculated from peptides that are present at least twice in replicate wild-type (WT) or knockout cells. The average coefficients of variation for peptides from histone H3 in WT and TXR1 knockout cells are 12.7% and 14.2%, respectively.
b PTM abbreviations: Ac, acetylation; Me, monomethylation; Me2, dimethylation; Me3, trimethylation; Pr, propionylation.
15N-labeled versus 14N-labeled Histone H3
(15NH4)2SO4 was used as the sole nitrogen source for metabolically labeling E. coli and, ultimately, Tetrahymena. Each protein or peptide incorporates a variable number of heavy nitrogen atoms, depending on the length and composition of amino acids present in the protein or peptide (49). We then separated Tetrahymena cells from the bacteria, collected nuclei, extracted bulk histones from the nuclei, and isolated and purified individual histones using reversed-phase HPLC as we did in wild-type and knockout cells. The ratios of heavy versus light peptides calculated using Mascot Distiller software were mostly over 99% (supplemental Fig. S2, supplemental Table S4), indicating that metabolic labeling was highly efficient. This was also confirmed by the Mascot search results, which indicated that very few peptides could be identified as unlabeled.
In the next step, wild-type histone H3 from Tetrahymena grown on light medium was mixed with an equal amount of 15N-labeled histone H3. The ratios of light/heavy histone H3 were evaluated in three biological replicates. Only peptides having p values less than 0.05 with a 20% threshold change from unity were considered as having changed significantly in the mutants. MS analysis of intact histone H3 also suggested that gross changes in histone modifications did not occur between heavy and light isotope forms of histone H3 (see supplemental Fig. S3). More important, most of the peptides were quantified with relatively small statistical variations (an average CV of ∼12.7% was observed for the wild-type cells). Note that two final ratios are provided in the tables to aid in validation: raw ratios and normalized ratios that compensate for small differences in histone levels between experiments (the normalized ratios are used in this discussion). Both the p values and the CVs are reported for each experiment. The former are useful for identifying ratios that have changed significantly, but they are less useful for those that have not; for those, the CVs are more useful. A few peptides had relatively large CVs, which may be ascribed to low intensity spectra or trypsin cleavage variability (e.g. at -RR- sites where the products may be -R, -RR, R-, and -).
Relative Quantification of Histone H3 PTMs in Wild-type and ΔTXR1 Cells
As illustrated in Fig. 2, the light forms of histone H3 purified from wild-type and ΔTXR1 cells were each equally mixed with the heavy form of histone H3. Both wild-type and ΔTXR1 cells were evaluated in three biological replicates. The statistical differences in the ratios of light/heavy peptides from each group can be inferred from the biological replicates, and the final ratios of peptides in each group can be directly converted from their ratios of light versus heavy forms. Most of the peptides in ΔTXR1 cells have ratios similar to their counterparts in wild-type cells (Table I, supplemental Table S2). Most of the peptides also exhibited small statistical variations within each group, with average CVs of 12.7% and 14.2% achieved for wild-type and ΔTXR1, respectively. Furthermore, the peptide ratios were quite consistent for different charge states or different degrees of propionylation such as KQLASKAAR, [Ac]KQLAS[Ac]KAAR, KQLAS[Ac]KAAR, KSAPATGGIKKPHR, [Me1]KSAPATGGIKKPHR, [Me2]KSAPATGGIKKPHR, [Me3]KSAPATGGIKKPHR, etc. On the whole, the use of uniformly labeled internal histone standards provides a robust, reproducible, MS-based quantitative proteomics method for studying histone PTMs in this species.
TXR1 Knockout Leads to Dramatic Reduction of H3K27Me1
The TXR1 gene encodes a putative HMT as suggested from sequence homology and domain structure (Fig. 1), but its substrate specificity was unproven. Quantitative analyses of the PTM patterns of histone H3 were performed in wild-type and ΔTXR1 cells in order to ascertain the relative effects of the TXR1 knockout on H3 modification patterns. We found that monomethylation of H3K27 (pr/me1K27SAPATGGIprK36prK37PHR)in the major H3 was significantly decreased (∼70%, p = 8.29× 10−5; see Fig. 4A). In addition, dimethylation of H3K27 (pr/me2K27SAPATGGIprK36prK37PHR) was decreased by ∼40% (Fig. 4B), and trimethylation of H3K27 (pr/Me3K27SAPATGGIprK36prK37PHR) was slightly depressed, by ∼10% (Fig. 4C). Note that the significant amount of trimethylation at K27 remaining in the TXR1 mutant indicates that although the monomethylation of K27 by Txr1p contributes to the pool, it is not the sole source of substrate for dimethyl- and trimethyl-H3K27. The corresponding unmodified peptides (prK27SAPATGGIprK36prK37PHR, 2prK27SAPATGGIprK36prK37PHR) increased in ratio in ΔTXR1 cells, consistent with under-methylation at K27. These results are summarized in Table I. Monomethylation of H3K36 (2prK27SAPATGGIpr/me1K36prK37PHR) also appeared to be reduced in ΔTXR1 cells (Fig. 4D). However, as this peptide had the same m/z ratio and partially co-eluted with the peptide monomethylated at K27 (pr/me1K27SAPATGGIprK36prK37PHR) on the C18 analytical column, we were not able to unambiguously conclude from this peptide that H3K36Me1 was also affected by TXR1 knockout. Nonetheless, we also observed that a peptide monomethylated at both K27 and K36 (2pr/me1K27SAPATGGIpr/me1K36prK37PHR) had the largest decrease in ΔTXR1 cells (with a ratio of 0.11; see Fig. 4E), which is consistent with H3K36Me1 also being a target of Txr1p. Monomethylation of H3K27 in the H3.3 variant was also significantly reduced (by 60%), as suggested from the diagnostic peptide pr/me1K27SAPVSGGVprK36prK37PHprK40FRPGTVALR, unique to H3.3 (see supplemental Table S2). The corresponding unmodified peptide (prK27SAPVSGGVprK36prK37PHprK40FRPGTVALR) ratio was increased, which is also consistent with under-methylation of H3.3 K27 in ΔTXR1 cells.
Fig. 4.

Analysis of Lys27 and Lys36 methylation via quantitative mass spectrometry. Monomethylation of H3K27 is significantly down-regulated by ∼70% in ΔTXR1 cells (A), dimethylation of H3K27 is decreased by ∼40% (B), and trimethylation is decreased by ∼10% (C). Monomethylation of H3K36 might also be significantly affected, as suggested in D and E.
Additional changes in PTMs were also observed and are summarized in Table I. Monomethylation of H3K4, H3K23, and H3K56 was increased in ΔTXR1 cells (1.8, 2.5, and 2.5, respectively). Increased off-site methylation was also observed for H3K4 and H3K23 in the ΔEZL2 cells (see below). All this might represent either a compensatory mechanism or an indirect response. Acetylation levels of H3 appeared to be essentially unchanged in both ΔTXR1 and ΔEZL2 cells.
Ezl2p Is a Histone Methyltransferase Specific for H3K27Me2/Me3, and Its Activity Is Not Tightly Coupled to Txr1p
Based on earlier studies (24, 25), Ezl2p is one of the homologues of the Drosophila enhancer of zeste, which is specific for H3K27 methylation. We applied the same strategy used for ΔTXR1 to ΔEZL2 for analysis of the global PTM profile changes in histone H3. In ΔEZL2 cells, a significant decrease in H3K27 di- and trimethylation (by >80%; p < 0.01) and a significant increase in H3K27 monomethylation (by >70%; p < 0.01) were observed (supplemental Table S5), presumably because the monomethylation state was no longer being converted to higher methylation states efficiently. Note that EZL2 knockout did not completely eliminate H3K27 di- and trimethylation, suggesting the presence of an alternative pathway. EZL2 knockout also led to increased monomethylation at K4 and K23, as we observed in ΔTXR1 cells (see above).
Principal component analysis (PCA) and a PCA biplot analysis of histone PTM data generated from wild-type and knockout cells revealed that ΔTXR1 and ΔEZL2 were not tightly correlated in their changes in H3 modification patterns (Fig. 5; note that cosines of angles between vectors reflect relationships between variables). This analysis is consistent with the observation that although Ezl2p may utilize monomethyl-H3K27 generated by Txr1p, this is not the sole feedstock for synthesis of dimethyl- and trimethyl-H3K27. Ezl2p might be the major source of trimethyl H3K27, but these observations are consistent with multiple pathways for the formation of trimethyl-H3K27. PCA analysis also revealed that di- and trimethylation of K27 were closely correlated, but they were less correlated with monomethylation of K27, suggesting that the three methylation states of K27 might be differentially regulated in Tetrahymena.
Fig. 5.
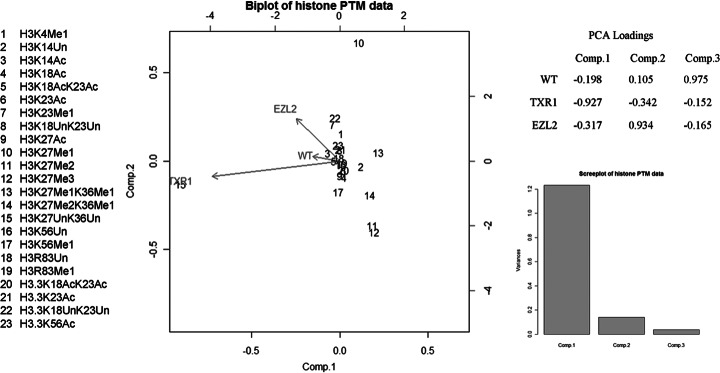
PCA biplot of histone PTM data. PCA and PCA biplot analysis of histone PTM data suggest that ΔTXR1 and ΔEZL2 act independently in the regulation of H3K27 mono-, di-, and trimethylation states. Di- and trimethylation of K27 are closely co-regulated and have a relationship with the monomethylation of K27.
Validation of the Changes in H3K27 Methylation States via Immunoblotting
Following LC/MS analysis, we performed immunoblotting analysis to validate the changes in H3K27 methylation states in wild-type, ΔTXR1, and ΔEZL2 cells with site-specific antibodies recognizing mono-, di-, and trimethylated H3K27 (Fig. 6). As controls, we also included antibodies against H3K27Ac and general H3 (no bias for modifications). The TXR1 knockout exhibited attenuation of mono-, di-, and trimethylation at H3K27, with the strongest effects being on monomethyl- and dimethyl-K27, and weak attenuation of trimethyl-K27. In contrast, the ELZ2 knockout showed greatly reduced H3K27Me2 and H3K27Me3 levels but significantly increased H3K27Me1 levels. Little effect was observed for H3K27Ac. The results are consistent with the MS-based quantification. Commercial antibodies against H3K36Me1 failed to detect the modification in Tetrahymena (data not shown), probably because of the significant sequence divergence around K36 in histone H3 of Tetrahymena and higher eukaryotes.
Fig. 6.
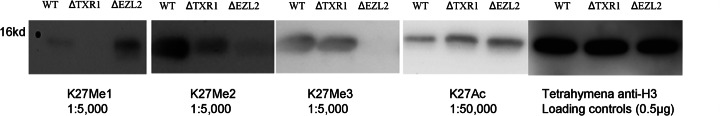
Validation of PTM expression by means of Western blot. Rabbit polyclonal antibodies to H3K27Me1 (1:5000), H3K27Me2 (1:5000), and H3K27Me3 (1:5000) and mouse monoclonal antibody to H3K27Ac (1:50,000) were used to validate the PTM changes in wild-type and knockout cells with a 10-min exposure. The amount of H3 loaded was 0.5 μg. The elimination of TXR1 shows a significant reduction of H3K27Me1/Me2, whereas the knockout of ELZ2 leads to a reduction in H3K27Me2 and an undetectable signal in H3K27Me3. These results are consistent with the data from mass spectrometric quantification.
It is noteworthy that all unmodified peptides in the variant H3.3 were found to be up-regulated in ΔTXR1 cells, and the increase in the H3.3 protein ratio in ΔTXR1 versus wild-type was around 2.5 (see supplemental Table S1), suggesting that knockout of TXR1 induces the overexpression of H3.3, possibly in an effort to compensate for the loss of H3K27Me1. However, no significant change in H3.3 was observed in ΔEZL2 cells. It is also worth noting, with regard to compensatory mechanisms, that ΔTXR1 cells also exhibit increases in H3 monomethylation at K4, K23, and K56.
Conclusions and Implications
In this study, we successfully characterized the global PTM changes in histone H3 of wild-type, ΔTXR1, and ΔEZL2 Tetrahymena cells. The application of our modified 15N uniform labeling technique allows the identification and quantification of a total of 64 H3-derived peptides, which covers 18 PTMs in the major H3 and its minor variant H3.3, with acceptable coefficients of variation. Our quantitative proteomics data, along with the immunoblotting validation, constitute a body of evidence strongly supporting the idea that Txr1p is an HMT mainly responsible for H3K27Me1 in Tetrahymena. The result is in line with Txr1p being a member of the ATXR5/ATXR6 subfamily of HMTs (23). Indeed, our work provides the first evidence of the ancient origin in evolution and functional conservation of the ATXR5/ATXR6 subfamily. Interestingly, TXR1 knockout also can lead to a significant reduction in H3K36Me1, potentially making H3K36 an alternative modification site for Txr1p and this subfamily of HMTs.
In addition, we have also defined the distinct roles of Txr1p and Ezl2p in regulating H3K27 methylation states. TXR1 knockout exhibits a significant effect on H3K27Me1 levels, which we assume is direct. The diminishing effects on H3K27Me2 and H3K27Me3, as well the increase in unmodified H3K27 levels, are most likely the indirect consequences of the inhibition of monomethylation of H3K27. In contrast to TXR1 knockout, EZL2 knockout greatly reduces levels of H3K27Me2 and H3K27Me3 while significantly increasing levels of H3K27Me1. Apparently, Ezl2p can build up H3K27Me2 and H3K27Me3 levels, utilizing H3K27Me1 deposited by Txr1p. Most likely, Ezl2p can also directly monomethylate H3K27, which would make it responsible for the remaining H3K27Me1 in ΔTXR1 cells. This also explains why the higher methylation states of H3K27 are not dramatically affected in the absence of Txr1p. However, H3K27Me2 and H3K27Me3 are not completely abolished in ΔEZL2 cells. This leaves the possibility that Txr1p might be responsible for at least part of the higher methylation states. All these results are consistent with a model in which the H3K27 methylation states are jointly regulated by Txr1p and Ezl2p: Txr1p is mainly responsible for H3K27Me1 and plays a minor role in H3K27Me2/Me3, and Ezl2p is mainly responsible for H3K27Me2/Me3 and plays a minor role in H3K27Me1. As a theoretical alternative, there might be some additional HMTs that can affect H3K27 methylation states in Tetrahymena.
The differential effects of Txr1p and Ezl2p on the methylation states of H3K27 strongly suggest that they function in different pathways, and H3K27Me1 might have biological functions distinct from those of H3K27Me2 and H3K27Me3. This is supported by the observation that ATXR5 and ATXR6 are involved in replication control in Arabidopsis (50). Furthermore, studies of histone modification turnover have revealed significant accumulation of H3K27Me1 on newly deposited H3 in the S-phase of the cell cycle (51). All this potentially points to a novel role for H3K27Me1 in DNA replication that is apparently separated from the well-established roles of H3K27Me2 and H3K27Me3 in transcriptional repression and heterochromatin formation.
The establishment of this uniform labeling method to identify the histone substrates and target sites for HMTs was the primary goal of our research, and our initial focus on the Txr1p and Ezl2p methyl transferases on H3 was intended to fill a gap in our functional knowledge and form a foundation for future studies. Methylation may occur via multiple routes, and the relationships among histone modifications are complex. The development of an accurate quantitative method is a necessary part of clarifying these relationships for H3 and other histones. Our approach will now allow us to explore functional crossover between HMTs and their relative relationships, as well as the in vivo relationships between methylation and other PTMs, which is a continuing goal of our research. In continuing studies, we will explore the global changes of PTMs across other core histones.
Supplementary Material
Footnotes
* This work was funded in part by NIH Grant Nos. #1P41RR018627 (P.C.A.) and R01GM087343 (Y.L.), and by a University of Michigan Rackham Graduate Student Research Grant to C.Z.
Raw data can be obtained from Proteome Commons using the hash code 0f360CLM7HrsVmtNTr%2B%2B2AicdjTmDYwv9iVer5gZo%2BQMLlR4uTEU%2FyRZWMVl9B8QCtb%2BvL3qhXfIATNArPK%2BSP0mv%2BUAAAAAAAAWFQ==.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- Ac
- acetylation
- ATXR
- Arabidopsis trithorax related
- CV
- coefficient of variation
- EZL2
- enhancer of zeste-like 2
- HMT
- histone methyltransferase
- Me1
- monomethylation
- Me2
- dimethylation
- Me3
- trimethylation
- PCA
- principal component analysis
- PHD
- plant homeodomain
- Pr
- propionylation
- PTM
- post-translational modification
- SET
- suppressor of variegation, enhancer of zeste, trithorax
- TXR1
- Tetrahymena trithorax related 1.
REFERENCES
- 1. Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 2. Campos E. I., Reinberg D. (2009) Histones: annotating chromatin. Annu. Rev. Genet 43, 559–599 [DOI] [PubMed] [Google Scholar]
- 3. Allis C. D., Jenuwein T., Reinberg D., Caparros M. L. (2007) Epigenetics, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 23–62 [Google Scholar]
- 4. Jenuwein T., Allis C. D. (2001) Translating the histone code. Science 293, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 5. Strahl B. D., Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 6. Klose R. J., Zhang Y. (2007) Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell. Biol. 8, 307–318 [DOI] [PubMed] [Google Scholar]
- 7. Zhang Y., Reinberg D. (2001) Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 15, 2343–2360 [DOI] [PubMed] [Google Scholar]
- 8. Chi P., Allis C. D., Wang G. G. (2010) Covalent histone modifications—miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 10, 457–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bannister A. J., Kouzarides T. (2005) Reversing histone methylation. Nature 436, 1103–1106 [DOI] [PubMed] [Google Scholar]
- 10. Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- 11. Loyola A., Tagami H., Bonaldi T., Roche D., Quivy J. P., Imhof A., Nakatani Y., Dent S. Y., Almouzni G. (2009) The HP1alpha-CAF1-SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep. 10, 769–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beck D. B., Oda H., Shen S. S., Reinberg D. (2012) PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 26, 325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schotta G., Lachner M., Sarma K., Ebert A., Sengupta R., Reuter G., Reinberg D., Jenuwein T. (2004) A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 18, 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zee B. M., Levin R. S., Xu B., LeRoy G., Wingreen N. S., Garcia B. A. (2010) In vivo residue-specific histone methylation dynamics. J. Biol. Chem. 285, 3341–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Albert M., Helin K. (2010) Histone methyltransferases in cancer. Semin. Cell. Dev. Biol. 21, 209–220 [DOI] [PubMed] [Google Scholar]
- 16. Zhang K., Dent S. Y. (2005) Histone modifying enzymes and cancer: going beyond histones. J. Cell. Biochem. 96, 1137–1148 [DOI] [PubMed] [Google Scholar]
- 17. Raynaud C., Sozzani R., Glab N., Domenichini S., Perennes C., Cella R., Kondorosi E., Bergounioux C. (2006) Two cell-cycle regulated SET-domain proteins interact with proliferating cell nuclear antigen (PCNA) in Arabidopsis. Plant J. 47, 395–407 [DOI] [PubMed] [Google Scholar]
- 18. Sanchez R., Zhou M. M., (2011) The PHD finger: a versatile epigenome reader. Trends Biochem. Sci. 36, 364–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H., Ilin S., Wang W., Duncan E. M., Wysocka J., Allis C. D., Patel D. J. (2006) Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 442, 91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peña P. V., Davrazou F., Shi X., Walter K. L., Verkhusha V. V., Gozani O., Zhao R., Kutateladze T. G. (2006) Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 442, 100–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shi X., Hong T., Walter K. L., Ewalt M., Michishita E., Hung T., Carney D., Peña P., Lan F., Kaadige M. R., Lacoste N., Cayrou C., Davrazou F., Saha A., Cairns B. R., Ayer D. E., Kutateladze T. G., Shi Y., Côté J., Chua K. F., Gozani O. (2006) ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maga G., Hubscher U. (2003) Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J. Cell Sci. 116, 3051–3060 [DOI] [PubMed] [Google Scholar]
- 23. Jacob Y., Feng S., LeBlanc C. A., Bernatavichute Y. V., Stroud H., Cokus S., Johnson L. M., Pellegrini M., Jacobsen S. E., Michaels S. D. (2009) ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat. Struct. Mol. Biol. 16, 763–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Y., Taverna S. D., Muratore T. L., Shabanowitz J., Hunt D. F., Allis C. D. (2007) RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes Dev. 21, 1530–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chung P. H., Yao M. C. (2012) Tetrahymena JMJD3 homolog regulates H3K27 methylation and nuclear differentiation. Eukaryot. Cell 11, 601–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang K., Tang H. (2003) Analysis of core histones by liquid chromatography-mass spectrometry and peptide mapping. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 783, 173–179 [DOI] [PubMed] [Google Scholar]
- 27. Britton L.-M. P., Gonzales-Cope M., Zee B. M., Garcia B. A. (2011) Breaking the histone code with quantitative mass spectrometry. Expert Rev. Proteomics 8, 631–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garcia B. A., Shabanowitz J., Hunt D. F. (2007) Characterization of histones and their post-translational modifications by mass spectrometry. Curr. Opin. Chem. Biol. 11, 66–73 [DOI] [PubMed] [Google Scholar]
- 29. Ueberheide B., Mollah S. (2007) Deciphering the histone code using mass spectrometry. Int. J. Mass Spectrom. 259, 46–56 [Google Scholar]
- 30. Su X., Ren C., Freitas M. A. (2007) Mass spectrometry-based strategies for characterization of histones and their post-translational modifications. Exp. Rev. Proteomics 4, 211–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beck H. C., Nielsen E. C., Matthiesen R., Jensen L. H., Sehested M., Finn P., Grauslund M., Hansen A. M., Jensen O. N. (2006) Quantitative proteomic analysis of post-translational modifications of human histones. Mol. Cell. Proteomics 5, 1314–1325 [DOI] [PubMed] [Google Scholar]
- 32. Plazas-Mayorca M. D., Bloom J. S., Zeissler U., Leroy G., Young N. L., DiMaggio P. A., Krugylak L., Schneider R., Garcia B. A. (2010) Quantitative proteomics reveals direct and indirect alterations in the histone code following methyltransferase knockdown. Mol. Biosyst. 6, 1719–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jung H. R., Pasini D., Helin K., Jensen O. N. (2010) Quantitative mass spectrometry of histones H3.2 and H3.3 in Suz12-deficient mouse embryonic stem cells reveals distinct, dynamic post-translational modifications at Lys-27 and Lys-36. Mol. Cell. Proteomics 9, 838–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ong S. E., Mann M. (2005) Mass spectrometry-based proteomics turns quantitative. Nat. Chem. Biol. 1, 252–262 [DOI] [PubMed] [Google Scholar]
- 35. Garcia B. A., Joshi S., Thomas C. E., Chitta R. K., Diaz R. L., Busby S. A., Andrews P. C., Ogorzalek Loo R. R., Shabanowitz J., Kelleher N. L., Mizzen C. A., Allis C. D., Hunt D. F. (2006) Comprehensive phosphoprotein analysis of linker histone H1 from Tetrahymena thermophila. Mol. Cell. Proteomics 5, 1593–1609 [DOI] [PubMed] [Google Scholar]
- 36. Syka J. E. P., Marto J. A., Bai D. L., Horning S., Senko M. W., Schwartz J. C., Ueberheide B., Garcia B. A., Busby S., Muratore T., Shabanowitz J., Hunt D. F. (2004) Novel linear quadrupole ion trap/FT mass spectrometer: performance characterization and use in the comparative analysis of histone H3 post-translational modifications. J. Proteome Res. 3, 621–626 [DOI] [PubMed] [Google Scholar]
- 37. Garcia B. A., Mollah S., Ueberheide B. M., Busby S. A., Muratore T. L., Shabanowitz J., Hunt D. F. (2007) Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2, 933–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ishihama Y., Sato T., Tabata T., Miyamoto N., Sagane K., Nagasu T., Oda Y. (2005) Quantitative mouse brain proteomics using culture-derived isotope tags as internal standards. Nat. Biotechnol. 23, 617–621 [DOI] [PubMed] [Google Scholar]
- 39. Wu C. C., MacCoss M. J., Howell K. E., Matthews D. E., Yates J. R., III (2004) Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Anal. Chem. 76, 4951–4959 [DOI] [PubMed] [Google Scholar]
- 40. Geiger T., Cox J., Ostasiewicz P., Wisniewski J. R., Mann M. (2010) Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods 7, 383–385 [DOI] [PubMed] [Google Scholar]
- 41. Loidl J., Mochizuki K. (2009) Tetrahymena meiotic nuclear reorganization is induced by a checkpoint kinase-dependent response to DNA damage. Mol. Biol. Cell 20, 2428–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cassidy-Hanley D., Bowen J., Lee J. H., Cole E., VerPlank L. A., Gaertig J., Gorovsky M. A., Bruns P. J. (1997) Germline and somatic transformation of mating Tetrahymena thermophila by particle bombardment. Genetics 146, 135–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gorovsky M. A. (1970) Studies on nuclear structure and function in Tetrahymena pyriformis. II. Isolation of macro- and micronuclei. J. Cell Biol. 47, 619–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gorovsky M. A. (1970) Studies on nuclear structure and function in Tetrahymena pyriformis. 3. Comparison of the histones of macro- and micronuclei by quantitative polyacrylamide gel electrophoresis. J. Cell Biol. 47, 631–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shechter D., Dormann H. L., Allis C. D., Hake S. B. (2007) Extraction, purification and analysis of histones. Nat. Protoc. 2, 1445–1457 [DOI] [PubMed] [Google Scholar]
- 46. Cui B., Liu Y., Gorovsky M. A. (2006) Deposition and function of histone H3 variants in Tetrahymena thermophila. Mol. Cell. Biol. 26, 7719–7730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yu L., Gorovsky M. A. (1997) Constitutive expression, not a particular primary sequence, is the important feature of the H3 replacement variant hv2 in Tetrahymena thermophila. Mol. Cell. Biol. 17, 6303–6310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Strahl B. D., Ohba R., Cook R. G., Allis C. D. (1999) Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc. Natl. Acad. Sci. U.S.A. 96, 14967–14972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Beynon R. J., Pratt J. M. (2005) Metabolic labeling of proteins for proteomics. Mol. Cell. Proteomics 4, 857–872 [DOI] [PubMed] [Google Scholar]
- 50. Jacob Y., Stroud H., Leblanc C., Feng S., Zhuo L., Caro E., Hassel C., Gutierrez C., Michaels S. D., Jacobsen S. E. (2010) Regulation of heterochromatic DNA replication by histone H3 lysine 27 methyltransferases. Nature 466, 987–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu M., Wang W., Chen S., Zhu B. (2011) A model for mitotic inheritance of histone lysine methylation. EMBO Rep. 13, 60–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.