

Abstract
Identifying the building blocks of mammalian tissues is a precondition for understanding their function. In particular, global and quantitative analysis of the proteome of mammalian tissues would point to tissue-specific mechanisms and place the function of each protein in a whole-organism perspective. We performed proteomic analyses of 28 mouse tissues using high-resolution mass spectrometry and used a mix of mouse tissues labeled via stable isotope labeling with amino acids in cell culture as a “spike-in” internal standard for accurate protein quantification across these tissues. We identified a total of 7,349 proteins and quantified 6,974 of them. Bioinformatic data analysis showed that physiologically related tissues clustered together and that highly expressed proteins represented the characteristic tissue functions. Tissue specialization was reflected prominently in the proteomic profiles and is apparent already in their hundred most abundant proteins. The proportion of strictly tissue-specific proteins appeared to be small. However, even proteins with household functions, such as those in ribosomes and spliceosomes, can have dramatic expression differences among tissues. We describe a computational framework with which to correlate proteome profiles with physiological functions of the tissue. Our data will be useful to the broad scientific community as an initial atlas of protein expression of a mammalian species.
The proteins expressed by a tissue are the principal active units that determine its function. Mapping tissue proteomes can therefore identify the molecular regulators and effectors of their physiological activity. A broad and quantitative proteome of a mammalian species has, however, not been determined yet. Instead, global analyses of mouse tissues have previously been performed mainly on the mRNA level using microarray or deep sequencing technologies (1, 2). Although these methods can provide a near-comprehensive view of the biological system at the transcriptional level, mRNA levels do not necessarily predict protein expression levels and therefore miss an important determinant of biological function. Recent studies comparing mRNA to protein levels showed a correlation of 0.4 to 0.6 (see, for example, Refs. 3 and 4), and it appears that the main process additionally determining protein levels is the regulation of translation rates (5). These observations highlight the importance of determining the proteome in addition to the transcriptome.
Mass spectrometry (MS)-based1 proteomics is the primary technology that enables a system-wide view of proteomes and their changes (6–8). In “shotgun” proteomics, proteins are digested to peptides, and the peptides are analyzed via liquid chromatography coupled to mass spectrometry (LC-MS/MS). The development of mass spectrometers with high resolution, high mass accuracy, and high sequencing speed now allows routine identification of large proportions of the mammalian proteome with high confidence (9). Previously, Kislinger et al. performed a proteomic analysis of six mouse tissues and identified ∼2,000 proteins per tissue using low-resolution MS (10). A recent deep phospho-proteomic study in mouse also analyzed the proteome of nine mouse tissues (11). These studies were only semi-quantitative, as they were based on spectral counting and restricted to a small number of tissues. Here we set out to correlate protein expression and tissue physiology with higher quantification accuracy on a much larger scale. Although we did not achieve complete proteome coverage, the accuracy and wide range of the study further enable a determination of tissue-specific functions.
MS became truly quantitative with the development of stable-isotope-based methods in which the ratios of “light” and “heavy” versions of the same peptides are accurately determined (12). Stable isotope labeling with amino acids in cell culture (SILAC) is generally considered as the most accurate technology for relative protein quantification. It relies on metabolic labeling, in which heavy amino acids, typically lysine and arginine, are incorporated into proteins during their synthesis (13). In the past, SILAC was limited to cells in culture, but it has now been expanded to whole organisms and human tissues (14–17). We have generated SILAC-labeled mice that were grown for more than two generations on a diet containing heavy lysine (Lys6-13C6) as their sole source of this amino acid, leading to complete labeling of their proteome (18, 19). SILAC mice provide a source of heavy tissues that can serve as a reference for relative quantification and which represent the full complexity of the tissue in vivo.
In this study we took advantage of the SILAC mouse technology to quantify 28 mouse tissues relative to a mix of these heavy tissues. We provide a deep proteomic map of the proteins and functions across these tissues and highlight key regulators of tissue specificity.
EXPERIMENTAL PROCEDURES
Sample Preparation
C57BL/6 mice were sacrificed via cervical dislocation, and 28 tissues and organs were dissected and snap-frozen in liquid nitrogen. For protein isolation, tissues were homogenized at 4 °C in denaturing buffer containing 1% N-octylglucoside, 6 m urea, and 2 m thiourea in 10 mm Tris buffer (pH 8.0). Homogenates were centrifuged at 45,000g, and the supernatant was precipitated overnight in five volumes of ice-cold acetone. Precipitated proteins were resuspended in denaturing buffer without N-octylglucoside, and Bradford assay was performed to determine the protein concentration. Samples from three animals belonging to each group (unlabeled and SILAC-labeled) were pooled to minimize individual variability. Equal amounts of protein extracts from all SILAC-labeled tissues were combined to create the heavy spike-in protein standard for all samples.
Protein Digestion and Peptide Separation
The SILAC spike-in standard was mixed with each of the unlabeled tissues in equal amounts (50 μg each) and digested in solution with endoprotease Lys-C. Peptides were fractionated via isoelectric focusing on an OffGel Fractionator (Agilent, Waldbronn, Germany) in a 12-well format as described elsewhere (20). Peptides from each of the 12 fractions were purified using C18 StageTips (21).
Liquid Chromatography–Mass Spectrometric Analysis
LC-MS/MS measurements were performed on an Easy-nano-LC (Thermo Fisher Scientific) coupled to an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific). Peptides were separated on a reverse-phase column (15 cm, 75 μm inner diameter and 3 μm Reprosil resin) using a 100-min gradient of water–acetonitrile. All MS measurements were performed in the positive ion mode. Precursor ions were measured in the Orbitrap analyzer at 60,000 resolution (at 400 m/z) and a target value of 106 ions. The five most intense ions from each MS scan (with a target value of 5,000 ions) were isolated, fragmented, and measured in the linear ion trap. Replicate analysis (Fig. 2) was performed in single runs on an EASY-nLC1000 (22) coupled to a Q-Exactive mass spectrometer (Thermo Fisher Scientific) (23). Peptides were separated on a reverse-phase column (30 cm, 75 μm inner diameter and 1.8 μm Reprosil resin) using a 200-min gradient of water–acetonitrile. All MS measurements were performed in the positive ion mode. Precursor ions were measured in the Orbitrap analyzer at 70,000 resolution (at 200 m/z) and a target value of 106 ions. The 10 most intense ions from each MS scan were isolated, fragmented, and measured in the Orbitrap with a resolution of 17,500.
Fig. 2.

Reproducibility of proteomic data. Comparison of triplicate analysis of lung and liver tissues shows high correlation between replicates. A, heat map of the Pearson correlations of ratios relative to super-SILAC. B, C, scatter plots comparing replicate lung (B) and liver (C) samples. Color code represents density as indicated in the bar at the bottom.
Data Analysis in MaxQuant
Raw mass spectrometric files were analyzed in the MaxQuant environment (24), version 1.3.0.5. MS/MS spectra were searched using the Andromeda search engine (25) against the decoy UniProt-mouse database (59,345 entries) supplemented with 262 frequently observed contaminants such as human keratins, bovine serum proteins, and proteases and containing forward and reverse sequences. Precursor mass and fragment mass were searched with initial mass tolerances of 6 ppm and 0.5 Da, respectively, in the LTQ-Orbitrap runs, and 6 ppm and 20 ppm in the Q Exactive runs. The search included variable modifications of methionine oxidation and N-terminal acetylation and a fixed modification of carbamidomethyl cysteine. The maximum number of missed cleavages was set at two, and the minimum peptide length was set at six amino acids. The false discovery rate was set at 0.01 for peptide and protein identifications. When identified peptides were all shared between two proteins, they were combined and reported as one protein group. In order to determine SILAC ratios, a minimum of two ratio counts between SILAC peptide pairs was required. We used the summed peptide intensities of the individual unlabeled proteins as a proxy for protein abundance, and then divided by the overall unlabeled intensity of each sample to calculate its relative abundance. For the analysis of protein intensities in the various tissues, only the light intensities were considered (non-SILAC tissue). The ratio values were used for relative comparison across the different tissues. Ratio values were normalized assuming that the most likely scenario is the one that minimizes overall changes in protein abundance between heavy and light samples.
Bioinformatic Analysis
All bioinformatic analysis was performed with Perseus software. Categorical annotation was supplied by Gene Ontology biological process, molecular function, and cellular component; the TRANSFAC database; the KEGG pathway database; and the Pfam-protein family database. All annotation except for TRANSFAC was extracted from the UniProt database.
For bioinformatic analysis, the data were filtered so as to have at least 14 out of 28 samples with valid ratio values (at least two ratio counts per individual value). Next, the data were imputed to fill missing data points by creating a Gaussian distribution of random numbers with a standard deviation of 30% relative to the standard deviation of the measured values and one standard deviation down-shift of the mean to simulate the distribution of low signal values.
Hierarchical clustering of proteins was performed in Perseus on logarithmized intensities after z-score normalization of the data, using Euclidean distances. Principal component analysis was done on logarithmized values without z-scoring. The implementation in Perseus utilizes singular value decomposition in order to find the principal components. The “annotation matrix algorithm” filters protein annotation terms by testing the difference among means for any protein annotation from the overall ratio distribution for all cells of the expression matrix. The statistical test is a two-dimensional non-parametric Mann–Whitney test, with a Benjamini–Hochberg multiple hypothesis testing correction that was controlled by using a false discovery rate threshold of 0.05. All categories that survived the test for at least one of the samples were converted to one row of the annotation matrix. The values for each row are the differences between groups of the Mann–Whitney test mentioned above.
Fisher's exact test was performed with a false discovery rate value of 0.02.
Tissue Processing
Tissue samples were fixed in formalin for 24 h at room temperature, followed by standard histoprocessing procedures. After fixation, tissues were dehydrated and paraffin impregnated using a vacuum infiltrating processor (Ventana Medical Systems, Tuscon, AZ). The processed tissues were embedded in paraffin, and 4-μm sections were cut and placed on glass slides (SuperFrost Plus, Menzel Gläser, Braunschweig, Germany) (26).
Immunohistochemistry
Tissue slides were deparaffinized in xylene, hydrated in graded series of alcohol, and blocked for endogenous peroxidase (0.3% H2O2 in 95% alcohol for 5 min). Heat-induced epitope retrieval was performed in citrate buffer (pH 6) (Thermo Fisher Scientific) for 4 min at 125 °C using a pressure boiler (decloaking chamber, Biocare Medical, Walnut Creek, CA). Automated immunohistochemical analysis (IHC) was performed using an Autostainer 480S instrument (Thermo Fisher Scientific). Primary antibodies and the associated horseradish peroxidase polymer visualization system (Thermo Fisher Scientific) were incubated for 30 min each at room temperature. Slides were developed in diaminobenzidine (Thermo Fisher Scientific) as chromogen. Counterstaining was performed in an Autostainer XL (Leica Biosystems, Wetzlar, Germany) using Mayers hematoxylin (Histolab, Gothenburg, Sweden), and the slides were coverslipped in an automated glass coverslipper (CV5030, Leica Biosystems) using PERTEX (Histolab). Slides were digitalized at 20× magnification using a ScanScope XT (Aperio Technologies, Vista, CA).
RESULTS
Proteomic Analysis of Mouse Tissues
To measure a quantitative map of the mouse proteome, we prepared protein extracts from 27 distinct adult tissues and from embryonic tissue of C57BL/6 mice (Fig. 1A). For accurate quantification, we took advantage of the SILAC mouse (18) and used it as a spike-in standard (Figs. 1B and 1C). We combined equal protein amounts of each of these 28 tissues from a SILAC mouse and mixed this standard with an equal amount of each of the light tissues. We digested each tissue proteome with endoprotease LysC and separated the peptides into 12 fractions according to their isoelectric point (see “Experimental Procedures”). Each of the fractions was analyzed via LC-MS/MS on a linear ion trap-Orbitrap mass spectrometer for a total of 336 runs. Analysis of the data with MaxQuant software (24) and the Andromeda search engine (25) identified 7,349 proteins at a false discovery rate of 1% and quantified 6,974 of them (supplemental Table S1). These data represent a catalogue of expressed mouse proteins and their profiles across the mouse tissues. Importantly, these proteomes reflect the proteins expressed in the indicated organ, and not necessarily in individual cell types in the tissue. Nevertheless, they highlight tissue-specific proteins that have high ratios toward the standard in one or only a few related tissues and can determine proteins with constant expression levels in the tissues. For example, Myelin A1 is highly expressed in the five brain tissues included in this study, but not in any other tissue (Fig. 1D). Pancreatic α amylase protein is expressed mainly in the pancreas, but lower levels are also seen in the ileum and duodenum. Presumably, this protein was secreted from the pancreas to the lumen of the digestive tract rather than being expressed locally. Arginase-1, an enzyme of the urea cycle, is almost exclusively expressed in the liver. In contrast, proteasomal subunits are co-regulated and expressed at similar levels in all tissues (Fig. 1E).
Fig. 1.

Proteomic analysis of mouse tissues. A, 28 tissues were isolated from C57BL/6 mice. B, the same tissues were isolated from SILAC mice and combined to create a SILAC mix that served as a spike-in standard. This SILAC reference was mixed with each of the “light” tissues prior to combined protein digestion with Lys-C. Peptides were separated into 12 fractions with an OffGel fractionator and analyzed via LC-MS/MS on the LTQ-Orbitrap. C, peptides from each tissue were quantified relative to the SILAC standard, as illustrated by peptides in the muscle and in the cerebellum that are accurately quantified relative to the same heavy standard. D, the data can serve as a catalogue of expressed mouse proteins across the tissues, as shown by the tissue distribution of three proteins with highly specific expression. E, three proteasomal subunits are similarly expressed in all tissues.
Our analysis consisted of one replicate of pooled proteomes from three mice. To examine the variability between mice, we performed non-pooled triplicate analyses of two tissues, liver and lung, which were all quantified relative to the super-SILAC standard. We identified a total of 4,906 proteins, and more than 2,800 proteins in each tissue (supplemental Table S1). We found very high correlation between the replicates of the same tissue (0.84–0.95; Fig. 2). These results show that our pooling approach in combination with super-SILAC-based quantification reveals highly reproducible quantitative data about thousands of proteins.
To corroborate mass spectrometric results with an independent method, we performed IHC of mouse liver, kidney, spleen, small intestine, brain cortex, and skeletal muscle. Because IHC is not a quantitative method, we selected proteins that showed the highest tissue specificity with over 30-fold higher expression in the liver, brain, intestine, or kidney relative to the other tissues. We selected antibodies from the Protein Atlas antibody resource (27), which has antibodies against more than 12,000 proteins. From a list of 69 tissue-specific proteins determined via proteomics, we found antibodies to 38 proteins and selected 18 antibodies (targeting nine proteins) for which the epitope region had high sequence similarity between mouse and human. We further added antibodies to two proteasome subunits that, according to the mass spectrometric data, are similarly expressed in the various tissues. Antibodies against 7 out of the 11 proteins reacted positively with the mouse proteins and were compared with the MS results. In agreement with the MS data (Fig. 1), the two proteasome subunits PSMA1 and -2 were highly expressed in all the tissues examined (Fig. 3; supplemental Fig. S1). IHC verified the liver specificity of Arginase 1 and of glycogen synthase 2 and the intestine specificity of Cant1 (Fig. 3). According to the MS data, Amacr is highly expressed in the kidney and the liver; in the IHC data, it stained mainly in the kidney, and only very low signal was seen in the liver. Possibly the high intensity in the MS resulted from the higher homogeneity of expression in the liver, as compared with stronger, but more scattered, expression in the kidney. PAK1 had the highest expression in the neuronal tissues and a medium expression in the intestine according to the MS data. The IHC staining identified high expression levels in the brain cortex and in the small intestine but was negative in the muscle and in the spleen. The discrepancy related to the expression in the intestine is probably related to the qualitative nature of IHC, which could not detect the more subtle differences in protein expression. Thus, except for small differences, the comparison of the proteomic data to IHC staining confirmed the MS results using completely independent methodology. Furthermore, our examples highlight how IHC addresses the internal morphology in the tissue, for instance, when the various cell types in the tissue have different expression levels.
Fig. 3.

Images showing protein expression profiles based on five antibodies, IHC, and bright field microscopy. Protein expression is shown in brown, and counterstaining in blue. All antibodies were obtained from the Human Protein Atlas project. A, the antibody HPA008188 staining PSMA2 shows general, moderate to strong cytoplasmic staining in all tissues. B, the antibody HPA024006 staining ARG1 shows cytoplasmic and nuclear staining of hepatocytes in liver. The remaining tissues were negative. C, the antibody HPA039482 staining GYS2 shows moderate to strong cytoplasmic staining of hepatocytes in liver and myocytes in skeletal muscle. A weaker luminal staining was observed in small intestine, and the remaining tissues were negative. D, the antibody HPA019639 staining CANT1 shows a general cytoplasmic staining of all tissues, with the strongest staining being of small intestine. E, the antibody HPA003565 staining PAK1 shows moderate to strong cytoplasmic staining of neuropil, glandular cells in small intestine, and a subset of the lymphoid cells in spleen.
The MS analysis identified 3,000 to 5,000 proteins in individual tissues (supplemental Fig. S2A). There was high overlap of the identified proteins between the various tissues, with 1,972 proteins identified in all of them and only 650 proteins that were detected in only one (supplemental Figs. S2B and S2C). The large differences in the numbers of identified proteins are partially because of the finite dynamic range of the MS analysis. A few highly abundant proteins in particular tissues can interfere with the identification of some of the lower abundance proteins, thereby reducing the overall number of identifications. To illustrate this, we plotted the contribution of the 100 most abundant proteins to the total protein mass of each tissue, according to the estimated individual protein intensities (the light intensities of the non-labeled tissues) (Fig. 4A). For the three muscle samples—skeletal muscle, diaphragm, and heart muscle—these 100 proteins contributed 85%, 74%, and 68% of the protein mass, respectively. In contrast, the 100 most abundant proteins constituted only 35% to 40% of the kidney cortex and the intestine parts. Plotting the accumulation of protein mass shows that in skeletal muscle the most abundant protein, parvalbumin alpha, already contributes 10% of the total protein mass, and only 10 proteins account for more than half of it (Fig. 4B). In contrast, reaching this percentage requires hundreds of proteins in other tissues. These very high-abundance proteins in the muscle presumably contribute to the relatively low number of identified proteins.
Fig. 4.

Protein abundance in the mouse tissues—the “top-100 proteome.” A, relative abundance of the 100 most abundant proteins in each tissue. Abundance calculation was based on protein intensity relative to the overall intensity of the “light” tissue. B, accumulation of protein mass in kidney cortex, midbrain, and muscle. These tissues represent the extremes and middle values from A. Proteins are rank-ordered according to their intensity. This shows that in muscle, only 10 proteins are responsible for over 50% of tissue mass. C, principal component analysis of the mouse tissues based on the top-100 proteome enables discrimination of the brain and muscle tissues and association of the intestine regions. D, principal component analysis based on the complete dataset shows the same tissue discrimination as the top-100 proteome.
“Top-100 Proteome” Discriminates between Tissues
Examination of the 100 most abundant proteins in each tissue, their “top-100 proteome,” revealed a high degree of specialization. Combining the top-100 proteomes yielded a list of 640 proteins (supplemental Table S2). This group of proteins included metabolic enzymes, cytoskeletal proteins, ribosomal proteins, and histones. The protein list also included albumin and globins, which presumably originate from residual blood in several highly vascularized tissues. However, these few proteins do not affect the overall proteome profiles. Despite the very high abundance of the top-100 proteins, none of them retained constant expression levels across tissues. Instead, all exhibited fold changes of at least 2.5 between the highest and lowest expressing tissues, and moreover, the SILAC ratios of 53% of them changed by more than 100-fold. Principal component analysis of the expression values of these highly abundant proteins readily discriminated between the tissues (Fig. 4C). In this analysis, the brain tissues clearly segregated from the others; muscle tissues were also discrete, whereas the intestine regions were associated with each other. This principal component analysis pattern was highly similar to that based on the complete dataset (Fig. 4D), showing that the most highly abundant proteins can be determinants of unique tissue characteristics. These abundant proteins have the highest ratios toward the other tissues and are drivers of the segregation between tissues. One implication from these findings is that many of the tissue-specific functions require high expression levels of the specialized proteins.
Co-regulation of Molecular Complexes
We and others have previously reported that there is tight regulation of protein levels for members of stable protein complexes (28–31). In agreement with these observations, our data indicate that the components of large molecular complexes are regulated in a concerted manner across the mouse tissues. For the core proteasome, all subunits had very similar ratios toward the internal SILAC standard in all tissues (Fig. 5A). The spliceosome subunits were also co-regulated, but with a higher spread of the ratios of the components, partially because of the large number and diversity of components annotated as “spliceosomal” in databases (Fig. 5B). The mean levels of spliceosomal proteins markedly changed between the various samples, with the embryonic tissue expressing the highest levels; similarly high levels in the thymus, spleen, uterus, and ovary; and the lowest levels in the muscle tissues. Ribosome levels varied dramatically between the tissues, with ∼50-fold more ribosomes in the pancreas than in the cerebellum (Fig. 5C). It has been known for decades that the pancreas has high levels of ribosomes (32, 33), and this is readily explained by the high translation rates that are necessary in order to support the secretory activity of this organ. Thus our data provide an indication of the translational load of each tissue. Last, the respiratory chain complexes showed similar patterns of co-regulation of their components (Fig. 5D). The brown fat tissue had the highest levels of these complexes, followed by heart muscle and diaphragm. These results are explained by the high energetic demands of these tissues—the brown fat for its thermogenic activity, and the muscle for contractile activity.
Fig. 5.

Co-regulation of molecular complexes in the mouse tissues. The graphs indicate the ratios of the subunits of the proteasome (A), spliceosome (B), ribosome (C), and respiratory chain complex (D) relative to the internal standard in each of the tissues. The subunits of the complexes are co-regulated with a few outliers in each complex, but the overall level of the complex varies between them.
Unsupervised Clustering of Tissue Proteomes
The quantitative nature of our data enabled us to investigate associations between proteins and tissues. Hierarchical clustering of the proteomic data recapitulated the similarities between related tissues (Fig. 6A). For example, the five brain regions tightly clustered together, as did the three muscle tissues, the various intestine regions, and kidney cortex and medulla. Interestingly, quantitative proteomics also linked anatomically distinct tissues that have related functions. For example, the pancreas and salivary gland co-clustered. These exocrine glands share the physiological requirement of high protein translation rates for protein secretion. Indeed, both of these tissues expressed high levels of proteins involved in the translation machinery, including ribosomes, as well as proteins from the endoplasmic reticulum and Golgi apparatus. The analysis placed embryonic tissue, which is known to have multiple unique functions and proteins, farthest from all other tissues.
Fig. 6.
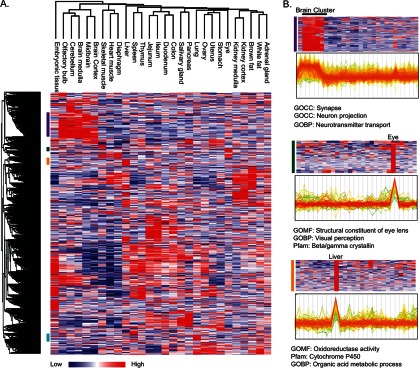
Unsupervised clustering of mouse tissue proteomes. A, hierarchical clustering of proteins and tissues shows that embryonic tissue is separated from the other tissues and functionally related tissues are co-clustered. B, profile plots show the normalized ratios (z-scored ratios toward the super-SILAC standard) of the brain protein cluster, the eye cluster, and the liver cluster (purple, green, and orange bars in A, respectively). Lines (indicating individual proteins) are colored according to the density of proteins with those ratios. Fisher's exact test (false discovery rate = 0.02) for enrichment analysis of protein annotations in each cluster highlights the tissue-specific annotations.
The correlation between the tissue-specific proteins (those predominantly expressed in that tissue) and the physiological functions was statistically analyzed for enrichment of gene ontologies, pathways, and protein families. Fisher's exact test of the protein cluster of the five brain regions highlighted “synapse,” “neuron projection,” and “neurotransmitter transport” as the most significant categories (Fig. 6B). In the eye cluster, “structural constituent of eye lens,” “visual perception,” and “beta/gamma crystalline” had the highest significance. The most prominent liver cluster was enriched for “oxidoreductase activity,” “cytochrome p450,” “organic acid metabolic process,” and peroxisomes, also conforming to key liver functions in metabolism and detoxification (Fig. 6B). These results demonstrate that our proteomic approach clearly captured the essential physiological activities of each tissue and the proteins responsible for them in an unbiased manner. Thus the distinct clusters of proteins that were highly expressed in one tissue or several related tissues can serve as a resource for the identification of novel tissue-specific functions of known proteins, and for proposing functions of unknown ones.
Global Analysis of Tissue-specific Annotations
After we analyzed the expression profiles of all quantified proteins across the tissues, we investigated the distribution of protein annotations themselves, such as protein functions, processes, and pathways. Specifically, we examined gene ontologies, KEGG pathways, TRANSFAC-transcription factor targets, and Pfam-protein families. Such an overview can potentially uncover the tissue specificity of each annotation category and can further reveal the co-regulation of several such annotations. We developed a bioinformatics tool that examines the enrichment of categories in each of the tissues and then creates an annotation matrix of categories versus tissues (see “Experimental Procedures”; supplemental Table S3). Unsupervised two-way hierarchical clustering of this annotation and tissue matrix resulted in a similar grouping of the tissues (supplemental Fig. S3). This again connected functionally related tissues, such as the brain tissues, muscles, and intestines; however, this time it was based on the quantitative distribution of the annotations, rather than individual proteins. In contrast to clustering based on proteins, here the liver was the most distant tissue, and the embryonic tissue was clustered near the lung, thymus, and spleen. The liver has a unique physiological function in metabolism and detoxification. The annotation matrix uncovered the liver-specific expression of proteins involved in amino acid biosynthesis and co-clustered them with targets of the liver-specific transcription factor HNF1 as annotated in the TRANSFAC database. In addition, the annotation matrix identified metabolic pathways that are shared between the liver and unrelated tissues. As an example, the liver and kidney tissues (medulla and cortex) did not co-cluster, but there was a group of annotations that were shared between them. These included vitamin biosynthesis, nucleotide biosynthesis, detoxification processes, and peroxisomes, all of which are necessary for proper function of these organs. Likewise, the liver shared fatty acid metabolism with the adipose tissues kidney and muscle tissues. In a similar manner, the lung and the kidney tissues, which are dominated by epithelial cells, shared cell–cell junction proteins, which were co-clustered with the protein family of FERM-domain-containing proteins.
The annotation-based approach enabled examination of the correlations between the annotations themselves, potentially adding another dimension to our understanding of the biological system and its regulation. We were particularly interested in links between transcription factors and biological processes that were changing in a tissue-specific manner. We used the TRANSFAC database of transcription factor targets and looked for co-clustering with other annotations in a tissue-specific manner (Fig. 7A). This identified targets of the steroidogenic factor SF1 in a cluster of the adrenal gland that were co-clustered with the biological processes of hormone biosynthesis and catecholamine biosynthesis, validating our approach. The muscle tissues and the eye were regulated by a large number of transcription factors, including MyoD, MEF2A, TEF1, and TEF5. These transcription factors were co-clustered with myofibril assembly, troponin, and sarcoplasmic reticulum. Regulation of the eye was additionally associated with Pax6 targets and was co-clustered with “apoptosis involved in morphogenesis” and with the “alpha-crystallin” category. Similarly, the targets of the liver-specific transcription factor HNF1 clustered with “histidine catabolic process,” “carbon nitrogen lyase activity,” and “ammonia lyase activity.”
Fig. 7.

Association of transcription factors and biological processes. We created an annotation matrix to globally look at the tissue distribution of annotations (supplemental Fig. S2). A, co-clustering of transcription factor targets from the TRANFAC database with protein families (Pfam) and gene ontology annotations: BP (biological processes), MF (molecular functions), and CC (cellular compartments). Tissue-specific associations are shown for the adrenal gland, muscle tissues, eye, and liver. B, association of transcription factor targets based on ChIP-Seq analysis highlights the tissue specificity of E2F, Zfx, and Myc with AAA proteins and ribosome biosynthesis.
The analysis based on the TRANSFAC database highlighted well-annotated transcription factors and linked them to their known tissue-specific functions via the quantitative proteomics data. Next we expanded this analysis to transcription factors that were not yet known to be associated with specific tissues. We used the transcription factor binding site annotation in Ensembl, which includes ChIP-seq data of 15 factors (CTCF, c-Myc, E2F1, Esrrb, Klf4, Nanog, Oct4, Stat3, Smad1, Sox2, Suz12, Tcfcp2l1, Zfx, n-Myc, and p300) (34). We linked these genomic loci to genes encoding proteins in our dataset using increasing window lengths around each transcription start site. After performing annotation matrix analysis as described above, we selected the smallest statistically significant window to define the protein annotation category (typically 500 bp). The resulting annotation matrix revealed new links between known transcription factors, biological processes, and tissue specificity. The ChIP-Seq experiments of the 15 factors were performed in several cell types, including mouse embryonic stem cells. Reflecting this, our data identified significant enrichment of four of them in the embryonic tissue. E2F1 and Zfx clustered with the AAA protein family of ATPases (Fig. 7B). Inspecting this protein family in our data, we found multiple proteins involved in DNA replication that were highly expressed in the embryonic tissue relative to the others. These included the replication factor C complex proteins, Trp13, Nvl, and Atad2. A nearby cluster, with high expression levels in the embryonic tissue as well as in the salivary gland and the pancreas, was enriched for n-Myc and c-Myc. These transcription factors were co-clustered with ribosome synthesis categories. Recent work has shown the importance of Myc in this process, in particular with regard to its role in oncogenesis (35). Our analysis expands this relationship to non-disease tissue and suggests a key role for Myc-regulated ribosome biogenesis in embryonic tissue and in other tissues that require high protein translation rates.
Next we moved from a global view of the tissues to a focused comparison of white and brown adipose tissues in order to identify their functional similarities and differences from a proteomic basis. Both tissues specialize in fat storage, and their differentiation is transcriptionally controlled by PPARγ and C/EBP (36), but only the brown adipose tissue has high fatty acid catabolism that enables its thermogenic properties (36). Plotting the annotation terms of two tissues against each other distinguishes among processes that are high in both, low in both, or specific to one tissue or the other (Fig. 8A). The TRANSFAC annotations of the major known regulators of lipid storage, PPARγ and C/EBPα and -β, were high in both tissues (Fig. 8B). In the “low-low” region of the plot we found various annotations that were clearly not associated with adipose function—for instance, actin binding, targets of Pax6, the regulator of eye development mentioned above, and PTF1, a pancreatic transcription factor. Our data did not show a distinct population of annotations that were specific for white fat, but there was a large population of brown-fat-specific annotations. These were related to the respiratory chain, reflecting the main functional difference between the tissues. Thus proteomic analysis can capture the main functional differences between tissues and can therefore be used as a valuable source of information on tissue specificities.
Fig. 8.

Protein annotation in brown and white fat tissues. A, schematic graph showing the different zones of the annotation scatter plot. The indicated areas distinguish the annotations that are relevant to both tissues (high-high), the ones that are low in both tissues (low-low), and the ones that are specific to only one of the tissues. B, comparison of annotations between brown and white adipose tissues indicates the prominent role of the transcription factors PPARγ and C/EBP in both tissues, and of metabolic activity specifically in the brown fat tissue.
Finally, we examined the correlation of the proteomic data to publically available mRNA expression data. Comparing our proteome data to a widely used microarray expression profile (1), we found low Spearman correlation coefficients of 0.2 to 0.3. However, correlation to recent RNA-sequencing-based expression profiles (37) was much higher at about 0.5 to 0.6 (supplemental Fig. S4), which is similar to values for cell lines obtained using high-resolution MS (4, 5). As the correlation of the RNA-seq data to the microarray data was also 0.5, the low correlation of microarray to our proteomics data likely reflects technical limitations of early microarray studies. As expected, much lower correlations were found between RNA-seq and the proteome of different and unrelated tissues (0.1–0.3). The good but not complete agreement between the RNA-seq and the proteomics data independently validates both studies because of the complete independence of the mRNA and protein analyses, while highlighting the utility of separate proteome measurements.
DISCUSSION
We performed a deep and quantitative proteome analysis of 27 adult mouse tissues, as well as embryonic tissue, using the SILAC mouse as a common reference. We identified a total of 7,349 proteins and 3,000 to 5,000 proteins in each tissue. These numbers of identifications per sample are similar to those obtained in previous studies that included multiple tissues (10, 11). However, our data represent by far the largest accurately quantified multi-tissue proteome. One of the main challenges in tissue analysis is the high abundance of extracellular matrix proteins that limit the dynamic range of the analysis. Furthermore, we found that tissues often have large quantities of non-protein contaminants (such as glycogen in the liver) that might interfere with appropriate peptide separation. As a result, tissue analyses result in a lower number of protein identifications. Therefore, our initial mouse proteome expression study also highlights the need for even more technological advances in shotgun proteomics.
Nevertheless, the broad scale of the analysis combined with SILAC-based quantification captured key protein determinants of tissue identity. Replicate analysis and comparison of the MS data to IHC and mRNA data further strengthens it with regard to the overall accuracy and reproducibility. At the same time, the correlation of 0.5 to 0.6 that we found between the mRNA and the protein levels emphasizes the need to study proteins and not solely the transcript levels. For thousands of proteins, we have provided expression profiles across the 28 tissues examined that can be used by the scientific community to allocate specific proteins of interest to tissues. The complete dataset has been deposited in the MaxQB database, which provides a user-friendly interface for detecting the levels of proteins of interest between tissues and the relative expression within each tissue (38). Such knowledge can assist in the prediction and interpretation of tissue-specific phenotypes of knock-out mice or suggest which non-diseased tissues might be affected by treatment aimed at specific proteins.
Interestingly, we found that tissue specificity is evident already in the most abundant proteins. This rather surprising result implies that the characteristic tissue functions are often the most dominant ones at the proteome level, highlighting the degree of specialization of differentiated tissues. These large differences in the high-abundance proteins do not imply a large number of strictly tissue-specific proteins. This is in agreement with a previous study on cell lines (3) and a previous tissue-based transcriptome study (2), which also found a very small proportion of genes expressed exclusively by one cell or tissue. Unlike the tissue proteome results reported here, the previous cell line data indicated high concordance at both qualitative and quantitative levels of gene expression. This discrepancy can be attributed to the nature of cancer cell lines, which are non-differentiated, highly proliferative, and adapted to cell culture conditions. Thus, the marked differences between the studies emphasize the large extent of proteome remodeling upon tissue differentiation, something we had also observed before by comparing the proteome of a cancer cell line to a primary cell (39).
In this work we have developed a framework for connecting protein expression patterns to protein annotations, which then points to the functions of the tissue. We combined the unbiased proteomic approach with unsupervised bioinformatic analysis, recapitulating numerous known tissue functions and regulators of tissue differentiation in remarkable detail. In addition, our data provide a source of multiple novel tissue-specific proteins and, conversely, associate proteins with unknown functions with protein annotations. For example, we uncovered the proteomic imprint of known and novel transcriptional regulators of tissue specificity. Most of these transcription factors were involved in muscle and embryonic tissues. In the muscle, this reflects the uniqueness of the function of this tissue, including its fibrillary structure and contractile activity. Embryonic tissue was strongly associated with targets of E2F, Zfx, and Myc, which are involved in cell cycle progression and protein translation. Co-clustering of Zfx and E2F suggests a novel functional association of these two transcription factors related to cell cycle progression and embryonic development. Beyond these mechanisms of transcriptional regulation, our proteomic data highlighted the differential involvement of other modes of regulation of protein expression in the different tissues. Elevated levels of ribosomes in the pancreas and salivary gland underscore the high significance of protein translation in these tissues. Similarly, the strong expression of spliceosomes in the embryonic tissue points to their importance in the regulation of embryonic development. In contrast, the relatively constant proteasome amounts might suggest that proteasomal degradation is influenced not so much by the amount of the complex components, but rather by specific regulation via processes such as protein ubiquitination. Alternatively, and in agreement with previous analyses (5), these constant levels point to the reduced significance of protein degradation mechanisms in the regulation of protein levels relative to protein translation. Last, constant proteasome levels might imply that these are very tightly controlled and that deregulation of these levels would be detrimental to cells and tissues.
In conclusion, our initial quantitative proteomic map of a mammal provides a useful resource for the community and connects protein expression, protein function, and tissue organization in a novel manner.
Supplementary Material
Acknowledgments
We thank the members of the Department of Proteomics and Signal Transduction for fruitful discussions. Special thanks to Igor Paron for the assistance in the MS runs and data analysis.
Footnotes
* This project was supported by the European Commission's 7th Framework Program PROteomics SPECificat ion in Time and Space (PROSPECTS, HEALTH-F4-2008-021,648).
The data associated with this manuscript may be downloaded from the Proteome Commons Tranche network using the following hash: ot60LLSjOCtuH5IoJRh8gCVsyj0HlsMTDgKe0C103Qqs3go0V7OhpuV/oJNouAn4lNyPNs9RP4PpHaEqMx7k21S0nbQAAAAAAACr0g==.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- IHC
- immunohistochemical analysis
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- MS
- mass spectrometry
- SILAC
- stable isotope labeling with amino acids in cell culture.
REFERENCES
- 1. Su A. I., Wiltshire T., Batalov S., Lapp H., Ching K. A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G., Cooke M. P., Walker J. R., Hogenesch J. B. (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U.S.A. 101, 6062–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ramskold D., Wang E. T., Burge C. B., Sandberg R. (2009) An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput. Biol. 5, e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lundberg E., Fagerberg L., Klevebring D., Matic I., Geiger T., Cox J., Algenas C., Lundeberg J., Mann M., Uhlen M. (2010) Defining the transcriptome and proteome in three functionally different human cell lines. Mol. Syst. Biol. 6, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nagaraj N., Wisniewski J. R., Geiger T., Cox J., Kircher M., Kelso J., Paabo S., Mann M. (2011) Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 6. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 7. Cox J., Mann M. (2011) Quantitative, high-resolution proteomics for data-driven systems biology. Annu. Rev. Biochem. 80, 273–299 [DOI] [PubMed] [Google Scholar]
- 8. Yates J. R., 3rd, Gilchrist A., Howell K. E., Bergeron J. J. (2005) Proteomics of organelles and large cellular structures. Nat. Rev. Mol. Cell Biol. 6, 702–714 [DOI] [PubMed] [Google Scholar]
- 9. Mann M., Kelleher N. L. (2008) Precision proteomics: the case for high resolution and high mass accuracy. Proc. Natl. Acad. Sci. U.S.A. 105, 18132–18138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kislinger T., Cox B., Kannan A., Chung C., Hu P., Ignatchenko A., Scott M. S., Gramolini A. O., Morris Q., Hallett M. T., Rossant J., Hughes T. R., Frey B., Emili A. (2006) Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell 125, 173–186 [DOI] [PubMed] [Google Scholar]
- 11. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villen J., Haas W., Sowa M. E., Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ong S. E., Mann M. (2005) Mass spectrometry-based proteomics turns quantitative. Nat. Chem. Biol. 1, 252–262 [DOI] [PubMed] [Google Scholar]
- 13. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 14. Geiger T., Cox J., Ostasiewicz P., Wisniewski J. R., Mann M. (2010) Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods 7, 383–385 [DOI] [PubMed] [Google Scholar]
- 15. Gouw J. W., Krijgsveld J., Heck A. J. (2010) Quantitative proteomics by metabolic labeling of model organisms. Mol. Cell. Proteomics 9, 11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sury M. D., Chen J. X., Selbach M. (2010) The SILAC fly allows for accurate protein quantification in vivo. Mol. Cell. Proteomics 9, 2173–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Larance M., Bailly A. P., Pourkarimi E., Hay R. T., Buchanan G., Coulthurst S., Xirodimas D. P., Gartner A., Lamond A. I. (2011) Stable-isotope labeling with amino acids in nematodes. Nat. Methods 8, 849–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kruger M., Moser M., Ussar S., Thievessen I., Luber C. A., Forner F., Schmidt S., Zanivan S., Fassler R., Mann M. (2008) SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell 134, 353–364 [DOI] [PubMed] [Google Scholar]
- 19. Zanivan S., Krueger M., Mann M. (2012) In vivo quantitative proteomics: the SILAC mouse. Methods Mol. Biol. 757, 435–450 [DOI] [PubMed] [Google Scholar]
- 20. Hubner N. C., Ren S., Mann M. (2008) Peptide separation with immobilized pI strips is an attractive alternative to in-gel protein digestion for proteome analysis. Proteomics 8, 4862–4872 [DOI] [PubMed] [Google Scholar]
- 21. Ishihama Y., Rappsilber J., Mann M. (2006) Modular stop and go extraction tips with stacked disks for parallel and multidimensional peptide fractionation in proteomics. J. Proteome Res. 5, 988–994 [DOI] [PubMed] [Google Scholar]
- 22. Nagaraj N., Kulak N. A., Cox J., Neuhauser N., Mayr K., Hoerning O., Vorm O., Mann M. (2012) System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell. Proteomics 11, M111.013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Michalski A., Damoc E., Hauschild J. P., Lange O., Wieghaus A., Makarov A., Nagaraj N., Cox J., Mann M., Horning S. (2011) Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell. Proteomics 10, M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 25. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 26. Paavilainen L., Edvinsson A., Asplund A., Hober S., Kampf C., Ponten F., Wester K. (2010) The impact of tissue fixatives on morphology and antibody-based protein profiling in tissues and cells. J. Histochem. Cytochem. 58, 237–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ponten F., Gry M., Fagerberg L., Lundberg E., Asplund A., Berglund L., Oksvold P., Bjorling E., Hober S., Kampf C., Navani S., Nilsson P., Ottosson J., Persson A., Wernerus H., Wester K., Uhlen M. (2009) A global view of protein expression in human cells, tissues, and organs. Mol. Syst. Biol. 5, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lam Y. W., Lamond A. I., Mann M., Andersen J. S. (2007) Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr. Biol. 17, 749–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Geiger T., Cox J., Mann M. (2010) Proteomic changes resulting from gene copy number variations in cancer cells. PLoS Genet. 6, e1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Torres E. M., Dephoure N., Panneerselvam A., Tucker C. M., Whittaker C. A., Gygi S. P., Dunham M. J., Amon A. (2010) Identification of aneuploidy-tolerating mutations. Cell 143, 71–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Torres E. M., Sokolsky T., Tucker C. M., Chan L. Y., Boselli M., Dunham M. J., Amon A. (2007) Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317, 916–924 [DOI] [PubMed] [Google Scholar]
- 32. Weiss S. B., Acs G., Lipmann F. (1958) Amino acid incorporation in pigeon pancreas fractions. Proc. Natl. Acad. Sci. U.S.A. 44, 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beeley J. A., Cohen E., Keller P. J. (1968) Canine pancreatic ribosomes. I. Preparation and some properties. J. Biol. Chem. 243, 1262–1270 [PubMed] [Google Scholar]
- 34. Flicek P., Amode M. R., Barrell D., Beal K., Brent S., Chen Y., Clapham P., Coates G., Fairley S., Fitzgerald S., Gordon L., Hendrix M., Hourlier T., Johnson N., Kahari A., Keefe D., Keenan S., Kinsella R., Kokocinski F., Kulesha E., Larsson P., Longden I., McLaren W., Overduin B., Pritchard B., Riat H. S., Rios D., Ritchie G. R., Ruffier M., Schuster M., Sobral D., Spudich G., Tang Y. A., Trevanion S., Vandrovcova J., Vilella A. J., White S., Wilder S. P., Zadissa A., Zamora J., Aken B. L., Birney E., Cunningham F., Dunham I., Durbin R., Fernandez-Suarez X. M., Herrero J., Hubbard T. J., Parker A., Proctor G., Vogel J., Searle S. M. (2011) Ensembl 2011. Nucleic Acids Res. 39, D800-D806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van Riggelen J., Yetil A., Felsher D. W. (2010) MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 10, 301–309 [DOI] [PubMed] [Google Scholar]
- 36. Tontonoz P., Spiegelman B. M. (2008) Fat and beyond: the diverse biology of PPARgamma. Annu. Rev. Biochem. 77, 289–312 [DOI] [PubMed] [Google Scholar]
- 37. Mortazavi A., Williams B. A., Mccue K., Schaeffer L., Wold B. (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 [DOI] [PubMed] [Google Scholar]
- 38. Schaab C., Geiger T., Stoehr G., Cox J., Mann M. (2012) Analysis of high accuracy, quantitative proteomics data in the MaxQB database. Mol. Cell. Proteomics 11, M111.014068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pan C., Kumar C., Bohl S., Klingmueller U., Mann M. (2009) Comparative proteomic phenotyping of cell lines and primary cells to assess preservation of cell type-specific functions. Mol. Cell. Proteomics 8, 443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.