

Abstract
We report a high quality and system-wide proteome catalogue covering 71% (3,542 proteins) of the predicted genes of fission yeast, Schizosaccharomyces pombe, presenting the largest protein dataset to date for this important model organism. We obtained this high proteome and peptide (11.4 peptides/protein) coverage by a combination of extensive sample fractionation, high resolution Orbitrap mass spectrometry, and combined database searching using the iProphet software as part of the Trans-Proteomics Pipeline. All raw and processed data are made accessible in the S. pombe PeptideAtlas. The identified proteins showed no biases in functional properties and allowed global estimation of protein abundances. The high coverage of the PeptideAtlas allowed correlation with transcriptomic data in a system-wide manner indicating that post-transcriptional processes control the levels of at least half of all identified proteins. Interestingly, the correlation was not equally tight for all functional categories ranging from rs >0.80 for proteins involved in translation to rs <0.45 for signal transduction proteins. Moreover, many proteins involved in DNA damage repair could not be detected in the PeptideAtlas despite their high mRNA levels, strengthening the translation-on-demand hypothesis for members of this protein class. In summary, the extensive and publicly available S. pombe PeptideAtlas together with the generated proteotypic peptide spectral library will be a useful resource for future targeted, in-depth, and quantitative proteomic studies on this microorganism.
In many respects Schizosaccharomyces pombe is a typical eukaryotic cell, and its genome contains several conserved genes necessary for eukaryotic cell organization such as cytoskeleton, compartmentalization, cell cycle control, proteolysis, protein phosphorylation, and RNA splicing (1). Yet it has one of the smallest numbers of protein-coding genes recorded to date for a self-sustaining eukaryote, and the cells are compatible with a range of experimental biology techniques, including genetic engineering (1). The evolution from the first prokaryote to the first eukaryote took about 2,300 million years. It then took another 500 million years for the first multicellular organisms to evolve from the first unicellular eukaryote. This time span suggests that not many new genes were required for the evolution from unicellular to multicellular eukaryotes, a fact that is supported by comparative genomics (2). Furthermore, fission yeast seems to be more closely related to mammalian cells than the budding yeast based on the degree of conservation of several cellular processes such as cell cycle control and heat shock response. It is estimated that fission yeast diverged from budding yeast around 330–420 million years ago (3). Hence, S. pombe has been a prime model organism for the study of numerous central biological processes such as cell cycle control, DNA repair, and recombination (4, 5). Because it shares many features with cells of multicellular eukaryotes, the proteome of S. pombe constitutes a “eukaryotic core proteome” with a high significance also for multicellular species.
Proteins carry out most biological functions. These functions are modulated through the dynamic adaptation of the cellular concentration of the corresponding proteins, their status of post-translational modification, and their subcellular distribution. Although transcriptomic studies indicate the potential for protein expression, they do not directly measure proteome characteristics, and the corresponding protein quantities cannot be precisely computed from the transcript levels due to poor correlation of mRNA and protein abundances. The type, site of attachment, and stoichiometry of post-translational modifications are even more difficult to infer from transcript profiles (6–8). Thus, comprehensive proteome analysis is essential for understanding the structure and control of physiological processes and for discovering new emergent properties of biological systems. To date, only few eukaryotic proteomes have been extensively catalogued. They include Saccharomyces cerevisiae with 67% coverage of the predicted ORFs (9, 10), Caenorhabditis elegans (54%) (11), Arabidopsis thaliana (50%) (12), Drosophila melanogaster (63%) (13), and more recently Mus musculus (50%) (14), and Homo sapiens (50%) (15, 16). Interestingly, the genomes of these species were also the first eukaryotic genomes to be sequenced and S. pombe became the sixth, signifying their paramount importance for biological studies.
The fact that there are only few comprehensive proteome datasets available from more than 40 eukaryotic species with completely sequenced genomes reflects that cataloging proteomes remains challenging. To date, all large scale proteome studies were carried out using mass spectrometry-based shotgun proteomics, which is currently the method of choice for whole proteome studies (17). In this method, the protein complement of a cell is extracted and digested by a protease, frequently trypsin, and the resulting peptides are identified by tandem mass spectrometry. In general, the proteome coverage achievable by this technique is largely dependent on sample complexity, dynamic range of the proteins in a sample, the mass spectrometric instrumentation and methods used, and algorithms for peptide identification and protein inference (18). Generally, multidimensional sample fractionation (9) or optimized analytical strategies such as analysis-driven experimentation (13) have been used to achieve saturation coverage of the expressed proteome (19). To date, less than half of all open reading frames of S. pombe have been identified by most previous proteome studies (20, 21). We recently published a dataset containing absolute protein abundances for 66% of the predicted ORFs in S. pombe determined using a MS intensity-based estimation approach (22). As only moderate sample fractionation was applied, proteome coverage has not been maximized in this dataset.
In this study, we provide an extensive peptide atlas of S. pombe using different growth conditions, an extensive fractionation strategy at the protein level, and a standard fractionation approach at the peptide level, respectively, followed by high resolution mass spectrometry, and we combined the peptides identified from the dataset by different search engines using the iProphet tool (23) in the Trans Proteomics Pipeline (TPP). We covered 71% of all predicted ORFs of S. pombe and, more importantly, more than twice the number of identified unique peptide sequences compared with previous studies. Therefore, this extensive peptide catalogue of S. pombe provides an excellent resource for the development of sensitively targeted proteomics workflows for high throughput quantification of selected sets of proteins. Moreover, the dataset allowed us to generate a spectral library to improve the interpretation of LC-MS/MS data in future proteomics S. pombe studies via the discovery of proteomic methods and also to estimate the cellular concentration of the identified proteins to infer primary features of a eukaryotic core proteome.
To publicly access and browse this MS dataset, a PeptideAtlas of all identified and statistically validated MS/MS spectra was generated and stored in the PeptideAtlas repository. The PeptideAtlas web interface has a rich set of visualization and data exploration tools, allowing users to interactively mine information about individual proteins and peptides, their genome mappings, and the supporting spectral evidence. Therefore, the PeptideAtlas has become the tool of choice for selecting proteotypic peptides (24) and to build methods for targeted proteomics and selected reaction monitoring (10).
EXPERIMENTAL PROCEDURES
Sample Preparation
Fission yeast (S. pombe) strain FY7056 was acquired from the Yeast Genetic Resource Center, Japan. The cells were grown in liquid medium (SD medium supplemented with 100 mg/liter lysine and 2% glucose) at 30°C until an A600 of 0.6–0.7 was reached. Cells were harvested by centrifugation at 300 × g for 5 min, resuspended in stop buffer (0.9% NaCl, 1 mm NaN3, 10 mm EDTA, 50 mm NaF), and centrifuged at 2,000 × g for 1 min. For the IEF fractionation experiment, the following aliquots were used: 1) rapidly growing cells in EMM (∼4 × 106 cells/ml); 2) cells grown in EMM to ∼4 × 106 cells/ml, then centrifuged, washed 1× in EMM-N, and incubated (nitrogen-starved) in EMM-N for 24 h, before harvesting; 3) cells grown in yeast extract medium (YE), ∼4 × 106 cells/ml, and 0.5 mm H2O2, harvested 1 h after H2O2 addition; 4) Cells grown in YE ∼4 × 106 cells/ml, rapidly shifted to 39°C, and harvested 30 min after temperature shift. Aliquots 3 and 4 were performed as described in more detail in Ref. 25. For aliquots 5 and 6 (the meiotic samples), strain 968 h90 was grown in EMM to ∼3 × 106 cells/ml, washed once in EMM + 0.5% glucose and without NH4Cl, resuspended in EMM + 0.5% glucose and without NH4Cl at 28°C to induce meiosis, and harvested after 12 and 15 h as described in Ref. 26. For samples 1–4, strain 972 h- was employed and grown at 30°C.
Protein Level Fractionation
Cells were washed with S-buffer (1.4 m sorbitol, 0.5 mm MgCl2, 40 mm HEPES, pH 6.5, 1 mm PMSF). Washed cells were resuspended with S-buffer containing additionally 10 mm DTT and incubated at 30°C for 10 min with gentle shaking. Pellets collected by spinning at 300 × g for 5 min were resuspended in S-buffer containing 1:3 (w/v) of lyticase (L4025, Sigma) and incubated at 30°C for 40 min with gentle shaking. The cell suspension was diluted with S-buffer, and the pellets (spheroplasts) were collected by centrifugation at 300 × g, 4°C for 5 min, followed by two washes with ice-cold S-buffer. The lysis of spheroplasts in F-buffer (18% Ficoll 400, 0.5 mm MgCl2, 20 mm PIPES, pH 6.5, 1 mm PMSF) was carried out by homogenization using 20 strokes of a loose-fitting Dounce homogenizer on ice. Unlysed material was removed by gentle centrifugation at 300 × g for 10 min. The supernatant was carefully layered on top of ice-cold GF-buffer (7% Ficoll 400, 20% glycerol, 0.5 mm MgCl2, 20 mm PIPES, pH 6.5, 1 mm PMSF) and was centrifuged at 4,000 × g, 4°C for 5 min. The pellet (pellet-1) was saved, and the supernatant was centrifuged at 55,000 × g, 4°C for 30 min (F2 pellet-2). The supernatant was mixed with ice-cold 5× RIPA buffer and centrifuged at 20,000 × g, 4°C for 20 min to obtain the supernatant as the first fraction named F1. Pellet-1 and pellet-2 were separately resuspended in ice-cold 1× RIPA. Sonicated (five times for 10 s) suspensions were centrifuged at 20,000 × g, 4°C for 20 min, to obtain the supernatant as the second fraction named F2 from pellet-1 and third fraction named F3 from pellet-2. All the pellets from F1 to F3 preparations were combined and lysed with 2% SDS, 6 m urea in PBS by boiling for 5 min. The lysate was centrifuged at 20,000 × g, 4°C for 20 min, to obtain the fourth fraction F4.
SDS-PAGE and In-gel Digestion
Protein amounts of all fractions were estimated by BCA assay (Thermo Fisher Scientific). 80 μg of protein lysate from each fraction was used for SDS-PAGE using a NuPAGE 4–12% BisTris1 gel, 1.0 mm, 10 wells (Invitrogen), and done following the manufacturers' instructions. The gel was cut into 20 equal lanes, and tryptic in-gel digestion was done as described previously (27).
Preparation of IEF-fractionated Peptide Samples
Aliquots of 108 cells of the six different S. pombe samples described above were lysed, respectively, with glass beads in 100 μl of lysis buffer (8 m urea, 0.1% RapiGest, and 0.1 m ammonium bicarbonate) using rigorous shaking six times for 30 s in a FastPrep instrument (Q-Biogene) and pooled. The protein mix obtained was reduced with 5 mm tris(2-carboxyethyl)phosphine for 60 min at 37°C and alkylated with 10 mm iodoacetamide for 30 min in the dark at 25°C. After quenching the reaction with 12 mm N-acetylcysteine, the samples were diluted with 100 mm ammonium bicarbonate buffer to a final urea concentration of 1.5 m. Proteins were digested by incubation with sequencing-grade modified trypsin (1:50, w/w; Promega, Madison, WI) overnight at 37°C. Then the samples were acidified with 2 m HCl to a final concentration of 50 mm and incubated for 15 min at 37°C, and the cleaved detergent removed by centrifugation at 10,000 × g for 5 min. Subsequently, peptides were desalted on C18 reversed-phase spin columns according to the manufacturer's instructions (Macrospin, Harvard Apparatus), dried under vacuum, and re-solubilized in Off-Gel electrophoresis buffer containing 6.25% glycerol and 1.25% IPG buffer (GE Healthcare). The peptides were separated on a 12-cm pH 3–10 IPG strip (GE Healthcare) with a 3100 OFFGEL fractionator (Agilent) using a protocol of 1 h of rehydration at maximum 500 V, 50 μA, and 200 milliwatts. Peptides were separated at maximum 8000 V, 100 μA, and 300 milliwatts until 20 kV-h were reached. Subsequently, the peptides in each of the 12 IEF fractions were desalted using C18 reversed-phase columns according to the manufacturer's instructions (Macrospin, Harvard Apparatus), dried under vacuum, and stored at −80°C until further use.
LC-MS/MS Analysis
LC-MS analysis was carried out as described previously (28). In brief, freeze-dried samples were reconstituted in 0.1% formic acid and analyzed using nanoHPLC coupled to a LTQ Orbitrap XL or classic or LTQ-FT-ICR Ultra (Thermo Fisher Scientific). Peptides were trapped onto a C18 pre-column and separated on an analytical column using a 2-h gradient ranging from 0 to 80% acetonitrile, 0.1% formic acid. MS scans were acquired with a resolution of 60,000 (100,000) Full Width at Half Maximum at 400 m/z in the Orbitrap (LTQ-FT-ICR). The 10 (five for the LTQ-FT-ICR) most intense ions were selected for collision-induced dissociation (CID) fragmentation and analyzed in the ion trap with enabled preview mode.
Data Processing and Analysis
The instrument data were first converted to the peak lists in the centroid mzXML file format. The conversion was performed with ReAdW.exe (version 4.0.2), which is part of the Trans-Proteomic Pipeline (TPP) (version 4.5.0) (29). The peak list files were searched against a target-decoy database (30) consisting of all S. pombe sequences downloaded from PomBase (version 17Feb2010) as well as known contaminants such as porcine trypsin and human keratins (Non-Redundant Protein Database, National Institutes of Health, NCI Advanced Biomedical Computing Center, 2004) with forward and reverse sequences. The final database, which contained 10,584 sequence entries, was compiled using Sequence Reverser (part of MaxQuant version 1.0.13.13) and was searched using three search engines Mascot (Matrix Science, London, UK version 2.3) (31), X! Tandem (version 2011.12.01.1, k-score plugin) (32), and OMSSA (version 2.1.9) (23), respectively. The database search parameters were set as follows: full tryptic specificity was required (cleavage after lysine or arginine residues at two peptide termini, unless followed by proline); no missed cleavages were allowed; carbamidomethylation was set as fixed modification; mass tolerance of the precursor ion and the fragment ions was set at 15 ppm and 0.4 Da, respectively. A statistical analysis of the identified peptides was performed through the TPP (version 4.5.0), a uniform proteomics MS/MS analysis platform utilizing open XML file formats. In brief, the database search output results were further validated using the PeptideProphet software and the results merged with iProphet (34) to generate a single list of peptide assignments. Only assignments with an FDR below 1% were used to perform the protein assembly with ProteinProphet (35). The FDR was also set below 1% for protein and peptide assignments corresponding to a probability cutoff of p ≥ 1 and p ≥ 0.98, respectively. Additionally, the data were uploaded into the PeptideAtlas database as described previously (10) and are available for browsing and downloading. There, all identified proteins and peptides can be viewed along with the corresponding MS/MS scans, protein coverage, precursor m/z, charge, and score. Importantly, we also included peptides with up to two missed cleavages and semi-tryptic in the on-line version of the PeptideAtlas. Relative protein abundances were calculated using the exponentially modified Protein Abundance Index (emPAI) algorithm as described by Ishihama et al. (36) only for proliferating cells from all peptide spectrum matches (including shared peptides) at an FDR of 1% of the corresponding sample dataset (Experiment 2, Fig. 1A and supplemental Table S6). In addition, a spectral library was created from the PSMs that matched the 1% FDR cutoff. The creation was done with SpectraST (37) in three steps. First, a raw library was created containing the raw PSMs matching the cutoff. Second, all PSMs that matched a decoy entry in the original search results were removed. In the final step, spectra assigned to a single peptide were assembled to consensus spectra, and decoy entries were added to the library (38, 39).
Fig. 1.

Experimental design of the S. pombe proteome analysis. A, proteomics workflow of identification of the S. pombe proteome consisted of two different fractionation approaches (Experiment 1 and 2), high performance LC-MS analysis, and protein identification using three database search engines (Mascot, X!tandem, and OMSSA) and the TPP for validation of MS data. The sample mix analyzed in Experiment 1 consisted of equal amounts of protein extracts obtained from cells in six different states, including proliferating and quiescent cells, cells under oxidative and heat stress, and cells during two stages of meiotic differentiation, whereas proteins from proliferating cells were analyzed and extensively fractionated in Experiment 2 (see “Experimental Procedures” for details). B, pie chart indicates the number (percentage) of proteins being identified by 1, 2, or more unique stripped peptides.
Analysis of Undetected Proteins
For S. cerevisiae, we used a PeptideAtlas built May 2009 (10), and mRNA concentrations were obtained as median values across several measurements under normal growth (40). In the case of S. pombe, mRNA expression measurements were not available for the protein samples. Thus, mRNA expression levels were derived from a previously published tiling array dataset obtained under similar experimental conditions (41). We selected 21 CEL files closely matching the experimental conditions of the protein samples, namely exponential growth, oxidative stress, heat shock, quiescent cells (nitrogen starvation), and meiotic cells. To map tiling array expression data to individual gene expression levels, we utilized a custom CDF file created specifically for the platform used in the study (Affymetrix GeneChip® S. pombe Tiling 1.0FR array; CDF file available upon request). Robust MultiArray Average (rma in Affy/Bioconductor) was used for background correction, normalization, and evaluation of the expression levels. Finally, measurements across the conditions were averaged yielding expression levels for 5489 S. pombe genes.
Orthologous genes were determined using a manually curated ortholog list (version 2.16) (42). Gene Ontology (GO) annotations were obtained from BioMart (43). The TopGO R package from Bioconductor was used for GO enrichment analysis (44). By default, TopGO takes into account the GO structure for significance analysis. We used the Fisher test to measure significance of enrichment. All GO terms with less than 20 or more than 1,000 annotated gene products were ignored. We grouped genes into three classes, namely high expression, medium expression, and low expression, according to their measured mRNA levels. Genes that are among the top 25% quantile with respect to their mRNA levels were included in high class, middle 50% quantile in medium class, and bottom 25% quantile in low class. The background set for the enrichment analysis of undetected proteins in a given class is set to be all genes in the respective class. For example, we used all genes in the high class as the background for the undetected proteins in the high class. For the undetected ortholog enrichment analysis, all genes that are known to have an ortholog were used as background.
RESULTS AND DISCUSSION
Generation of an Extensive S. pombe PeptideAtlas
The workflow for the identification of the S. pombe proteome is summarized in Fig. 1A. For protein level separation (Fig. 1A, Experiment 2), we used fractionation methods based on density centrifugation and differential solubility in a range of buffer systems as described under “Experimental Procedures.” The aim was to reduce dynamic range of proteins in each fraction (F) by depleting highly abundant proteins from fraction to fraction. The basis of fractionation was to isolate mainly cytosolic proteins in F1 and nuclear and other organelle proteins in F2 and F3. In the last fraction, we used a buffer containing denaturating agents (2% SDS and 6 m urea) to solubilize proteins with low solubility in nonionic detergents. The band intensities and patterns of SDS-polyacrylamide gels of these fractions were somewhat different, indicating that each fraction contained overlapping but different protein populations and/or abundances (supplemental Fig. S1A). GO-slim cellular component analysis with the protein abundances (emPAI values) of each fraction indicated that overall proteins of cytosolic, nuclear, and organelle were enriched in F1, F2 (also in F4), and F3, respectively (supplemental Fig. S1B). However, we observed that the proteins of some organelles, such as mitochondria, were also enriched in F2, suggesting F2 is a mixture of nuclear and some organelle proteins, but were less in highly abundant cytosolic proteins that interfered with protein identification of low abundant nuclear/organelle proteins. In a separate experiment (Fig. 1A, Experiment 1), cell extracts from cells in six different states, including proliferating and quiescent cells, cells under oxidative and heat stress, and cells during two stages of meiotic differentiation (see “Experimental Procedures” for details) were combined to maximize the number of expressed proteins, and the combined sample was proteolyzed and separated using off gel electrophoretic fractionation followed by LC-MS/MS analysis of each fraction. All MS/MS data sets were subjected to the Trans Proteomics Pipeline (TPP) software for MS data analysis and validation (29). To maximize the number of peptides identifiable from the acquired fragment ion spectra, we searched the data sequentially with the search engines Mascot (31), X!Tandem (32), and OMSSA (23) and combined the results using iProphet (34). The generated list of peptide identifications was used to determine the probability value that matches 1% FDR threshold. All peptides matching this cutoff were then used to assemble the list of proteins with ProteinProphet.
Overall, the combined data set of the gel-based and IEF fractionation experiment consisted of 384 raw MS files with 4,770,221 MS/MS spectra of which 3,252,093 spectra could be assigned to peptide sequences. From these spectra, we identified 40,947 unique peptide sequences at a false-discovery rate (FDR) of 1% from which we inferred 3,542 unique proteins at an FDR of 1% (45). All identified proteins and peptides together with their human orthologs can be found in supplemental Table S1. The identified proteins cover 71% of the genes predicted from the S. pombe genome annotation (pompep_17Feb2010). Importantly, 90% of the proteins were identified with three or more peptides and, on average, 11.4 uniquely stripped peptides were identified per protein (Fig. 1B), indicating that extensive proteome coverage was achieved and suggesting that the dataset will be very valuable as a source of proteotypic peptides (46) for the generation of targeted MS assays. To facilitate proteotypic peptide selection, we provide a table (supplemental Table S2) of all identified peptides ranked by their MS detectability (number of PSMs) for each protein. Additionally, all PSMs are shown in supplemental Table S3. The instrumental raw files as well as the search results are also publicly available through PeptideAtlas.
The extensive MS sequencing and global peptide coverage of the PeptideAtlas also allowed us to generate an extensive spectral library of the most frequently observed peptides to increase the sensitivity, speed, and reliability of MS/MS spectra interpretation in future LC-MS studies. The library of consensus spectra that was created with SpectraST (47) is also publicly available via PeptideAtlas. To demonstrate the utility of this library, we assigned all MS/MS spectra generated from three independent LC-MS/MS analyses of the same sample to peptide sequences using spectral library and database searching. As is apparent from supplemental Fig. S2, a considerable increase in PSMs could be achieved using a combination of both search approaches compared with standard database searching.
Usability of the S. pombe PeptideAtlas for Selected Reaction Monitoring Assay Generation
We also evaluated the usability of the MS/MS spectra, which were acquired in fast scanning linear ion trap instruments, for generating sensitive SRM assays that are typically applied to triple quadrupole (QQQ) LC-MS platforms. Because this is a critical issue affecting most current large LC-MS datasets, several publications have assessed the similarity of MS/MS spectra acquired across different MS platforms (48, 49). In both studies, the authors found a strong correlation between the y-ion peak rank order and relative intensity for QQQ and ion trap data. To evaluate if these observations are also true for our S. pombe PeptideAtlas, we extracted all MS/MS spectra with matching sequence assignments from the latest SRM atlas build of S. cerevisiae. We found around 80 matching MS/MS spectra acquired on QTOF and QQQ instruments (supplemental Table S4), respectively, and we compared their similarity to our dataset (supplemental Fig. S3). In line with previous results, we found a good correlation of y-ion peak ranks across the different platforms also for this subset of identified S. pombe peptides. From this, we conclude that our S. pombe PeptideAtlas provides a useful resource for SRM assay development using different LC-MS platforms.
Proteome Coverage Evaluation
To assess the extent of proteome coverage achieved by the S. pombe PeptideAtlas, we performed bias analysis of physicochemical protein properties and Gene Ontology (GO) terms. By comparing the ratio of identified and unidentified proteins in these classes, we detected biases against protein groups that are notoriously difficult to identify by LC-MS/MS due to their physicochemical properties such as very short proteins (<100 amino acids), proteins with very basic isoelectric point (pI) values, and multiple transmembrane domains (supplemental Fig. S4) (11, 50). Importantly, no large biases against specific GO categories could be detected in our dataset (supplemental Fig. S5 and supplemental Table S5). The bias against short proteins could also be explained, at least in part, by the commonly difficult annotation of small genes in S. pombe (1, 51). Taken together, the available data suggest near complete extraction and identification of most proteins in S. pombe expressed under the multiple conditions employed. However, we cannot rule out that some proteins, such as poorly soluble or very short proteins, are missing in the dataset.
Next, we compared the list of identified proteins with the predicted proteome according to the annotations defined in S. pombe GeneDB. The results of this analysis are shown in Table I and supplemental Table S1 (protein details). We observed only one dubious protein and have not observed any “pseudogenes” and “unknown” proteins in our PeptideAtlas, which highlights the globally good annotation of the S. pombe genome. Intriguingly, our PeptideAtlas covers 59% of the conserved hypothetical proteins and 78% of proteins with a predicted role from homology.
Table I. Summary of annotation status.
| Annotation status | Predicted | Detected |
|---|---|---|
| Conserved hypothetical | 586 | 345 |
| Sequence orphan | 294 | 90 |
| S. pombe-specific | 60 | 4 |
| Dubious | 55 | 1 |
| Transposable element | 22 | 1 |
| Role from homology | 2,158 | 1,685 |
| Experiment characterized | 1,911 | 1,416 |
Estimation of Protein Abundances
Because the PeptideAtlas was generated from a set of different S. pombe samples that would complicate the interpretation of the calculated protein abundances, we estimated relative abundance levels only for proteins identified in experiment 2 from proliferating cells (Fig. 1A). Notably, this data subset accounted for the large majority of the MS data used to build the PeptideAtlas, and therefore most identified proteins could be quantified from this LC-MS dataset (supplemental Table S6). We used the emPAI calculation approach (36) and grouped these proteins according to their cellular concentrations. A good correlation (R2 = 0.78, see supplemental Table S7) with recently published absolute protein concentrations determined by fluorescence microscopy of 28 fission yeast proteins fused to yellow fluorescent protein in exponentially growing cells was observed (52). This demonstrates the following: (i) that we covered a protein concentration range of at least 3.5 orders of magnitude from 1.43 × 106 copies per cell (act1) to 600 copies per cell (cdc12) (52), and (ii) that the calculated emPAI scores can be employed as a good estimate of protein abundances in S. pombe. Additionally, we also compared the spectral counting-based emPAI values of this study with those obtained recently using more accurate MS intensity-based protein abundance estimation (22). The high number of overlapping proteins (supplemental Table S6) and the good correlation of protein abundances between the two datasets (supplemental Fig. S6, r = 0.7) further strengthen the validity of our calculated protein levels. This allowed us to cluster the quantified proteins according to their cellular concentrations into five categories as very high (proteins with abundance values greater than 90th percentile of the log (emPAI)), high (80–90% quantile of log emPAI), medium (20–80% quantile of log emPAI), low (10–20% quantile of log emPAI), and very low (abundance values less than 10th percentile of the log (emPAI)). The relative protein abundance distribution and cluster information of all quantified proteins are shown in Fig. 2, A and B.
Fig. 2.

Analysis of S. pombe protein abundance clusters and their associated physicochemical and functional properties. Protein abundances for proliferating cells (Experiment 2) were assessed using emPAI value calculation. A, all identified proteins were clustered into five major abundance categories based on protein density distribution against log emPAI value. B, number of proteins of the different abundance clusters. Bias analysis of protein length (C), pI (D), and GO-slim (E) for the high and low abundant protein clusters. *, frequency = (number of detected proteins associated with corresponding GO term within the cluster/number of total proteins in the cluster)/(number of total detected proteins associated with corresponding GO term/number of total detected proteins).
Functional Analysis of Protein Abundance Clusters
Physicochemical Properties
First we carried out bias analysis for physicochemical properties for the extreme protein abundance clusters (Fig. 2A) to assess enriched features of those protein groups (either low or high). This analysis revealed that highly abundant proteins were consistently smaller than proteins expressed at low levels (Fig. 2C), a characteristic that was also observed in a system-wide study of the S. cerevisiae proteome using a genetics approach (53) and that is in accordance with the biosynthetic cost minimization hypothesis (54). Along that line, Lackner et al. (55) reported that short mRNAs are more efficiently translated, are more stable (with longer poly(A) tails), and are more efficiently transcribed. This underpins the observation that short proteins are more abundant due to higher gene expression at multiple levels (transcription, RNA turnover, and translation). Additionally, distributing the protein abundance clusters according to their pI value showed that abundant proteins generally have more extreme pI values, which reduces their aggregation potential (56). Furthermore, the large majority of proteins with high pI values consist of very abundant (and small) proteins, a result from the high number of basic histones and ribosomal proteins present in this cluster (Fig. 2D).
GO-slim Analysis
Next, we performed GO-slim analysis using the on-line tool GO term mapper to evaluate the functional bias in each abundance category. To simplify this analysis, we combined the protein clusters of less than 20% quantile of log emPAI (very low and low clusters) and more than 80% of quantile of log emPAI (very high and high clusters), respectively. Among the identified proteins (proliferating cells from experiment 2), 3,504 proteins (99%) were found to be associated with at least one GO term (Fig. 2E). Overall, 120 out of 127 GO terms were included. We observed that proteins involved in metabolic and biosynthetic processes were expressed over the entire range of abundance clusters (supplemental Table S8). All 678 proteins in the very low + low cluster and 710 out of 711 (except SPCC757.15) in the very high + high cluster were found to be annotated. The top three under- and over-represented GOs in these two abundance clusters are shown in Fig. 2E. For better comparison, we ignored GO terms with a small number of proteins in Fig. 2E, e.g. “cell death” proteins (only three genes in total, supplemental Table S8). Interestingly, some categories of proteins were under-represented in one cluster, whereas the same protein groups were over-represented in the other abundance cluster, additionally validating the observations. For example, structure molecular activity and ribosomal activity were the most strongly under-represented categories in low abundant clusters and the most highly over-represented classes in high abundant clusters (Fig. 2E).
KEGG Pathway Analysis
To further investigate differences of low and high abundance proteins in relation to their biological activities, we carried out enrichment analysis of pathways as annotated in the KEGG database. Because S. pombe has been thoroughly used as a model organism to study cell cycle control, we selected genes present in this pathway and mapped the five protein abundance clusters to annotated cell cycle proteins. We identified 55 (74%) of the 74 annotated KEGG pathway cell cycle proteins, underlining the high coverage of our PeptideAtlas (supplemental Table S9). For comparison, the coverage of these cell cycle proteins with existing datasets was only 22% (21). With the exception of three abundant proteins, all proteins in this functional class were in the categories of medium or low abundance (Fig. 3). This result is consistent with the exponentially growing, unsynchronized cell population used in these analyses where the fraction of mitotic cells is low. In particular, more than 50% of the G2 phase proteins detected in our analysis were found to be low or very low abundant (Fig. 3). We observed that the majority of the detected proteins of anaphase-promoting complex-cyclosome complex such as Cut4, Cut9, and Apc2 were very low abundant. By contrast, Skp1-Cullin-F-box E3 ligase complex consisted of relatively high abundant proteins.
Fig. 3.
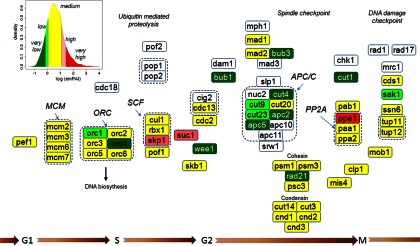
Abundance distribution of cell cycle proteins. Proteins were mapped to the cell cycle pathway according to the KEGG database together with their abundance classes determined from the analysis of unsynchronized proliferating cells (Fig. 1A, Experiment 2). Proteins not identified in the PeptideAtlas are indicated in white.
Proteome and Transcriptome Comparison
We compared our relative protein abundances in proliferating cells with published gene expression data using microarrays of S. pombe at the vegetative stage (www.ncbi.nlm.nih.gov). Overall, the population of mRNA transcripts quantified in this data source largely overlapped with the population of corresponding proteins quantified in this study (3,497 out of 3,509 protein abundances, see supplemental Table S10). We observed a correlation (Spearman rank correlation) of rs = 0.58 between protein abundance and mRNA abundance (Fig. 4) that is comparable with the correlation of mRNA-protein abundance for several organisms reported so far (rs (S. cerevisiae) = 0.57, rs (C. elegans) = 0.59, and rs (D. melanogaster) = 0.66) (11) indicating that post-transcriptional processes control the levels of around half of all identified proteins. Moreover, the analysis revealed that proteins in the high abundance cluster showed a stronger correlation than those in the low abundance cluster (Fig. 4). To understand the correlation between protein and mRNA abundance in specific functional categories, we determined rs values for each GO-slim category (supplemental Fig. S7). This analysis showed that the overall protein abundance correlations were not equally tight across functional categories. For example, we found that the correlation between transcript and protein levels is particularly poor for genes involved in signal transduction (rs = 0.43, supplemental Fig. S7). The same observation was reported for C. elegans and D. melanogaster (11) and S. cerevisiae (57), suggesting that post-transcriptional regulation in these functional classes is conserved through eukaryotes. Conversely, proteins involved in translation show a high correlation between mRNA and protein levels (rs = 0.80, supplemental Fig. S7), which was also observed previously (57), indicating that these processes are tightly controlled primarily at the level of mRNA synthesis (transcriptional control). It is important to note that the mRNA and protein abundances were not determined from the same proliferating cells, which might have an impact on the correlation of highly variable transcripts/proteins. A thorough comparison of absolute mRNA and protein abundances on a copy per cell level determined in the same proliferating and quiescence S. pombe cells, respectively, was published recently (22).
Fig. 4.

Correlation of protein and mRNA levels. Scatterplot of normalized mRNA abundances and normalized protein abundances (emPAI values) for proliferating S. pombe cells is shown, and both values are presented in log scale. The Spearman rank correlation of rs = 0.58 is indicated.
Proteins Missing in the PeptideAtlas
To characterize proteins that were not identified in the PeptideAtlas (data from all experiments) but predicted from genome annotation, we conducted functional enrichment analysis and analyzed the mRNA levels for those proteins in matching conditions (see “Experimental Procedures”). First, we tested if undetected proteins in the fission yeast PeptideAtlas were not identified because the corresponding genes were not transcribed. We split genes into three classes based on their mRNA levels as follows: high expressed (upper 25% percentile), low expressed (lower 25% percentile), and medium expressed (middle 50%). Interestingly, proteins of many genes belonging to the “high” and “medium” abundance classes were not detected in our experiments (supplemental Tables S11.1–3). As expected, we found genes that were difficult to identify, due to their physiochemical properties, enriched in the undetected protein fraction (e.g. plasma membrane proteins) independent of their mRNA abundance levels. In contrast, we did find a significant enrichment of DNA-binding proteins and proteins involved in DNA damage for genes with high but not low mRNA levels (supplemental Table S11.2.). We conducted the same analysis for proteins not detected in the S. cerevisiae PeptideAtlas, which resulted in the enrichment of similar terms (supplemental Tables S11.4–6). Thus, there are functionally or biochemically related proteins that are consistently difficult to identify across species, a notion that was also confirmed when specifically analyzing orthologous genes between the two species (supplemental Table S11.7).
The enrichment of transmembrane proteins among undetected proteins is most likely due to technical limitations (50) rather than a biological phenomenon. We expected that proteins that are needed only under specific stress conditions would not be expressed in normal growth and thus remain undetected. To improve the coverage of the proteome, we therefore created the PeptideAtlas from a pool of cells grown under various conditions. However, DNA damage was not among the conditions included in this pool, thus explaining why undetected proteins were enriched for this function.
We have previously shown that stress- and signaling-related proteins in S. cerevisiae are often subjected to translation on demand (40), i.e. the mRNA of these proteins is transcribed even under “normal” conditions. Upon induction of stress, those proteins can quickly be synthesized, which constitutes an energy-efficient yet fast means of controlling abundance of these proteins. The fact that transcriptional regulators and stress-related proteins were enriched among the high and medium classes suggests that many undetected proteins in S. pombe are also subjected to translation on demand.
Cross-species Comparison of Protein Abundances
Finally, we compared S. pombe protein abundances (emPAI values) to the published data sets of S. cerevisiae and H. sapiens (15). First we performed ortholog mapping (58) that resulted in 2,695 pairs of S. cerevisiae and 2,142 pairs of H. sapiens orthologs, respectively, with protein abundances (supplemental Table S12). These pairs were obtained after removing duplicates, i.e. if several S. pombe protein IDs match to one ortholog, we considered only one of the IDs and if a group of orthologs matches to one S. pombe ID, only the leading ortholog was considered. Next, we determined overall Spearman correlations (rs) for the three datasets and observed good protein abundance correlations between all species, particular between the two yeast datasets (rs = 0.66, supplemental Fig. S8).
In total, we found 1,758 genes with abundance values that map to all three species (supplemental Table S13) and compared their protein levels by hierarchical clustering (Fig. 5). We observed several protein clusters with common or specific protein expression patterns for the different species. Using enrichment analysis, we could identify four clusters with significantly enriched GO terms. As expected, proteins involved in cell cycle and nucleotide binding (cluster 1) are low abundant across all species, whereas the opposite is true for ribosomal proteins (cluster 4). Because S. cerevisiae has the lowest number of introns of the three organisms compared (0.04 introns/gene (1)), the expression of proteins involved in splicing is expected to be lowest in budding yeast, which can be confirmed in cluster 2. Accordingly, the expression of these proteins is higher in S. pombe (1 intron/gene (1)) and highest in the human cells (7.8 introns/gene (33)). Interestingly, proteins of the proteasome are specifically highly expressed in the human U2OS cell line compared with both yeast strains (cluster 3). This suggests that the human U2OS cells have higher proteasome activity and protein degradation rates compared with the two yeast strains.
Fig. 5.

Hierarchical clustering of protein levels of S. pombe genes and their orthologous genes in S. cerevisiae and humans. Protein clusters were subjected to GO-term enrichment analysis using DAVID (david.abcc.ncifcrf.gov). Only clusters with significant terms (p < 0.05) are displayed.
CONCLUSION
In conclusion, the proteomics information reported here together with the S. pombe peptide library, which is now available to the public via PeptideAtlas, will greatly alleviate the study of S. pombe as a prime model, particularly using targeted proteomics approaches. Our proteome analysis underlines the necessity and usefulness of proteome analyses to understand the features and traits of eukaryotic core proteomes together with technical aspects of proteome analysis and abundance of the actual effectors of biological processes.
Supplementary Material
Footnotes
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- emPAI
- exponentially modified Protein Abundance Index
- PSM
- peptide spectrum match
- FDR
- false discovery rate
- EMM
- EMM, Edinburgh minimal medium
- TPP
- Trans Proteomic Pipeline
- F
- fraction
- GO
- Gene Ontology.
REFERENCES
- 1. Wood V., Gwilliam R., Rajandream M. A., Lyne M., Lyne R., Stewart A., Sgouros J., Peat N., Hayles J., Baker S., Basham D., Bowman S., Brooks K., Brown D., Brown S., Chillingworth T., Churcher C., Collins M., Connor R., Cronin A., Davis P., Feltwell T., Fraser A., Gentles S., Goble A., Hamlin N., Harris D., Hidalgo J., Hodgson G., Holroyd S., Hornsby T., Howarth S., Huckle E. J., Hunt S., Jagels K., James K., Jones L., Jones M., Leather S., McDonald S., McLean J., Mooney P., Moule S., Mungall K., Murphy L., Niblett D., Odell C., Oliver K., O'Neil S., Pearson D., Quail M. A., Rabbinowitsch E., Rutherford K., Rutter S., Saunders D., Seeger K., Sharp S., Skelton J., Simmonds M., Squares R., Squares S., Stevens K., Taylor K., Taylor R. G., Tivey A., Walsh S., Warren T., Whitehead S., Woodward J., Volckaert G., Aert R., Robben J., Grymonprez B., Weltjens I., Vanstreels E., Rieger M., Schäfer M., Müller-Auer S., Gabel C., Fuchs M., Düsterhöft A., Fritzc C., Holzer E., Moestl D., Hilbert H., Borzym K., Langer I., Beck A., Lehrach H., Reinhardt R., Pohl T. M., Eger P., Zimmermann W., Wedler H., Wambutt R., Purnelle B., Goffeau A., Cadieu E., Dréano S., Gloux S., Lelaure V., Mottier S., Galibert F., Aves S. J., Xiang Z., Hunt C., Moore K., Hurst S. M., Lucas M., Rochet M., Gaillardin C., Tallada V. A., Garzon A., Thode G., Daga R. R., Cruzado L., Jimenez J., Sanchez M., del Rey F., Benito J., Domínguez A., Revuelta J. L., Moreno S., Armstrong J., Forsburg S. L., Cerutti L., Lowe T., McCombie W. R., Paulsen I., Potashkin J., Shpakovski G. V., Ussery D., Barrell B. G., Nurse P. (2002) The genome sequence of Schizosaccharomyces pombe. Nature 415, 871–880 [DOI] [PubMed] [Google Scholar]
- 2. Sipiczki M. (2000) Where does fission yeast sit on the tree of life? Genome Biol. 1, REVIEWS1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Remacle J. E., Albrecht G., Brys R., Braus G. H., Huylebroeck D. (1997) Three classes of mammalian transcription activation domain stimulate transcription in Schizosaccharomyces pombe. EMBO J. 16, 5722–5729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davis L., Smith G. R. (2001) Meiotic recombination and chromosome segregation in Schizosaccharomyces pombe. Proc. Natl. Acad. Sci. U.S.A. 98, 8395–8402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Humphrey T. (2000) DNA damage and cell cycle control in Schizosaccharomyces pombe. Mutat. Res. 451, 211–226 [DOI] [PubMed] [Google Scholar]
- 6. Anderson L., Seilhamer J. (1997) A comparison of selected mRNA and protein abundances in human liver. Electrophoresis 18, 533–537 [DOI] [PubMed] [Google Scholar]
- 7. Greenbaum D., Colangelo C., Williams K., Gerstein M. (2003) Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 4, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gygi S. P., Rochon Y., Franza B. R., Aebersold R. (1999) Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19, 1720–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Godoy L. M., Olsen J. V., Cox J., Nielsen M. L., Hubner N. C., Fröhlich F., Walther T. C., Mann M. (2008) Comprehensive mass spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254 [DOI] [PubMed] [Google Scholar]
- 10. Deutsch E. W., Lam H., Aebersold R. (2008) PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 9, 429–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schrimpf S. P., Weiss M., Reiter L., Ahrens C. H., Jovanovic M., Malmström J., Brunner E., Mohanty S., Lercher M. J., Hunziker P. E., Aebersold R., von Mering C., Hengartner M. O. (2009) Comparative functional analysis of the Caenorhabditis elegans and Drosophila melanogaster proteomes. PLoS Biol. 7, e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baerenfaller K., Grossmann J., Grobei M. A., Hull R., Hirsch-Hoffmann M., Yalovsky S., Zimmermann P., Grossniklaus U., Gruissem W., Baginsky S. (2008) Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 320, 938–941 [DOI] [PubMed] [Google Scholar]
- 13. Brunner E., Ahrens C. H., Mohanty S., Baetschmann H., Loevenich S., Potthast F., Deutsch E. W., Panse C., de Lichtenberg U., Rinner O., Lee H., Pedrioli P. G., Malmstrom J., Koehler K., Schrimpf S., Krijgsveld J., Kregenow F., Heck A. J., Hafen E., Schlapbach R., Aebersold R. (2007) A high-quality catalog of the Drosophila melanogaster proteome. Nat. Biotechnol. 25, 576–583 [DOI] [PubMed] [Google Scholar]
- 14. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beck M., Schmidt A., Malmstroem J., Claassen M., Ori A., Szymborska A., Herzog F., Rinner O., Ellenberg J., Aebersold R. (2011) The quantitative proteome of a human cell line. Mol. Syst. Biol. 7, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nagaraj N., Wisniewski J. R., Geiger T., Cox J., Kircher M., Kelso J., Pääbo S., Mann M. (2011) Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 18. Nesvizhskii A. I., Aebersold R. (2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 4, 1419–1440 [DOI] [PubMed] [Google Scholar]
- 19. Reiter L., Claassen M., Schrimpf S. P., Jovanovic M., Schmidt A., Buhmann J. M., Hengartner M. O., Aebersold R. (2009) Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol. Cell. Proteomics 8, 2405–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lackner D. H., Schmidt M. W., Wu S., Wolf D. A., Bähler J. (2012) Regulation of transcriptome, translation, and proteome in response to environmental stress in fission yeast. Genome Biol. 13, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmidt M. W., Houseman A., Ivanov A. R., Wolf D. A. (2007) Comparative proteomic and transcriptomic profiling of the fission yeast Schizosaccharomyces pombe. Mol. Syst. Biol. 3, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marguerat S., Schmidt A., Codlin S., Chen W., Aebersold R., Bähler J. (2012) Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 151, 671–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 24. Vizcaíno J. A., Foster J. M., Martens L. (2010) Proteomics data repositories: providing a safe haven for your data and acting as a springboard for further research. J. Proteomics 73, 2136–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen D., Toone W. M., Mata J., Lyne R., Burns G., Kivinen K., Brazma A., Jones N., Bähler J. (2003) Global transcriptional responses of fission yeast to environmental stress. Mol. Biol. Cell 14, 214–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mata J., Wilbrey A., Bähler J. (2007) Transcriptional regulatory network for sexual differentiation in fission yeast. Genome Biol. 8, R217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 28. Schmidt A., Beck M., Malmström J., Lam H., Claassen M., Campbell D., Aebersold R. (2011) Absolute quantification of microbial proteomes at different states by directed mass spectrometry. Mol. Syst. Biol. 7, 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deutsch E. W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J. K., Martin D. B., Nesvizhskii A. I., Aebersold R. (2010) A guided tour of the Trans-Proteomic Pipeline. Proteomics 10, 1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 31. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 32. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 33. Sakharkar M. K., Chow V. T., Kangueane P. (2004) Distributions of exons and introns in the human genome. In Silico Biol. 4, 387–393 [PubMed] [Google Scholar]
- 34. Shteynberg D., Deutsch E. W., Lam H., Eng J. K., Sun Z., Tasman N., Mendoza L., Moritz R. L., Aebersold R., Nesvizhskii A. I. (2011) iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics 10, M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Keller A., Eng J., Zhang N., Li X. J., Aebersold R. (2005) A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 1, 2005.0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ishihama Y., Oda Y., Tabata T., Sato T., Nagasu T., Rappsilber J., Mann M. (2005) Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 4, 1265–1272 [DOI] [PubMed] [Google Scholar]
- 37. Lam H. (2011) Spectral archives: a vision for future proteomics data repositories. Nat. Methods 8, 546–548 [DOI] [PubMed] [Google Scholar]
- 38. Lam H., Deutsch E. W., Aebersold R. (2010) Artificial decoy spectral libraries for false discovery rate estimation in spectral library searching in proteomics. J. Proteome Res. 9, 605–610 [DOI] [PubMed] [Google Scholar]
- 39. Lam H., Deutsch E. W., Eddes J. S., Eng J. K., Stein S. E., Aebersold R. (2008) Building consensus spectral libraries for peptide identification in proteomics. Nat. Methods 5, 873–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brockmann R., Beyer A., Heinisch J. J., Wilhelm T. (2007) Posttranscriptional expression regulation: what determines translation rates? PLoS Comput. Biol. 3, e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wilhelm B. T., Marguerat S., Watt S., Schubert F., Wood V., Goodhead I., Penkett C. J., Rogers J., Bähler J. (2008) Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 453, 1239–1243 [DOI] [PubMed] [Google Scholar]
- 42. Wood V. (2006) Schizosaccharomyces pombe comparative genomics; from sequence to systems. Comparative Genomics 15, 233–285 Available at: DOI: 10.1007/4735–97 [Google Scholar]
- 43. Zhang J., Haider S., Baran J., Cros A., Guberman J. M., Hsu J., Liang Y., Yao L., Kasprzyk A. (2011) BioMart: a data federation framework for large collaborative projects. Database 2011, bar038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alexa A., Rahnenführer J., Lengauer T. (2006) Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607 [DOI] [PubMed] [Google Scholar]
- 45. Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 12 [Google Scholar]
- 46. Kuster B., Schirle M., Mallick P., Aebersold R. (2005) Scoring proteomes with proteotypic peptide probes. Nat. Rev. Mol. Cell Biol. 6, 577–583 [DOI] [PubMed] [Google Scholar]
- 47. Lam H., Deutsch E. W., Eddes J. S., Eng J. K., King N., Stein S. E., Aebersold R. (2007) Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 7, 655–667 [DOI] [PubMed] [Google Scholar]
- 48. de Graaf E. L., Altelaar A. F., van Breukelen B., Mohammed S., Heck A. J. (2011) Improving SRM assay development: a global comparison between triple quadrupole, ion trap, and higher energy CID peptide fragmentation spectra. J. Proteome Res. 10, 4334–4341 [DOI] [PubMed] [Google Scholar]
- 49. Sherwood C. A., Eastham A., Lee L. W., Risler J., Mirzaei H., Falkner J. A., Martin D. B. (2009) Rapid optimization of MRM-MS instrument parameters by subtle alteration of precursor and product m/z targets. J. Proteome Res. 8, 3746–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Savas J. N., Stein B. D., Wu C. C., Yates J. R., 3rd (2011) Mass spectrometry accelerates membrane protein analysis. Trends Biochem. Sci. 36, 388–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bitton D. A., Wood V., Scutt P. J., Grallert A., Yates T., Smith D. L., Hagan I. M., Miller C. J. (2011) Augmented annotation of the Schizosaccharomyces pombe genome reveals additional genes required for growth and viability. Genetics 187, 1207–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu J. Q., Pollard T. D. (2005) Counting cytokinesis proteins globally and locally in fission yeast. Science 310, 310–314 [DOI] [PubMed] [Google Scholar]
- 53. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 54. Warringer J., Blomberg A. (2006) Evolutionary constraints on yeast protein size. BMC Evol. Biol. 6, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lackner D. H., Beilharz T. H., Marguerat S., Mata J., Watt S., Schubert F., Preiss T., Bähler J. (2007) A network of multiple regulatory layers shapes gene expression in fission yeast. Mol. Cell 26, 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ishihama Y., Schmidt T., Rappsilber J., Mann M., Hartl F. U., Kerner M. J., Frishman D. (2008) Protein abundance profiling of the Escherichia coli cytosol. BMC Genomics 9, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Beyer A., Hollunder J., Nasheuer H. P., Wilhelm T. (2004) Post-transcriptional expression regulation in the yeast Saccharomyces cerevisiae on a genomic scale. Mol. Cell. Proteomics 3, 1083–1092 [DOI] [PubMed] [Google Scholar]
- 58. Wood V., Harris M. A., McDowall M. D., Rutherford K., Vaughan B. W., Staines D. M., Aslett M., Lock A., Bähler J., Kersey P. J., Oliver S. G. (2012) PomBase: a comprehensive online resource for fission yeast. Nucleic Acids Res. 40, D695–D699 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.