

Abstract
An acute increase in the international normalized ratio (INR; a comparison of prothrombin time to monitor the effects of warfarin) over 3 in patients with chronic kidney disease (CKD) is often associated with an unexplained acute increase in serum creatinine (SC) and an accelerated progression of CKD. Kidney biopsy in a subset of these patients showed obstruction of the renal tubule by red blood cell casts, and this appears to be the dominant mechanism of the acute kidney injury. We termed this warfarin-related nephropathy (WRN), and previously reported cases of WRN only in patients with CKD. We now assess whether this occurs in patients without CKD, its risk factors, and consequences. In 15,258 patients who initiated warfarin therapy during a 5-year period, 4006 had an INR over 3 and SC measured at the same time; however, the large data set precluded individual patient clinical assessment. A presumptive diagnosis of WRN was made if the SC increased by over 0.3 mg/dl within 1 week after the INR exceeded 3 with no record of hemorrhage. WRN occurred in 20.5% of the entire cohort, 33.0% of the CKD cohort, and 16.5% of the no-CKD cohort. Other risk factors included age, diabetes mellitus, hypertension, and cardiovascular disease. The 1-year mortality was 31.1% with compared with 18.9% without WRN, an increased risk of 65%. Thus, WRN may be a common complication of warfarin therapy in high-risk patients and CKD doubles this risk. The mechanisms of these risks are unclear.
Keywords: acute kidney injury, mortality, warfarin
Recently, we reported a renal biopsy study of nine patients on warfarin therapy with unexplained acute kidney injury (AKI) associated with increased international normalized ratio (INR). Based on the renal biopsy findings, we concluded that the AKI was caused by glomerular hemorrhage and renal tubular obstruction by red blood cell casts.1 Each of these patients had clinical and renal biopsy evidence of chronic kidney disease (CKD) that predated the episode of AKI. Because this form of AKI appeared to be associated with CKD, we next undertook a retrospective analysis of 103 patients from our CKD population receiving warfarin therapy. Of these, 49 patients developed an INR >3.0. Of these, 18 patients (37%) experienced an unexplained increase in serum creatinine (SC) ≥0.3 mg/dl (mean increase 0.61±0.44 mg/dl) within 1 week of the INR >3.0.2 These patients also showed accelerated progression of their CKD, as compared with those who did not experience an increase in SC in relation to the INR >3.0. We have termed the unexplained increase in SC associated with INR >3.0, warfarin-related nephropathy (WRN).
The current study was undertaken to investigate further the prevalence, risk factors, and consequence of WRN, with emphasis on the extent to which WRN develops in CKD compared with no-CKD patients. The large data set of the present study precluded detailed individual patient clinical assessment to assess whether an increase in SC acutely related to an INR >3.0 was ‘unexplained’, as we were able to do in our previous reports.1,2 However, we did require that the patient’s medical record at the time of an INR >3.0 not describe hemorrhage or blood transfusion. Thus, the present work describes ‘presumptive’ WRN.
RESULTS
Identification of the study patients in the Ohio State University Medical Center information warehouse database
The algorithm used to identify patients for this analysis is provided in Materials and Methods. Briefly, the initial cohort consisted of 15,258 patients who, based on their information warehouse (IW) electronic medical record, received warfarin therapy as an inpatient or outpatient at the Ohio State University Medical Center (OSUMC) between 1 January 2005 and 31 December 2009 (cohort 1). Out of those, 6019 patients had at least one episode of INR >3.0 recorded during follow-up (cohort 2). If multiple episodes of INR >3.0 occurred, we used the first one for this analysis. In the cohort 2 patients, SC was recorded within 1 week after the first INR >3.0 in 4848. They became cohort 3. Out of those, 4816 patients also had at least one SC recorded within 3 months before the first abnormal INR >3.0. They became cohort 4. The final cohort (cohort 5) was selected by application of the inclusion and exclusion criteria described in Materials and Methods. This cohort consisted of 4006 patients. Cohort 5 consisted of 821 patients with presumptive WRN (increase in SC ≥0.3 mg/dl within 1 week of INR >3.0, 20.5% of cohort 5) and 3185 patients with no WRN (no increase in SC ≥0.3 mg/dl within 1 week of INR >3.0, 79.5% of cohort 5). An increase in SC ≥0.3 was taken as the evidence of AKI based on the guidelines of the Acute Kidney Injury Network.3 A patient was classified as a CKD patient if one of the International Classification of Diseases-9 (ICD-9) codes 585.1 through 585.5 or 585.9 was recorded in the patient’s record. Patients with ICD-9 code 585.6 (end-stage renal disease) were excluded from the final analysis.
INR and sequential changes in SC and estimated glomerular filtration rate in WRN patients compared with no-WRN patients
Figure 1a shows the INR values at the first episode of INR >3.0 for the WRN and no-WRN patients. The INR values were significantly higher in the WRN than no-WRN patients in cohort 5 (4.44±2.46 versus 4.15±2.15 IU, P = 0.0009) and for the no-CKD subset (4.57±2.69 versus 4.13±2.13 IU, P < 0.0001). However, for the CKD subset, there was no difference in mean INR values at the first episode of INR >3.0 between the WRN versus no-WRN patients (4.22±2.01 versus 4.22±2.224 IU P = 0.9569). There was no significant correlation between the magnitude of SC changes and the degree of elevation of INR >3.0 (data not shown).
Figure 1. International normalized ratio (INR) and changes in serum creatinine (SC) levels associated with INR increase ≥3.0 in patients with and without warfarin-related nephropathy (WRN).

Panel a shows that the INR for the first episode of INR >3.0 was significantly higher in WRN patients than the no-WRN patients for cohort 5 (all patients) and for the no-CKD (chronic kidney disease) subset but not the CKD subset. Panel b shows the changes in SC for cohort 5, stratified by the patient’s status as WRN (square) or no WRN (circle). The mean SC±1 s.d. is shown for the following intervals: 0–3 months before and at the onset of INR >3.0 (arbitrarily shown as 1 month before and at the onset of INR >3.0), 0–6 days after the INR >3.0 (arbitrarily shown as 6 days after the onset of INR >3.0), and 2–4 months after the INR >3.0 (arbitrarily shown as 3 months after the INR >3.0). The format for panels c and d is the same as that of panel b, except that panel c shows the no-CKD cohort and panel d shows the CKD cohort. As shown, mean SC increased significantly at onset of INR >3.0 in the WRN cohorts and tended to remain elevated 3 months later. SC values were available for all patients within 6 days after the INR >3.0. For the period 0–3 months after the INR >3.0, SC values were available for 1917 of 2780 (69%) of the surviving no-CKD and 611 of 722 (85%) of the surviving CKD patients. Longer-follow-up data are not shown because they were not consistently available across the indicated cohorts. To convert SC in mg/dl to mol/l, multiply by 88.4. *P<0.05 compared with no-WRN group, #P<0.05 compared with 0–3 months before INR >3.0. IU, international unit.
Figure 1b shows the sequential changes in mean SC in the cohort 5 patients, stratified by WRN, no-WRN status. As shown, the mean SC at the onset of INR >3.0 was significantly higher in the WRN patients than the no-WRN patients (2.75±1.65 versus 1.17±0.71 mg/dl, P<0.0001). By 3 months after the onset of WRN, the mean SC in the WRN patients remained higher than that of the no-WRN patients (1.80±1.24 versus 1.13±0.67 mg/dl, P<0.0001). Some of these differences are, however, attributable to a higher mean SC in the period preceding the INR >3.0 in those that developed WRN.
Figure 1c shows the sequential changes in mean SC in the no-CKD patients, stratified by their WRN, no-WRN status. As shown, at the onset of INR >3.0, mean SC was significantly higher in the WRN patients than the no-WRN patients (2.45±1.57 versus 1.01±0.49 mg/dl, P< 0.0001). By 3 months after the onset of WRN, SC decreased in the WRN patients but remained higher than that in the no-WRN patients (1.52±1.08 versus 1.00±0.51 mg/dl, P<0.0001).
Figure 1d shows the sequential changes in mean SC in the CKD patients, stratified by their WRN, no-WRN status. As shown, at the onset of INR >3.0, mean SC was significantly higher in the WRN patients than the no-WRN patients (3.25±1.67 versus 1.79±1.09 mg/dl, P<0.0001). By 3 months after the onset of WRN, mean SC in the WRN patients remained higher than that of the no-WRN patients (2.29±1.33 versus 1.65±0.94 mg/dl, P<0.0001).
Histograms showing the distribution of INR and changes in SC are depicted in Figure 2. As shown, using SC ≥0.3 mg/dl decisively separates WRN from no-WRN patients (Figure 2a, c and e). Regarding INR distribution, the INR values were slightly higher in WRN versus no-WRN patients for the entire cohort (cohort 5); however, this finding was not consistent across the CKD and no-CKD cohorts, as discussed above (Figure 2b, d and f).
Figure 2. Histograms of the international normalized ratio (INR) and changes in serum creatinine (SC) distributions in patients with and without warfarin-related nephropathy (WRN).

Panels a, c, and e show the distribution of changes in SC (delta SC) in all patients (cohort 5, panel a), patients without WRN (no WRN, Panel c), and WRN patients (Panel e). Panels b, d, and f show the distribution of INR in all patients (cohort 5, panel b), patients without WRN (no WRN, panel d), and WRN patients (panel f). IU, international unit.
Figure 3a shows changes in estimated glomerular filtration rate (eGFR) in WRN and no-WRN patients; eGFR was calculated based on the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation.4 Figure 3b and c show eGFR changes in CKD and no-CKD patients with and without WRN, respectively. The dynamics in eGFR changes were similar to SC changes—significant eGFR decrease in WRN patients with only a partial recovery by 3 months after INR >3.0 in CKD patients. In no-CKD patients, eGFR was not significantly different from pre-INR >3.0 levels (Figure 3b and c).
Figure 3. Changes in estimated glomerular filtration rate (eGFR) associated with international normalized ratio (INR) increase ≥3.0 in patients with and without warfarin-related nephropathy (WRN).

eGFR was calculated based on the chronic kidney disease (CKD)-EPI equation. Panel a shows the changes in eGFR for cohort 5, stratified by the patient’s status as WRN (square) or no WRN (circle). The mean eGFR±1 s.d. is shown for the following intervals: 0–3 months before and at the onset of INR >3.0 (arbitrarily shown as 1 month before and at the onset of INR >3.0), 0–6 days after the INR >3.0 (arbitrarily shown as 6 days after the onset of INR >3.0), and 2–4 months after the INR >3.0 (arbitrarily shown as 3 months after the INR >3.0). The format for panels b and c is the same as that of panel a, except that panel b shows the no-CKD cohort and panel c shows the CKD cohort. As shown, mean eGFR decreased significantly at onset of INR >3.0 in the WRN cohorts and tended to remain reduced 3 months later. eGFR was calculated based on serum creatinine (SC) values, which were available for all patients within 6 days after the INR >3.0. For the period 0–3 months after the INR>3.0, SC values were available for 1917 of 2780 (69%) of the surviving no-CKD and 611 of 722 (85%) of the surviving CKD patients. Longer-follow-up data are not shown because they were not consistently available across the indicated cohorts. *P<0.05 compared with no-WRN group, #P<0.05 compared with 0–3 months before INR >3.0.
Baseline clinical characteristics of the WRN patients and the no-WRN patients
Table 1 shows baseline clinical characteristics of cohort 5, stratified by WRN, no-WRN status. As shown, WRN patients did not differ from no-WRN patients with respect to sex and race. However, the risk of WRN was significantly increased in older patients and in those with CKD, diabetes, diabetic nephropathy, hypertension, and heart failure. Patients with an ICD-9 code for a specific kidney disease but without an ICD-9 code for CKD were stratified into the no-CKD cohort. These patients were more prevalent in WRN group, but the overall number of these patients was small.
Table 1.
Baseline clinical characteristics of cohort 5 (the final cohort) stratified by WRN or no WRN
| Variable | Level | WRN: ΔSC ≥0.3, n=821 (%) |
No WRN: ΔSC <0.3, n=3185 (%) |
P-value |
|---|---|---|---|---|
| Age at INR >3.0 | Mean±s.d. | 63.6±14.7 | 61.7±15.6 | 0.0049 |
| Sex | Female | 356 (43) | 1534 (48) | 0.0840 |
| Male | 465 (57) | 1651 (52) | ||
| Racea | White | 625 (78) | 2503 (79) | |
| Black | 173 (21) | 607 (19) | ||
| Asian | 0 (0) | 17 (1) | 0.2102 | |
| Native | 0 (0) | 4 (0) | ||
| American | ||||
| Other | 8 (1) | 19 (1) | ||
| CKD | No | 516 (63) | 2580 (81) | <0.0001 |
| Yes | 305 (37) | 605 (19) | ||
| Diabetes | No | 433 (53) | 2004 (63) | <0.0001 |
| Yes | 388 (47) | 1181 (37) | ||
| Diabetic nephropathy |
No | 741 (90) | 3054 (96) | <0.0001 |
| Yes | 80 (10) | 131 (4) | ||
| Hypertension | No | 153 (19) | 880 (28) | <0.0001 |
| Yes | 668 (81) | 2305 (72) | ||
| Heart failure | No | 315 (38) | 1848 (58) | <0.0001 |
| Yes | 506 (62) | 1337 (42) | ||
| Glomerulo- nephritis |
No | 786 (96) | 3134 (98) | <0.0001 |
| Yes | 35 (4) | 51 (2) | ||
| Nephrotic syndrome |
No | 801 (98) | 3159 (99) | 0.0008 |
| Yes | 20 (2) | 26 (1) | ||
| Polycystic kidneys |
No | 795 (97) | 3109 (98) | 0.3248 |
| Yes | 26 (3) | 76 (2) |
Abbreviations: CKD, chronic kidney disease; INR, international normalized ratio; SC, serum creatinine; WRN, warfarin-related nephropathy.
Data is not available for 50 patients.
Table 2 shows baseline clinical characteristics of the WRN and no-WRN cohorts, stratified by their CKD, no-CKD status. Within the no-CKD cohort, there was no significant association of age, sex, or race with WRN. However, diabetes, diabetic nephropathy, hypertension, and heart failure were significantly associated with WRN. For the patients with specific kidney disease diagnoses, there was no clear association with WRN.
Table 2.
Baseline clinical characteristics of WRN and no-WRN patients according to CKD or no-CKD status
| No CKD (n3096=3096) |
CKD (n=910) |
||||||
|---|---|---|---|---|---|---|---|
| Variable | Level | WRN: ΔSC ≥0.3, n=516 (%) |
No WRN: ΔSC<0.3, n=2580 (%) |
P-value | WRN: ΔSC ≥0.3, n=305 (%) |
No WRN: ΔSC<0.3, n=605 (%) |
P-value |
| Age at INR spike | Mean±s.d. | 61.9±14.8 | 60.9±15.7 | 0.1837 | 66.5±14.0 | 54.9±14.9 | 0.1093 |
| Gender | Female | 236 (46) | 1280 (50) | 0.1079 | 120 (39) | 254 (42) | 0.4450 |
| Male | 280 (54) | 1300 (50) | 185 (61) | 351 (58) | |||
| Racea | White | 412 (81) | 2093 (82) | 0.0947 | 213 (71) | 410 (68) | 0.6550 |
| Black | 87 (17) | 420 (16) | 86 (29) | 187 (31) | |||
| Asian | 0 (0) | 16 (1) | 0 (0) | 1 (0) | |||
| Native American | 0 (0) | 4 (0) | 0 (0) | 0 (0) | |||
| Other | 8 (2) | 18 (1) | 0 (0) | 1 (0) | |||
| Diabetes | No | 305 (59) | 1757 (68) | <0.0001 | 128 (42) | 247 (41) | 0.7414 |
| Yes | 211 (41) | 823 (32) | 177 (58) | 358 (59) | |||
| Diabetic | No | 495 (96) | 2551 (99) | <0.0001 | 246 (81) | 503 (83) | 0.3538 |
| nephropathy | Yes | 21 (4) | 29 (1) | 59 (19) | 102 (17) | ||
| Hypertension | No | 126 (24) | 827 (32) | 0.0006 | 27 (9) | 53 (9) | 0.9630 |
| Yes | 390 (76) | 1753 (68) | 278 (91) | 552 (91) | |||
| Heart failure | No | 257 (50) | 1647 (64) | <0.0001 | 58 (19) | 201 (33) | <0.0001 |
| Yes | 259 (50) | 933 (36) | 247 (81) | 404 (67) | |||
| Glomerular nephritis | No | 502 (97) | 2558 (99) | 0.0003 | 284 (93) | 576 (95) | 0.1911 |
| Yes | 14 (3) | 22 (1) | 21 (7) | 29 (5) | |||
| Nephrotic syndrome | No | 511 (99) | 2570 (100) | 0.0825 | 290 (95) | 589 (97) | 0.0743 |
| Yes | 5 (1) | 10 (0) | 15 (5) | 16 (3) | |||
| Polycystic kidneys | No | 504 (98) | 2535 (98) | 0.3698 | 291 (95) | 574 (95) | 0.7259 |
| Yes | 12 (2) | 45 (2) | 14 (5) | 31 (5) | |||
Abbreviations: CKD, chronic kidney disease; INR, international normalized ratio; SC, serum creatinine; WRN, warfarin-related nephropathy.
Data is not available for 50 patients.
Within the CKD cohort, there was no significant association of WRN with sex or race; however, older age was more prevalent in the WRN cohort. The diagnosis of diabetes or diabetic nephropathy did not increase the risk of WRN in the CKD cohort. However, heart failure did increase the risk of WRN. Specific renal disease diagnoses were not obviously different between the WRN, no-WRN status of the CKD patients.
Hematuria and WRN
Figure 4 shows changes in dipstick hematuria in relation to the onset of WRN. The method of comparison is described in the figure legend. As can be seen from panel a, there were no important differences in the extent of dipstick hematuria from 1 week after the INR >3.0 compared with 0–3 months before the INR >3.0. As shown in panel b, there was no important difference in the change of dipstick hematuria 0–3 months after the INR >3.0 compared with 0–3 months before the INR >3.0.
Figure 4. Changes in the hematuria grade in patients with and without warfarin-related nephropathy (WRN).
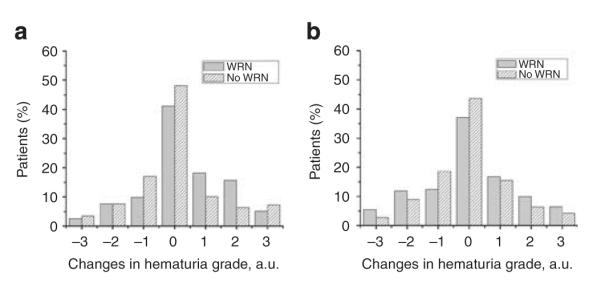
Hematuria was graded using a semiquantitative scale of 0 to 3+. Grade 0, no hematuria; 1+, mild hematuria; 2+, moderate hematuria; and 3+, large hematuria. Panel a shows changes in the hematuria grade in the WRN and no-WRN patients. The difference in hematuria grade at 1 week post-INR >3.0 and 0–3 months before the INR >3.0 was calculated, and the percentage of patients whose hematuria grade was changed is shown. Consecutive data were available in only 732 patients. Panel b shows changes in the hematuria grade in the WRN and no-WRN patients at 0–3 months after INR >3.0 and 0–3 months before INR >3.0. Consecutive data were available for only 771 patients. None of the differences shown in panel a or b was significant. a.u., arbitrary unit.
Concurrent medication and WRN
We assessed whether the medications that were being prescribed at the onset of the INR >3.0 might represent risk factors for WRN. The hypothesis is that the risk of WRN is increased by medications that increase glomerular hydrostatic pressure (PG), increase glomerular permeability, or increase coagulopathy. Table 3 shows the association of such medications with the risk of WRN. For this exploratory analysis, we did not stratify by CKD, no-CKD status, and did not account for whether the patient received medications in more than one-drug category.
Table 3.
Association of concurrent therapy with the patient’s status as WRN or no WRN
| Medication group | WRN: ΔSC ≥0.3, n=821 (%) |
No WRN: ΔSC <0.3, n=3185 (%) |
P-value |
|---|---|---|---|
| Drugs that affect blood pressure | 555 (68) | 1787 (56) | <0.001 |
| Drugs that may lower glomerular hydrostatic pressure (e.g., ACE inhibitors, angiotensin receptor blockers, β-blockers, diuretics, non-dihydropyridine CCB, clonidine) |
484 (59) | 1653 (52) | 0.002 |
| Drugs that may raise glomerular hydrostatic pressure (e.g., dihydropyridine CCB, direct-acting smooth muscle relaxants (hydralazine), potassium channel agonists (minoxidil), β-2-adrenergic receptor agonist (albuterol), erythropoietin, endothelin receptor antagonist (bosentan), dobutamine) |
311 (38) | 780 (24) | <0.001 |
| Antilipemic drugs | 167 (20) | 550 (17) | 0.243 |
| Statins | 145 (18) | 473 (15) | 0.243 |
| All other classes of antilipemic drugs | 32 (4) | 125 (4) | 0.972 |
| Drugs that affect the coagulation system | 513 (62) | 1882 (59) | 0.307 |
| Aspirin | 287 (35) | 897 (28) | 0.001 |
| Non-steroidal anti-inflammatory drugs | 36 (4) | 153 (5) | 0.307 |
| Clopidogrel, ticlopidine | 63 (8) | 214 (7) | 0.307 |
| Heparin | 387 (47) | 1627 (51) | 0.001 |
Abbreviations: ACE, angiotensin-converting enzyme; CCB, calcium channel blockers; SC, serum creatinine; WRN, warfarin-related nephropathy.
The drugs that affect blood pressure were arbitrarily classified as those that may lower or raise PG. As shown in Table 3, the use of drugs that affect blood pressure was more common in WRN than no-WRN patients. This may reflect greater use of antihypertensive drugs in the CKD cohort, which had a higher risk of WRN than the no-CKD cohort.
Consistent with our hypothesis that drugs that increase PG increase the risk of WRN is that use of the drugs in this class was 58% higher in WRN patients than the no-WRN patients (38 versus 25%, P<0.001). However, the use of drugs that may lower PG was also higher in WRN than no-WRN patients (59 versus 52%, P = 0.002). But the use was only 13% higher, and this could be explained by the greater use of this category of drugs in CKD, and CKD carries a greater risk of WRN.
Statins decrease glomerular capillary permeability (decrease proteinuria) and, for this reason, might protect against WRN. However, statin use was not significantly associated with the risk of WRN.
Among drugs that affect coagulation, aspirin use was significantly greater in WRN than no-WRN patients (35 versus 28%, P = 0.001). This is consistent with the notion that aspirin therapy contributes to coagulopathy and, therefore, the risk of WRN. However, heparin use was less in WRN than no-WRN patients (47 versus 51%, P = 0.001). This paradox could be explained if the heparin use in those initiating warfarin therapy was discontinued just before the onset of the INR >3.0.
Survival rate and WRN
Figure 5a shows that 5-year Kaplan–Meier survival rate was significantly lower in WRN than no-WRN patients (58 versus 73%, respectively; P<0.001), with survival differences being more pronounced shortly after the episode of INR >3.0 (1-year survival was 68.9% in WRN versus 81.1% in no-WRN patients, P = 0.049). Figure 5b shows survival rate stratified according to CKD, no-CKD status. The trend was the same: WRN CKD patients had decreased survival rate compared with no-WRN CKD patients (P = 0.064). Figure 5c used the univariate Cox model with survival as a time-varying covariate to estimate the hazard ratio for death in the WRN versus no-WRN cohorts, with 95% confidence intervals at selected early time points. The hazard for death (WRN versus no WRN) was highest within the first weeks after the INR >3.0 (hazard ratio at 1 week = 3.65, 95% confidence interval: 2.81, 4.75). Thereafter the hazard rate decreased progressively until it reached non-significant levels 6 months later.
Figure 5. Survival analysis and hazard ratio in patients with and without warfarin-related nephropathy (WRN) and chronic kidney disease (CKD).

Panel a: Kaplan–Meier plot (log-rank P-value <0.0001) for WRN patients (N = 3179, black dotted line) and no-WRN patients (N = 810, black solid line). Panel b: Kaplan–Meier plot (log-rank P-value <0.0001) for patients with WRN and no WRN, stratified by CKD, no-CKD status. Patients with WRN and CKD (black dotted line) had the lowest survival rate. Patients with WRN but no CKD had better survival rates (gray solid line). Patients without WRN, but with CKD had better survival rates compared with WRN patients with CKD (black dashed line). Patients without both WRN and CKD had the best survival rate (black solid line). Panel c: the univariate Cox model with survival as a time-varying covariate was used to estimate the hazard ratio for death in the WRN versus no-WRN cohorts, with 95% confidence intervals (CIs) at selected early time points. Because violation of the assumption of proportional hazards, WRN and no-WRN status was included as a time-varying covariate in the model. As shown, the hazard ratio for death (WRN versus no WRN) was highest within the first week after international normalized ratio (INR) >3.0. Thereafter it decreased progressively until it reached non-significant levels 6 months later. Estimated hazard ratio (solid black line) with 95% CIs (gray lines) is shown. Panel d shows the hazard ratio described in panel c but adjusted for the covariates that were significantly different between the WRN and no-WRN cohorts: age at INR >3.0, CKD, diabetes mellitus, heart failure, atrial fibrillation, and glomerulonephritis. The adjusted hazard ratio is similar to that of the unadjusted hazard ratio. Also, there was no evidence of an interaction between WRN, no-WRN status, and CKD status (P = 0.2102). Estimated hazard ratio (solid black line) with 95% CIs (gray lines) is shown.
Figure 5d shows the significance of WRN versus no WRN as a predictor of survival after controlling for covariates that were significantly associated with WRN: age at INR >3.0, CKD, diabetes mellitus, heart failure, and glomerulonephritis. There were no significant two-way interactions between the WRN and no-WRN patients with any of the covariates. Thus, WRN was a significant predictor of death after controlling for all of these covariates. Similar to the non-adjusted model, the maximal hazard ratio for the adjusted model was at the first week after INR >3.0 (hazard ratio: 3.19, 95% confidence interval: 2.45, 4.15) and steadily declined over time.
DISCUSSION
To the best of our knowledge, this is the first and the only large-scale study of the newly recognized syndrome, WRN. Our first study of WRN was a renal biopsy study of patients with warfarin coagulopathy who developed unexplained AKI. The kidney biopsy findings indicated that the AKI was attributable to widespread glomerular hemorrhage causing obstructive red blood cell cast formation.1 Next, we undertook a retrospective analysis of 103 warfarin-treated CKD patients followed up in our nephrology practice.2 We found that 37% of the patients who experienced an INR >3.0 acutely developed an unexplained acute increase in SC ≥0.3 mg/dl (mean increase 0.61±0.44 mg/dl). Thereafter, these patients showed accelerated progression of their CKD.2 None of these patients underwent kidney biopsy. The present study was undertaken to obtain a clearer understanding of WRN’s prevalence, risk factors, and outcomes in both CKD and no-CKD patients. The present study considered all of the patients at the OSUMC who received warfarin therapy during the 5-year period starting January 2005. From this cohort of 15,258 patients, we selected 4006 patients who met the study’s inclusion and exclusion criteria. The large number of patients in the study cohort precluded a detailed assessment of whether an acute increase in SC in relation to an acute increase in INR to >3.0 was ‘unexplained’. Thus, the present study describes ‘presumptive’ WRN.
We found that of the 4006 patients who experienced an INR >3.0, 20.5% developed presumptive WRN (an increase in SC to ≥0.3 mg/dl coincident with INR >3.0 in absence of evidence of hemorrhage). Among the CKD patients, the incidence of presumptive WRN was 33%, which is comparable to the 37% incidence of WRN that we previously reported in our warfarin-treated CKD patients.2 In those patients, a detailed assessment of each patient’s clinical record allowed a determination as to whether the increase in SC was unexplained. The good agreement between the present study and our previous study with regard to the incidence of WRN suggests that the presumptive WRN of the present study largely represents WRN.
Among the no-CKD patients, the incidence of presumptive WRN was 16.5%. Thus, presumptive WRN (and by inference WRN) is remarkably common. This begs the question as to why such a common complication of warfarin therapy has been unrecognized until just recently. The reasons could include the following: first, WRN is not part of the lexicon of causes of AKI. Although we5 and others6 had reported single cases of acute renal failure associated with severe warfarin glomerulo-pathy in which the renal biopsy confirmed extensive red blood cell cast formation, there was no compelling reason to believe from these case reports that lesser degrees of warfarin coagulopathy could cause AKI. That notion, however, was dispelled by our renal biopsy study of nine cases of AKI associated with relatively mild warfarin coagulopathy1 and by our retrospective analysis of the warfarin-treated CKD patient followed up in our nephrology program.2 Second, although WRN can occur in the same patient,1 the retrospective analysis of our warfarin-treated CKD patients indicated that WRN usually occurs early in the course of warfarin therapy. Thus, at any given time, the prevalence of acute WRN among all warfarin-treated patients is relatively low. Third, as demonstrated in the present study, WRN is associated with a substantial increase in mortality rate. Thus, patients susceptible to WRN are underrepresented in the population of warfarin-treated patients. Fourth, as demonstrated in the present study, the risk of WRN is particularly great in high-risk patients who have multiple risk factors for AKI. Because of the multiplicity of these risks, the presence of WRN was not easily recognized. Fifth, nephrologists might be reluctant to perform a kidney biopsy in patients receiving warfarin, because of the increased risk of hemorrhage.
The present work helps confirm our previous work that WRN does not require severe warfarin coagulopathy. Indeed, each of our three WRN studies shows that the average INR in the WRN cohorts were in the low to mid-4 range, with no clinically important differences in INR level between the CKD and no-CKD cohorts. The higher risk of WRN in CKD patients may be due to their higher likelihood of having a supratherapeutic INR.7 Indeed, in our study, we found INR to be slightly but significantly higher in WRN patients than in no-WRN patients. However, among CKD patients, INR levels were similar in WRN and no-WRN patients. There was no significant correlation between the degree of INR elevation and the magnitude of SC increase in any of the study groups. Also, we did not find a difference in the INR levels between WRN and no-WRN patients in our previous study (103 patients with CKD).
The present work is also the first to show that WRN is associated with a substantially decreased survival rate. This finding is consistent with the previous reports of increased mortality rate in warfarin-treated chronic hemodialysis patients.8,9 Although it is clear that the increased mortality rate associated with WRN is related to the comorbidities of diabetes, hypertension, and cardiovascular disease, how these comorbidities are related to the mechanisms of death in these patients remain to be elucidated.
The present work also provides new insights into the possible mechanisms of the AKI of WRN. Specifically, in exploratory studies, we show that therapies that tend to increase glomerular hydrostatic pressure are associated with an increased risk of WRN. In addition, concomitant aspirin therapy is associated with an increased risk of WRN. Both of these findings are consistent with the notion that glomerular hemorrhage causing tubular obstruction may be the dominant mechanism of the AKI associated with WRN. However, there are other possible mechanisms by which warfarin therapy could promote AKI, including atheroembolism,10 interstitial nephritis,11 and direct effects of warfarin on the glomerulus.12 Consistent with this notion is the finding that coagulopathy alone is not responsible for WRN in that gross hematuria is unusual, as suggested by this and our previous study.2
The limitations of the present work are those of a retrospective study in which the testing protocol was not prespecified. Thus, this work suffers from ascertainment bias. Of particular concern is that in order to be included in our analysis, we required a recorded SC by 1 week after the onset of INR >3.0. Frequent measurement of SC is more likely in sicker patients. Thus, our study may have identified the sickest patients with INR >3.0. Consistent with that notion is the high percent of the WRN and no-WRN cohort that had associated diabetes and cardiovascular disease. Nevertheless, even if all of the cases of WRN and its mortality in our study were identified because of ascertainment bias, WRN is still a major problem because cohort 5 was 26% of cohort 1. Also, our analysis of comorbidities and the risk of WRN indicate that WRN is an independent risk factor for increased mortality. Another limitation of this retrospective study is that there was no consistent testing of proteinuria around the time of the INR spike. Thus, we could not broadly test whether proteinuria itself was a risk factor for WRN.
To clearly establish the risk factors for WRN and its consequences will require a prospective study. We suggest that the present work provides compelling reasons to proceed with the prospective study.
In summary, warfarin is the most widely used anticoagulant to treat or prevent thrombotic complications. Currently, more than 30 million prescriptions for warfarin are filled annually in the United States.13 The present work provides clear evidence that warfarin coagulopathy is associated with a substantial increase in risk of AKI and acute mortality, especially in CKD patients. This AKI is independent of systemic hemorrhage. The mechanisms of these risks and how they might be mitigated will require further study. Nevertheless, we suggest that the evidence is sufficiently compelling to justify special precautions in managing warfarin therapy to minimize the risks of WRN. This recommendation is especially relevant to the CKD patients.
MATERIALS AND METHODS
We analyzed the de-identified data of consecutive patients who had initiated warfarin therapy during the period 1 January 2005 to 31 December 2009 at the OSUMC. The data were obtained from the OSUMC IW with approval of the OSU Institutional Review Board. A multistep algorithm was utilized to identify patients with presumptive WRN. The analysis reported here was performed on cohort 5 (the final cohort). The algorithm used to select cohort 5 is as follows:
Cohort 1 (N = 15,258). This consisted of all patients who had at least one order for warfarin, Coumadin, Jantoven, Marevan, Lawarin, or Waran with an order activation date between 1 January 2005 and 12 December 2009 based on the records of the OSUMC IW (prisoners and patients <18 years of age at discharge were excluded).
Cohort 2 (N = 6019). This consisted of all of the patients who had at least one episode of INR >3.0 recorded. If multiple episodes of INR >3.0 occurred, we used the first one for the present analysis.
Cohort 3 (N = 4848). This consisted of all the patients who had SC recorded within 1 week after the first INR >3.0.
Cohort 4 (N = 4816). This consisted of all the patients who had SC recorded within 3 months before the first abnormal INR >3.0. From cohort 4, we censored those who had, based on ICD-9 codes, end-stage renal disease (ICD-9 code 585.6) or evidence of clinically relevant hemorrhage (gastrointestinal tract hemorrhage, esophageal varices hemorrhage, rectal hemorrhage, placenta previa hemorrhage, associated with coagulation deficiency hemorrhage) within the first week after INR >3.0 (ICD-9 codes 456.0, 459.0, 569.3, 578.9, 641.1, and 641.30). These steps yielded cohort 5.
Cohort 5 (final cohort; N = 4006). This cohort was the object of the present study. This cohort was stratified into CKD or no CKD using the ICD-9 codes 581.1, 581.2, 581.3, 581.4, and 581.5, which identify the five stages of CKD according to the National Kidney Foundation Kidney Disease Outcomes Quality Initiative.14
Analytical studies
All laboratory testing was performed in the clinical laboratories of the OSUMC. The clinical pathology laboratory at the OSUMC is using a Beckman Unicel D×C 800 Chemistry Analyzer (Brea, CA) to measure SC levels. The SYNCHRON System(s) (Brea, CA) determine creatinine concentration by means of the Jaffe rate method. A precise volume of sample (16.5 ml serum or 5.5 ml urine) is injected in a reaction cup containing an alkaline picrate solution. The ratio used is one part sample to 35 parts reagent for serum, and one part sample to 105 parts reagent for urine. Creatinine from the sample combines with the reagent to produce a red color complex. Absorbance readings are taken at 520 nm between 19 and 25 s after sample injection. The absorbance rate has been shown to be a direct measure of the concentration of creatinine in the sample. The calibration method used for the creatinine assay is certified by the manufacturer to be traceable to isotope dilution–mass spectrometry as the gold standard.
Hematuria was graded using the dipstick semiquantitative scale of 0–3 +. Grade 0, no hematuria; 1 +, mild hematuria; 2 +, moderate hematuria; and 3 +, large hematuria.
Statistical methods
Demographics were first summarized for groups 1 and 2 separately. The χ2- tests were used to assess differences in categorical variables between groups; a two-sample t-test was used for age. Holm’s P-value adjustment was used to control type I error when testing for differences in INR, SC, and eGFR between WRN and no WRN shown in Figures 1 and 2 (ref. 15). Logistic regression models were used to assess the significance of the interaction between group and CKD status for all binary variables (diabetes, diabetic nephropathy, hypertension, heart failure, glomerular nephritis, nephrotic syndrome, polycystic kidneys, and atrial fibrillation). Similarly, a linear regression model was used to assess the interaction between group and CKD status for age at INR spike (Table 2). For the exploratory lab analyses, repeated measures models were used to assess trends in measurements over time (Figures 1 and 2). For the survival analysis, Kaplan–Meier plots were first produced, and then a Cox model was fitted to the data. The model-building process was performed as described in Moeschberger and Klein,16 Chapters 8 and 9. The model fit was evaluated as described in Chapter 11 of the same book. Because of violation of the assumption of proportional hazards, group was included as a time-varying covariate in the model. The estimated hazard ratio plot is shown in Figure 4d. We checked for the significance of group as a predictor of survival, controlling for the following covariates: age at INR spike, CKD, diabetes, diabetic nephropathy, heart failure, atrial fibrillation, and glomerular nephritis. None of the interactions of these variables and group was significant. These covariates were all significant in the final model as well as group.
Two-sample t-tests were performed to compare INR between WRN and no WRN as explorative analyses for the overall sample and CKD/non-CKD subgroups.
All analyses were performed using SAS/STAT software, version 9.2 (SAS Institute, Cary, NC).
ACKNOWLEDGMENTS
This study was supported in a part by a start-up fund provided to SVB by the Department of Pathology, Ohio State University.
Footnotes
DISCLOSURE All the authors declared no competing interests.
REFERENCES
- 1.Brodsky SV, Satoskar A, Chen J, et al. Acute kidney injury during warfarin therapy associated with obstructive tubular red blood cell casts: a report of 9 cases. Am J Kidney Dis. 2009;54:1121–1126. doi: 10.1053/j.ajkd.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 2.Brodsky SV, Collins M, Park E, et al. Warfarin therapy that results in an international normalization ratio above the therapeutic range is associated with accelerated progression of chronic kidney disease. Nephron Clin Pract. 2010;115:c142–c146. doi: 10.1159/000312877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mehta RL, Kellum JA, Shah SV, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11:R31. doi: 10.1186/cc5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kabir A, Nadasdy T, Nadasdy G, et al. An unusual cause of gross hematuria and transient ARF in an SLE patient with warfarin coagulopathy. Am J Kidney Dis. 2004;43:757–760. doi: 10.1053/j.ajkd.2003.08.050. [DOI] [PubMed] [Google Scholar]
- 6.Abt AB, Carroll LE, Mohler JH. Thin basement membrane disease and acute renal failure secondary to gross hematuria and tubular necrosis. Am J Kidney Dis. 2000;35:533–536. doi: 10.1016/s0272-6386(00)70209-5. [DOI] [PubMed] [Google Scholar]
- 7.Wysowski DK, Nourjah P, Swartz L. Bleeding complications with warfarin use: a prevalent adverse effect resulting in regulatory action. Arch Intern Med. 2007;167:1414–1419. doi: 10.1001/archinte.167.13.1414. [DOI] [PubMed] [Google Scholar]
- 8.Chan KE, Lazarus JM, Thadhani R, et al. Anticoagulant and antiplatelet usage associates with mortality among hemodialysis patients. J Am Soc Nephrol. 2009;20:872–881. doi: 10.1681/ASN.2008080824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan KE, Lazarus JM, Thadhani R, et al. Warfarin use associates with increased risk for stroke in hemodialysis patients with atrial fibrillation. J Am Soc Nephrol. 2009;20:2223–2233. doi: 10.1681/ASN.2009030319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moll S, Huffman J. Cholesterol emboli associated with warfarin treatment. Am J Hematol. 2004;77:194–195. doi: 10.1002/ajh.20210. [DOI] [PubMed] [Google Scholar]
- 11.Kapoor KG, Bekaii-Saab T. Warfarin-induced allergic interstitial nephritis and leucocytoclastic vasculitis. Intern Med J. 2008;38:281–283. doi: 10.1111/j.1445-5994.2008.01646.x. [DOI] [PubMed] [Google Scholar]
- 12.Yanagita M. Gas6, warfarin, and kidney diseases. Clin Exp Nephrol. 2004;8:304–309. doi: 10.1007/s10157-004-0305-z. [DOI] [PubMed] [Google Scholar]
- 13.Limdi NA, Beasley TM, Baird MF, et al. Kidney function influences warfarin responsiveness and hemorrhagic complications. J Am Soc Nephrol. 2009;20:912–921. doi: 10.1681/ASN.2008070802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eknoyan G, Levin NW, Eschbach JW, et al. Continuous quality improvement: DOQI becomes K/DOQI and is updated. National Kidney Foundation’s Dialysis Outcomes Quality Initiative. Am J Kidney Dis. 2001;37:179–194. doi: 10.1016/s0272-6386(01)80074-3. [DOI] [PubMed] [Google Scholar]
- 15.Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- 16.Moeschberger ML, Klein JP. Survival Analysis: Techniques for Censored and Truncated Data. 2nd edn Springer; New York, NY: 2005. [Google Scholar]