

Abstract
Rationale
High-myofilament Ca2+-sensitivity has been proposed as trigger of disease pathogenesis in familial hypertrophic cardiomyopathy (HCM) based on in vitro and transgenic mice studies. However, myofilament Ca2+-sensitivity depends on protein phosphorylation and muscle length, and at present, data in human are scarce.
Objective
To investigate whether high-myofilament Ca2+-sensitivity and perturbed length-dependent activation are characteristics for human HCM with mutations in thick- and thin-filament proteins.
Methods and Results
Cardiac samples from patients with HCM harboring mutations in genes encoding thick (MYH7, MYBPC3) and thin (TNNT2, TNNI3, TPM1) filament proteins were compared with sarcomere mutation-negative HCM and nonfailing donors. Cardiomyocyte force measurements showed higher myofilament Ca2+-sensitivity in all HCM samples and low phosphorylation of protein kinase A (PKA)-targets compared with donors. After exogenous PKA treatment, myofilament Ca2+-sensitivity was either similar (MYBPC3mut, TPM1mut, sarcomere mutation-negative HCM), higher (MYH7mut, TNNT2mut), or even significantly lower (TNNI3mut) compared with donors. Length-dependent activation was significantly smaller in all HCM than in donor samples. PKA treatment increased phosphorylation of PKA-targets in HCM myocardium and normalized length-dependent activation to donor values in sarcomere mutation-negative HCM and HCM with truncating MYBPC3 mutations, but not in HCM with missense mutations. Replacement of mutant by wild-type troponin in TNNT2mut and TNNI3mut corrected length-dependent activation to donor values.
Conclusions
High-myofilament Ca2+-sensitivity is a common characteristic of human HCM and partly reflects hypophosphorylation of PKA-targets compared with donors. Length-dependent sarcomere activation is perturbed by missense mutations, possibly via post-translational modifications other than PKA-hypophosphorylation or altered protein–protein interactions, and represents a common pathomechanism in HCM.
Keywords: calcium, cardiomyopathy, contractility, hypertrophy, myocardium
Myofilament contraction is initiated by interaction between the thin actin and thick myosin filaments. This actin–myosin interaction (ie, thin-filament-myosin cross-bridge binding) and the magnitude of myofilament force generation are tightly regulated by muscle length, Ca2+-binding, and protein phosphorylation.1 Defective proteins as a result of mutations in genes encoding sarcomeric proteins may directly impair regulation of muscle contraction and manifest themselves as phenotypic aberrations of the heart. Hypertrophic cardiomyopathy (HCM) reflects the pathological phenotype associated with sarcomeric gene mutations.2,3 Affecting 1:500 individuals worldwide, HCM is the most common cause of sudden death in young people.2 Genotyping studies have identified a disease-causing mutation in ≈70% of all patients with HCM.4 Mutations in thick-filament–encoding genes, MYH7 (β-myosin heavy chain [β-MyHC]) and MYBPC3 (cardiac myosin–binding protein-C [cMyBP-C]), account for ≈80% of all identified sarcomere mutations, whereas ≈18% of the mutations are found in thin-filament–encoding genes, TNNI3 (cardiac troponin I [cTnI]), TNNT2 (cardiac troponin T [cTnT]), TNNC1 (cardiac troponin C), TPM1 (α–tropomyosin), and ACTC1 (α-cardiac actin).5 The remaining 2% is attributed to incidental mutations in the thick-filament genes, MYL3 and MYL2, encoding the regulatory and essential myosin light chain, and the sarcomere-associated gene TTN, which encodes titin.5 Based on the numerous sarcomeric gene mutations, HCM is referred to as disease of the sarcomere.
Both animal6,7 and clinical8–10 studies have shown that carriers of HCM-causing mutations demonstrate early signs of cardiac dysfunction, even before a hypertrophic phenotype is observed. Additionally, sudden cardiac death occurs in young individuals in the absence of clinically detectable cardiac hypertrophy.11 This suggests that the initial defects in cardiac performance are triggered by mutant sarcomeric proteins rather than remodeling of the heart. Sarcomere mutations may directly impair myofilament function and contractile performance of the heart,3 or indirectly via changes in intracellular Ca2+-handling.12 Because the myofilaments represent a major intracellular buffer of Ca2+, any perturbation in myofilament Ca2+-sensitivity may provide a substrate for cardiac arrhythmias. Only recently, the group of Knollmann13 showed that TNNT2 mutations alter intracellular Ca2+-handling via myofilament Ca2+-sensitization in transgenic mice models, which was associated with altered action potential regulation and occurrence of arrhythmias. This implies that myofilament Ca2+-sensitivity is central in HCM pathology.
Whether or not myofilament sensitization to Ca2+ is of relevance in human HCM is a matter of ongoing research.14–16 Our recent studies in manifest HCM with MYBPC3 mutations showed higher myofilament Ca2+-sensitivity compared with nonfailing donor myocardium.15,16 However, high Ca2+-sensitivity coincided with low phosphorylation of target proteins of the β-adrenergic signaling pathway, cTnI, and cMyBP-C, and was normalized to donor values by exogenous protein kinase A (PKA) treatment.15 This suggests that the high-myofilament Ca2+-sensitivity observed in human HCM with MYBPC3 mutations is attributable to secondary disease–related post-translational modifications rather than the mutation itself. In addition to post-translational protein modifications, muscle length represents an important determinant of myofilament Ca2+-sensitivity. Recently, we observed impairment of length-dependent sarcomere activation evident from a blunted length–dependent increase in myofilament Ca2+-sensitivity in HCM with MYBPC3 mutations.15 Under normal conditions, intracellular Ca2+ buffering will increase with increased myofilament Ca2+-sensitivity on myofilament lengthening.17,18 Perturbations in length-dependent myofilament activation will alter Ca2+ buffering by the sarcomeres and may provide a basis for altered Ca2+-handling in HCM.
In the present study, we investigated whether perturbed length–dependent activation rather than high-myofilament Ca2+-sensitivity is a common pathomechanism in human HCM with mutations in thick- and thin-filament genes. Measurements in sarcomere mutation-positive HCM were compared with sarcomere mutation-negative HCM (HCMsmn) and nonfailing donors. Our study shows that mutation-related changes in myofilament Ca2+-sensitivity are diverse and depend on the affected gene. We observed impaired length–dependent sarcomere activation in all HCM samples. The blunted length–dependence of sarcomeres was attributable to low phosphorylation of PKA-targets compared with donor myocardium in patients with MYBPC3 truncating mutations and HCMsmn, but a direct consequence of HCM missense mutations. Using troponin exchange in HCM cardiomyocytes with a homozygous TNNT2 and a heterozygous TNNI3 mutation, we provide evidence that <50% of poison peptide is sufficient to impair length-dependent activation of the sarcomeres. Because most patients carry a heterozygous sarcomere mutation that may result in relatively low levels of mutant (poison) peptide, impaired length–dependent sarcomere activation represents a pathomechanism in HCM.
Methods
An expanded version of the methods section can be found in the online-only Data Supplement.
Myocardial Samples
Left ventricular septum tissue was obtained from patients with HCM harboring thick- and thin-filament gene mutations during myectomy surgery to relieve left ventricular outflow obstruction. Hypertrophic obstructive cardiomyopathy was evident from the high-septal thickness (normal value <13 mm) and high-left ventricular outflow tract pressure gradient (normal value <30 mm Hg). Clinical characteristics and mutation information of patients with HCM are listed in Table 1. Our study included patients carrying heterozygous mutations in MYBPC3 (n=21; MYBPC3mut), MYH7 (n=6; MYH7mut), TNNI3 (n=2; TNNI3mut), and TPM1 (n=1; TPM1mut). The MYBPC3mut group consisted of patients with truncating (n=17) and missense (n=4) mutations. Data for these 2 MYBPC3mut groups are presented separately. Septum tissue was also obtained from 1 end-stage failing patient with HCM carrying a homozygous TNNT2 mutation (TNNT2mut). Septum myectomy tissue from 7 patients with HCM in whom no mutation was found after screening of 8 genes (HCMsmn) and cardiac tissue from 12 nonfailing donors served as controls. Donors (age ranged 14–65 years; mean 39±5 years; 9/3 male/female, respectively) had no history of cardiac abnormalities, normal ECG, and normal ventricular function on echocardiography within 24 hours of heart transplantation. Samples were obtained after written informed consent, and the study protocol was approved by the local ethical committees.
Table 1. Mutations and Clinical Characteristics of Patients.
| Mutation | Type | Age | Sex | LVOT | ST | |
|---|---|---|---|---|---|---|
|
| ||||||
| MYBPC3mut | Truncating mutations | |||||
| 1 | c.927-2A>G | Splice site | 37 | M | 61 | 19 |
| 2 | c.927-2A>G | Splice site | 48 | M | 82 | 18 |
| 3 | c.927-2A>G | Splice site | 22 | M | 71 | 30 |
| 4 | c.1458-1G>C | Splice site | 41 | F | 92 | 22 |
| 5 | c.2373duplicationG | Insertion | 32 | F | 100 | 30 |
| 6 | c.2373duplicationG | Insertion | 39 | F | 60 | 20 |
| 7 | c.2373duplicationG | Insertion | 45 | F | 94 | 20 |
| 8 | c.2373duplicationG | Insertion | 62 | M | 64 | 23 |
| 9 | c.2373duplicationG | Insertion | 44 | F | 100 | 17 |
| 10 | c.2373duplicationG | Insertion | 69 | M | 74 | 19 |
| 11 | c.2373duplicationG | Insertion | 57 | F | 74 | 24 |
| 12 | c.2373duplicationG | Insertion | 32 | M | 88 | 23 |
| 13 | c.2373duplicationG | Insertion | 60 | M | 77 | 23 |
| 14 | c.2864.2865delCT | Deletion | 42 | M | 116 | 23 |
| 15 | c.2864.2865delCT | Deletion | 45 | M | 100 | 20 |
| 16 | c.2864.2865delCT | Deletion | 62 | F | 67 | 15 |
| 17 | c.3407.3409del | Deletion | 55 | M | UK | UK |
|
| ||||||
| MYBPC3mut | Missense mutations | |||||
|
| ||||||
| 18* | p.E258K | Missense | 35 | M | 4 | 27 |
| 19 | p.E258K | Missense | 45 | M | 72 | 24 |
| 20 | p.G531R | Missense | 51 | M | 110 | 22 |
| 21 | p.R597Q | Missense | 47 | F | 85 | 20 |
|
| ||||||
| MYH7mut | ||||||
|
| ||||||
| 1 | p.R403Q | Missense | 25 | M | 85 | 34 |
| 2 | p.V606M | Missense | 46 | F | 79 | 17 |
| 3 | p.S782R | Missense | 30 | F | 128 | 29 |
| 4 | p.R787H | Missense | 61 | M | UK | UK |
| 5 | p.T1377M | Missense | 58 | F | 100 | 20 |
| 6 | p.T1377M | Missense | 43 | M | UK | UK |
|
| ||||||
| TNNT2mut | ||||||
|
| ||||||
| 1 | p.K280N | Missense | 26 | M | UK | UK |
|
| ||||||
| TNNI3mut | ||||||
|
| ||||||
| 1 | p.R145W | Missense | 46 | M | 100 | 23 |
| 2 | p.R145W | Missense | 66 | M | 100 | 16 |
|
| ||||||
| TPM1mut | ||||||
|
| ||||||
| 1 | p.M281T | Missense | 65 | M | 100 | 16 |
|
| ||||||
| HCMsmn | Sarcomere mutation | Negative | ||||
|
| ||||||
| 1 | 46 | F | 105 | 24 | ||
| 2 | 57 | M | 117 | 25 | ||
| 3 | 75 | F | 137 | 20 | ||
| 4 | 72 | F | 88 | 24 | ||
| 5 | 65 | F | 85 | 19 | ||
| 6 | 49 | M | 61 | 20 | ||
| 7 | 46 | M | 81 | 19 | ||
c indicates codon region; del, deletion; LVOT, left ventricular outflow tract pressure gradient in mm Hg; p, protein amino residue; ST, septal thickness in mm; and UK, unknown.
Patient was operated because of extreme hypertrophy and small LV cavity.
Isometric Force Measurements and Protein Analysis
Analysis of single cardiomyocyte force measurements and sarcomeric protein phosphorylation was performed as described previously.15,19,20
Exchange of Recombinant Human Troponin Complex
Expression, purification, and reconstitution of recombinant human wild-type cardiac troponin in single cardiomyocytes from the TNNT2mut heart and 1 of the TNNI3mut hearts were performed as described previously.21 Evaluation of the degree of exchange into cardiomyocytes was determined by labeling cTnT of wild-type recombinant cardiac troponin with a myc-tag to allow differentiation between endogenous and recombinant wild-type cTnT in Western blot analysis.
Data Analysis
Data are presented as mean±SEM of all single cardiomyocytes per patient/donor group. To take into account the repeated sample assessments within patient/donor groups, multilevel analysis was performed. Comparison between all groups was performed for Ca2+-sensitivity at 2.2 μm sarcomere length and length-dependent activation of cardiomyocytes before and after PKA. Paired-group comparisons were performed for maximal developed force (Fmax) at 1.8 and 2.2 μm sarcomere length before and after PKA.
Detailed explanation of the data analyses, significance levels (exact P values), and 95% confidence intervals for all comparisons are given in the online-only Data Supplement file.
Results
Myofilament Ca2+-Sensitivity in HCM Compared With Nonfailing Donors
To assess myofilament Ca2+-sensitivity, force-Ca2+ relations were constructed for all cardiac samples at a sarcomere length of 2.2 μm. Ca2+-sensitivity for all HCM samples was significantly higher compared with donors, based on their lower EC50 values (Figure 1A). Because the high Ca2+-sensitivity may be attributable to low-phosphorylation levels of PKA-target proteins, measurements were repeated after PKA treatment. The PKA-induced reduction in Ca2+-sensitivity (ΔEC50) was larger in all HCM groups compared with donors (with significant changes in MYBPC3mut, MYH7mut, and TNNI3mut), except for the TNNT2mut, which only showed a minor nonsignificant change (Figure 1B). After PKA treatment, myofilament Ca2+-sensitivity was close to donor values in MYBPC3mut, TPM1mut, and HCMsmn, whereas Ca2+-sensitivity remained significantly higher compared with donor values in MYH7mut and TNNT2mut (Figure 1C). Interestingly, after PKA treatment, Ca2+-sensitivity was significantly lower in TNNI3mut compared with HCMsmn and donor values.
Figure 1. Myofilament Ca2+-sensitivity at a sarcomere length of 2.2 μm.
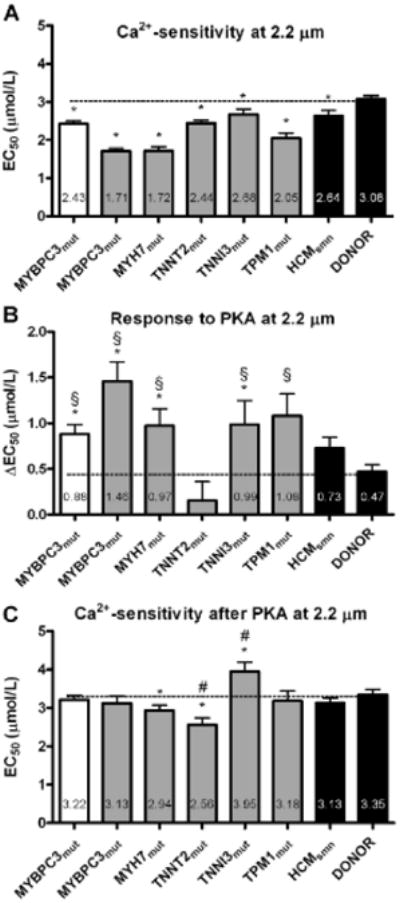
A, Myofilament Ca2+-sensitivity (EC50) was significantly higher in all hypertrophic cardiomyopathy (HCM) groups compared with donors. B, The protein kinase A (PKA)-induced reduction (ΔEC50) in myofilament Ca2+-sensitivity was larger in HCM groups compared with donors, except in the TNNT2mut sample, in which PKA had no significant effect. C, Myofilament Ca2+-sensitivity was similar in MYBPC3mut, TPM1mut, sarcomere mutation-negative HCM (HCMsmn), and donor after treatment with PKA, whereas it was higher than donor in MYH7mut and TNNT2mut. PKA-treated TNNI3mut cells showed a lower myofilament Ca2+-sensitivity compared with HCMsmn and donor. Open bar graph represents MYBPC3 truncating mutation; closed gray bar graphs represent missense mutations. *P<0.05 vs donor; §P<0.05 vs TNNT2mut; #P<0.05 vs HCMsmn.
Fmax at a sarcomere length of 2.2 μm was significantly lower in all HCM groups compared with donors and was not corrected by PKA treatment (Online Table I). In addition, the steepness of the force-Ca2+ relation was significantly lower and not corrected by PKA in all HCM groups compared with donors (data not shown).
Length-Dependent Myofilament Force Characteristics
Force development was measured at various [Ca2+] and 2 sarcomere lengths to determine length-dependent activation of myofilaments (Figure 2A). After a sarcomere length increase from 1.8 to 2.2 μm at maximal Ca2+-activation, donor samples showed a significant increase in Fmax (Table 2), which is in accordance with the well-known effects of length on force development.22 A significant length–dependent increase in Fmax was observed in MYBPC3mut, MYH7mut, TPM1mut, and HCMsmn heart samples, whereas no significant increase in Fmax was found for both TNNT2mut and TNNI3mut. As illustrated in Table 2, the ΔFmax (difference in Fmax between sarcomere lengths of 1.8 and 2.2 μm) was much lower in HCM with missense mutations compared with the other groups.
Figure 2. Myofilament length–dependent activation.
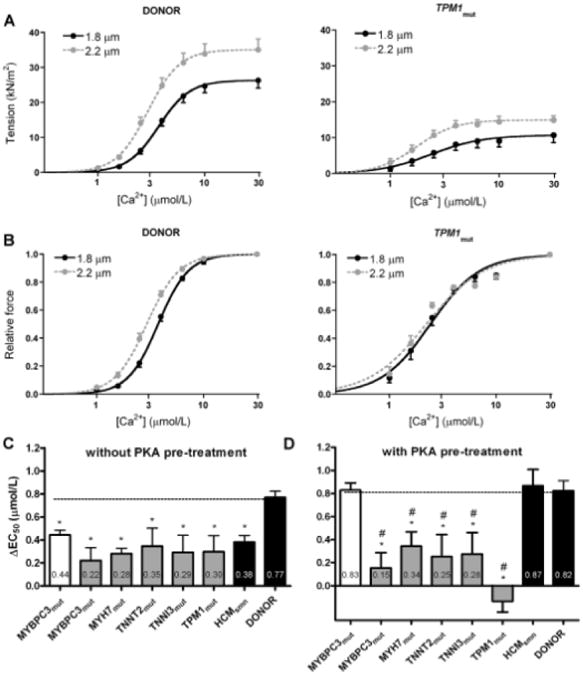
A, Tension development as a function of [Ca2+] at short (1.8 μm) and long (2.2 μm) sarcomere lengths for donor (left) and TPM1mut(right) heart samples. B, Normalized force–Ca2+ relationships for donor (left) and TPM1mut(right) heart samples. C, The length-dependent increase in myofilament Ca2+-sensitivity was lower in all sarcomere mutation-positive hypertrophic cardiomyopathy (HCMmut) compared with donors before protein kinase A (PKA) treatment. D, PKA pretreatment restored length-dependent activation to donor in HCM with truncating MYBPC3mut and sarcomere mutation-negative HCM (HCMsmn), but not in HCMmut with missense mutations. Open bar graph represents MYBPC3 truncating mutations; closed gray bar graphs represent missense mutations.*P<0.05 vs donor; #P<0.05 vs HCMsmn.
Table 2. Effects of Sarcomere Length Increase on Fmax Before and After PKA.
| No PKA Pretreatment | PKA Pretreatment | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||
| Fmax, kN/m2 | Fmax, kN/m2 | ||||||||
|
|
|||||||||
| Sample | 1.8 μm | 2.2 μm | ΔFmax | N/n | 1.8 μm | 2.2 μm | ΔFmax | N/n | |
| Truncating | |||||||||
| MYBPC3mut | 19.6±1.5 | 29.8±2.4** | 10.2±1.2 | N=14/n=47 | 27.6±1.5 | 37.0±2.0** | 9.4±1.3 | N=4/n=19 | |
| Missense | |||||||||
| MYBPC3mut | 13.0±1.3 | 15.5±1.7** | 2.5±0.8* | N=4/n=12 | 11.4±1.0 | 13.9±1.5** | 2.5±1.0* | N=4/n=12 | |
| MYH7mut | 15.1±1.6 | 19.1±1.8** | 3.9±1.0* | N=6/n=32 | 18.8±2.6 | 22.6±3.1** | 3.8±1.1* | N=4/n=15 | |
| TNNT2mut | 21.5±3.2 | 24.4±6.0 | 3.0±4.4 | N=1/n=6 | 13.7±2.1 | 15.2±2.4 | 1.5±1.3* | N=1/n=4 | |
| TNNI3mut | 9.8±2.5 | 10.1±2.8 | 0.3±1.1* | N=2/n=8 | 8.4±1.4 | 9.5±1.9 | 0.83±0.8* | N=2/n=10 | |
| TPM1mut | 10.7±2.1 | 17.0±3.0** | 6.3±1.5 | N=1/n=6 | 9.1±2.3 | 9.6±1.7 | 0.58±0.7*# | N=1/n=4 | |
| HCMsmn | 18.2±1.4 | 28.3±2.6** | 10.1±1.7 | N=7/n=31 | 26.8±2.3 | 38.7±2.7** | 12.0±1.4 | N=3/n=12 | |
| DONOR | 26.3±2.2 | 35.1±3.1** | 8.8±2.1 | N=9/n=32 | 26.2±1.4 | 36.2±1.9** | 10.0±1.6 | N=3/n=12 | |
ΔFmax indicates difference in Fmax between sarcomere lengths of 1.8 and 2.2 μm; HCM, hypertrophic cardiomyopathy; N, no. of samples; n, no. of cardiomyocytes; and PKA, protein kinase A.
P<0.05 was considered significant;
vs donor;
1.8 μm vs 2.2 μm;
ΔFmax (no PKA pretreatment) vs ΔFmax (PKA pretreatment).
To investigate the length-dependent increase in myofilament Ca2+-sensitivity, force at submaximal [Ca2+] was normalized to the Fmax to obtain normalized force-Ca2+ relations at both sarcomere lengths (Figure 2B). For all HCM and donor groups, the normalized force-Ca2+ relation shifted to the left as sarcomere length increased, indicative for increased Ca2+-sensitivity. However, the increase in myofilament Ca2+-sensitivity on an increase in sarcomere length (ΔEC50) was significantly lower in all sarcomere mutation-positive HCM and HCMsmn samples compared with donors (Figure 2C).
Stimulation of the β-adrenergic receptor pathway has been shown to enhance the length-dependent shift in the force-Ca2+ relation, suggesting a modulating role for PKA-mediated protein phosphorylation in length-dependent sarcomere activation.23,24 As previous studies showed lower phosphorylation levels of myofilament PKA-target proteins in HCM compared with donors,15,16 the blunted length–dependent activation in HCM compared with donors may be explained by a difference in protein phosphorylation level. Indeed, analysis of protein phosphorylation showed lower phosphorylation levels of both cTnI and cMyBP-C in all HCM groups compared with nonfailing donors, except in TNNT2mut, in which phosphorylation of the PKA-target proteins did not differ from donor (Figure 3A).
Figure 3. Phosphorylation of protein kinase A (PKA)-target proteins before and after PKA treatment.
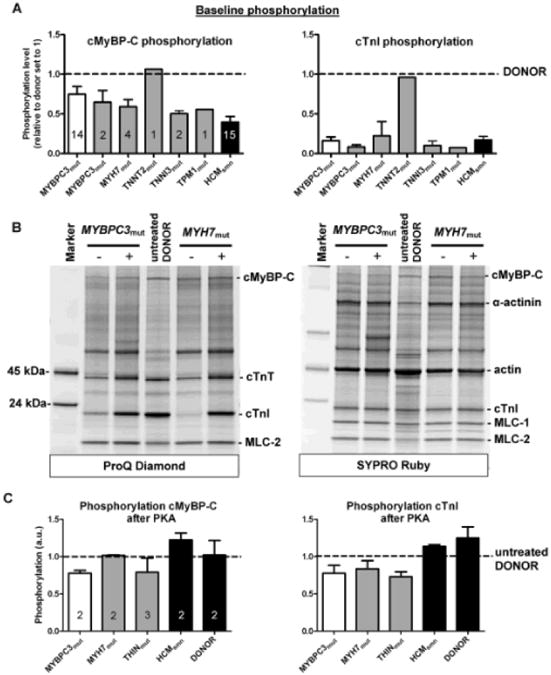
A, Protein phosphorylation values were corrected by the corresponding SYPRO-stained protein bands and normalized to the values found in donors, which were set to 1 (dotted line). Phosphorylation of cardiac myosin–binding protein-C (cMyBP-C; left) and cardiac troponin I (cTnI; right) was lower in all hypertrophic cardiomyopathy (HCM) samples compared with donors, except for the TNNT2mut sample, which showed relatively high phosphorylation. Open bar graph represents truncating MYBPC3 mutations; closed gray bar graphs represent missense mutations. B, Cardiac samples before (−) and after (+) PKA treatment separated by 1-dimensional gel electrophoresis and stained with ProQ-Diamond (phosphorylation) and SYPRO Ruby (total protein stain). C, Thin-filament mutations were clustered in a single group (THINmut). ProQ-stained protein phosphorylation values of PKA-treated samples were corrected by the corresponding SYPRO-stained protein bands and normalized to values in nontreated donor samples, which were included on the gel and set to 1 (dotted line). PKA increased phosphorylation of both target proteins in all HCM groups. Numbers of samples included in the analyses are indicated in the bar graphs.
To test whether the blunted length–dependent change in Fmax and myofilament Ca2+-sensitivity was corrected by PKA-mediated phosphorylation of cMyBP-C and cTnI, force measurements were performed after pretreatment with exogenous PKA in a subset of HCM and donor samples. PKA pretreated MYBPC3mut, MYH7mut, HCMsmn, and donor cells showed a significant increase in maximal force after a sarcomere length increase, similar to that observed in nontreated cardiomyocytes (Table 2). PKA did not restore the blunted length–dependent increase in Fmax evident from the significantly lower ΔFmax in HCM with missense mutations compared with donor (Table 2). Pretreatment with exogenous PKA significantly enhanced the length-dependent shift in myofilament Ca2+-sensitivity in HCM with truncating MYBPC3mut and HCMsmn samples to values observed in donors, but did not correct the blunted length–dependent change in EC50 in all other HCM mutant groups harboring missense mutations (Figure 2D).
Because the MYBPC3mut and MYH7mut groups consisted of different mutations, we averaged functional data for each HCM mutation separately. Data are shown in the online-only Data Supplement material (Online Table II).
PKA-Mediated Protein Phosphorylation
To determine whether absence of an effect of PKA on sarcomere functional properties is attributable to a defect in PKA-mediated protein phosphorylation in HCM myocardium, phosphorylation status of PKA-target proteins was analyzed in HCM and donor samples incubated with exogenous PKA. Figure 3B shows HCM samples incubated without and with PKA separated by 1-dimensional gel electrophoresis and stained with ProQ-Diamond and SYPRO Ruby. PKA increased phosphorylation of both PKA-targets, cMyBP-C and cTnI, in HCM samples. Phosphorylation levels (normalized to untreated donor samples, which were included on the gel and set to 1; dotted line) are depicted in Figure 3C and show that phosphorylation of cMyBP-C after PKA was close to values observed in donor samples in all HCM samples. Analysis of phosphorylation at specific PKA sites (Ser275 and Ser284) on cMyBP-C confirmed increased PKA–mediated phosphorylation in HCM samples (Figure 4). In addition to cMyBP-C, phosphorylation of cTnI increased by PKA in all HCM samples (Figure 3B), although values did not reach the level found in donor myocardium (Figure 3C). PKA treatment of donor samples increased cTnI phosphorylation by ≈25%. Similar data were obtained when cTnI phosphorylation was analyzed by Phos-Tag gel electrophoresis creating a pattern of un, mono-, and bisphosphorylated cTnI (Online Figure II). PKA-treated HCM samples showed increased bisphosphorylated cTnI levels, but some monophosphorylated cTnI remained. Previous Phos-Tag analysis of the donor samples (n=12) revealed a distribution pattern of 7% unphosphorylated, 27% monophosphorylated, and 66% bisphosphorylated cTnI.15 On treatment of donor samples (n=2) with PKA, cTnI was mostly bisphosphorylated (Online Figure II).
Figure 4. Site-specific phosphorylation of protein kinase A (PKA)-target sites of cardiac myosin–binding protein-C (cMyBP-C).
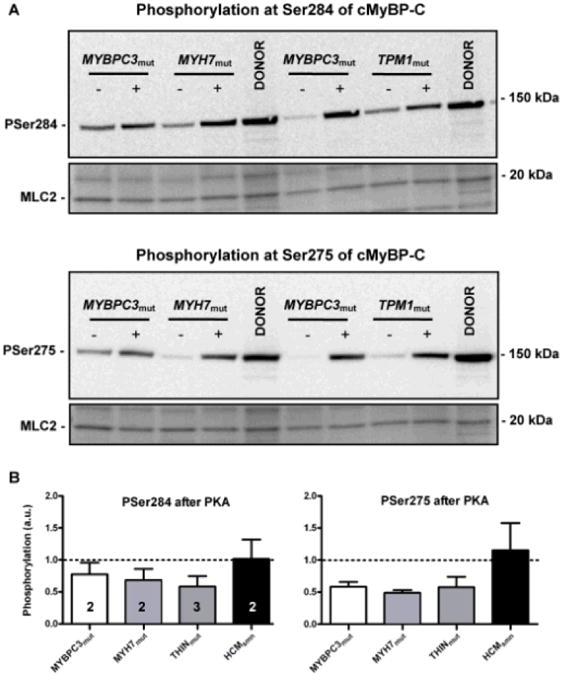
A, Western blot analysis of cMyBP-C phosphorylation with specific antibodies for PKA sites Ser284 (top) and Ser275 (bottom) before (−) and after (+) PKA treatment (phosphorylation values were corrected for minor differences in protein loading by Ponceau-stained MLC2, myosin light chain 2). B, Protein phosphorylation values were normalized to the values found in untreated donor samples, which were included on the blot and set to 1 (dotted line). Thin-filament mutations were clustered in a single group (THINmut). Numbers of samples included in the analyses are indicated in the bar graphs. All samples showed an increased phosphorylation at both sites on PKA treatment.
Correction of Length-Dependent Activation in HCM With Mutant cTnT and Mutant cTnI by Human Recombinant Wild-Type Troponin
To determine whether mutant sarcomeric protein is the direct cause of the blunted length–dependent increase in myofilament Ca2+-sensitivity, we performed troponin exchange experiments in cardiomyocytes from the TNNT2mut sample. The homozygous TNNT2mut necessarily results in 100% mutant cTnT and as such represents a unique tool to assess the level at which mutant protein perturbs sarcomere function. Exchange with increasing concentrations of wild-type human troponin complex (0.25, 0.5, and 1 mg/mL in the exchange solution) resulted in 62±2%, 78±1%, and 86±1% troponin exchange based on Western blot analyses of endogenous and myc-tag labeled wild-type cTnT (Figure 5A; left blot). In exchanged cells without PKA pretreatment, replacement of mutant troponin with unphosphorylated recombinant wild-type troponin did not restore the reduced length–dependent activation to donor values (Figure 5B). However, replacement of endogenous mutant troponin by unphosphorylated recombinant troponin reduces cTnI phosphorylation in the TNNT2mut cells. Therefore, measurements were also performed in the troponin-exchanged cells after PKA treatment to increase cTnI phosphorylation to donor levels. In cells in which 62% of mutant troponin was replaced by wild-type troponin, the length-dependent increase in myofilament Ca2+-sensitivity was still significantly lower compared with donor values, indicating that 38% of mutant protein is sufficient to impair length-dependent activation. However, the blunted length–dependent activation was restored to donor values in TNNT2mut-exchanged cells, harboring 22% and 14% endogenous mutant cTnT (Figure 5C). Similarly, exchange of mutant cTnI in 1 of the HCM samples harboring the R145W missense mutation in TNNI3 by ≈90% unphosphorylated wild-type troponin complex (Figure 5A; right blot) corrected length-dependent activation only after PKA treatment (Figure 5D and 5E). Hence, normalization of length-dependent activation to donor values on exchange with wild-type cTn complex is only evident after PKA treatment of troponin-exchanged cells.
Figure 5. Replacement of endogenous mutant cardiac troponin by recombinant human wild-type troponin complex.
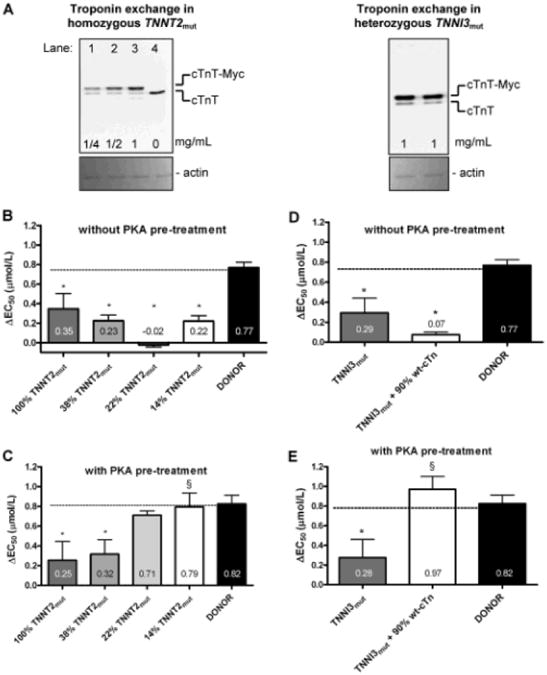
A, Quantification of troponin exchange in cardiomyocytes from a TNNT2mut and a TNNI3mut heart. Immunoblots stained with an antibody against cardiac troponin T (cTnT) that recognizes both endogenous cTnT (lower band) and recombinant myc-tag labeled cTnT (cTnT-myc; upper band). Left, An example is shown of a suspension of cardiomyocytes from a TNNT2mut heart exchanged with increasing concentrations of wild-type human recombinant troponin complex. Exchange with 0.25 mg/mL (lane 1), 0.5 mg/mL (lane 2), and 1 mg/mL (lane 3) troponin complex. TNNT2mut heart without added recombinant troponin complex (lane 4). Right, A suspension of cardiomyocytes from a TNNI3mut heart exchanged with 1 mg/mL wild-type human recombinant troponin complex. Similar amounts were loaded on the blots (shown by Ponceau-stained actin) to allow cTnT analysis within the linear detection range. B, The length-dependent activation was significantly lower compared with donor in all TNNT2mut cells harboring varying amounts of mutant cTnT (100% without exchange and 38%, 22%, and 14% in cells exchanged with increasing concentrations of unphosphorylated recombinant wild-type troponin). C, Measurements were also performed in exchanged cardiomyocytes, which were subsequently treated with protein kinase A (PKA) to normalize cTnI phosphorylation. Pretreatment with exogenous PKA was not able to recover the blunted length–dependent activation in TNNT2mut cells with 38% of mutant cTnT, but did recover the reduced length–dependence of TNNT2mut exchanged with 78% and 86% wild-type troponin. D, The length-dependent activation was significantly lower in TNNI3mut cells exchanged with ≈90% unphosphorylated recombinant wild-type troponin (wt-cTn) compared with donor cells. E, Pretreatment with exogenous PKA restored the blunted length–dependent activation in TNNI3mut cells exchanged with wt-cTn complex to donor values. *P<0.05 vs donor; §P<0.05 vs TNNT2mut or TNNI3mut.
Discussion
Our study shows that high-myofilament Ca2+-sensitivity in human HCM myocardium is independent of the presence of a sarcomere mutation and at least partly explained by protein hypophosphorylation. Sarcomere mutations may modify Ca2+-sensitivity, but the direction and magnitude of the change depend on the affected gene. Impaired length–dependent activation of sarcomeres represents a common pathomechanism underlying HCM, and could not be corrected by PKA treatment in HCM with missense mutations in genes encoding thick- and thin-filament proteins. Moreover, our troponin exchange experiments provide direct proof that mutant troponin impairs length-dependent activation. Our data indicate that mutant proteins resulting from missense mutations could perturb length-dependent sarcomere activation and underlie cardiac dysfunction observed at early stages of HCM disease development.
Myofilament Ca2+-Sensitivity and Phosphorylation Background
Our studies in human HCM with mutations in both thick- and thin-filament proteins showed high-myofilament Ca2+-sensitivity at a sarcomere length of 2.2 μm (Figure 1A). This is in agreement with previous studies in transgenic mouse models and in vitro studies with mutant proteins, which indicate that HCM sarcomere mutations sensitize myofilaments to calcium.3,12,13,25 However, the higher Ca2+-sensitivity in sarcomere mutation-positive HCM groups compared with nonfailing donors coincided with lower phosphorylation levels of cMyBP-C and cTnI (Figure 3A). The difference in myofilament Ca2+-sensitivity between sarcomere mutation-positive HCM and nonfailing donor can thus be partly explained by hypophosphorylation of sarcomeric proteins rather than the sarcomere gene mutation itself.
A possible explanation for the low-phosphorylation levels may reside in a blunted β–adrenergic response in patients with HCM. A blunted response to isoproterenol, a β-adrenoreceptor agonist, has been reported in transgenic mice harboring TPM1 and MYH7 mutations.26,27 Moreover, reduced β–adrenoreceptor density has been reported in patients with HCM.28 Treatment of HCM samples with PKA increased phosphorylation of the PKA-target proteins (Figures 3 and 4 and Online Figure II) and normalized myofilament Ca2+-sensitivity in MYBPC3mut, TPM1mut, and HCMsmn to values observed in nonfailing donors. In contrast, after PKA, higher myofilament Ca2+-sensitivity was still present in MYH7mut and TNNT2mut, suggesting a Ca2+-sensitizing effect by these mutations. Interestingly, PKA treatment significantly lowered Ca2+-sensitivity of the TNNI3mut R145W compared with donor. This observation contrasts with transgenic animal models and reconstituted thin-filaments using recombinant human mutant cTnI.29–31 It has been suggested that the Ca2+-sensitizing action of mutations in the inhibitory region of cTnI (residues 137–148) directly impairs the intrinsic inhibitory activity of cTnI.29,30 A possible explanation for the contradicting results compared with our TNNI3mut samples may reside in the amount of endogenous mutant protein expression. Using adenovirus gene transfection to incorporate the cTnI R145W mutant into adult rat cardiomyocytes, Davis et al32 observed no elevation of myofilament Ca2+-sensitivity, which was attributed to poor incorporation (≈35%) of mutant protein into the sarcomeric structure compared with wild-type cTnI. In addition, the phosphorylation background of the sarcomeres may have been different among studies.
Overall, our data show that high Ca2+-sensitivity is not a specific characteristic of human sarcomere mutation-positive HCM, as a similarly high-myofilament Ca2+-sensitivity was found in HCMsmn compared with donors, which may be ascribed to reduced β–adrenergic signaling as part of cardiomyopathy development. Although PKA is the archetypical kinase involved in modulating Ca2+-sensitivity through cTnI and cMyBP-C phosphorylation, it is by no means the only kinase that phosphorylates myofilament proteins. Both cTnI and cMyBP-C are targets for a whole range of kinases.33–37 cMyBP-C phosphorylation is thought to mainly affect cross-bridge cycling kinetics,38 although a role in mediating Ca2+-sensitivity of force has been suggested.39 Cardiac TnI is considered to be the key regulator of Ca2+-sensitivity, and it is mainly through phosphorylation of Ser23 and Ser24 that PKA exerts its Ca2+-desensitizing effect, although many other phosphorylation sites have been identified.40 Phosphorylation is but one of many possible post-translational modifications. Recent reports have hinted at possible involvement of other post-translational modifications in the regulation of sarcomere function, such as oxidation and S-glutathionylation41 or O-GlcNAcylation.42 It would thus be an oversimplification to propose that a reduction in PKA-phosphorylation of sarcomere proteins is solely responsible for the myofilament changes in HCM. However, the baseline Ca2+-sensitivity seems to be dominated by the relatively low-phosphorylation levels of PKA myofilament target proteins compared with donors. Higher phosphorylation levels mimicked by exogenous PKA treatment, as would be induced during increased cardiac stress (eg, exercise), unveils a higher, similar, or even lower myofilament Ca2+-sensitivity in HCM dependent on the affected gene. Our findings suggest diverse mutation–induced changes in Ca2+-sensitivity, whereas high-myofilament Ca2+-sensitivity is partly explained by secondary disease–related changes in protein phosphorylation.
Impairment of Length-Dependent Sarcomere Activation in HCM
Our data indicate that mutant sarcomeric proteins in HCM perturb length-dependent activation of myofilaments, which may contribute to early cardiac dysfunction observed in sarcomere mutation carriers. Studies in transgenic mouse models and troponin-exchange techniques with mouse tissue harboring HCM mutations were not conclusive as a reduced or normal length–dependent activation was found compared with controls.27,43–46 The recent study of Ford et al46 may shed some light on these previous observations as the authors studied length-dependent differences in mice expressing TNNT2 mutations either in an α-MyHC (predominant in murines) or in a β-MyHC (predominant in healthy adult human hearts) background. It was observed that mice expressing the R92L mutation in the physiological α-MyHC background presented a normal length–dependent Ca2+-activation, whereas in the presence of the slow cycling β-MyHC isoform, length dependence was lost.46 Defects in length-dependent sarcomere properties were not similar in all HCM samples, because the blunted length–dependent increase in myofilament Ca2+-sensitivity was corrected to donor values by exogenous PKA in truncating MYBPC3mut and HCMsmn, whereas it remained defective in HCM with missense mutations in MYBPC3, MYH7, TNNT2, TNNI3, and TPM1. Moreover, the increase in maximal force generating capacity on an increase in sarcomere length was almost entirely absent in HCM with troponin mutations (Table 2). Although PKA increased phosphorylation of cTnI and cMyBP-C and reduced myofilament Ca2+-sensitivity at 2.2 μm in all HCM samples (except in the homozygous TNNT2mut), it did not restore length-dependent activation in HCM samples with missense mutations. Intriguingly, a negative myofilament length–dependent activation was observed after PKA in the TPM1mut sample (Figure 2D), suggesting that length-dependent activation is even more impaired during increased cardiac stress.
The blunted length–dependent activation may be partly related to the relatively low phosphorylation of PKA-targets and high-baseline myofilament Ca2+-sensitivity. The suggestion that PKA-mediated myofilament protein phosphorylation has a modulatory role in length-dependent activation comes from studies in ferret papillary muscles,23 in which isoprenaline, a stimulator of the β-adrenergic receptor pathway, enhanced the length-dependent change in the force-Ca2+ relation. Studies in cardiac tissue in which cTnI was replaced by skeletal TnI, which lacks the PKA-target serines (Ser23/24), showed higher myofilament Ca2+-sensitivity, but a significantly reduced length–dependent activation,24,47 indicating a role for cTnI phosphorylation in length-dependent activation. A study by Cazorla et al44 in transgenic mice lacking cMyBP-C demonstrated lower length–dependent activation than wild-type mice that could not be restored by exogenous PKA treatment, which suggests that cMyBP-C is needed for proper length–dependent sarcomere activation. In our human samples with truncating MYBPC3 mutations, impaired length–dependent activation was corrected to donor values by PKA. Our previous study in HCM with truncating mutations in MYBPC3 showed reduced expression of full-length cMyBP-C to ≈70% compared with donor values (ie, haploinsufficiency). Overall, our studies in human HCM with MYBPC3 truncation mutations indicate that the presence of ≈70% of full-length cMyBP-C protein in the sarcomere is sufficient to preserve the length-dependent properties of the sarcomeres.15
The perturbations in length-dependent activation in HCM with mutations in thin-filament genes may be explained by the 3-state model of filament transition, in which the troponin–tropomyosin complex has a central regulatory role. Myofilament contraction and force production are tightly regulated by the troponin–tropomyosin complex that regulates the interaction between the actin and myosin filaments. It is believed that the myofilaments exist in a dynamic equilibrium between 3 biochemical transitions (Figure 6) that reflect different interactions between actin and myosin, termed the blocked (B-state), closed (C-state), and open (M-state) states of thin-filament regulation.48,49 In the B-state Ca2+ is not bound to cardiac troponin C, and tropomyosin sterically blocks myosin-binding sites on F-actin (Figure 6A). In the C-state, Ca2+ binds to cardiac troponin C, which changes conformation of the troponin–tropomyosin complex, resulting in nontension-generating cross-bridges, which bind weakly to F-actin (ie, weakly bound cross-bridges; Figure 6B).49–51 The M-state involves strong binding of tension-generating cross-bridges, which results in myofilament contraction and force development (Figure 6C).49–51 Because of the central roles of cTnT and cTnI in the transition from the B-state to the C-state,52–54 it is likely that mutation-induced irregularities in protein interactions may translate into thin-filament abnormalities.
Figure 6. Schematic drawing of the thin-filament functional unit.
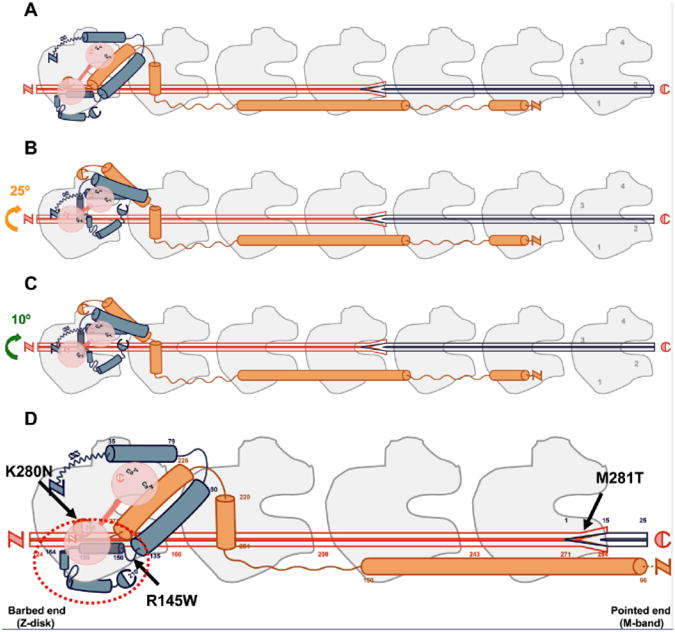
Seven actin monomers (gray) spanned by 1 tropomyosin dimmer (red) and 1 troponin complex: cardiac troponin C (cTnC; pink), cardiac troponin I (cTnI; blue), and cardiac troponin T (cTnT; orange). Capitals N and C depict N- and C-terminal protein ends, respectively. Dark-blue tropomyosin depicts near-neighbor tropomyosin dimmer interaction.69,70 The orientation of thin-filament proteins is as follows: the N-terminal region of cTnT points toward the pointed end (M-band), whereas the core domain of the troponin complex is oriented to the barbed end (Z-disk).71 Interacting sites and structural location of actin–tropomyosin–troponin proteins are matched in accordance with available literature.25,50,51,55,70,72 Cardiac TnI residues 1 to 34 are arbitrarily positioned. Our drawing follows the proposed mechanism for Ca2+-regulation of contraction proposed by Murakami et al.55 A, B-state (blocked); when ATP is present and cytoplasmic [Ca2+] is low such that Ca2+ is not bound to cTnC, tropomyosin sterically blocks the myosin-binding sites on F-actin. B, C-state (Ca2+-induced); cytoplasmic [Ca2+] rises such that Ca2+ binds to cTnC inducing conformational changes of the troponin complex, resulting in an ≈25° movement of tropomyosin on the thin filament, thereby exposing most of the myosin-binding sites on F-actin. Note the movement of tropomyosin away from subdomains 1 and 2 of F-actin. In the C-state, the myofilament is not yet activated because nontension-generating cross-bridges bind weakly to F-actin. C, M-state (myosin-induced); involves the strong binding of tension-generating cross-bridges that induce an additional ≈10° movement of tropomyosin on F-actin, resulting in myofilament activation and contraction. Note the transition of tropomyosin into subdomains 3 and 4 of F-actin. D, Solid arrows depict the location of mutation sites on thin-filament proteins present in our human HCM samples. Cardiac TnT residues 1 to 65 are shortened to fit the enlarged scale.
The C-terminal half of cTnI docks the troponin–tropomyosin complex onto the outer domain of F-actin at low-cytoplasmic [Ca2+],55 stabilizing the formation of the B-state. Interestingly, ≈86% of cTnI HCM–causing mutations (dashed red circle in Figure 6D), including the one present in our study, are found in the C-terminal half of cTnI (residues 137–210), a region responsible for actin binding.25,56 Indeed, disruption of the B-state has been suggested in HCM-causing mutations not only affecting cTnI,57 but also cTnT.58,59 The relevance of the transition from the B- to the C-state for proper length–dependent activation has been shown by Smith and Fuchs60 who were the first to provide evidence for a length-sensitive step in the transitions of thin-filament activation. A reduction in ionic strength (<0.05 mol/L), known to shift the B-state equilibrium toward a stable C-state,61 coincided with impairment of length-dependent activation.60 The blunted length–dependent increase in myofilament Ca2+-sensitivity observed in the thin-filament mutation groups can thus be explained by disruption of the B-state and an increased number of weakly bound cross-bridges in the C-state.
Eleven HCM mutations have been identified in α–tropomyosin.56 To the best of our knowledge, the present study is the first to analyze the effects of an HCM-causing mutation (M281T) in the overlap region of tropomyosin, which has a central role in cooperative activation of the thin filament.62,63 The steepness of the force-Ca2+ relation, which is an indicator of the relative number of near-neighbor interacting sites, was significantly lower in TPM1mut compared with donors (1.98±0.37 and 3.33±0.11, respectively; Figure 2B), indicative for impairment of the cooperative response in activating the thin filament. Palm et al64 demonstrated that tropomyosin overlap regions are required for proper formation of a ternary complex with the N-terminal tail of cTnT. Because the N-terminal region of cTnT is needed to maintain the thin filament in the B-state,53,54 it is likely that mutations in the overlap region of α–tropomyosin structurally impair formation of the B-state and thereby impair length-dependent activation of myofilaments.
Previous studies indicated that myosin is not involved in the formation of the first 2 equilibrium states, that is, B- and C-states.49–51 However, myosin is crucial for formation of the M-state (myosin-induced), because strong binding of tension-generating cross-bridges are required for thin-filament activation and force production.49–51 Because 5 of the 6 samples used in our study have mutations in the myosin S1 domain, responsible for actin-binding,65 it is likely that the deleterious effects of MYH7 mutations occur via perturbation of the M-state. A recent study by Farman et al66 highlights the essential role of the orientation of the myosin heads that precede thin-filament activation for proper lattice spacing and length-dependent activation. Thus, altered myosin head orientation, as a result of mutations, may impair formation of the M-state and affect length-dependent activation.
The possible involvement of cMyBP-C in the modulation of thin-filament activity has been only recently addressed. Electron microscopy and 3-dimensional reconstruction of thin filaments with cMyBP-C suggest that the N-terminal extension of cMyBP-C could modulate the movement of tropomyosin on F-actin and interfere with actomyosin interactions, possibly involved in the regulation of thin-filament activation.67 The MYBPC3 missense mutations in our study are located along the N-terminal extension of cMyBP-C and may alter the tropomyosin–actin interaction and thereby impair length-dependent activation.
Study Limitations and Clinical Implications
Although we were able to study a large collection of human HCM samples with different mutations in thick- and thin-filament proteins, care must be taken to extrapolate our findings to all patients with HCM. Our HCM population consisted of patients with left ventricular outflow tract obstruction, and the patient with the homozygous TNNT2 mutation had end-stage heart failure. Our data do highlight that mutation-induced changes in myofilament Ca2+-sensitivity and length-dependent sarcomere activation are diverse and depend on the affected gene, and most likely location and type of the mutation in the affected protein. We provide evidence that mutant protein may impair sarcomere function at ≈38% expression (Figure 5C), which emphasizes the importance of studying the mutant protein level at which cardiac performance is impaired. Future studies in transgenic mice models and human myectomy samples are warranted to extend our findings to a broader set of sarcomere mutations and assess the toxic dose of mutant proteins.
Although sarcomere mutation-negative patients are commonly used as control group, we cannot completely rule out the presence of rare mutations. However, as our cardiomyocyte analyses revealed no functional impairments (PKA normalized length-dependent activation in HCMsmn), the likelihood of the presence of a rare mutation is low.
Our study revealed perturbed sarcomere length–dependent activation as a common mechanism underlying cardiac dysfunction in HCM. Pathological hypertrophy presumably reflects the compensatory response of the heart to counteract impaired sarcomere defects, such as the blunted length–dependent myofilament activation. The relatively low-force generating capacity of cardiomyocytes and the inability to increase force on an increase in sarcomere length may in part underlie cardiac dysfunction and initiate compensatory hypertrophy. Pak et al68 showed a blunted end-systolic pressure volume relation in patients with HCM, suggesting that the hearts were unable to properly recruit preload to augment contractility. The latter observation may be explained by cardiac remodeling. However, likewise, the mutation-induced blunted length–dependent sarcomere activation may limit preload-mediated contractile reserve of the heart in patients with HCM. Our study in combination with others6–10 supports the hypothesis that defective sarcomere function as a result of gene mutations is central to early stages of HCM disease and precedes development of hypertrophy. Moreover, the blunted increase in myofilament Ca2+-sensitivity on an increase in length may alter Ca2+-buffering in the cardiomyocytes and provide a substrate for arrhythmias.
Supplementary Material
Novelty and Significance.
What Is Known?
Hypertrophic cardiomyopathy (HCM) is commonly caused by mutations in genes encoding sarcomeric e proteins.
Previous studies demonstrated early signs of myocardial dysfunction, in the absence of clinically detectable cardiac hypertrophy.
In vitro and transgenic animal models expressing mutant thick- and thin-filament proteins in the heart implicate elevated myofilament Ca2+-sensitivity as a major mechanism for the pathogenesis of HCM.
What New Information Does This Article Contribute?
Cardiac myocytes isolated from the interventricular septal tissues of patients with hypertrophic obstructive cardiomyopathy show diverse and mutation-related changes in myofilament Ca2+-sensitivity, which are dependent on the affected gene.
This increase in myofilament Ca2+-sensitivity could be in part attributable to hypophosphorylation of protein kinase A-targets.
Myofilament length–dependent activation is impaired in cardiac myocytes isolated from patients with HCM harboring missense mutations, and represents a mechanism underlying the development of HCM.
Data on myofilament length–dependent activation in transgenic animal models harboring HCM mutations are conflicting, and studies in human HCM are scarce. High-myofilament Ca2+-sensitivity partly reflects hypophosphorylation of protein kinase A-targets, indicative for secondary disease–related alterations. Reduced length–dependent sarcomere activation is a common feature of human HCM caused by mutations in genes encoding thick- and thin-filament proteins. Pretreatment with protein kinase A did not totally correct impaired length–dependent sarcomere activation in HCM samples carrying missense mutations. Mutation-induced impaired length–dependent activation may limit the preload-mediated contractile reserve of the heart. Our findings, in combination with clinical studies, support the notion that sarcomere mechanical dysfunction is central to the pathogenesis of HCM and precedes the development of cardiac hypertrophy.
Acknowledgments
Sources of Funding: We acknowledge support from the seventh Framework Program of the European Union (BIG-HEART, grant agreement 241577), the National Institutes of Health (NIH; grant agreement R01 HL63038), and from the Netherlands organization for scientific research (NWO; VIDI grant 91711344).
Nonstandard Abbreviations and Acronyms
- B-state
blocked state
- cMyBP-C
cardiac myosin–binding protein-C
- C-state
Ca2+-induced state
- cTnI
cardiac troponin I
- cTnT
cardiac troponin T
- Fmax
maximal developed force
- HCM
hypertrophic cardiomyopathy
- HCMsmn
sarcomere mutation-negative HCM
- MyHC
myosin heavy chain
- M-state
myosin-induced state
- PKA
protein kinase A
Footnotes
Disclosures: None.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.111.300436/-/DC1.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Tombe PP, Mateja RD, Tachampa K, Ait Mou Y, Farman GP, Irving TC. Myofilament length dependent activation. J Mol Cell Cardiol. 2010;48:851–858. doi: 10.1016/j.yjmcc.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults: echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation. 1995;92:785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 3.Tardiff JC. Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev. 2005;10:237–248. doi: 10.1007/s10741-005-5253-5. [DOI] [PubMed] [Google Scholar]
- 4.Richard P, Charron P, Carrier L, et al. EUROGENE Heart Failure Project. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 5.Santos S, Marques V, Pires M, Silveira L, Oliveira H, Lança V, Brito D, Madeira H, Esteves JF, Freitas A, Carreira IM, Gaspar IM, Monteiro C, Fernandes AR. High resolution melting: improvements in the genetic diagnosis of hypertrophic cardiomyopathy in a Portuguese cohort. BMC Med Genet. 2012;13:17. doi: 10.1186/1471-2350-13-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagueh SF, Kopelen HA, Lim DS, Zoghbi WA, Quiñones MA, Roberts R, Marian AJ. Tissue Doppler imaging consistently detects myocardial contraction and relaxation abnormalities, irrespective of cardiac hypertrophy, in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2000;102:1346–1350. doi: 10.1161/01.cir.102.12.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fraysse B, Weinberger F, Bardswell SC, Cuello F, Vignier N, Geertz B, Starbatty J, Krämer E, Coirault C, Eschenhagen T, Kentish JC, Avkiran M, Carrier L. Increased myofilament Ca2+ sensitivity and diastolic dysfunction as early consequences of Mybpc3 mutation in heterozygous knock-in mice. J Mol Cell Cardiol. 2012;52:1299–1307. doi: 10.1016/j.yjmcc.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagueh SF, Bachinski LL, Meyer D, Hill R, Zoghbi WA, Tam JW, Quiñones MA, Roberts R, Marian AJ. Tissue Doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation. 2001;104:128–130. doi: 10.1161/01.cir.104.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho CY, Carlsen C, Thune JJ, Havndrup O, Bundgaard H, Farrohi F, Rivero J, Cirino AL, Andersen PS, Christiansen M, Maron BJ, Orav EJ, Køber L. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:314–321. doi: 10.1161/CIRCGENETICS.109.862128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Germans TRI, Götte MJ, Spreeuwenberg MD, Doevendans PA, Pinto YM, van der Geest RJ, van der Velden J, Wilde AA, van Rossum AC. How do hypertrophic cardiomyopathy mutations affect myocardial function in carriers with normal wall thickness? Assessment with cardiovascular magnetic resonance. J Cardiovas Mag Res. 2010;12:13. doi: 10.1186/1532-429X-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O'Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG, Seidman CE. Mutations in the genes for cardiac troponin T and α-tropomyosin in hypertrophic cardiomyopathy. New Eng J Med. 1995;332:1058–1065. doi: 10.1056/NEJM199504203321603. [DOI] [PubMed] [Google Scholar]
- 12.Knollmann BC, Kirchhof P, Sirenko SG, Degen H, Greene AE, Schober T, Mackow JC, Fabritz L, Potter JD, Morad M. Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circ Res. 2003;92:428–436. doi: 10.1161/01.RES.0000059562.91384.1A. [DOI] [PubMed] [Google Scholar]
- 13.Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ, Knollmann BC. Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ Res. 2012;111:170–179. doi: 10.1161/CIRCRESAHA.112.270041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoskins AC, Jacques A, Bardswell SC, McKenna WJ, Tsang V, dos Remedios CG, Ehler E, Adams K, Jalilzadeh S, Avkiran M, Watkins H, Redwood C, Marston SB, Kentish JC. Normal passive viscoelasticity but abnormal myofibrillar force generation in human hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2010;49:737–745. doi: 10.1016/j.yjmcc.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Dijk SJ, Paalberends ER, Najafi A, Michels M, Sadayappan S, Carrier L, Boontje NM, Kuster DW, van Slegtenhorst M, Dooijes D, dos Remedios C, ten Cate FJ, Stienen GJ, van der Velden J. Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ Heart Fail. 2012;5:36–46. doi: 10.1161/CIRCHEARTFAILURE.111.963702. [DOI] [PubMed] [Google Scholar]
- 16.van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJ, van der Velden J. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 17.Hofmann PA, Fuchs F. Effect of length and cross-bridge attachment on Ca2+ binding to cardiac troponin C. Am J Physiol. 1987;253:C90–C96. doi: 10.1152/ajpcell.1987.253.1.C90. [DOI] [PubMed] [Google Scholar]
- 18.Wannenburg T, Heijne GH, Geerdink JH, Van Den Dool HW, Janssen PM, De Tombe PP. Cross-bridge kinetics in rat myocardium: effect of sarcomere length and calcium activation. Am J Physiol Heart Circ Physiol. 2000;279:H779–H790. doi: 10.1152/ajpheart.2000.279.2.H779. [DOI] [PubMed] [Google Scholar]
- 19.Zaremba R, Merkus D, Hamdani N, Lamers JM, Paulus WJ, Dos Remedios C, Duncker DJ, Stienen GJ, van der Velden J. Quantitative analysis of myofilament protein phosphorylation in small cardiac biopsies. Proteomics Clin Appl. 2007;1:1285–1290. doi: 10.1002/prca.200600891. [DOI] [PubMed] [Google Scholar]
- 20.Hamdani N, Borbély A, Veenstra SP, Kooij V, Vrydag W, Zaremba R, Dos Remedios C, Niessen HW, Michel MC, Paulus WJ, Stienen GJ, van der Velden J. More severe cellular phenotype in human idiopathic dilated cardiomyopathy compared to ischemic heart disease. J Muscle Res Cell Motil. 2010;31:289–301. doi: 10.1007/s10974-010-9231-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wijnker PJM, Foster DB, Tsao AL, Frazier AH, dos Remedios C, Murphy AM, Stienen GJM, van der Velden J. Impact of site-specific phosphorylation of the protein kinase A sites ser23 and ser24 of cardiac troponin in human cardiomyocytes. Am J Physiol. 2013;304:H260–H268. doi: 10.1152/ajpheart.00498.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen DG, Kentish JC. The cellular basis of the length-tension relation in cardiac muscle. J Mol Cell Cardiol. 1985;17:821–840. doi: 10.1016/s0022-2828(85)80097-3. [DOI] [PubMed] [Google Scholar]
- 23.Komukai K, Kurihara S. Length dependence of Ca(2+)-tension relationship in aequorin-injected ferret papillary muscles. Am J Physiol. 1997;273:H1068–H1074. doi: 10.1152/ajpheart.1997.273.3.H1068. [DOI] [PubMed] [Google Scholar]
- 24.Konhilas JP, Irving TC, Wolska BM, Jweied EE, Martin AF, Solaro RJ, de Tombe PP. Troponin I in the murine myocardium: influence on length-dependent activation and interfilament spacing. J Physiol (Lond) 2003;547:951–961. doi: 10.1113/jphysiol.2002.038117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tardiff JC. Thin filament mutations: developing an integrative approach to a complex disorder. Circ Res. 2011;108:765–782. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Freeman K, Colon-Rivera C, Olsson MC, Moore RL, Weinberger HD, Grupp IL, Vikstrom KL, Iaccarino G, Koch WJ, Leinwand LA. Progression from hypertrophic to dilated cardiomyopathy in mice that express a mutant myosin transgene. Am J Physiol Heart Circ Physiol. 2001;280:H151–H159. doi: 10.1152/ajpheart.2001.280.1.H151. [DOI] [PubMed] [Google Scholar]
- 27.Prabhakar R, Boivin GP, Grupp IL, Hoit B, Arteaga G, Solaro RJ, Wieczorek DF. A familial hypertrophic cardiomyopathy alpha-tropomyosin mutation causes severe cardiac hypertrophy and death in mice. J Mol Cell Cardiol. 2001;33:1815–1828. doi: 10.1006/jmcc.2001.1445. [DOI] [PubMed] [Google Scholar]
- 28.Choudhury L, Guzzetti S, Lefroy DC, Nihoyannopoulos P, McKenna WJ, Oakley CM, Camici PG. Myocardial beta adrenoceptors and left ventricular function in hypertrophic cardiomyopathy. Heart. 1996;75:50–54. doi: 10.1136/hrt.75.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi-Yanaga F, Morimoto S, Harada K, Minakami R, Shiraishi F, Ohta M, Lu QW, Sasaguri T, Ohtsuki I. Functional consequences of the mutations in human cardiac troponin I gene found in familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2001;33:2095–2107. doi: 10.1006/jmcc.2001.1473. [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi T, Patrick SE, Kobayashi M. Ala scanning of the inhibitory region of cardiac troponin I. J Biol Chem. 2009;284:20052–20060. doi: 10.1074/jbc.M109.001396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng Y, Schmidtmann A, Redlich A, Westerdorf B, Jaquet K, Thieleczek R. Effects of phosphorylation and mutation R145G on human cardiac troponin I function. Biochemistry. 2001;40:14593–14602. doi: 10.1021/bi0115232. [DOI] [PubMed] [Google Scholar]
- 32.Davis J, Wen H, Edwards T, Metzger JM. Allele and species dependent contractile defects by restrictive and hypertrophic cardiomyopathy-linked troponin I mutants. J Mol Cell Cardiol. 2008;44:891–904. doi: 10.1016/j.yjmcc.2008.02.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuster DW, Bawazeer AC, Zaremba R, Goebel M, Boontje NM, van der Velden J. Cardiac myosin binding protein C phosphorylation in cardiac disease. J Muscle Res Cell Motil. 2012;33:43–52. doi: 10.1007/s10974-011-9280-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bardswell SC, Cuello F, Kentish JC, Avkiran M. cMyBP-C as a promiscuous substrate: phosphorylation by non-PKA kinases and its potential significance. J Muscle Res Cell Motil. 2012;33:53–60. doi: 10.1007/s10974-011-9276-3. [DOI] [PubMed] [Google Scholar]
- 35.Haworth RS, Cuello F, Herron TJ, Franzen G, Kentish JC, Gautel M, Avkiran M. Protein kinase D is a novel mediator of cardiac troponin I phosphorylation and regulates myofilament function. Circ Res. 2004;95:1091–1099. doi: 10.1161/01.RES.0000149299.34793.3c. [DOI] [PubMed] [Google Scholar]
- 36.Kuster DWD, Sequeira V, Najafi A, Boontje N, J M, Wijnker P, Witjas-Paalberends R, Marston S, dos Remedios CG, Carrier L, Demmers JAA, Redwood CS, Sadayappan S, van der Velden J. GSK3β phosphorylates newly identified site in the Pro-Ala rich region of cardiac myosin binding protein C and alters cross-Bridge cycling kinetics in human. Circ Res. 2013;112:633–639. doi: 10.1161/CIRCRESAHA.112.275602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jideama NM, Noland TA, Jr, Raynor RL, Blobe GC, Fabbro D, Kazanietz MG, Blumberg PM, Hannun YA, Kuo JF. Phosphorylation specificities of protein kinase C isozymes for bovine cardiac troponin I and troponin T and sites within these proteins and regulation of myofilament properties. J Biol Chem. 1996;271:23277–23283. doi: 10.1074/jbc.271.38.23277. [DOI] [PubMed] [Google Scholar]
- 38.Stelzer JE, Patel JR, Moss RL. Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ Res. 2006;99:884–890. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- 39.Chen PP, Patel JR, Rybakova IN, Walker JW, Moss RL. Protein kinase A-induced myofilament desensitization to Ca(2+) as a result of phosphorylation of cardiac myosin-binding protein C. J Gen Physiol. 2010;136:615–627. doi: 10.1085/jgp.201010448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang P, Kirk JA, Ji W, dos Remedios CG, Kass DA, Van Eyk JE, Murphy AM. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation. 2012;126:1828–1837. doi: 10.1161/CIRCULATIONAHA.112.096388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avner BS, Shioura KM, Scruggs SB, Grachoff M, Geenen DL, Helseth DL, Jr, Farjah M, Goldspink PH, Solaro RJ. Myocardial infarction in mice alters sarcomeric function via post-translational protein modification. Mol Cell Biochem. 2012;363:203–215. doi: 10.1007/s11010-011-1172-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramirez-Correa GA, Jin W, Wang Z, Zhong X, Gao WD, Dias WB, Vecoli C, Hart GW, Murphy AM. O-linked GlcNAc modification of cardiac myofilament proteins: a novel regulator of myocardial contractile function. Circ Res. 2008;103:1354–1358. doi: 10.1161/CIRCRESAHA.108.184978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang B, Chung F, Qu Y, Pavlov D, Gillis TE, Tikunova SB, Davis JP, Tibbits GF. Familial hypertrophic cardiomyopathy-related cardiac troponin C mutation L29Q affects Ca2+ binding and myofilament contractility. Physiol Genomics. 2008;33:257–266. doi: 10.1152/physiolgenomics.00154.2007. [DOI] [PubMed] [Google Scholar]
- 44.Cazorla O, Szilagyi S, Vignier N, Salazar G, Krämer E, Vassort G, Carrier L, Lacampagne A. Length and protein kinase A modulations of myocytes in cardiac myosin binding protein C-deficient mice. Cardiovasc Res. 2006;69:370–380. doi: 10.1016/j.cardiores.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 45.Chandra M, Rundell VLM, Tardiff JC, Leinwand LA, de Tombe PP, Solaro RJ. Ca2+ activation of myofilaments from transgenic mouse hearts expressing R92Q mutant cardiac troponin T. Am J Physiol. 2001;280:H705–H713. doi: 10.1152/ajpheart.2001.280.2.H705. [DOI] [PubMed] [Google Scholar]
- 46.Ford SJ, Mamidi R, Jimenez J, Tardiff JC, Chandra M. Effects of R92 mutations in mouse cardiac troponin T are influenced by changes in myosin heavy chain isoform. J Mol Cell Cardiol. 2012;53:542–551. doi: 10.1016/j.yjmcc.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arteaga GM, Palmiter KA, Leiden JM, Solaro RJ. Attenuation of length dependence of calcium activation in myofilaments of transgenic mouse hearts expressing slow skeletal troponin I. J Physiol (Lond) 2000;526(pt 3):541–549. doi: 10.1111/j.1469-7793.2000.t01-1-00541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lehman W, Hatch V, Korman V, Rosol M, Thomas L, Maytum R, Geeves MA, Van Eyk JE, Tobacman LS, Craig R. Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J Mol Biol. 2000;302:593–606. doi: 10.1006/jmbi.2000.4080. [DOI] [PubMed] [Google Scholar]
- 50.Vibert P, Craig R, Lehman W. Steric-model for activation of muscle thin filaments. J Mol Biol. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- 51.Pirani A, Xu C, Hatch V, Craig R, Tobacman LS, Lehman W. Single particle analysis of relaxed and activated muscle thin filaments. J Mol Biol. 2005;346:761–772. doi: 10.1016/j.jmb.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 52.Schaertl S, Lehrer SS, Geeves MA. Separation and characterization of the two functional regions of troponin involved in muscle thin filament regulation. Biochemistry. 1995;34:15890–15894. doi: 10.1021/bi00049a003. [DOI] [PubMed] [Google Scholar]
- 53.Tobacman LS, Nihli M, Butters C, Heller M, Hatch V, Craig R, Lehman W, Homsher E. The troponin tail domain promotes a conformational state of the thin filament that suppresses myosin activity. J Biol Chem. 2002;277:27636–27642. doi: 10.1074/jbc.M201768200. [DOI] [PubMed] [Google Scholar]
- 54.Gollapudi SK, Mamidi R, Mallampalli SL, Chandra M. The N-terminal extension of cardiac troponin T stabilizes the blocked state of cardiac thin filament. Biophys J. 2012;103:940–948. doi: 10.1016/j.bpj.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murakami K, Yumoto F, Ohki SY, Yasunaga T, Tanokura M, Wakabayashi T. Structural basis for Ca2+-regulated muscle relaxation at interaction sites of troponin with actin and tropomyosin. J Mol Biol. 2005;352:178–201. doi: 10.1016/j.jmb.2005.06.067. [DOI] [PubMed] [Google Scholar]
- 56.Harvard-Medical-School-Genetic-database. Sarcomere protein gene mutation database. Cardiogenomics Harvard Medical School database [Google Scholar]
- 57.Kobayashi T, Solaro RJ. Increased Ca2+ affinity of cardiac thin filaments reconstituted with cardiomyopathy-related mutant cardiac troponin I. J Biol Chem. 2006;281:13471–13477. doi: 10.1074/jbc.M509561200. [DOI] [PubMed] [Google Scholar]
- 58.El-Mezgueldi M, Wazeer Z. Transient kinetic analysis of hypertrophic and dilated cardiomyopathy linked mutations in troponin T; The 40th European muscle conference; 2011. Abstract. [Google Scholar]
- 59.Burhop J, Rosol M, Craig R, Tobacman LS, Lehman W. Effects of a cardiomyopathy-causing troponin t mutation on thin filament function and structure. J Biol Chem. 2001;276:20788–20794. doi: 10.1074/jbc.M101110200. [DOI] [PubMed] [Google Scholar]
- 60.Smith SH, Fuchs F. Effect of ionic strength on length-dependent Ca(2+) activation in skinned cardiac muscle. J Mol Cell Cardiol. 1999;31:2115–2125. doi: 10.1006/jmcc.1999.1043. [DOI] [PubMed] [Google Scholar]
- 61.Head JG, Ritchie MD, Geeves MA. Characterization of the equilibrium between blocked and closed states of muscle thin filaments. Eur J Biochem. 1995;227:694–699. doi: 10.1111/j.1432-1033.1995.tb20190.x. [DOI] [PubMed] [Google Scholar]
- 62.Rao VS, Marongelli EN, Guilford WH. Phosphorylation of tropomyosin extends cooperative binding of myosin beyond a single regulatory unit. Cell Motil Cytoskeleton. 2009;66:10–23. doi: 10.1002/cm.20321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pan BS, Gordon AM, Luo ZX. Removal of tropomyosin overlap modifies cooperative binding of myosin S-1 to reconstituted thin filaments of rabbit striated muscle. J Biol Chem. 1989;264:8495–8498. [PubMed] [Google Scholar]
- 64.Palm T, Greenfield NJ, Hitchcock-DeGregori SE. Tropomyosin ends determine the stability and functionality of overlap and troponin T complexes. Biophys J. 2003;84:3181–3189. doi: 10.1016/S0006-3495(03)70042-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rayment I, Holden HM, Sellers JR, Fananapazir L, Epstein ND. Structural interpretation of the mutations in the beta-cardiac myosin that have been implicated in familial hypertrophic cardiomyopathy. Proc Natl Acad Sci USA. 1995;92:3864–3868. doi: 10.1073/pnas.92.9.3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Farman GP, Gore D, Allen E, Schoenfelt K, Irving TC, de Tombe PP. Myosin head orientation: a structural determinant for the Frank-Starling relationship. Am J Physiol Heart Circ Physiol. 2011;300:H2155–H2160. doi: 10.1152/ajpheart.01221.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mun JY, Gulick J, Robbins J, Woodhead J, Lehman W, Craig R. Electron microscopy and 3D reconstruction of F-actin decorated with cardiac myosin-binding protein C (cMyBP-C) J Mol Biol. 2011;410:214–225. doi: 10.1016/j.jmb.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pak PH, Maughan WL, Baughman KL, Kieval RS, Kass DA. Mechanism of acute mechanical benefit from VDD pacing in hypertrophied heart: similarity of responses in hypertrophic cardiomyopathy and hypertensive heart disease. Circulation. 1998;98:242–248. doi: 10.1161/01.cir.98.3.242. [DOI] [PubMed] [Google Scholar]
- 69.Greenfield NJ, Huang YJ, Swapna GV, Bhattacharya A, Rapp B, Singh A, Montelione GT, Hitchcock-DeGregori SE. Solution NMR structure of the junction between tropomyosin molecules: implications for actin binding and regulation. J Mol Biol. 2006;364:80–96. doi: 10.1016/j.jmb.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 70.Murakami K, Stewart M, Nozawa K, Tomii K, Kudou N, Igarashi N, Shirakihara Y, Wakatsuki S, Yasunaga T, Wakabayashi T. Structural basis for tropomyosin overlap in thin (actin) filaments and the generation of a molecular swivel by troponin-T. Proc Natl Acad Sci USA. 2008;105:7200–7205. doi: 10.1073/pnas.0801950105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Paul DM, Morris EP, Kensler RW, Squire JM. Structure and orientation of troponin in the thin filament. J Biol Chem. 2009;284:15007–15015. doi: 10.1074/jbc.M808615200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takeda S, Yamashita A, Maeda K, Maéda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.