

Abstract
Introduction
In the last 10 years the field of mitochondrial genetics has widened, shifting the focus from rare sporadic, metabolic disease to the effects of mitochondrial DNA (mtDNA) variation in a growing spectrum of human disease. The aim of this review is to guide the reader through some key concepts regarding mitochondria before introducing both classic and emerging mitochondrial disorders.
Sources of data
In this article, a review of the current mitochondrial genetics literature was conducted using PubMed (http://www.ncbi.nlm.nih.gov/pubmed/). In addition, this review makes use of a growing number of publically available databases including MITOMAP, a human mitochondrial genome database (www.mitomap.org), the Human DNA polymerase Gamma Mutation Database (http://tools.niehs.nih.gov/polg/) and PhyloTree.org (www.phylotree.org), a repository of global mtDNA variation.
Areas of agreement
The disruption in cellular energy, resulting from defects in mtDNA or defects in the nuclear-encoded genes responsible for mitochondrial maintenance, manifests in a growing number of human diseases.
Areas of controversy
The exact mechanisms which govern the inheritance of mtDNA are hotly debated.
Growing points
Although still in the early stages, the development of in vitro genetic manipulation could see an end to the inheritance of the most severe mtDNA disease.
Keywords: mitochondria, genetics, mitochondrial DNA, mitochondrial disease, mtDNA
Mitochondria
The mitochondrion is a highly specialized organelle, present in almost all eukaryotic cells and principally charged with the production of cellular energy through oxidative phosphorylation (OXPHOS). In addition to energy production, mitochondria are also key components in calcium signalling, regulation of cellular metabolism, haem synthesis, steroid synthesis and, perhaps most importantly, programmed cell death (apoptosis).1 However, the simplistic elegance of biochemical ATP production belies a, complex, synergistic relationship between two genomes: the mitochondrial genome (mtDNA) and the nuclear genome (nDNA). The aim of this review is to introduce these two genomes and shed light on the clinical problems arising when communication breaks down. The emphasis is on the basic science underpinning mitochondrial diseases. Clinical aspects are not considered in detail because they have recently been reviewed elsewhere in open-access publications.2–4
mtDNA
MtDNA is the only source of critical cellular proteins outside of the eukaryotic nucleus. In the majority of eukaryotes, mtDNA is organizsed as a circular, double-stranded DNA molecule (Fig. 1).5 The strands are distinguished by their nucleotide composition: heavy (H-strand) is guanine rich, compared with the cytosine-rich light strand (L-strand). The length varies between species (15 000–17 000 bp), but is fairly consistent in humans (∼16 569 bp).5 MtDNA is a multi-copy DNA, with cells containing between 100 and 10 000 copies of mtDNA (dependent upon cellular energy demand).
Fig. 1.
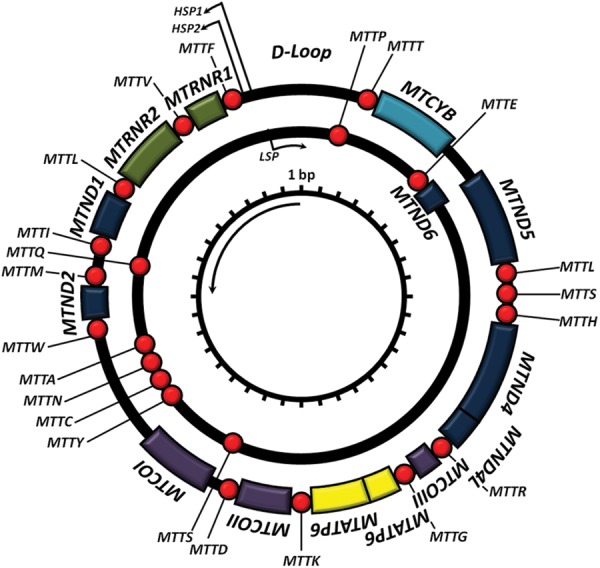
Mitochondrial DNA. Schematic diagram of the 16.6-kb, circular, double-stranded mtDNA molecule, where the outer circle represents the heavy strand and the inner circle the light strand. Shown are the genes encoding the mitochondrial RC: MTND1–6, MTCOI–II, MTATP6 and 8 and MTCYB; the two ribosomal RNAs (green boxes) and each of the 22 tRNAs (red spheres).
Structure
MtDNA contains 37 genes, 28 on the H-strand and 9 on the L-strand. Thirteen of the genes encode one polypeptide component of the mitochondrial respiratory chain (RC), the site of cellular energy production through OXPHOS. Twenty-four genes encode a mature RNA product: 22 mitochondrial tRNA molecules, a 16 s rRNA (large ribosomal subunit) and a 12 s rRNA (small ribosomal subunit).5 Unlike its nDNA counterpart, mtDNA is extremely efficient with ∼93% representing a coding region. Unlike nDNA, mtDNA genes lack intronic regions and some genes, notably MTATP6 and MTATP8, have overlapping regions. Most genes are contiguous, separated by one or two non-coding base pairs. mtDNA contains only one significant non-coding region, the displacement loop (D-loop).5 The D-loop contains the site of mtDNA replication initiation (origin of heavy strand synthesis, OH) and is also the site of both H-strand transcription promoters (HSP1 and HSP2).
The mitochondrial genetic code differs slightly from nuclear DNA (nDNA). MtDNA uses only two stop codons: ‘AGA’ and ‘AGG’6 (compared with ‘UAA’, ‘UGA’ and ‘UAG’ in nDNA), conversely ‘UGA’ encodes tryptophan. To compensate UAA codons have to be introduced at the post-transcriptional level. In addition ‘AUA’, isoleucine in nDNA, encodes for methionine in mtDNA.
Inheritance
Prevailing theory suggests that mtDNA is maternally inherited, with mtDNA nucleoids the unit of inheritance. During mammalian zygote formation, sperm mtDNA is removed by ubiquitination, likely occurring during transport through the male reproductive tract.7 Consequently, the mtDNA content of the zygote is determined exclusively by the previously unfertilized egg.
To date only a single case of paternal transmission in humans has been recorded.8 However, paternal transmission in other animals is both common and recurring. Theory suggests that the lack of paternal inheritance is due to either (i) a dilution effect; sperm contain only 100 copies of mtDNA, compared with 100 000 in the unfertilized egg, (ii) selective ubiquitination of paternal mtDNA or (iii) the ‘mtDNA bottleneck’ excludes the ‘minor’ paternal alleles.7 The advent of deep, next generation sequencing, allowing mtDNA can be sequenced at great depths (>20 000 fold) may enable researchers to revisit this phenomenon.
Homoplasmy and heteroplasmy
Cells contain thousands of molecules of mtDNA;9 and in the majority of cases their sequence is identical, known as homoplasmy. However, an inefficient mtDNA repair, a localized oxidative environment and increased replication10 make mtDNA mutation frequent. The polyploid nature of mtDNA means that mutations often co-exist with their wild-type counterpart in various proportions (termed heteroplasmy). The proportion of mutant has important consequences in understanding mitochondrial disease (discussed later).11
nDNA and mitochondrial function
According to recent data the mitochondrial proteome is estimated at ∼1500 proteins.12 Mitochondria are dependent upon the nuclear genome for the majority of the OXPHOS system and also for maintaining and replicating mtDNA as well as organelle network proliferation and destruction (Fig. 2).
Fig. 2.
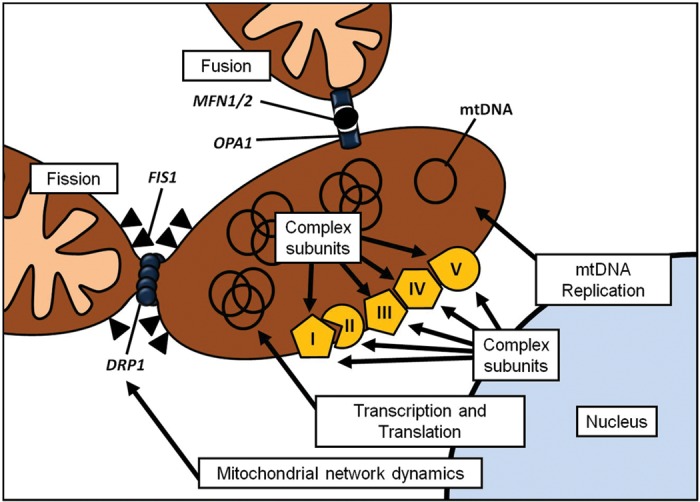
Interaction between nDNA and mtDNA. Cartoon demonstrating the complex interaction between genes encoded by nDNA and the processes they control in the mitochondrion.
OXPHOS system
To date, 92 structural OXPHOS subunit genes have been identified: 13 encoded by mtDNA (Fig. 1) and 79 encoded by the nuclear genome. Briefly, complex I (NADH:ubiquinone oxidoreductase), the largest of the RC components, consists of 44 subunits: 14 enzymatic ‘core subunits’ (7 from mtDNA and 7 from nDNA)13 and a further 30 nDNA accessory subunits thought to maintain complex stability.14 Complex II (succinate:ubiquinone oxidoreductase) is encoded entirely by nDNA (four subunits). Complex III (ubiquinol:cytochrome c oxidoreductase) contains 11 subunits, 1 encoded by mtDNA (MTCYB) and 10 encoded by nDNA.15 Complex IV (cytochrome c oxidase) consists of three mtDNA-encoded subunits and a further 11 nDNA-encoded subunits. Finally, complex V (F0F1-ATP synthase) comprises 19 subunits, 2 encoded by mtDNA and the remaining 17 encoded by nDNA.
In addition, nDNA encodes over 35 proteins required for the RC assembly: complex I = 11 nDNA assembly factors,16 complex III = 2,15 complex IV = 1817 and complex V = 4.18
mtDNA replication
Unlike nDNA, mtDNA replication is not governed by the cell cycle (eukaryotic cell division) and is continuously recycled. MtDNA replication and integrity maintenance is handled entirely by the nDNA. In eukaryotes, mtDNA is replicated in a ‘replisome’ (a DNA/protein complex making up the replication machinery) by a trimeric protein complex composed of a catalytic subunit: polymerase gamma, a 140 kDa DNA polymerase encoded by POLG and two 55 kDa accessory subunits, encoded by POLG2.19 This enzyme complex performs three activities, DNA polymerase activity, 3′-5′ exonuclease/proofreading activity and a 5′dRP lyase activity (required for enzymatic DNA repair).
In addition, the replisome also includes the mitochondrial single-stranded binding protein (encoded by mtSSB), which is involved in stabilizing single-stranded regions of mtDNA at replication forks, enhancing polymerase gamma activity. Twinkle is a 5′-3′ DNA helicase, which unwinds double-stranded mtDNA, facilitating mtDNA synthesis, as well as acting as a mtDNA primase (an enzyme required to prime nucleotide synthesis).19 Several topoisomerases have been indentified in humans, including the mitochondrial topoisomerases 1 (encoded by TOP1mt) and IIIα (encoded by TOP3a). Finally, the synergy between mitochondrial transcription factor A (encoded by TFAM) and mtDNA copy number suggests that TFAM may act as an mtDNA chaperone (a protein that assists the function of another protein) protecting it against oxidative damage.
mtDNA arrangement
Like its nDNA counterpart, mtDNA is also packaged in protein–DNA complexes, known as nucleoids.20 MtDNA nucleoids are associated with the inner mitochondrial membrane, spaced evenly along the cristae. In addition to a single mtDNA molecule,21 mtDNA nucleoids contain a number of proteins.20 Principally the site of mtDNA replication, it is unsurprising that mtDNA nucleoids contain the protein machinery required for DNA replication, transcription, repair and packaging, including the mtDNA polymerase POLG, its accessory subunit POLG2, the activator of mtDNA transcription (encoded by TFAM) as well as mtDNA helicases and binding proteins (twinkle and mtSSB, respectively).20 In addition, mtDNA nucleoids contain chaperone proteins (HSP90-β and HSP70) required for mtDNA stability.
Transcription and translation
Transcription of mtDNA is ‘prokaryotic like’ and was thought of a two-component system involving a protein complex containing the mitochondrial RNA polymerase (POLRMT) and two transcription factors (TFB1M and 2M).22,23 However, recent research indicates that TFB1M does not modulate mtDNA transcription in the presence of TFB2M, rather it acts as a dimethyltransferase which stabilizes the small subunit of the mitochondrial ribosome. RNA transcription is regulated by mitochondrial transcription factor A (TFAM).24
Briefly, each strand is transcribed as a polycistronic precursor mRNA molecule (i.e. the mRNA contains all of the genes in one molecule). Light-strand transcription is initiated from the light-strand promoter; however, heavy-strand transcription initiates from two heavy strand promoters: HSP1 and HSP2 (Fig. 1).25 Transcript elongation is performed by POLRMT, enhanced by both ‘transcription elongation factor mitochondrial’ (TEFM) and termination of mature transcripts is carried out by mitochondrial termination factor 1 (MTERF1).25
Translation of the 13 mtDNA protein coding genes occurs in the mitochondria. The mitoribosomes are partly coded by mtDNA (MTRNR1 and MTRNR2, Fig. 1), but require a further 81 nDNA proteins. Translation is initiated by two mitochondrial initiation factors: mtIF1 and mtIF3.26,27 mtIF3 begins initiation by dissociating the ‘mitoribosome’ (the mitochondrial ribosomes) allowing assembly of the initiation complex.28 MRNA is then bound to the small subunit, aligning the start codon to the peptidyl site of the mitoribosome. Peptide elongation is controlled by a number of nuclear-encoded genes, including mitochondrial elongation factor Tu (mtEFTu),29,30 which binds the tRNA to the mitoribosome and mitochondrial elongation factor G1 (mtEFG1), required to move the newly added amino acid along one position and allowing amino acid inclusion.31 Translation termination is carried out solely by mitochondrial release factor 1a (mtRF1a),32 which recognizes the stop codons (UAA and UAG)33 and triggers hydrolysis of the bond between the terminal tRNA and the nascent peptide.
Controlling mitochondrial network dynamics
Mitochondria are often depicted as distinct organelles; however, they actually form a complex reticulum that is undergoing continual fusion and fission (Fig. 2).34 It is likely that fusion has evolved as a mechanism to promote intermictochondrial cooperation—allowing the sharing and dissemination of mtDNA and mitochondrial proteins. Fission promotes mitochondrial compartmentalization,34 a mechanism that is needed to distribute mitochondria during cell division. Mitochondrial network dynamics, much like mtDNA replication, is controlled completely by nDNA, although likely involves mtDNA–nDNA communication.34
Mitochondrial fusion
The principle player in mitochondrial fusion is mitofusin (Mfn) and mammalian mitochondria contain two similar mitofusin proteins: Mfn1 and Mfn2 (Fig. 2),34 sharing 80% sequence homology. Studies indicate that both Mfn1 and Mfn2 uniformly localize to the mitochondrial outer membrane and overexpression leads to peri-nuclear clustering on mitochondria.34 Mitochondrial fusion is also dependent upon OPA1 expression (Fig. 2),34 where inhibition of gene expression causes an increase in mitochondrial fragmentation, conversely the overexpression of OPA1 breaks the network into spheres.
Mitochondrial fission
DNM1L, dynamin 1 like, controls mitochondrial fission in mammalian cells (Fig. 2).34 DNM1L codes for a principally cytosolic protein; however, it also localizes to fission sites on the mitochondria. Similar to Mfn1, the overexpression of ‘mutant’ DNM1L results in a breakdown of mitochondrial networks. Due to its dynamin similarity, two different functions have been proposed for DNM1L. It has been hypothesized that DNM1L may mechanically mediate membrane fission through GTP hydrolysis; alternatively, it may act as a signalling molecule, conscripting and activating separate fission enzymes such as Dnm1: the yeast homologue of Drp1.
Areas of agreement
Mitochondrial disease
An area where all mitochondrial researchers would agree is the capacity for mitochondrial dysfunction to manifest as disease. Mitochondrial disease is principally a chronic loss of cellular energy, where a failure to meet cellular energy demand results in a clinical phenotype. The clinical spectrum of mitochondrial disease is diverse (Fig. 3); however, tissues where there is a high metabolic demand, such as the central nervous system (CNS) or heart, are typically affected.
Fig. 3.

Clinical spectrum of mitochondrial disease. Schematic diagram showing the organ and corresponding disease affected by mitochondrial dysfunction.
The broad clinical spectrum of mitochondrial dysfunction, coupled with the heterogeneity of mtDNA variation, makes the prevalence of mitochondrial DNA (mtDNA) difficult to calculate. Estimates, based on clinical observations, indicate that as many as 1 in 5000 people in the North East of England have manifested mitochondrial disease,35 with similar figures reported in other parts of the world.36–38
Identifying and diagnosing mitochondrial genetic disease: general principles
Mitochondrial dysfunction should be considered in the differential diagnosis of any progressive, multisystem, disorder. However, clinical diagnosis can be difficult if patients do not present with ‘classical mitochondrial’ disease (see later).
A detailed family history is important; a clear maternal inheritance (without male transmission) indicates a primary mtDNA defect, whilst an autosomal inheritance pattern is indicative of nDNA interaction. In many cases blood and/or CSF lactate concentration,39 neuroimaging,40,41 cardiac evaluation and muscle biopsy for histological or histochemical evidence can indicate mitochondrial disease. However, establishing a molecular genetic diagnosis is preferred.
Molecular genetic testing can be carried out on DNA extracted from blood (useful for the identification of some mtDNA and nDNA mutations),42,43 but DNA extracted from the affected tissue is preferred (as pathogenic mtDNA mutations are often not detectable in blood).44 Southern blot analysis can be used to identify mtDNA rearrangements and ‘common’ mutations can be targeted by Sanger sequencing of either mtDNA or nDNA.
The genetics of mitochondrial disease
The complex interaction between the two cellular genomes means mitochondrial disease can arise through either (i) a primary mtDNA defect or (ii) a defect in a nuclear-encoded mitochondrial protein.
mtDNA and disease
Understanding mtDNA variation
mtDNA integrity is constantly attacked by mitochondrial reactive oxygen species (ROS) generated during cellular OXPHOS.45 ROS are potent genotoxic agents, which cause mutagenic and cytotoxic effects. The proximity of mtDNA to the site of mitochondrial ROS production (principally complexes I and III of the RC) is the major cause of oxidative lesions and mtDNA instability and is directly responsible for the higher nucleotide instability when compared with nDNA.
Despite being packaged in mitochondrial nucleoids and possessing DNA repair pathways evolved to cope with oxidative damage, including base excision repair mechanisms,46 mtDNA mutation rates are significantly higher than nDNA. Mutation creates two distinct classes of mtDNA variant: (i) single-base-pair variants and (ii) mtDNA rearrangements (deletions and insertions). Single-base-pair variants are typically inheritable and are either common in the populace (as proposed neutral variants) or enriched in individuals with disease (as mtDNA mutations). Understanding the complex nature of mtDNA variation is critical to understanding its affect on disease and there are a few key points that must be understood before assessing an mtDNA variant.
Consequences of mtDNA heteroplasmy
MtDNA heteroplasmy (described earlier) has a complex relationship with disease. The clinical expression of a heteroplasmic pathogenic mtDNA mutation is directly correlatable with the relative proportion of wild-type and mutant genomes.47 For common point mutations, a typical threshold of 80–90% mutant is required to manifest as disease at the cellular level,48,49 and tissue levels correlate loosely with the severity of the clinical phenotype. However, there is emerging evidence that mutation levels can change over time, increasing in post-mitotic tissues, such as brain and muscle and decreasing in mitotic tissues including blood. This can present a challenge when interpreting some clinical molecular genetic tests.44,50,51
Common mtDNA variation
Evolutionarily, common inherited mtDNA mutations have created stable population subgroups separated by common sequence variation known as haplogroups. Many of the major sub-divisions occurred over 10 000 years ago, developing as humans migrated into new geographic areas. Over 95% of Europeans belong to 1 of 10 major haplogroups, H, J, T, U, K (a subgroup of U), M, I, V, W and X, with each haplogroup defined by specific sequence variants within the population.52 These common, inherited, mtDNA variants are usually not heteroplasmic, and due to their selection neutrality have become fixed in the population. However, different haplogroups have been associated with a variety of human diseases, including primary mitochondrial disorders such as Leber's hereditary optic neuropathy (LHON, an age-related loss of vision), where background mitochondrial haplogroup has a direct, functional, effect on the RC protein complex assembly;53 but has expanded to include age-related neurodegenerative disorders such as Parkinson's disease (PD)54 Alzheimer's disease55,56 and age-related macular degeneration.57
Rare mtDNA variation
Rare, inherited, point mutations are a major cause of disease in humans, with an estimated incidence of 1 in 5000.58 They primarily occur in protein coding and tRNA genes and ultimately result in a reduction of cellular energy, through either a reduction in mitochondrial RC enzyme activity or an impairment of mitochondrial protein synthesis.59 Unlike common inherited variants, rare point mutations are often heteroplasmic.
In contrast to point mutations, primary mitochondrial rearrangements of mtDNA are not inheritable; they are primarily, sporadic, large-scale deletions, typically heteroplasmic and usually result in disease. To date around 120 different mtDNA deletions have been identified in patients with mitochondrial disease.60 Similarly to mtDNA point mutations, the ratio of deleted versus ‘wild-type’ molecules is critical to disease aetiology, with mtDNA deletions manifesting disease at a lower heteroplasmic threshold (∼50–60%).61 The exact mechanism of deletion formation is under debate and current research indicates two likely models of deletion formation: (i) replication error and (ii) mtDNA repair inefficiency.62,63
‘Classical’ mtDNA diseases
LHON is a common cause of inherited blindness that typically presents with bilateral, painless, sub-acute visual failure in young adult males. LHON was the first maternally inherited disease to be associated with an mtDNA point mutation.64 Today, clinical diagnosis is usually confirmed by molecular genetic analysis for one of three ‘common’ mtDNA mutations, which all affect genes coding for complex I subunits of the RC: m.3460G>A, m.11778G>A and m14484T>C.65 Mitochondrial dysfunction causes a specific loss of retinal ganglion cells,66 whilst preserving the remaining retinal layers. The optic nerve also shows characteristic degeneration and an accumulation of mitochondria suggesting an impairment of axoplasmic transport. LHON mutations are typically homoplasmic; however, not all patients harbouring a pathogenic LHON mtDNA mutation develop visual failure. Studies of LHON have identified common mtDNA variants that may modulate LHON expression;67,68 additionally environmental factors, such as cigarette smoke69 and oestrogen levels may play a role.70 However, the majority of research has focused on the identification of a nuclear-encoded susceptibility allele.67,71–74
Non-syndromic and aminoglycoside-induced sensorineuronal hearing loss is associated with m.1555A>G, a homoplasmic point mutation in the 12sRNA gene.75 The variant alters a highly conserved region of 12sRNA, mutating the molecule to more closely resemble its bacterial homologue. In vitro experiments on m.1555A>G mutant cell lines demonstrated that exposure to aminoglycoside would impair growth; however, not all symptomatic individuals have been exposed to aminoglycoside.75
Surprisingly, given that they make up only 5% of mtDNA, the vast majority of pathogenic mtDNA point mutations occur in the tRNA genes (Fig. 1).76,77 In addition, pathogenic tRNA mutations are typically heteroplasmic.
Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) is typically a childhood, multisystem disorder. Patients commonly manifest with generalized tonic-clonic seizures, recurrent headaches, anorexia with recurrent vomiting and postlingual hearing loss,78–80 but can manifest with impaired: motor ability, vision and mental acuity due to the cumulative effect of multiple stroke-like episodes. MELAS is commonly (80% of cases) caused by a A>G transition at m.3243 in MTTL1,81 but is also associated with variants in MTND5.82 Biochemically, MELAS manifests as defects of complex I and IV activity; however, care must be taken when interpreting the findings as biochemical results can often appear normal.
Myoclonus epilepsy with ragged red fibres (MERRF) is a neuromuscular disorder primarily caused by m.8344A>G in MTTK.83 Clinically, patients with m.8344A>G present with myoclonus, epilepsy, muscle weakness, cerebellar ataxia and dementia, although neurological symptoms can develop with age.83 Clinical severity is correlated with patient heteroplasmy with high levels of mutant mtDNA often causing, severe complex I or IV deficiency and occasionally a combined complex I and IV deficiency. Much like MELAS, the genotype–phenotype correlation of m.8344A>G can be extended beyond MERRF. M.8344A>G has been identified is diverse mitochondrial phenotypes such as Leigh's syndrome.
m.7472insC, affecting MTTS (Fig. 1), was first identified in a large Italian family presenting with hearing loss, ataxia and myoclonus. This mutation was later found in several unrelated families, all showing a wide clinical spectrum, including isolated hearing loss, ataxia and MERRF. This mutation has been found at increasing frequencies in families presenting with maternally inherited hearing loss.
Pathogenic rearrangements of mtDNA are typically large-scale deletions and to date over 120 different pathogenic mtDNA deletions have been identified.60 As described previously, mtDNA deletions are typically sporadic and not inheritable. Clinical severity is directly correlatable with the level and tissue distribution of the rearrangement and mitochondrial dysfunction is simply a result of the removal of key mitochondrial genes. Homoplasmic tRNA gene loss is particularly detrimental as mitochondria cannot synthesize a functional OXPHOS system. mtDNA deletions are associated with three main clinical phenotypes: Kearns–Sayre syndrome (KSS),84 sporadic progressive external ophthalmoplegia (PEO)85 and Pearson's syndrome.86
KSS is an early onset, sporadic, disorder characterized by PEO and pigmentary retinopathy; however, cases can also present with cerebellar syndrome, heart block, diabetes and shortness of stature. Mitochondrial dysfunction manifests as ragged red fibres (RRFs), an accumulation of dysfunctional mitochondria in the sub-sarcolemmal region of a muscle fibre (detectable when a muscle section is stained with Gomori trichrome stain).85
Large-scale deletions and duplications of mtDNA are a known cause of Pearson's bone-marrow–pancreas syndrome, a rare infant disorder characterized by infantile sideroblastic anaemia and occasionally including severe exocrine pancreatic insufficiency.86
nDNA variation and mitochondrial disease
Nuclear–mitochondrial disease can be classified into four distinct groups: (i) disorders resulting from a reduction in mtDNA stability; (ii) disorders resulting from mutations in nuclear-encoded components or assembly factors of the OXPHOS system; (iii) disorders resulting from mutations affecting mitochondrial translation and (iv) disorders due to defects in genes controlling mitochondrial network dynamics.
Disorders resulting from a reduction in mtDNA stability
A growing number of disorders have become associated with mtDNA instability, primarily a result of impaired mtDNA replication. Mutations in POLG, the gene encoding the only mtDNA polymerase, are by far the commonest cause of mtDNA stability disorders. Mutations in the POLG gene can cause either point mutations (through impaired mtDNA proofreading) or deletions (through impaired polymerase activity) in mtDNA.19 The first pathogenic mutations in POLG were identified in families with autosomal dominant PEO (adPEO); however, the spectrum of disease associated with POLG mutations has been expanded to include autosomal recessive PEO, adult onset ataxia, Alpers' syndrome, parkinsonism and premature ovarian failure.87
adPEO, characterized by multiple mtDNA deletions, is caused by mutations in PEO1, which encodes ‘twinkle’ the putative mitochondrial helicase.88 It is thought that twinkle mutations result in an accumulation of replication intermediates, causing replication stalling and eventually depletion. adPEO is also associated with mutations in ANT1,89 the gene coding adenine nucleotide translocase. Mutations in ANT1 impair ADP–ATP exchange through the mitochondrial membrane, causing a nucleotide imbalance (affecting replication) and a severe reduction in cellular energy.
In addition to structurally altering mtDNA, several disorders have been identified that are caused by a reduction in mtDNA copy number.19 Alpers syndrome, characterized by diffuse and progressive cerebral atrophy,90 has been associated with mutations in POLG,91,92 which cause impairment of the replicative machinery.93
Recessive mutations in thymidine phosphorylase cause mitochondrial neurogastrointestinal encephalopathy, characterized by mtDNA depletion, multiple deletions and point mutations. mtDNA depletion has also been identified in early onset hypotonia with myopathy and hepatic involvement, caused by mutations in either thymidine kinase (TK2) or deoxyguanosine kinase (DGUOK).94 Mutations in both of these genes cause a reduction in the mtDNA nucleotide pooling, reducing replication efficiency.
Disorders resulting from mutations in nuclear-encoded components or assembly factors of the OXPHOS system
Isolated complex I deficiency is by far the commonest biochemical defect found in mitochondrial disorders; however, it is also the most complex aetiology and clinical spectrum.95 Complex I deficiency is associated with a broad range of clinical phenotypes ranging from lethal neonatal disease to adult onset neurodegenerative disorders.96,97 A high level of genetic heterogeneity, coupled with weak genotype–phenotype correlations, makes it difficult to predict the genetic basis on pure clinical grounds.95 This is important because of the different inheritance patterns and different natural histories of the different genetic causes. However, some patterns are starting to emerge.
There are at least 46 nuclear-encoded subunits of complex I (compared with 7 mtDNA encoded subunits) and so it is unsurprising that nDNA mutations have been identified in 14 of the structural subunits. Pathogenic mutations in NDUFS1,98 NDUFS3,95,99 NDUFS4,100 NDUFS7,101 NDUFS8,102 NDUFV1,98,103 NDUFA10,104 NDUFB395 and NDUFA2105 typically manifest as Leigh or Leigh-like syndromes.60,106 Conversely, mutations in NDUFS2,107 NDUFS6,108 NDUFV2,109 NDUFA1, NDUFA11110 and ACAD9111 are typically associated with hypertrophic cardiomyopathy and encephalopathy. In addition, mutations in complex I assembly proteins can manifest as disease: Leigh syndrome (NDUFAF2 and NDUFAF5),112,113 encephalopathy (NDUFAF4)114 and cardioencephalomyopathy (NDUFAF1).115
Complex II is completely encoded by nDNA and is composed of four polypeptide subunits: SHD-A, -B, -C and -D. Mutations in SHD-A are rare, but are associated with Leigh's syndrome. Surprisingly, mutations in SHD-B, -C and -D appear to be a common cause of inherited paragagliomas and phaeochromocytomas.116
Complex III deficiency typically causes a severe multisystem early onset disorder, which is recessively inherited and rare.117,118 identified mutations in BCS1l, a complex III assembly protein, presenting with neonatal proximal tubulopathy, hepatic involvement and encephalopathy. Subsequently, a deletion in human ubiquinone–cytochrome c reductase binding protein of complex III (UQCRB) was identified in a consanguineous family presenting with hypoglycaemia and lactic acidosis;119 and a missense mutation was identified in UQCRC, a ubiquinone-binding protein, in a large consanguineous Israeli-Bedoiun kindred.120 More recently, a mutation in TTC19 (a complex III structural subunit gene) was identified in individuals with a progressive neurodegenerative disorder in late infancy,121 expanding the phenotype of complex mutations beyond early infant disorders.
Mutations in complex IV result in severe, typically fatal, infantile disease and to date mutations in four complex IV structural subunits have been identified. A homozygous mutation in COX6BI, identified in brothers from a consanguineous Saudi Arabian family, presented with gait instabilities visual disturbances, progressive neurological deterioration and leukodystrophic brain changes.122 Mutations in COX10, a homologue of yeast haem A:farneslytransferase, are associated with Leigh syndrome123,124 and a multisystem disorder.123 Atypically, mutations in COX7B125 are associated with facial dysmorphisms and congenital abnormalities,126 and a single mutation in the structural subunit gene, COX4I2, was identified in adult Arab Muslim patients with exocrine pancreatic insufficiency, dyserythropoietic anaemia and calvarial hyperostosis.127
In contrast, a number of mutations have been identified in complex IV assembly factors. Complex IV assembly gene disorders include SURF1 (Surfeit locus protein 1), associated with Leigh Syndrome;128,129 C12ORF62 (chromosome 12 open reading frame 62), associated with fatal, neonatal, mitochondrial IV deficiency;130 COA5 (cytochrome c oxidase assembly factor 5), associated with neonatal hypertrophic cardiomyopathy131 and FASTKD2, associated with cytochrome c oxidase-defective encephalomyopathy.132
Mutations in nDNA-encoded complex V subunit genes also appear very rare. A mutation in ATP5E (ATP synthase, H+ transporting, mitochondrial F1 complex, epsilon subunit) was identified in an Austrian woman with complex V deficiency,133 and a single gene defect has been identified in the complex V assembly factor gene ATPAF2, resulting in impaired complex V activity.134
Disorders resulting from mutations affecting mitochondrial translation
Several nDNA mutations have been identified which influence the efficiency of mitochondrial translation. Mitochondrial ribosomal protein S16 (MRPS16) and mitochondrial ribosomal protein S22 (MRPS22) are components of the mitoribosome. Mutations in these genes are known to cause severe, infantile, lactic acidosis, developmental defects in the brain, and facial dysmorphisms (MRPS16) and fatal neonatal hypertrophic cardiomyopathy and kidney tubulopathy (MRPS22).135
Mutations in PUS1, peudorine synthase 1, have been shown to cause myopathy, lactic acidosis and sideroblastic anaemia.136 The mutation, in the catalytic core of the protein, is thought to disrupt the conversion of uridine to pseudouridine, required for tRNA synthesis.
Disorders due to defects in genes controlling mitochondrial network dynamics
Mutations in OPA1 are primarily a cause of optic atrophy,66 but additional phenotypes, such as deafness and neuromuscular disease, have also been seen. Interestingly, mutations in OPA1 also appear to cause the formation of mtDNA deletions, indicating that Opa1 is also important to mtDNA maintenance.
Much like OPA1, defects in MFN2 cause a disturbance of mtDNA maintenance through impairment of mitochondrial network dynamics.66 Mutations in MFN2 are typically associated with Charcot-Marie-Tooth disease (CMT2A) and hereditary motor and sensory neuropathy (CMT with HMSN type VI).66
DNM1L (dynamin 1-like), another GTPase, is required for fission of mitochondria.137 To date, only a single DNM1L has been identified in an infant presenting with both defective mitochondrial and peroxisomal fission.138 The patient presented in the first days of life with severe microcephaly, abnormal brain development, optic atrophy with hyperplasia and lactic acidemia.138
Areas of controversy?
The mitochondrial bottleneck
Mutations in mtDNA are often heteroplasmic, with severity correlating with increasing percentage of mutant. Observations indicate that the amount of a variant inherited from a heteroplasmic mother varies between offspring.139,140 This is important when investigating disease aetiology, as an asymptomatic mother, with a sub-clinical heteroplasmy level, can give birth to children with significantly higher levels of an mtDNA mutation.
The ‘mitochondrial bottleneck theory’ attempts to explain this phenomenon.140 Briefly, the reduction of mtDNA during early development ‘redistributes’ mtDNA to daughter cells (effectively sharing mtDNA content amongst daughter cells). Oocyte maturation is associated with the rapid replication of mtDNA. This reduction-amplification leads to a purportedly random shift in mtDNA mutational load between cells. Researchers agree that the bottleneck is due to a rapid reduction in mtDNA levels during embryonic development; however, the exact mechanism of segregation is hotly debated. There are currently three leading theories of the mtDNA bottleneck mechanism:140 (i) variation in heteroplasmy is due to an unequal segregation of mtDNA during cell division, (ii) variation in heteroplasmy is due to an unequal segregation of mtDNA nucleoids during cell division and (iii) variation in heteroplasmy is due to the selective replication of a specific sub-population of mtDNA.
Growing points
Assigning variant causality
Optimal mitochondrial function requires the synergistic cooperation of both mtDNA and nDNA; hence, the investigation of dysfunction requires the interrogation of both genomes. Correctly determining the pathogenicity of potential mutants (in either genome) is critical to understanding mitochondrial disease. This underpins the genetic counselling and subsequent prenatal diagnosis of mitochondrial disorders.
Despite the complexity of both mtDNA point mutations and deletions, as well as the potential for heteroplasmy, assigning pathogenicity to mtDNA variants is analogous to nDNA mutations and is comprehensively described by DiMauro and Schon.141 Briefly, the mutation must be present in cases significantly more than asymptomatic controls; if heteroplasmic, the proportion of mutated mtDNA must be higher in patients compared with controls (and subsequently higher in clinically affected tissues compared with unaffected tissues). More importantly, the mutated mtDNA must segregate with defined clinical outcome (described previously). Other criteria, such as evolutionary conservation must be interpreted with care, as very rare neutral variants (so-called ‘private polymorphisms’) or homoplasmic changes (such as in LHON) may be wrongly miss-classified using this approach.141 Assigning pathogenicity to tRNA mutations is slightly more challenging; tRNA variants are common; however, a small number of tRNA mutations are responsible for a disproportionate majority of mitochondrial disease.77 McFarland et al.77 provide a comprehensive scoring system which can be used to accurately determine tRNA mutation pathogenicity.
Whole-exome sequencing (WES)142 has emerged as the preferred method for identifying Mendelian disease genes, and is proving valuable in the diagnostic evaluation of phenotypically and genetically heterogeneous disorders such as mitochondrial disease.95,143 Initially, candidate mutations can be identified by prioritizing known mitochondrial genes, such as the 1500 proposed in ‘MitoCarta’144 or Mitop2.145 Secondly, WES can drive the discovery of novel mitochondrial disease genes or provide a link to previous disease genes that demonstrate an overlapping clinical phenotype.146–151 However, as with all new technologies, care must be taken when interpreting WES data in novel disease genes. Variants identified in poorly characterized genes will require extensive biochemical and functional laboratory analysis to assign causality. Additionally, WES is not wholly comprehensive, not capturing non-coding or regulatory regions and often failing to sequence large portions of the exome.142,152 However, as technology improves and bioinformatic analysis becomes streamlined, WES is likely to become a major facet in indentifying nuclear genes that affect mitochondrial function.
Managing mitochondrial disease
There are limited treatment options for patients with mitochondrial diseases. The main emphasis is on disease prevention and the management of complications. Effective genetic counselling, especially given a family history of mitochondrial disease, is crucial. However, the clinical variability, coupled with the unpredictable inheritance of a heteroplasmic ‘mutant dose’ (through the bottleneck), makes a definite diagnosis difficult.153,154
Empiric recurrence risks are available for common homoplasmic mutations (i.e. for LHON), but genetic counselling for heteroplasmic mutations is difficult because of the genetic bottleneck (described earlier). Increased knowledge of the natural history of specific mitochondrial disorders has informed clinical practice. Particular attention to cardiac, ophthalmological and endocrine complications (especially diabetes), can lead to prompt supportive management.155 However, there are no specific disease-modifying treatments at present, although some drugs show promise.156
An area that has had some in vitro and pre-clinical success is the development of ‘gene therapies’.157 There are currently three strategies for applying gene therapy to mitochondrial disease: (i) the rescue of an RC defect by expression of a ‘replacement’ gene product from the nucleus (so-called allotopic and xenotpoic expression,158,159 (ii) the rescue of a primary mitochondrial defect by importing ‘wild-type’ mtDNA into mitochondria (so-called mtDNA transfection) and (iii) manipulation of the heteroplasmic mtDNA balance (i.e. adjusting the wild-type:mutant type ratio), which can be achieved by improving a patients exercise regime.160
More recently, and although in very early stages, allogenic haematopoietic stem cell therapy has been successfully used to treat mitochondrial neurogastrointestinal encephalomyopathy, but associated with high mortality.161 Similarly, liver transplants in patients (typically children) suffering from MPV17-associated hepatocerebral mitochondrial depletion syndrome have a poor prognosis.162
Pre-implantation genetic diagnosis can assist female heteroplasmic mtDNA mutation carriers in determining the risk to their offspring, assisting by preventing transmission of deleterious mtDNA.163,164 Briefly, embryos obtained after in vitro fertilization are analysed and only those with very low-level mutant levels are transferred to the uterus. However, these techniques are of little help to woman harbouring intermediate-level heteroplasmic mtDNA mutations, where uncertainty regarding the clinical mutation threshold remains.163
Advances, harnessing ‘pro-nuclear transfer’, have made significant steps towards treating primary mitochondrial disease at a mtDNA level.165 Briefly, the technique involves the transfer of nDNA from a donor zygote (from the mtDNA mutation carrier mother) to an enucleated recipient zygote via fusion. The new ‘reconstructed zygote’ retains the nDNA from the mother, but the mtDNA from a donor. More recently, a competing group has attempted a similar technique, utilizing ‘spindle transfer’ of nDNA to an enucleated donor.166 Unlike pro-nuclear transfer, nDNA isolation occurs pre-fertilization, meaning once the technique is approved it can be integrated into established in vitro fertilization techniques. However, caution is advised, as both pro-nuclear transfer and spindle transfer would only benefit a minority of female mtDNA mutation carriers, whereas prenatal diagnostic testing can be utilized for both all Mendelian mitochondrial disorders and the majority of mtDNA mutations.163,167
Funding
Funding to pay the Open Access publication charges for this article was provided by The Wellcome Trust.
References
- 1.van der Giezen M, Tovar J. Degenerate mitochondria. EMBO Rep. 2005;6:525–30. doi: 10.1038/sj.embor.7400440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012;13:878–90. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schapira AH. Mitochondrial diseases. Lancet. 2012;379:1825–34. doi: 10.1016/S0140-6736(11)61305-6. [DOI] [PubMed] [Google Scholar]
- 4.Koopman WJ, Distelmaier F, Smeitink JA, et al. OXPHOS mutations and neurodegeneration. EMBO J. 2013;32:9–29. doi: 10.1038/emboj.2012.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andrews RM, Kubacka I, Chinnery PF, et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 6.Temperley R, Richter R, Dennerlein S, et al. Hungry codons promote frameshifting in human mitochondrial ribosomes. Science. 2010;327:301. doi: 10.1126/science.1180674. [DOI] [PubMed] [Google Scholar]
- 7.Sutovsky P. Ubiquitin-dependent proteolysis in mammalian spermatogenesis, fertilization, and sperm quality control: killing three birds with one stone. Microsc Res Tech. 2003;61:88–102. doi: 10.1002/jemt.10319. [DOI] [PubMed] [Google Scholar]
- 8.Vissing JSaM. Paternal Inheritance if mitochondrial DNA. N Engl J Med. 2002:2081–2. [PubMed] [Google Scholar]
- 9.Miller FJ, Rosenfeldt FL, Zhang C, et al. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: lack of change of copy number with age. Nucleic Acids Res. 2003;31:e61. doi: 10.1093/nar/gng060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birky CW., Jr The inheritance of genes in mitochondria and chloroplasts: laws, mechanisms, and models. Annu Rev Genet. 2001;35:125–48. doi: 10.1146/annurev.genet.35.102401.090231. [DOI] [PubMed] [Google Scholar]
- 11.Payne BA, Wilson IJ, Yu-Wai-Man P, et al. Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet. 2013;22:384–90. doi: 10.1093/hmg/dds435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calvo S, Jain M, Xie X, et al. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat Genet. 2006;38:576–82. doi: 10.1038/ng1776. [DOI] [PubMed] [Google Scholar]
- 13.Hirst J. Why does mitochondrial complex I have so many subunits? Biochem J. 2011;437:e1–3. doi: 10.1042/BJ20110918. [DOI] [PubMed] [Google Scholar]
- 14.Angerer H, Zwicker K, Wumaier Z, et al. A scaffold of accessory subunits links the peripheral arm and the distal proton-pumping module of mitochondrial complex I. Biochem J. 2011;437:279–88. doi: 10.1042/BJ20110359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith PM, Fox JL, Winge DR. Biogenesis of the cytochrome bc(1) complex and role of assembly factors. Biochim Biophys Acta. 2012;1817:276–86. doi: 10.1016/j.bbabio.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogel RO, Smeitink JA, Nijtmans LG. Human mitochondrial complex I assembly: a dynamic and versatile process. Biochim Biophys Acta. 2007;1767:1215–27. doi: 10.1016/j.bbabio.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Mick DU, Fox TD, Rehling P. Inventory control: cytochrome c oxidase assembly regulates mitochondrial translation. Nat Rev Mol Cell Biol. 2011;12:14–20. doi: 10.1038/nrm3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang ZG, White PS, Ackerman SH. Atp11p and Atp12p are assembly factors for the F(1)-ATPase in human mitochondria. J Biol Chem. 2001;276:30773–8. doi: 10.1074/jbc.M104133200. [DOI] [PubMed] [Google Scholar]
- 19.Copeland WC. Defects in mitochondrial DNA replication and human disease. Crit Rev Biochem Mol Biol. 2012;47:64–74. doi: 10.3109/10409238.2011.632763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bogenhagen DF. Mitochondrial DNA nucleoid structure. Biochim Biophys Acta. 2012;1819:914–20. doi: 10.1016/j.bbagrm.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Kukat C, Wurm CA, Spahr H, et al. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci USA. 2011;108:13534–9. doi: 10.1073/pnas.1109263108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCulloch V, Seidel-Rogol BL, Shadel GS. A human mitochondrial transcription factor is related to RNA adenine methyltransferases and binds S-adenosylmethionine. Mol Cell Biol. 2002;22:1116–25. doi: 10.1128/MCB.22.4.1116-1125.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCulloch V, Shadel GS. Human mitochondrial transcription factor B1 interacts with the C-terminal activation region of h-mtTFA and stimulates transcription independently of its RNA methyltransferase activity. Mol Cell Biol. 2003;23:5816–24. doi: 10.1128/MCB.23.16.5816-5824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Metodiev MD, Lesko N, Park CB, et al. Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab. 2009;9:386–97. doi: 10.1016/j.cmet.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Gaspari M, Larsson NG, Gustafsson CM. The transcription machinery in mammalian mitochondria. Biochim Biophys Acta. 2004;1659:148–52. doi: 10.1016/j.bbabio.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Ma J, Farwell MA, Burkhart WA, et al. Cloning and sequence analysis of the cDNA for bovine mitochondrial translational initiation factor 2. Biochim Biophys Acta. 1995;1261:321–4. doi: 10.1016/0167-4781(95)00041-e. [DOI] [PubMed] [Google Scholar]
- 27.Koc EC, Spremulli LL. Identification of mammalian mitochondrial translational initiation factor 3 and examination of its role in initiation complex formation with natural mRNAs. J Biol Chem. 2002;277:35541–9. doi: 10.1074/jbc.M202498200. [DOI] [PubMed] [Google Scholar]
- 28.Christian B, Haque E, Spremulli L. Ribosome shifting or splitting: it is all up to the EF-G. Mol Cell. 2009;35:400–2. doi: 10.1016/j.molcel.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 29.Hammarsund M, Wilson W, Corcoran M, et al. Identification and characterization of two novel human mitochondrial elongation factor genes, hEFG2 and hEFG1, phylogenetically conserved through evolution. Hum Genet. 2001;109:542–50. doi: 10.1007/s00439-001-0610-5. [DOI] [PubMed] [Google Scholar]
- 30.Ling M, Merante F, Chen HS, et al. The human mitochondrial elongation factor tu (EF-Tu) gene: cDNA sequence, genomic localization, genomic structure, and identification of a pseudogene. Gene. 1997;197:325–36. doi: 10.1016/s0378-1119(97)00279-5. [DOI] [PubMed] [Google Scholar]
- 31.Smits P, Smeitink J, van den Heuvel L. Mitochondrial translation and beyond: processes implicated in combined oxidative phosphorylation deficiencies. J Biomed Biotechnol. 2010;2010:737385. doi: 10.1155/2010/737385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Spremulli LL. Identification and cloning of human mitochondrial translational release factor 1 and the ribosome recycling factor. Biochim Biophys Acta. 1998;1443:245–50. doi: 10.1016/s0167-4781(98)00223-1. [DOI] [PubMed] [Google Scholar]
- 33.Soleimanpour-Lichaei HR, Kuhl I, Gaisne M, et al. mtRF1a is a human mitochondrial translation release factor decoding the major termination codons UAA and UAG. Mol Cell. 2007;27:745–57. doi: 10.1016/j.molcel.2007.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–5. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schaefer AM, McFarland R, Blakely EL, et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35–9. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- 36.Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126(Pt 8):1905–12. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- 37.Majamaa K, Moilanen JS, Uimonen S, et al. Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: prevalence of the mutation in an adult population. Am J Hum Genet. 1998;63:447–54. doi: 10.1086/301959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Darin N, Oldfors A, Moslemi AR, et al. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA anbormalities. Ann Neurol. 2001;49:377–83. [PubMed] [Google Scholar]
- 39.Magner M, Szentivanyi K, Svandova I, et al. Elevated CSF-lactate is a reliable marker of mitochondrial disorders in children even after brief seizures. Eur J Paediatr Neurol. 2011;15:101–8. doi: 10.1016/j.ejpn.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 40.Barkovich AJ, Good WV, Koch TK, et al. Mitochondrial disorders: analysis of their clinical and imaging characteristics. AJNR Am J Neuroradiol. 1993;14:1119–37. [PMC free article] [PubMed] [Google Scholar]
- 41.Lin DD, Crawford TO, Barker PB. Proton MR spectroscopy in the diagnostic evaluation of suspected mitochondrial disease. AJNR Am J Neuroradiol. 2003;24:33–41. [PMC free article] [PubMed] [Google Scholar]
- 42.Leonard JV, Schapira AH. Mitochondrial respiratory chain disorders I: mitochondrial DNA defects. Lancet. 2000;355:299–304. doi: 10.1016/s0140-6736(99)05225-3. [DOI] [PubMed] [Google Scholar]
- 43.Leonard JV, Schapira AH. Mitochondrial respiratory chain disorders II: neurodegenerative disorders and nuclear gene defects. Lancet. 2000;355:389–94. doi: 10.1016/s0140-6736(99)05226-5. [DOI] [PubMed] [Google Scholar]
- 44.Rahman S, Poulton J, Marchington D, et al. Decrease of 3243 A–>G mtDNA mutation from blood in MELAS syndrome: a longitudinal study. Am J Hum Genet. 2001;68:238–40. doi: 10.1086/316930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–7. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 46.Hegde ML, Mantha AK, Hazra TK, et al. Oxidative genome damage and its repair: implications in aging and neurodegenerative diseases. Mech Ageing Dev. 2012;133:157–68. doi: 10.1016/j.mad.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Howell N. Origin, cellular expression, and cybrid transmission of mitochondrial CAP-R, PYR-IND, and OLI-R mutant phenotypes. Somat Cell Genet. 1983;9:1–24. doi: 10.1007/BF01544045. [DOI] [PubMed] [Google Scholar]
- 48.Chinnery PF, Howell N, Lightowlers RN, et al. Molecular pathology of MELAS and MERRF. The relationship between mutation load and clinical phenotypes. Brain. 1997;120:1713–21. doi: 10.1093/brain/120.10.1713. [DOI] [PubMed] [Google Scholar]
- 49.White SL, Collins VR, Wolfe R, et al. Genetic counseling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am J Hum Genet. 1999;65:474–82. doi: 10.1086/302488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weber K, Wilson JN, Taylor L, et al. A new mtDNA mutation showing accumulation with time and restriction to skeletal muscle. Am J Hum Genet. 1997;60:373–80. [PMC free article] [PubMed] [Google Scholar]
- 51.Poulton J, O'Rahilly S, Morten KJ, et al. Mitochondrial DNA, diabetes and pancreatic pathology in Kearns-Sayre syndrome. Diabetologia. 1995;38:868–71. doi: 10.1007/s001250050366. [DOI] [PubMed] [Google Scholar]
- 52.Torroni A, Huoponen K, Francalacci P, et al. Classification of European mtDNAs from an analysis of three European populations. Genetics. 1996;144:1835–50. doi: 10.1093/genetics/144.4.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pello R, Martin MA, Carelli V, et al. Mitochondrial DNA background modulates the assembly kinetics of OXPHOS complexes in a cellular model of mitochondrial disease. Hum Mol Genet. 2008;17:4001–11. doi: 10.1093/hmg/ddn303. [DOI] [PubMed] [Google Scholar]
- 54.Pyle A, Foltynie T, Tiangyou W, et al. Mitochondrial DNA haplogroup cluster UKJT reduces the risk of PD. Ann Neurol. 2005;57:564–7. doi: 10.1002/ana.20417. [DOI] [PubMed] [Google Scholar]
- 55.Santoro A, Balbi V, Balducci E, et al. Evidence for sub-haplogroup h5 of mitochondrial DNA as a risk factor for late onset Alzheimer's disease. PLoS One. 2010;5:e12037. doi: 10.1371/journal.pone.0012037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ridge PG, Maxwell TJ, Corcoran CD, et al. Mitochondrial genomic analysis of late onset Alzheimer's disease reveals protective haplogroups H6A1A/H6A1B: the Cache County Study on Memory in Aging. PLoS One. 2012;7:e45134. doi: 10.1371/journal.pone.0045134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Udar N, Atilano SR, Memarzadeh M, et al. Mitochondrial DNA haplogroups associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:2966–74. doi: 10.1167/iovs.08-2646. [DOI] [PubMed] [Google Scholar]
- 58.Chinnery PF, Elliott HR, Hudson G, et al. Epigenetics, epidemiology and mitochondrial DNA diseases. Int J Epidemiol. 2012;41:177–87. doi: 10.1093/ije/dyr232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ghezzi D, Zeviani M. Assembly factors of human mitochondrial respiratory chain complexes: physiology and pathophysiology. Adv Exp Med Biol. 2012;748:65–106. doi: 10.1007/978-1-4614-3573-0_4. [DOI] [PubMed] [Google Scholar]
- 60.MITOMAP. A human mitochondrial genome database. 2000 doi: 10.1093/nar/24.1.177. http://www.gen.emory.edu/mitomap.html . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rossignol R, Faustin B, Rocher C, et al. Mitochondrial threshold effects. Biochem J. 2003;370(Pt 3):751–62. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schon EA, Rizzuto R, Moraes CT, et al. A direct repeat is a hotspot for large-scale deletion of human mitochondrial DNA. Science. 1989;244:346–9. doi: 10.1126/science.2711184. [DOI] [PubMed] [Google Scholar]
- 63.Krishnan KJ, Reeve AK, Samuels DC, et al. What causes mitochondrial DNA deletions in human cells? Nat Genet. 2008;40:275–9. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- 64.Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242:1427–30. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 65.Carelli V, Ghelli A, Ratta M, et al. Leber's hereditary optic neuropathy: biochemical effect of 11778/ND4 and 3460/ND1 mutations and correlation with the mitochondrial genotype. Neurology. 1997;48:1623–32. doi: 10.1212/wnl.48.6.1623. [DOI] [PubMed] [Google Scholar]
- 66.Carelli V, La Morgia C, Valentino ML, et al. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta. 2009;1787:518–28. doi: 10.1016/j.bbabio.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 67.Hudson G, Keers S, Yu Wai Man P, et al. Identification of an X-chromosomal locus and haplotype modulating the phenotype of a mitochondrial DNA disorder. Am J Hum Genet. 2005;77:1086–91. doi: 10.1086/498176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Achilli A, Iommarini L, Olivieri A, et al. Rare primary mitochondrial DNA mutations and probable synergistic variants in Leber's hereditary optic neuropathy. PLoS One. 2012;7:e42242. doi: 10.1371/journal.pone.0042242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kirkman MA, Yu-Wai-Man P, Korsten A, et al. Gene-environment interactions in Leber hereditary optic neuropathy. Brain. 2009;132(Pt 9):2317–26. doi: 10.1093/brain/awp158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hudson G, Carelli V, Spruijt L, et al. Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am J Hum Genet. 2007;81:228–33. doi: 10.1086/519394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Carvalho MR, Muller B, Rotzer E, et al. Leber's hereditary optic neuroretinopathy and the X-chromosomal susceptability factor: no linkage to DXS7. Hum Hered. 1992;42:316–20. doi: 10.1159/000154089. [DOI] [PubMed] [Google Scholar]
- 72.Chen JD, Cox I, Denton MJ. Prelimanary exclusion of an X-linked gene in Leber optic atrophy by linkage analysis. Hum Genet. 1989;82:302–7. doi: 10.1007/BF00291154. [DOI] [PubMed] [Google Scholar]
- 73.Abu-Amero K, Jaber M, Hellani A, et al. Genome-wide expression profile of LHON patients with the 11778 mutation. Br J Ophthalmol. 2009 doi: 10.1136/bjo.2009.165571. 2009/09/04 ed. [DOI] [PubMed] [Google Scholar]
- 74.Phasukkijwatana N, Kunhapan B, Stankovich J, et al. Genome-wide linkage scan and association study of PARL to the expression of LHON families in Thailand. Hum Genet. doi: 10.1007/s00439-010-0821-8. 2010/04/22 ed. [DOI] [PubMed] [Google Scholar]
- 75.Prezant TR, Agapian JV, Bohlman MC, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet. 1993;4:289–94. doi: 10.1038/ng0793-289. [DOI] [PubMed] [Google Scholar]
- 76.Zifa E, Giannouli S, Theotokis P, et al. Mitochondrial tRNA mutations: clinical and functional perturbations. RNA Biol. 2007;4:38–66. doi: 10.4161/rna.4.1.4548. [DOI] [PubMed] [Google Scholar]
- 77.McFarland R, Elson JL, Taylor RW, et al. Assigning pathogenicity to mitochondrial tRNA mutations: when ‘definitely maybe’ is not good enough. Trends Genet. 2004;20:591–6. doi: 10.1016/j.tig.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 78.Pavlakis SG, Phillips PC, DiMauro S, et al. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol. 1984;16:481–8. doi: 10.1002/ana.410160409. [DOI] [PubMed] [Google Scholar]
- 79.Hirano M, Ricci E, Koenigsberger MR, et al. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord. 1992;2:125–35. doi: 10.1016/0960-8966(92)90045-8. [DOI] [PubMed] [Google Scholar]
- 80.Manwaring N, Jones MM, Wang JJ, et al. Population prevalence of the MELAS A3243G mutation. Mitochondrion. 2007;7:230–3. doi: 10.1016/j.mito.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 81.Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651–3. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- 82.Santorelli FM, Tanji K, Kulikova R, et al. Identification of a novel mutation in the mtDNA ND5 gene associated with MELAS. Biochem Biophys Res Commun. 1997;238:326–8. doi: 10.1006/bbrc.1997.7167. [DOI] [PubMed] [Google Scholar]
- 83.Shoffner JM, Lott MT, Lezza AM, et al. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell. 1990;61:931–7. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- 84.Zeviani M, Moraes CT, DiMauro S, et al. Deletions of mitochondrial DNA in Kearns-Sayre syndorme. Neurology. 1988;38:1339–46. doi: 10.1212/wnl.38.9.1339. [DOI] [PubMed] [Google Scholar]
- 85.Moraes CT, DiMauro S, Zeviani M, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med. 1989;320:1293–9. doi: 10.1056/NEJM198905183202001. [DOI] [PubMed] [Google Scholar]
- 86.Rotig A, Cormier V, Blanche S, et al. Pearson's marrow-pancreas syndrome. A multisystem mitochondrial disorder in infancy. J Clin Invest. 1990;86:1601–8. doi: 10.1172/JCI114881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Horvath R, Hudson G, Ferrari G, et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain. 2006;129(Pt 7):1674–84. doi: 10.1093/brain/awl088. [DOI] [PubMed] [Google Scholar]
- 88.Spelbrink JN, Li FY, Tiranti V, et al. Human mitochondrial DNA deletios associated with mutations in the gene encoding Twinkle, a phage T7 gene4-like protein localized in mitochondria. Nat Genet. 2001;28:223–31. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- 89.Agostino A, Valletta L, Chinnery PF, et al. Mutations of ANT1, Twinkle, and POLG1 in sporadic progressive external ophthalmoplegia (PEO) Neurology. 2003;60:1354–6. doi: 10.1212/01.wnl.0000056088.09408.3c. [DOI] [PubMed] [Google Scholar]
- 90.Wolf A, Cowen D. The cerebral atrophies and encephalomalacias of infancy and childhood. Res Publ Assoc Res Nerv Ment Dis. 1955;34:199–330. [PubMed] [Google Scholar]
- 91.Kollberg G, Moslemi AR, Darin N, et al. POLG1 mutations associated with progressive encephalopathy in childhood. J Neuropathol Exp Neurol. 2006;65:758–68. doi: 10.1097/01.jnen.0000229987.17548.6e. [DOI] [PubMed] [Google Scholar]
- 92.Naviaux RK, Nguyen KV. POLG mutations associated with Alpers' syndrome and mitochondrial DNA depletion. Ann Neurol. 2004;55:706–12. doi: 10.1002/ana.20079. [DOI] [PubMed] [Google Scholar]
- 93.Clayton DA. Replication of animal mitochondrial DNA. Cell. 1982;28:693–705. doi: 10.1016/0092-8674(82)90049-6. [DOI] [PubMed] [Google Scholar]
- 94.Spinazzola A, Zeviani M. Disorders of nuclear-mitochondrial intergenomic communication. Biosci Rep. 2007;27:39–51. doi: 10.1007/s10540-007-9036-1. [DOI] [PubMed] [Google Scholar]
- 95.Haack TB, Haberberger B, Frisch EM, et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J Med Genet. 2012;49:277–83. doi: 10.1136/jmedgenet-2012-100846. [DOI] [PubMed] [Google Scholar]
- 96.Loeffen JL, Smeitink JA, Trijbels JM, et al. Isolated complex I deficiency in children: clinical, biochemical and genetic aspects. Hum Mutat. 2000;15:123–34. doi: 10.1002/(SICI)1098-1004(200002)15:2<123::AID-HUMU1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 97.Lebre AS, Rio M, Faivre d'Arcier L, et al. A common pattern of brain MRI imaging in mitochondrial diseases with complex I deficiency. J Med Genet. 2011;48:16–23. doi: 10.1136/jmg.2010.079624. [DOI] [PubMed] [Google Scholar]
- 98.Benit P, Chretien D, Kadhom N, et al. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am J Hum Genet. 2001;68:1344–52. doi: 10.1086/320603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Benit P, Slama A, Cartault F, et al. Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome. J Med Genet. 2004;41:14–7. doi: 10.1136/jmg.2003.014316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.van den Heuvel L, Ruitenbeek W, Smeets R, et al. Demonstration of a new pathogenic mutation in human complex I deficiency: a 5-bp duplication in the nuclear gene encoding the 18-kD (AQDQ) subunit. Am J Hum Genet. 1998;62:262–8. doi: 10.1086/301716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Smeitink J, van den Heuvel L. Human mitochondrial complex I in health and disease. Am J Hum Genet. 1999;64:1505–10. doi: 10.1086/302432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Loeffen J, Smeitink J, Triepels R, et al. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am J Hum Genet. 1998;63:1598–608. doi: 10.1086/302154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schuelke M, Smeitink J, Mariman E, et al. Mutant NDUFV1 subunit of mitochondrial complex I causes leukodystrophy and myoclonic epilepsy. Nat Genet. 1999;21:260–1. doi: 10.1038/6772. [DOI] [PubMed] [Google Scholar]
- 104.Hoefs SJ, van Spronsen FJ, Lenssen EW, et al. NDUFA10 mutations cause complex I deficiency in a patient with Leigh disease. Eur J Hum Genet. 2011;19:270–4. doi: 10.1038/ejhg.2010.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hoefs SJ, Dieteren CE, Distelmaier F, et al. NDUFA2 complex I mutation leads to Leigh disease. Am J Hum Genet. 2008;82:1306–15. doi: 10.1016/j.ajhg.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shoubridge EA. Nuclear genetic defects of oxidative phosphorylation. Hum Mol Genet. 2001;10:2277–84. doi: 10.1093/hmg/10.20.2277. [DOI] [PubMed] [Google Scholar]
- 107.Loeffen J, Elpeleg O, Smeitink J, et al. Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann Neurol. 2001;49:195–201. doi: 10.1002/1531-8249(20010201)49:2<195::aid-ana39>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 108.Kirby DM, Salemi R, Sugiana C, et al. NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I deficiency. J Clin Invest. 2004;114:837–45. doi: 10.1172/JCI20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Benit P, Beugnot R, Chretien D, et al. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum Mutat. 2003;21:582–6. doi: 10.1002/humu.10225. [DOI] [PubMed] [Google Scholar]
- 110.Berger I, Hershkovitz E, Shaag A, et al. Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann Neurol. 2008;63:405–8. doi: 10.1002/ana.21332. [DOI] [PubMed] [Google Scholar]
- 111.Haack TB, Danhauser K, Haberberger B, et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet. 2010;42:1131–4. doi: 10.1038/ng.706. [DOI] [PubMed] [Google Scholar]
- 112.Calvo SE, Tucker EJ, Compton AG, et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat Genet. 2010;42:851–8. doi: 10.1038/ng.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gerards M, Sluiter W, van den Bosch BJ, et al. Defective complex I assembly due to C20orf7 mutations as a new cause of Leigh syndrome. J Med Genet. 2010;47:507–12. doi: 10.1136/jmg.2009.067553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Saada A, Edvardson S, Rapoport M, et al. C6ORF66 is an assembly factor of mitochondrial complex I. Am J Hum Genet. 2008;82:32–8. doi: 10.1016/j.ajhg.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dunning CJ, McKenzie M, Sugiana C, et al. Human CIA30 is involved in the early assembly of mitochondrial complex I and mutations in its gene cause disease. EMBO J. 2007;26:3227–37. doi: 10.1038/sj.emboj.7601748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Baysal BE. Hereditary paraganglioma targets diverse paraganglia. J Med Genet. 2002;39:617–22. doi: 10.1136/jmg.39.9.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Petruzzella V, Tiranti V, Fernandez P, et al. Identification and characterization of human cDNAs specific to BCS1, PET112, SCO1, COX15, and COX11, five genes involved in the formation and function of the mitochondrial respiratory chain. Genomics. 1998;54:494–504. doi: 10.1006/geno.1998.5580. [DOI] [PubMed] [Google Scholar]
- 118.de Lonlay P, Valnot I, Barrientos A, et al. A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat Genet. 2001;29:57–60. doi: 10.1038/ng706. [DOI] [PubMed] [Google Scholar]
- 119.Haut S, Brivet M, Touati G, et al. A deletion in the human QP-C gene causes a complex III deficiency resulting in hypoglycaemia and lactic acidosis. Hum Genet. 2003;113:118–22. doi: 10.1007/s00439-003-0946-0. [DOI] [PubMed] [Google Scholar]
- 120.Barel O, Shorer Z, Flusser H, et al. Mitochondrial complex III deficiency associated with a homozygous mutation in UQCRQ. Am J Hum Genet. 2008;82:1211–6. doi: 10.1016/j.ajhg.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ghezzi D, Arzuffi P, Zordan M, et al. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat Genet. 2011;43:259–63. doi: 10.1038/ng.761. [DOI] [PubMed] [Google Scholar]
- 122.Massa V, Fernandez-Vizarra E, Alshahwan S, et al. Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am J Hum Genet. 2008;82:1281–9. doi: 10.1016/j.ajhg.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Antonicka H, Leary SC, Guercin GH, et al. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum Mol Genet. 2003;12:2693–702. doi: 10.1093/hmg/ddg284. [DOI] [PubMed] [Google Scholar]
- 124.Coenen MJ, van den Heuvel LP, Ugalde C, et al. Cytochrome c oxidase biogenesis in a patient with a mutation in COX10 gene. Ann Neurol. 2004;56:560–4. doi: 10.1002/ana.20229. [DOI] [PubMed] [Google Scholar]
- 125.Indrieri A, van Rahden VA, Tiranti V, et al. Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am J Hum Genet. 2012;91:942–9. doi: 10.1016/j.ajhg.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zvulunov A, Kachko L, Manor E, et al. Reticulolinear aplasia cutis congenita of the face and neck: a distinctive cutaneous manifestation in several syndromes linked to Xp22. Br J Dermatol. 1998;138:1046–52. doi: 10.1046/j.1365-2133.1998.02277.x. [DOI] [PubMed] [Google Scholar]
- 127.Shteyer E, Saada A, Shaag A, et al. Exocrine pancreatic insufficiency, dyserythropoeitic anemia, and calvarial hyperostosis are caused by a mutation in the COX4I2 gene. Am J Hum Genet. 2009;84:412–7. doi: 10.1016/j.ajhg.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhu Z, Yao J, Johns T, et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet. 1998;20:337–43. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 129.Tiranti V, Jaksch M, Hofmann S, et al. Loss-of-function mutations of SURF-1 are specifically associated with Leigh syndrome with cytochrome c oxidase deficiency. Ann Neurol. 1999;46:161–6. doi: 10.1002/1531-8249(199908)46:2<161::aid-ana4>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 130.Weraarpachai W, Sasarman F, Nishimura T, et al. Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am J Hum Genet. 2012;90:142–51. doi: 10.1016/j.ajhg.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Huigsloot M, Nijtmans LG, Szklarczyk R, et al. A mutation in C2orf64 causes impaired cytochrome c oxidase assembly and mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88:488–93. doi: 10.1016/j.ajhg.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ghezzi D, Saada A, D'Adamo P, et al. FASTKD2 nonsense mutation in an infantile mitochondrial encephalomyopathy associated with cytochrome c oxidase deficiency. Am J Hum Genet. 2008;83:415–23. doi: 10.1016/j.ajhg.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mayr JA, Havlickova V, Zimmermann F, et al. Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 epsilon subunit. Hum Mol Genet. 2010;19:3430–9. doi: 10.1093/hmg/ddq254. [DOI] [PubMed] [Google Scholar]
- 134.De Meirleir L, Seneca S, Lissens W, et al. Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12. J Med Genet. 2004;41:120–4. doi: 10.1136/jmg.2003.012047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Saada A, Shaag A, Arnon S, et al. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet. 2007;44:784–6. doi: 10.1136/jmg.2007.053116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zeharia A, Fischel-Ghodsian N, Casas K, et al. Mitochondrial myopathy, sideroblastic anemia, and lactic acidosis: an autosomal recessive syndrome in Persian Jews caused by a mutation in the PUS1 gene. J Child Neurol. 2005;20:449–52. doi: 10.1177/08830738050200051301. [DOI] [PubMed] [Google Scholar]
- 137.Smirnova E, Shurland DL, Ryazantsev SN, et al. A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol. 1998;143:351–8. doi: 10.1083/jcb.143.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Waterham HR, Koster J, van Roermund CW, et al. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–41. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- 139.Cree LM, Samuels DC, de Sousa Lopes SC, et al. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat Genet. 2008;40:249–54. doi: 10.1038/ng.2007.63. [DOI] [PubMed] [Google Scholar]
- 140.Carling PJ, Cree LM, Chinnery PF. The implications of mitochondrial DNA copy number regulation during embryogenesis. Mitochondrion. 2011;11:686–92. doi: 10.1016/j.mito.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 141.DiMauro S, Schon EA. Mitochondrial DNA mutations in human disease. Am J Med Genet. 2001;106:18–26. doi: 10.1002/ajmg.1392. [DOI] [PubMed] [Google Scholar]
- 142.Clark MJ, Chen R, Lam HY, et al. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol. 2011;29:908–14. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.McCormick E, Place E, Falk MJ. Molecular Genetic Testing for Mitochondrial Disease: From One Generation to the Next. Neurotherapeutics. 2013;10:251–61. doi: 10.1007/s13311-012-0174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pagliarini DJ, Calvo SE, Chang B, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–23. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Prokisch H, Andreoli C, Ahting U, et al. MitoP2: the mitochondrial proteome database–now including mouse data. Nucleic Acids Res. 2006;34(database issue):D705–11. doi: 10.1093/nar/gkj127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pierson TM, Adams D, Bonn F, et al. Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. 2011;7:e1002325. doi: 10.1371/journal.pgen.1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Vedrenne V, Gowher A, De Lonlay P, et al. Mutation in PNPT1, which encodes a polyribonucleotide nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency. Am J Hum Genet. 2012;91:912–8. doi: 10.1016/j.ajhg.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Janer A, Antonicka H, Lalonde E, et al. An RMND1 Mutation causes encephalopathy associated with multiple oxidative phosphorylation complex deficiencies and a mitochondrial translation defect. Am J Hum Genet. 2012;91:737–43. doi: 10.1016/j.ajhg.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Casey JP, McGettigan P, Lynam-Lennon N, et al. Identification of a mutation in LARS as a novel cause of infantile hepatopathy. Mol Genet Metab. 2012;106:351–8. doi: 10.1016/j.ymgme.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 150.Galmiche L, Serre V, Beinat M, et al. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum Mutat. 2011;32:1225–31. doi: 10.1002/humu.21562. [DOI] [PubMed] [Google Scholar]
- 151.Gotz A, Tyynismaa H, Euro L, et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88:635–42. doi: 10.1016/j.ajhg.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pfeffer G, Elliott HR, Griffin H, et al. Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain. 2012;135(Pt 6):1695–713. doi: 10.1093/brain/aws102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chinnery PF, Thorburn DR, Samuels DC, et al. The inheritance of mitochondrial DNA heteroplasmy: random drift, selection or both? Trends Genet. 2000;16:500–5. doi: 10.1016/s0168-9525(00)02120-x. [DOI] [PubMed] [Google Scholar]
- 154.Monnot S, Gigarel N, Samuels DC, et al. Segregation of mtDNA throughout human embryofetal development: m.3243A>G as a model system. Hum Mutat. 2011;32:116–25. doi: 10.1002/humu.21417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pfeffer G, Majamaa K, Turnbull DM, et al. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2012;4:CD004426. doi: 10.1002/14651858.CD004426.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Klopstock T, Yu-Wai-Man P, Dimitriadis K, et al. A randomized placebo-controlled trial of idebenone in Leber's hereditary optic neuropathy. Brain. 2011;134:2677–86. doi: 10.1093/brain/awr170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kyriakouli DS, Boesch P, Taylor RW, et al. Progress and prospects: gene therapy for mitochondrial DNA disease. Gene Ther. 2008;15:1017–23. doi: 10.1038/gt.2008.91. [DOI] [PubMed] [Google Scholar]
- 158.Nagley P, Farrell LB, Gearing DP, et al. Assembly of functional proton-translocating ATPase complex in yeast mitochondria with cytoplasmically synthesized subunit 8, a polypeptide normally encoded within the organelle. Proc Natl Acad Sci USA. 1988;85:2091–5. doi: 10.1073/pnas.85.7.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bonnet C, Kaltimbacher V, Ellouze S, et al. Allotopic mRNA localization to the mitochondrial surface rescues respiratory chain defects in fibroblasts harboring mitochondrial DNA mutations affecting complex I or v subunits. Rejuvenation Res. 2007;10:127–44. doi: 10.1089/rej.2006.0526. [DOI] [PubMed] [Google Scholar]
- 160.Taivassalo T, Gardner JL, Taylor RW, et al. Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain. 2006;129(Pt 12):3391–401. doi: 10.1093/brain/awl282. [DOI] [PubMed] [Google Scholar]
- 161.Halter J, Schupbach WM, Casali C, et al. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach. Bone Marrow Transplant. 2011;46:330–7. doi: 10.1038/bmt.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wong LJ, Brunetti-Pierri N, Zhang Q, et al. Mutations in the MPV17 gene are responsible for rapidly progressive liver failure in infancy. Hepatology. 2007;46:1218–27. doi: 10.1002/hep.21799. [DOI] [PubMed] [Google Scholar]
- 163.Hellebrekers DM, Wolfe R, Hendrickx AT, et al. PGD and heteroplasmic mitochondrial DNA point mutations: a systematic review estimating the chance of healthy offspring. Hum Reprod Update. 2012;18:341–9. doi: 10.1093/humupd/dms008. [DOI] [PubMed] [Google Scholar]
- 164.Thorburn D, Wilton L, Stock-Myer S. Healthy baby girl born following pre-implantation Genetic diagnosis for mitochondrial DNA m.8993t>g Mutation. Mol Genet Metab. 2009;98:5–6. [Google Scholar]
- 165.Craven L, Tuppen HA, Greggains GD, et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature. 2010;465:82–5. doi: 10.1038/nature08958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tachibana M, Amato P, Sparman M, et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature. 2013;493:627–31. doi: 10.1038/nature11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Poulton J, Oakeshott P. Nuclear transfer to prevent maternal transmission of mitochondrial DNA disease. BMJ. 2012;345:e6651. doi: 10.1136/bmj.e6651. [DOI] [PubMed] [Google Scholar]