

Abstract
Oestrogen protects cardiovascular health partially via an up-regulation of NO• (NO radical) production. Its synthetic analogue DES (diethylstilbestrol), used as a potent androgen deprivation therapy for patients with prostate cancer, is however associated with high incidence of thromboembolic events. Exposure of BAECs (bovine aortic endothelial cells) to pharmacologically relevant dosage (12.5 μmol/l, 24 h) of DES resulted in a marked reduction in endothelial NO• bioavailability determined by ESR (electron spin resonance), while 17β-oestradiol instead increased NO• production as expected. Intriguingly, endothelial O2•− (superoxide anion) production was up-regulated by DES in vitro and in vivo, which was, however, attenuated by the ER (oestrogen receptor) antagonist ICI 182780, the XO (xanthine oxidase) inhibitor oxypurinol or the NOX (NADPH oxidase) inhibitor NSC23766. These agents also restored NO• production. DES alone in a cell-free system did not produce any ESR-sound O2•− signal. Of note, eNOS (endothelial NO synthase) mRNA and protein remained unchanged in response to DES. These results suggest that receptor-dependent activation of XO or NOX, and subsequent production of O2•−, mediate DES-induced NO• deficiency. This could represent a previously unrecognized mechanism that is responsible for cardiovascular complications of DES administration. Importantly, DES-induced suppression of LNCaP cell invasion and apoptosis were not affected by XO or NOX inhibitor. Therefore combinatorial therapy of DES and XO/NOX inhibitor may prove to be an innovative and useful therapeutic option in eliminating cardiovascular complications of DES, while preserving its anti-cancer effects, benefiting patients with advanced cancer who do not respond well to any other treatments but DES.
Keywords: combinatorial therapy, diethylstilbestrol (DES), LNCaP cell, NADPH oxidase (NOX), nitric oxide, reactive oxygen species, superoxide anion, xanthine oxidase (XO)
INTRODUCTION
In postmenopausal women, the incidence of CVD (cardiovascular disease) is much higher than in premenopausal women, implicating a potential impact of oestrogen in protecting cardiovascular health. Even though oestrogen therapy in treating CVD remains controversial [1], 17β-oestradiol supplementation to postmenopausal women has been shown to have beneficial effects on vascular function [2–4]. These include improvement in NO• (NO radical)-dependent endothelium-dependent vasodilation as a result of eNOS (endothelial nitric oxide synthase) activation and/or up-regulation of its protein expression [5,6]. Previous findings demonstrated that oestrogen-induced vascularization in response to injury was impaired in eNOS-knockout mice, further implicating a role of eNOS in oestrogen-dependent vascular protection [7]. On the other hand, Wagner et al. [8] and Miller et al. [9] have shown that 17β-oestradiol inhibits expression of NOX (NADPH oxidase) to reduce endothelial production of O2•− (superoxide anion). Oestrogen has also been shown to inhibit Rac 1 activation, which is required for NOX activation [10,11]. H2O2-induced endothelial cell apoptosis in rats was found attenuated by oestrogen [12]. Taken together, these data suggest that 17β-oestradiol is vascular protective via activation of the NO• pathway and attenuation of ROS (reactive oxygen species) signalling.
DES (diethylstilbestrol), on the other hand, is a synthetic non-steroid oestrogen. DES has been used as a highly effective agent for androgen deprivation therapy in patients with prostate cancer [13], particularly in those who do not respond to other treatments, or those who are castrate-resistant. It is, however, no longer used as a hormone replacement in postmenopausal women, at least in the U.S.A., due to its carcinogenic properties in pregnant women and their daughters (DES was prescribed to pregnant women to stabilize pregnancy). Nonetheless, it is still actively used worldwide, though to a less extent due to the worries from the findings in females, and the cardiovascular side effects. DES not only inhibits DNA synthesis and RNA polymerase in tumour tissues of the prostate but also blocks testosterone production through suppression of LH (luteinizing hormone) secretion, all of which make it highly effective in treating prostate cancer. DES has been reported to induce cytotoxicity and inhibit cell proliferation by cell-cycle arrest in androgen-dependent prostate LNCaP cells [14,15], and induce apoptosis in androgen-independent prostate cancer [16]. A study on gene expression profiling following DES treatment of androgen-dependent prostate LNCaP cells and PC-3 cells showed that expression of many genes responsible for invasion, proliferation and survival were down-regulated more than 5-fold [17], indicating the extensive signalling impact of DES on prostate cancer cells. More importantly, DES is often the last solution when patients are not responding to other alternative treatments [18]. However, DES at the dosage of 5 mg/day has exerted modest to severe thromboembolic cardiovascular events such as myocardial infarction [19]. Although lower doses of DES have been found to be effective for prostate cancer with fewer side effects, thromboembolic toxicity remained [19]. It has been suggested that DES-induced hypercoagulability is associated with different mechanisms including increased coagulation factors, diminished levels of anticoagulators, decreased fibrinolysis and altered platelet function [20]. Due to therapeutic advantages of DES including its low cost and efficacy, combinatorial therapy with other preventive drugs, which may reduce cardiovascular side effects, has received great attention. Indeed, warfarin sodium or aspirin, anti-coagulators, have been tested but shown to be ineffective in preventing cardiovascular complications [19,21], and these treatments are associated with high risk of bleeding.
In the present study, we investigated whether and how DES induces endothelial NO• deficiency, which could be pro-thrombotic. Of note, NO• attenuates expression of pro-thrombotic proteins such as PAI-1 (plasminogen-activator inhibitor-1) [22], and up-regulates antithrombotic proteins such as thrombomodulin [23]. Interestingly, we found that DES is able to activate endothelial NOX or XO (xanthine oxidase), leading to increased production of O2•−, which could in turn inactivate NO• to result in a hypercoagulable state. Combinatorial therapy with DES and NOX/XO inhibitor may therefore prove to be effective in restoring NO• production, while ideally preserving the anti-cancer effects of DES. Indeed, we found that the combination of DES with either the NOX inhibitor NSC23766 or the XO inhibitor oxypurinol was highly effective in restoring NO• production without affecting the reducing effect of DES on the viability of LNCaP cells and PC-3 cells, and the reducing effect of DES on the invasion of LNCaP cells. These effects are specific for prostate cancer cells as these responses were not observed in BAECs (bovine aortic endothelial cells), which served as a control non-cancer cell line. Therefore these findings provide new hope for potentially revitalizing DES for prostate cancer treatment by improving NO• bioavailability to eliminate cardiovascular side effects, which are often associated with NO•-deficiency related hypercoagulability. It may ultimately benefit a large population of patients with advanced prostate cancer who have to use DES for chemotherapy.
MATERIALS AND METHODS
Materials
DES, oxypurinol and PEG–SOD {PEG [poly(ethylene glycol)]-conjugated SOD (superoxide dismutase)} were purchased from Sigma. NSC23766 and ICI 182780 were purchased from Calbiochem and Tocris Bioscience respectively. DES (7-days release) and 17β-oestradiol tablets were obtained from Innovative Research of America.
Cell culture
BAECs were cultured in M199 containing 10% FBS (fetal bovine serum) and antibiotics as described previously [27]. LNCaP and PC-3 cells were purchased from A.T.C.C., and maintained in RPMI 1640 medium containing 10% FBS. HeLa cells were maintained in DMEM (Dulbecco's modified Eagle's medium) containing 10% FBS. Upon confluence, cells were starved with 0.1% FBS-containing medium overnight. Before stimulation with 12.5 μmol/l of DES for 24 h, cells were pre-treated in the presence or absence of 50 μmol/l oxypurinol, 10 μmol/l NSC23766 or 10 μmol/l ICI 182780 for 30 min.
In vivo treatment with DES or 17β-oestradiol
C57BL6 mice were implanted subcutaneously with DES and 17β-oestradiol tablets (controlled even release, 0.5 mg over 7 days). On day 7, freshly harvested aortas were subjected to DHE (dihydroethidium) staining as we described previously [24]. The use of experimental animals and procedures were approved by the Institutional Animal Care and Usage Committee at University of California Los Angeles.
DHE detection of ROS
Endothelial cells were cultured on glass cover slips till confluence, serum deprived and then treated with DES (12.5 μmol/l) for 24 h. Cells were then incubated with fresh DHE solution (2 μmol/l) in the dark for 30 min. After washing with PBS, cells were mounted on glass slides and the fluorescent images were captured using a Zweiss Axioskop inverted fluorescent microscope. Some cells were pre-incubated with PEG–SOD (100 units/ml) for 30 min before DHE incubation. In additional experiments, fresh aortic OCT sections from DES or 17β-oestradiol-treated mice were incubated with DHE for 1 h before analysis of fluorescent images.
ESR (electron spin resonance) detection of endothelial NO• production
Bioavailable NO• produced by confluent endothelial cells was detected using ESR as we described previously [27]. In brief, endothelial cells were rinsed with modified Kreb's/Hepes buffer and incubated with freshly prepared NO•-specific spin trap Fe2+ (DETC)2 colloid (0.5 mmol/l) for 60 min at 37°C. Gently collected cell suspensions were snap-frozen in liquid nitrogen and loaded into a finger Dewar for analysis with an e-Scan ESR spectrophotometer (Bruker Biospin) at the following settings: static field 3498.98, field sweep 100, resolution 512, microwave frequency 9.72 GHz, modulation amplitude 9.82, number of X-scan 20, reaction gain 3560.
ESR detection of O2•− production
Gently collected endothelial cells were suspended in modified Kreb's/Hepes buffer containing deferoxamine (25 μmol/l, metal chelator). Approximately 106 cells were mixed with O2•− -specific spin trap CMH (1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine; 1 mmol/l) in the presence or absence of PEG–SOD (100 units/ml) [25]. The cell mixture loaded into glass capillaries was immediately analysed for O2•− production kinetically for 10 min. The ESR settings used were centre field, 3475; sweep width, 9G; static field, 3484.981; microwave frequency, 9.75 GHz; microwave power, 21.02 mW; modulation frequency, 86 kHz; modulation amplitude, 2.47 G; resolution in X, 512; and number of x-scans, 10. The SOD-inhibitable O2•− signals at 10 min time point, normalized by protein concentrations, were compared among different experimental groups.
Amplex Red assay for H2O2
Post-stimulation, endothelial cell suspensions were assessed for H2O2 production using a fluorometric HRP (horseradish peroxidase) assay (Amplex Red assay; Molecular Probes). Fluorescence was measured at λex and λem of 530 nm and 590 nm respectively, after 1 h incubation at 37°C in the dark. The PEG–CAT (PEG-conjugated catalase; 300 units/ml)-inhibitable fraction, reflective of a specific H2O2 signal, was analysed and quantified using a calibration curve.
Northern blotting for eNOS
Total RNA was isolated using TRI reagent. RNA (20 μg) was subjected to gel electrophoresis on 1% agarose gels containing 2.2 mol/l formaldehyde, transferred to nitrocellulose membrane (Optitran), UV immobilized and hybridized with cDNA probe labelled by random primer kit (Stratagene). After hybridization, the membranes were washed twice at 65°C with 1×SSC (0.15 M NaCl/0.015 M sodium citrate) and 0.5% SDS, followed by autoradiography at −80°C.
Western blotting for eNOS
BAECs were lysed in ice-cold Tris buffer (50 mmol/l Tris/HCl, pH 7.4, 2 mmol/l EDTA and 1 mmol/l EGTA) containing 1% Triton X-100, 0.1% SDS, 50 mmol/l NaF, 10 mmol/l Na4P2O7, 1 mmol/l Na3VO4, 1 mmol/l DTT (dithiothreitol), 1 mmol/l PMSF and 10 μl/ml of the protease inhibitor cocktail (Sigma). Lysate proteins were subjected to SDS/PAGE (7.5% gel) and transferred to NC membrane (Amersham Biosciences). Blots were subjected to immunostaining with anti-eNOS antibody (1:1000 dilution) overnight at 4°C, and subsequent incubation with peroxidase-conjugated secondary antibody for 1 h at room temperature, and proteins were visualized using an enhanced chemiluminescence technique.
Invasion assay
Transwell invasion chambers (8 μm pore size) in 24 well plates (Corning) were used for invasion analyses. Confluent cells were starved with serum-free medium overnight before plating to upper chamber. Transwell was coated with BME (basal membrane extract) overnight. Cells were pre-treated with NSC23766, oxypurinol and ICI 182780 for 30 min before loading to transwell. Cells were loaded into coated transwell at 2.5×105 cells/well in serum-free medium. The lower chamber was filled with medium containing 5% FBS in the presence or absence of DES. After incubation at 37°C for 24 h, cells that traversed through the BME-coated membrane were subsequently detached using cell detachment buffer containing calcein-AM (acetoxymethyl ester) fluorescent dye. The fluorescence was measured in a fluorescence plate reader set at an λex of ~485 nm and an λem of ~520 nm.
Apoptosis detection
Floating or adherent cells were collected after treatment. Apoptosis was assessed with the apo-BrdU (bromodeoxyuridine) TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling) Assay kit (Invitrogen), according to manufacturer's instructions. Fragmentation of DNA in apoptotic cells was measured by BrdU incorporation, which was visualized by conjugation to an AlexaFluor® 488 dye-labelled anti-BrdU antibody.
Statistical Analysis
All results are means±S.E.M. from three to eight independent experiments. ANOVA was used to compare means of different experimental groups, and Tukey's multiple test was used as a post-test. A P value <0.05 was considered significant.
RESULTS
DES increases endothelial production of ROS
In cultured BAECs, DES (12.5 μmol/l, 24 h) induced a dramatic increase in ROS production (detected by DHE fluorescence), which was attenuated by SOD (Figure 1A). Chronic treatment of C57BL6 mice with subcutaneously implanted DES tablets (controlled release, 0.5 mg over 7 days) also resulted in a striking increase in aortic ROS production. In contrast, the identical treatment of mice with 17β-oestradiol attenuated aortic ROS production.
Figure 1. DES stimulates endothelial and vascular production of ROS detected by DHE staining.

(A) DES stimulates endothelial O2•− production. Endothelial cells stimulated with DES (12.5 μmol/l) for 24 h were incubated with DHE (2 μmol/l) for 30 min in the presence or absence of PEG–SOD pre-incubation (100 units/ml for 30 min). (B) DES stimulates aortic O2•− production. Mice received subcutaneously released DES or 17β-oestradiol (0.5 mg over 7 days). Fresh aortic OCT sections were stained with DHE for 1 h. The fluorescent images were captured with a Zweiss Axioskop inverted fluorescent microscope. CTRL, control.
DES induces NO deficiency
Next, bioavailable NO was measured by ESR in cultured aortic endothelial cells exposed to DES. As shown in Figure 2, DES (12.5 μmol/l, 24 h) induced a marked reduction in NO• bioavailability (0.534-fold of control; P<0.001). To investigate whether this response is dependent on ER (oestrogen receptor), endothelial cells were pre-treated with receptor antagonist ICI 182780 before DES stimulation. It turned out that ICI 182780 prevented DES-induced endothelial NO• deficiency (0.914±0.184-fold compared with 0.522±0.04-fold for ICI 182780 compared with DES). Either pre-treatment with the XO inhibitor oxypurinol or the NOX inhibitor NSC23766 significantly alleviated DES-induced endothelial NO• deficiency (0.677±0.044-and 0.683±0.063-fold compared with 0.522±0.04-fold for oxypurinol and NSC23766 compared with DES respectively), indicating that DES induction of NO• deficiency involves ER and activation of XO and NOX.
Figure 2. DES induces endothelial NO• deficiency that is attenuated by inhibition of NOX or XO.

DES-stimulated (12.5 μmol/l, 24 h) endothelial cells were subjected to ESR detection of NO•. Some cells were pre-incubated with the XO inhibitor oxypurinol (50 μmol/l, 30 min), the NOX inhibitor NSC23766 (10 μmol/l, 30 min) or the ER antagonist ICI 182780 (10 μmol/l, 30 min) before DES stimulation. (A) Representative ESR spectra of bioavailable NO•. (B) Grouped data from six independent experiments. Results are means±S.E.M. ***P<0.001 compared with control; #P<0.05 compared with DES.
DES increases superoxide production via NOX and XO
To further confirm whether DES stimulates NOX or XO-dependent O2•− production that can inactivate NO•, we measured O2•− production from endothelial cells pre-treated with the NOX inhibitor NSC23766 or the XO inhibitor oxypurinol before being exposed to DES stimulation. DES induced a 2-fold increase in O2•− production in endothelial cells (Figure 3A; P<0.05). Either NOX or XO inhibitor abolished DES-induced O2•− production to near control levels of 1.16- and 1.14-fold respectively. The inhibitors themselves did not affect the O2•− signal. The spatial distribution of XO and NOX is different within intracellular compartments. NOX is located at the membrane, whereas XO exists in the cytosol. It has been known that O2•− generated by NOX mediates conversion of xanthine dehydrogenase into XO via oxidation-dependent cleavage of the protein [26].
Figure 3. DES stimulates superoxide production via activation of NOX and XO.
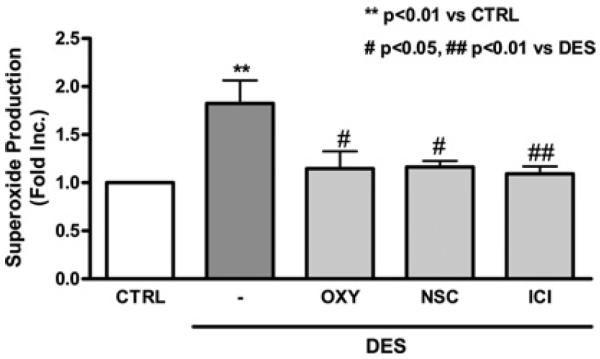
Endothelial cells were pre-treated with the XO inhibitor oxypurinol (50 μmol/l, 30 min), the NOX inhibitor NSC23766 (10 μmol/l, 30 min) or the ER antagonist ICI 182780 (10 μmol/l, 30 min) before DES stimulation. Superoxide production was determined by ESR. Results are means±S.E.M. **P<0.01 compared with control (CTRL); #P<0.05 and ##P<0.01 compared with DES.
DES induces superoxide production via ER
The presence of ERα in endothelial cells has been well established. It is known to mediate an oestrogen-induced increase in NO• bioavailability partially via attenuation of ROS. Thus we investigated whether DES increases O2•− production in a receptor-dependent manner. Intriguingly, the ER antagonist ICI 182780 attenuated DES stimulation of O2•− production (Figure 3).
DES stimulates endothelial H2O2 production
Endothelial H2O2 was detected specifically using an Amplex Red assay. DES treatment led to a 2.4-fold increase in H2O2 levels relative to vehicle-treated control cells (Figure 4). Pre-treatment with either NSC23766 or oxypurinol decreased DES-induced H2O2 production (1.365±0.226- and 1.495±0.369-fold for NSC23766 and oxypurinol respectively). ER antagonist ICI 182780 also attenuated DES-induced increase in H2O2 production (1.342±0.33-fold). These results indicate that DES-induced H2O2 production is also dependent on XO and NOX.
Figure 4. DES-induced H2O2 production is attenuated by inhibition of XO, NOX or ER.
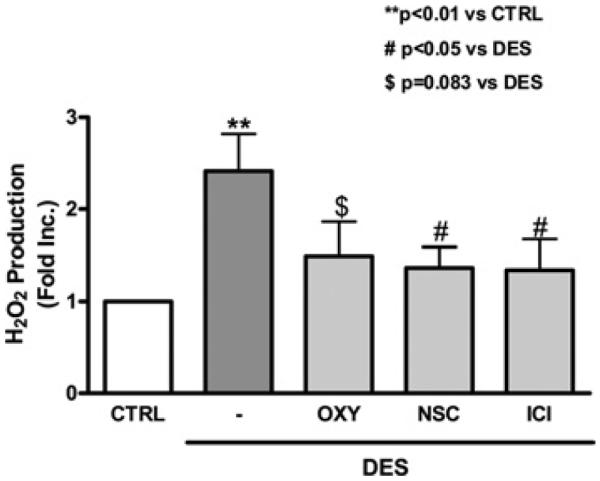
Endothelial cells were pre-treated with the ER antagonist ICI 182780 (10 μmol/l), the XO inhibitor oxypurinol (50 μmol/l) or the NOX inhibitor NSC23766 (10 μmol/l) for 30 min before DES stimulation (12.5 μmol/l, 24 h). H2O2 production was analysed using an Amplex Red assay (Molecular Probes). Results are means±S.E.M. **P<0.01 compared with control; #P<0.05 compared with DES; $P=0.083 compared with DES.
DES up-regulates eNOS gene expression
We investigated whether DES treatment affects eNOS gene expression in addition its effects on NO• inactivation via ROS production. In the case of 17β-oestradiol, it up-regulates eNOS expression as well as activating eNOS via receptor-dependent eNOS phosphorylation. As shown by representative Northern and Western blots in Figure 5, DES treatment modestly up-regulates eNOS mRNA and protein expression. However, this may not be beneficial when excessive ROS is produced by DES. eNOS has been shown to become uncoupled to produce O2•− when its cofactor tetrahydrobiopterin becomes deficient due to oxidation [27,28].
Figure 5. DES up-regulates eNOS mRNA and protein expression.

(A) Representative Northern blot of eNOS mRNA expression. (B) Representative Western blot and grouped densitometric data of eNOS protein expression. Results are means±S.E.M. *P<0.05 compared with control.
DES invasion of LNCaP cells is not affected by NOX/XO inhibitor
Before proposing the use of combinatorial therapy, it is important to examine whether the anti-cancer effects of DES are preserved when NOX or XO is inhibited. As shown in Figure 6(A), the invasive ability of LNCaP cells was significantly reduced by DES treatment. This inhibitory effect of DES on cell invasion was not altered by pre-treatment with either NSC23766 or oxypurinol. Of note, the effect of DES on invasion was not obvious in PC-3 cells, which are known to be highly metastatic (Figure 6B), there was also no effect on endothelial cells as a non-cancer cell control (Figure 6C). In HeLa cells, DES treatment led to a significant reduction on invasion (Figure 6D). Taken together, these results suggest that combinational therapy of NOX/XO inhibitor and DES is able to preserve the anti-cancer effects of DES.
Figure 6. Combined treatment of DES with NOX/XO inhibitor has no effect on DES inhibition of invasion in prostate cancer LNCaP cells.

LNCaP, PC-3, HeLa cells or BAECs were placed in BME-coated upper chamber for invasion assay. Cells were pre-treated with the ER antagonist ICI 182780 (10 μmol/l), the XO inhibitor oxypurinol (50 μmol/l) or the NOX inhibitor NSC23766 (10 μmol/l) for 30 min before DES treatment (12.5 μmol/l, 24 h). Invasion ability was determined by BME-coated transwell invasion assay in (A) LNCap cells, (B) PC-3 cells, (C) BAECs and (D) HeLa cells. Results are means±S.E.M. *P<0.05, **P<0.01 and ***P<0.001 compared with untreated cells. CTRL, control.
DES-induced apoptosis of LNCaP and PC-3 cells is not affected by NOX/XO inhibitor
As shown in Figures 7(A) and 7(B), DES induced a significant increase in apoptotic death in both prostate cancer cell line LNCaP and PC-3 cells. These anti-cancer effects were specific for prostate cancer cell lines as there were no effect on endothelial cells (Figure 7C) and non-prostate cancer HeLa cells (Figure 7D). In addition, the apoptosis-inducing effect of DES was not altered by pre-treatment with NSC23766 or oxypurinol. This result is consistent with previous findings that DES-induced ROS does not mediate apoptosis of PC-3 cells [29].
Figure 7. Combined treatment of DES with NOX/XO inhibitor has no effect on DES induction of apoptosis in prostate cancer LNCaP and PC-3 cells.

LNCaP, PC-3, BAEC and HeLa cells were pre-treated with the ER antagonist ICI 182780 (10 μmol/l), the XO inhibitor oxypurinol (50 μmol/l) or the NOX inhibitor NSC23766 (10 μmol/l) for 30 min before DES treatment (12.5 μmol/l, 24 h). After treatment, floating and adherent cells were collected and subjected to apo-BrdU TUNEL labelling as described in the Materials and methods section. (A) LNCaP cell, (B) PC-3 cell, (C) BAEC and (D) HeLa cell. ***P<0.001 compared with untreated cells; #P<0.05 compared with DES.
DISCUSSION
The major findings of the present study include: (i) DES induces endothelial NO• deficiency, which is mediated by activation of ROS production; (ii) the source of DES-induced ROS production appears to be endothelial NOX and XO; and (iii) combinatorial treatment of DES and NOX/XO inhibitor restores NO• bioavailability without affecting the anti-prostate cancer properties of DES. These results demonstrate that DES-induced cardiovascular side effects may be attributed to oxidative-stress-induced NO• deficiency and consequent endothelial dysfunction [30].
A growing body of evidence has shown that physiological concentration of oestrogen up-regulates eNOS expression and activation. In addition, it has been reported that oestrogen decreases the generation of ROS and subsequently maintains NO• bioavailability to protect endothelial function [31,32]. In the present study, however, the oestrogen analogue DES induced a NO• deficiency, which was not mediated by alteration of eNOS expression, but by an increase in ROS production. We demonstrated further that DES induces ROS production via activation of NOX and XO in endothelial cells. This is interesting as usually one oxidase system is predominantly responsible for agonist-stimulated ROS production. The detailed mechanisms as to cross-talk between the two systems warrant future investigation for a better understanding of vascular oxidant signalling in general.
Of note, DES-induced oxidative stress occurs via ER. Receptor-dependent signalling of DES has been reported in various cells, including endothelial cells, testicular cells and aortic smooth muscle cells [33,34]. In the presence of ICI 182720, DES-induced changes in NO• and O2•− levels were reversed. However, another antagonist of ER, tamoxifen, did not affect any of these responses (results not shown). This discrepancy might derive from known differences in properties of these inhibitors. Tamoxifen has been reported to act as a partial agonist accompanied by its own effect on Akt or eNOS activation, whereas ICI 182780 has been shown to have a complete blocking effect of ER [35]. It has been shown previously that the modulatory effects of oestrogen on ROS reduction were completely blocked by ICI 182780, implying an ER-dependent response [36]. In contrast, the effect of tamoxifen on the cardiovascular system is controversial. Although vasodilation induced by tamoxifen has been considered beneficial in postmenopausal women [37], its clinical usage has been linked to increased risk of venous thrombosis [38].
Our present findings provide the first evidence that DES-induced reduction in LNCaP cell invasion is ROS-independent. Even though a detailed mechanism by which DES affects LNCaP cell migration or invasion has not been described, it was shown that DES suppresses expression of several genes, including cadherin, catenein b1, integrin and metalloproteinase domain 9V, all of which are necessary for attachment and invasion [17]. A clinical report has demonstrated that DES therapy might restrict metastasis of prostate cancer [39]. The suppressive ability of DES on PSA (prostate-specific antigen) might be associated with regression of metastasis [40], since PSA has been known to facilitate invasion of prostate cancer cells by degradation of extracellular matrix glycoproteins [41]. Therefore our observation that anti-invasion effects of DES in LNCaP cells was unaffected by NOX or XO inhibitor could prove a valuable foundation for combinatorial therapy enabling protection of the cardiovascular system while preserving the anti-cancer effects of DES. Indeed, DES-induced LNCaP and PC-3 cell apoptosis was not affected by NOX or XO inhibition.
Our results suggest the possibilities of using oxypurinol or NSC23766 for combinatorial therapy with DES, since the combination of these agents with DES improved endothelial NO• bioavailability while retaining anti-invasion and apoptosis-inducing activities of DES in prostate cancer cells. Chronic therapy with NSC23766 for myelogenous leukaemia in a murine model did not show any toxic effect [42], although its clinical safety remains to be evaluated in humans. Oxypurinol has been approved by the FDA (Food and Drug Administration) and subjected to a Phase II clinical trial for symptomatic heart failure [43]. In this trial, oxypurinol, however, showed neither a beneficial effect nor side effects at the dosage of 5 mg/kg of body weight per day [43]. It may not prove to be a good agent for heart failure, but its safety makes it a good candidate for combinatorial therapy with DES for patients with prostate cancer.
In summary, the results of the present study have demonstrated that DES induces endothelial NO• deficiency that is mediated by ROS production derived from activated endothelial NOX and XO. How XO and NOX interact to be both involved in DES stimulation of O2•−, however, requires further investigation. Nonetheless, our findings provide novel insights into molecular mechanisms underlying DES's thromboembolic complications that often occur in prostate patient taking DES. This may ultimately encourage investigation of potential combinatorial therapy of DES and NOX/XO inhibitor, for safer prostate cancer treatment, particularly in those with advanced cancer who do not respond to any other treatments but DES.
Acknowledgments
FUNDING This study was supported by the National Heart, Lung and Blood Institute (NHLBI) [grant numbers HL077440 and HL088975 (to H.C.), HL101228 (to P.P. Ping, J.N. Weiss and H.C.), HL108701 (to H.C. and D.G. Harrison)] and an American Heart Association Established Investigator Award [grant number 12EIA8990025 (to H.C.)].
Abbreviations
- BAEC
bovine aortic endothelial cell
- BME
basal membrane extract
- BrdU
bromodeoxyuridine
- CMH
1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine
- CVD
cardiovascular disease
- DES
diethylstilbestrol
- DHE
dihydroethidium
- eNOS
endothelial NO synthase
- ER
oestrogen receptor
- ESR
electron spin resonance
- FBS
fetal bovine serum
- NO•
NO radical
- NOX
NADPH oxidase
- O2•−
superoxide anion
- PEG
poly(ethylene glycol)
- PSA
prostate-specific antigen
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling
- XO
xanthine oxidase
Footnotes
AUTHOR CONTRIBUTION Ji-Youn Youn contributed to data collection, data analysis and preparation of the paper. Andrew Nguyen contributed to data collection. Hua Cai contributed to study design, data analysis, data interpretation and preparation of the paper.
REFERENCES
- 1.Hodgin JB, Knowles JW, Kim HS, Smithies O, Maeda N. Interactions between endothelial nitric oxide synthase and sex hormones in vascular protection in mice. J. Clin. Invest. 2002;109:541–548. doi: 10.1172/JCI14066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N. Engl. J. Med. 1999;340:1801–1811. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- 3.Cid MC, Schnaper HW, Kleinman HK. Estrogens and the vascular endothelium. Ann. N.Y. Acad. Sci. 2002;966:143–157. doi: 10.1111/j.1749-6632.2002.tb04211.x. [DOI] [PubMed] [Google Scholar]
- 4.Wagner JD, Clarkson TB. The applicability of hormonal effects on atherosclerosis in animals to heart disease in postmenopausal women. Semin. Reprod. Med. 2005;23:149–156. doi: 10.1055/s-2005-869482. [DOI] [PubMed] [Google Scholar]
- 5.Schulz E, Anter E, Zou MH, Keaney JF., Jr Estradiol-mediated endothelial nitric oxide synthase association with heat shock protein 90 requires adenosine monophosphate-dependent protein kinase. Circulation. 2005;111:3473–3480. doi: 10.1161/CIRCULATIONAHA.105.546812. [DOI] [PubMed] [Google Scholar]
- 6.Li HL, Zhuo ML, Wang D, Wang AB, Cai H, Sun LH, Yang Q, Huang Y, Wei YS, Liu PP, et al. Targeted cardiac overexpression of A20 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circulation. 2007;115:1885–1894. doi: 10.1161/CIRCULATIONAHA.106.656835. [DOI] [PubMed] [Google Scholar]
- 7.Iwakura A, Luedemann C, Shastry S, Hanley A, Kearney M, Aikawa R, Isner JM, Asahara T, Losordo DW. Estrogen-mediated, endothelial nitric oxide synthase-dependent mobilization of bone marrow-derived endothelial progenitor cells contributes to reendothelialization after arterial injury. Circulation. 2003;108:3115–3121. doi: 10.1161/01.CIR.0000106906.56972.83. [DOI] [PubMed] [Google Scholar]
- 8.Wagner AH, Schroeter MR, Hecker M. 17β-estradiol inhibition of NADPH oxidase expression in human endothelial cells. FASEB J. 2001;15:2121–2130. doi: 10.1096/fj.01-0123com. [DOI] [PubMed] [Google Scholar]
- 9.Miller AA, Drummond GR, Mast AE, Schmidt HH, Sobey CG. Effect of gender on NADPH-oxidase activity, expression, and function in the cerebral circulation: role of estrogen. Stroke. 2007;38:2142–2149. doi: 10.1161/STROKEAHA.106.477406. [DOI] [PubMed] [Google Scholar]
- 10.Laufs U, Adam O, Strehlow K, Wassmann S, Konkol C, Laufs K, Schmidt W, Bohm M, Nickenig G. Down-regulation of Rac-1 GTPase by Estrogen. J. Biol. Chem. 2003;278:5956–5962. doi: 10.1074/jbc.M209813200. [DOI] [PubMed] [Google Scholar]
- 11.Jayachandran M, Sanzo A, Owen WG, Miller VM. Estrogenic regulation of tissue factor and tissue factor pathway inhibitor in platelets. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H1908–H1916. doi: 10.1152/ajpheart.01292.2004. [DOI] [PubMed] [Google Scholar]
- 12.Sudoh N, Toba K, Akishita M, Ako J, Hashimoto M, Iijima K, Kim S, Liang YQ, Ohike Y, Watanabe T, et al. Estrogen prevents oxidative stress-induced endothelial cell apoptosis in rats. Circulation. 2001;103:724–729. doi: 10.1161/01.cir.103.5.724. [DOI] [PubMed] [Google Scholar]
- 13.Perlmutter MA, Lepor H. Androgen deprivation therapy in the treatment of advanced prostate cancer. Rev. Urol. 2007;9(Suppl. 1):S3–S8. [PMC free article] [PubMed] [Google Scholar]
- 14.Kalach JJ, Joly-Pharaboz MO, Chantepie J, Nicolas B, Descotes F, Mauduit C, Benahmed M, Andre J. Divergent biological effects of estradiol and diethylstilbestrol in the prostate cancer cell line MOP. J. Steroid Biochem. Mol. Biol. 2005;96:119–129. doi: 10.1016/j.jsbmb.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 15.Montgomery RB, Bonham M, elson PS, Grim J, Makary E, Vessella R, Stahl WL. Estrogen effects on tubulin expression and taxane mediated cytotoxicity in prostate cancer cells. Prostate. 2005;65:141–150. doi: 10.1002/pros.20246. [DOI] [PubMed] [Google Scholar]
- 16.Robertson CN, Roberson KM, Padilla GM, O'Brien ET, Cook JM, Kim CS, Fine RL. Induction of apoptosis by diethylstilbestrol in hormone-insensitive prostate cancer cells. J. Natl. Cancer Inst. 1996;88:908–917. doi: 10.1093/jnci/88.13.908. [DOI] [PubMed] [Google Scholar]
- 17.Koike H, Ito K, Takezawa Y, Oyama T, Yamanaka H, Suzuki K. Insulin-like growth factor binding protein-6 inhibits prostate cancer cell proliferation: implication for anticancer effect of diethylstilbestrol in hormone refractory prostate cancer. Br. J. Cancer. 2005;92:1538–1544. doi: 10.1038/sj.bjc.6602520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Debruyne F. Hormonal therapy of prostate cancer. Semin. Urol. Oncol. 2002;20:4–9. doi: 10.1053/suro.2002.35051. [DOI] [PubMed] [Google Scholar]
- 19.Malkowicz SB. The role of diethylstilbestrol in the treatment of prostate cancer. Urology. 2001;58:108–113. doi: 10.1016/s0090-4295(01)01252-3. [DOI] [PubMed] [Google Scholar]
- 20.Scherr DS, Pitts WR., Jr The nonsteroidal effects of diethylstilbestrol: the rationale for androgen deprivation therapy without estrogen deprivation in the treatment of prostate cancer. J. Urol. 2003;170:1703–1708. doi: 10.1097/01.ju.0000077558.48257.3d. [DOI] [PubMed] [Google Scholar]
- 21.Farrugia D, Ansell W, Singh M, Philp T, Chinegwundoh F, Oliver RT. Stilboestrol plus adrenal suppression as salvage treatment for patients failing treatment with luteinizing hormone-releasing hormone analogues and orchidectomy. B.J.U. Int. 2000;85:1069–1073. doi: 10.1046/j.1464-410x.2000.00673.x. [DOI] [PubMed] [Google Scholar]
- 22.Bouchie JL, Hansen H, Feener EP. Natriuretic factors and nitric oxide suppress plasminogen activator inhibitor-1 expression in vascular smooth muscle cells. Role of cGMP in the regulation of the plasminogen system. Arterioscler., Thromb., Vasc Biol. 1998;18:1771–1779. doi: 10.1161/01.atv.18.11.1771. [DOI] [PubMed] [Google Scholar]
- 23.Shi J, Wang J, Zheng H, Ling W, Joseph J, Li D, Mehta JL, Ponnappan U, Lin P, Fink LM, Hauer-Jensen M. Statins increase thrombomodulin expression and function in human endothelial cells by a nitric oxide-dependent mechanism and counteract tumor necrosis factor alpha-induced thrombomodulin downregulation. Blood Coagul. Fibrinol. 2003;14:575–585. doi: 10.1097/00001721-200309000-00010. [DOI] [PubMed] [Google Scholar]
- 24.Oak JH, Cai H. Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes. 2007;56:118–126. doi: 10.2337/db06-0288. [DOI] [PubMed] [Google Scholar]
- 25.Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, Harrison DG. Role of p47phox in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–515. doi: 10.1161/01.hyp.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meneshian A, Bulkley GB. The physiology of endothelial xanthine oxidase: from urate catabolism to reperfusion injury to inflammatory signal transduction. Microcirculation. 2002;9:161–175. doi: 10.1038/sj.mn.7800136. [DOI] [PubMed] [Google Scholar]
- 27.Chalupsky K, Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 2005;102:9056–9061. doi: 10.1073/pnas.0409594102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ. Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 29.Kikuchi E, Nakashima J, Horiguchi Y, Oya M, Ohigashi T, Murai M. Enhancement of diethylstilbestrol induced cytotoxicity by Bcl-2 antisense oligodeoxynucleotides and a glutathione depletor for prostate cancer. J. Urol. 2003;169:730–734. doi: 10.1097/01.ju.0000037786.60544.9e. [DOI] [PubMed] [Google Scholar]
- 30.Stagliano NE, Dietrich WD, Prado R, Green EJ, Busto R. The role of nitric oxide in the pathophysiology of thromboembolic stroke in the rat. Brain Res. 1997;759:32–40. doi: 10.1016/s0006-8993(97)00200-x. [DOI] [PubMed] [Google Scholar]
- 31.Shehata M, Kamel MA. Protective effect of antioxidant adjuvant treatment with hormone replacement therapy against cardiovascular diseases in ovariectomized rats. Endocr. Regul. 2008;4:69–75. [PubMed] [Google Scholar]
- 32.Billon A, Lehoux S, Lam Shang Leen L, Laurell H, Filipe C, Benouaich V, Brouchet L, Dessy C, Gourdy P, Gadeau AP, et al. The estrogen effects on endothelial repair and mitogen-activated protein kinase activation are abolished in endothelial nitric-oxide (NO) synthase knockout mice, but not by NO synthase inhibition by N-nitro-l-arginine methyl ester. Am. J. Pathol. 2008;172:830–838. doi: 10.2353/ajpath.2008.070439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raddino R, Pela G, Uberti D, Portera C, Ferrari R, Scarabelli TM, Cooper TJ, Manca C. Estrogen derivative relaxes rabbit aorta via the endothelial receptor system. Ital. Heart J. 2001;2:49–54. [PubMed] [Google Scholar]
- 34.Lassurguere J, Livera G, Habert R, Jegou B. Time- and dose-related effects of estradiol and diethylstilbestrol on the morphology and function of the fetal rat testis in culture. Toxicol. Sci. 2003;73:160–169. doi: 10.1093/toxsci/kfg065. [DOI] [PubMed] [Google Scholar]
- 35.Florian M, Lu Y, Angle M, Magder S. Estrogen induced changes in Akt-dependent activation of endothelial nitric oxide synthase and vasodilation. Steroids. 2004;69:637–645. doi: 10.1016/j.steroids.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 36.Wassmann S, Laufs U, Stamenkovic D, Linz W, Stasch JP, Ahlbory K, Rosen R, Bohm M, Nickenig G. Raloxifene improves endothelial dysfunction in hypertension by reduced oxidative stress and enhanced nitric oxide production. Circulation. 2002;105:2083–2091. doi: 10.1161/01.cir.0000014618.91633.67. [DOI] [PubMed] [Google Scholar]
- 37.Stamatelopoulos KS, Lekakis JP, Poulakaki NA, Papamichael CM, Venetsanou K, Aznaouridis K, Protogerou AD, Papaioannou TG, Kumar S, Stamatelopoulos SF. Tamoxifen improves endothelial function and reduces carotid intima-media thickness in postmenopausal women. Am. Heart J. 2004;147:1093–1099. doi: 10.1016/j.ahj.2003.12.029. [DOI] [PubMed] [Google Scholar]
- 38.Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, Bevers TB, Fehrenbacher L, Pajon ER, Jr, Wade JL, III, et al. Effects of tamoxifen compared with raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. J. Am. Med. Assoc. 2006;295:2727–2741. doi: 10.1001/jama.295.23.joc60074. [DOI] [PubMed] [Google Scholar]
- 39.Heffner JE, Duffey DJ, Schwarz MI. Massive pleural effusions from prostatic lymphangitic carcinomatosis: resolution with endocrine therapy. Arch. Intern. Med. 1982;142:375–376. [PubMed] [Google Scholar]
- 40.Hoshi S, Ohyama C, Hagisawa S, Ono K, Satoh M, Saito S, Fukuzaki A, Arai Y. Complete regression of bone metastases on super bone scan, by low-dose cisplatin, UFT, diethylstilbestrol diphosphate, and dexamethasone in a patient with hormone-refractory prostate cancer. Int. J. Clin. Oncol. 2003;8:118–120. doi: 10.1007/s101470300021. [DOI] [PubMed] [Google Scholar]
- 41.Webber MM, Waghray A, Bello D. Prostate-specific antigen, a serine protease, facilitates human prostate cancer cell invasion. Clin. Cancer Res. 1995;1:1089–1094. [PubMed] [Google Scholar]
- 42.Quintas-Cardama A, Cortes J. Therapeutic options against BCR-ABL1 T315I-positive chronic myelogenous leukemia. Clin. Cancer Res. 2008;14:4392–4399. doi: 10.1158/1078-0432.CCR-08-0117. [DOI] [PubMed] [Google Scholar]
- 43.Hare JM, Mangal B, Brown J, Fisher C, Jr, Freudenberger R, Colucci WS, Mann DL, Liu P, Givertz MM, Schwarz RP. Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT-CHF study. J. Am. Coll. Cardiol. 2008;51:2301–2309. doi: 10.1016/j.jacc.2008.01.068. [DOI] [PubMed] [Google Scholar]