

Abstract
The potential of hydrocarbon biodegradation in marine sediments was determined through the detection of a functional biomarker, the bssA gene, coding for benzylsuccinate synthase, the key enzyme of anaerobic toluene degradation. Eight bssA clone libraries (409 sequences) were constructed from polluted sediments affected by the Prestige oil spill in the Atlantic Islands National Park and from hydrocarbon-amended sediment microcosms in Mallorca. The amplified products and database-derived bssA-like sequences grouped into four major clusters, as determined by phylogenetic reconstruction, principal coordinate analysis (PCoA), and a subfamily prediction tool. In addition to the classical bssA sequences that were targeted, we were able to detect sequences homologous to the naphthylmethylsuccinate synthase gene (nmsA) and the alkylsuccinate synthase gene (assA), the bssA homologues for anaerobic 2-methylnaphthalene and alkane degradation, respectively. The detection of bssA-like variants was determined by the persistence and level of pollution in the marine samples. The observed level of gene diversity was lower in the Mallorca sediments, which were dominated by assA-like sequences. In contrast, the Atlantic Islands samples, which were highly contaminated with methylnaphthalene-rich crude oil, showed a high proportion of nmsA-like sequences. Some of the detected genes were phylogenetically related to Deltaproteobacteria communities, previously described as the predominant hydrocarbon degraders at these sites. Differences between all detected bssA-like genes described to date indicate separation between marine and terrestrial sequences and further subgrouping according to taxonomic affiliation. Global analysis suggested that bssA homologues appeared to cluster according to substrate specificity. We observed undetected divergent gene lineages of bssA homologues, which evidence the existence of new degrader groups in these environments.
INTRODUCTION
Marine habitats are continually exposed to hydrocarbon pollutants that are released through natural seepage and also generated by anthropogenic activities. Many bacteria have adapted the ability to degrade low-molecular-weight hydrocarbons to carbon dioxide and water. Thus, biodegradation of contaminants mediated by autochthonous microorganisms has become the basis for bioremediation strategies in contaminated sites. Application of such technologies requires preceding efforts to identify the relevant players in the remediation process and assessment of the optimal attenuation conditions. Until recently, these strategies were based primarily on the exploitation of aerobic processes performed by aerobic bacteria, but many exposed ecological niches, such as marine sediments, are under permanent anoxic conditions (1, 2). Thus, the anaerobic catabolism of hydrocarbons is of crucial importance for natural attenuation (3, 4).
Investigations of the biochemistry, genetics, and physiology of anaerobic hydrocarbon degradation are essentially centered on the degradation of monoaromatic compounds and n-alkanes (5, 6), naphthalene (7), and 2-methylnaphthalene (2MN) (8, 9). The initial steps in the anaerobic degradation of hydrocarbons are considerably diverse between microorganisms (6). Still, activation of a broad range of aromatic hydrocarbons converges into a few major central metabolites, which are further dearomatized and channeled to the central cell metabolism. Fumarate addition, the initial and key step in toluene degradation, is a general activation strategy in many anaerobic aromatic activation pathways. The reaction is catalyzed by the glycyl radical enzyme benzylsuccinate synthase (Bss), and the reaction product is further converted in several steps to the final key metabolite benzoyl coenzyme A (CoA) (6). Benzylsuccinate synthase is composed of three different subunits. The large subunit (bssA gene) included in the pyruvate formate-lyase (PFL) family of glycyl-radical enzymes contains the active-site determinants that characterize this family, which consists of a conserved glycine motif located near the C-terminal end of the subunit and a conserved cysteine residue in the middle of the protein sequence (10). Benzylsuccinate synthase catalyzes the addition of the methyl group of toluene to a fumarate cosubstrate to produce (R)-benzylsuccinate (11–13).
Several hydrocarbons have been shown to undergo a similar activation reaction as the main step in their degradation. For example, anaerobic degradation of o- and m-xylenes, m- and p-cresols, and ethylbenzene is initiated by BssA-like activity (12, 14–17). A similar fumarate addition catalyzed by a 2-naphthylmethylsuccinate synthase, encoded by the nmsA gene, was shown to be the first step in the degradation of 2MN by sulfate-reducing bacteria (SRB) (9, 18). SRB and nitrate-reducing bacteria (NRB) also activate n-alkane degradation in a reaction resembling BssA-dependent toluene activation, rendering 1-methylalkyl-succinates (19, 20). The masD and assA genes, encoding the large subunit of this enzyme, were simultaneously described in the denitrifier Azoarcus sp. strain HxN1 (21) and the SRB Desulfatibacillum alkenivorans AK-01 (22), respectively.
PCR amplification of catabolic genes has been proposed as a means to assess anaerobic BTEX (benzene, toluene, ethylbenzene, and xylene) metabolisms in situ (23). A limited number of bssA sequences from pure cultures are available to date (13, 24–29), but many bssA-like sequences have been detected in enrichment cultures and consortia able to degrade hydrocarbons (30–32). The strong conservation of bssA sequences and the recurring detection of this gene in hydrocarbon-contaminated environments have promoted its use as an indicator of hydrocarbon degradation at these sites (23, 33–37). Sequence analysis revealed greater-than-expected gene diversity, likely related to local distribution, substrate specificity, and dominant respiration strategies (35). Four nmsA sequences retrieved from SRB isolates or sulfate-reducing enrichment cultures (8, 9, 18) clustered together with nmsA sequences recently detected in environmental samples of different origins (38). Additionally, orthologues of assA and masD genes were recently found in pure cultures of sulfate-reducing and denitrifying Proteobacteria (22, 39) and in a thermophilic methanogenic community (40). Analysis of retrieved sequences revealed that assA sequences grouped into a distinct cluster that was separated from canonical bssA sequences. The majority of the environmental sequences were retrieved from hydrocarbon-contaminated aquifers and river sediments; very little is known regarding bssA-like genes present in marine environments, and studies targeting the effect of pollution on the microbial communities in subtidal sediments are scarce.
Indigenous microbial communities play a significant role in hydrocarbon degradation of oil-polluted environments, as evidenced by a great repertoire of metabolic genes that have evolved to oxidize these hydrocarbons. The presence and diversity of the functional genes in the environment could identify the biodegradative potential of microbial communities. In this study, we analyzed for the first time the bssA-like gene diversity of subtidal marine sediment samples collected from a heavily polluted beach affected by the Prestige oil spill (Atlantic Islands National Park, Spain), where the oxidative capacities of microbial communities have already been demonstrated (41, 42). To determine the time-exposure effect on functional diversity dynamics, crude oil or naphthalene contamination was simulated in microcosms from sediments collected in Alcúdia Bay (Mallorca, Mediterranean Sea). The resulting bssA gene diversity was compared with all available sequences to provide a comprehensive analysis of the global bssA gene distribution. Our data suggested that contamination had a positive influence on the functional gene diversity. The observed grouping of BssA-like homologues into consistent clusters was apparently related to substrate specificity (toluenes, methylnaphthalenes, and alkanes).
MATERIALS AND METHODS
Site description and microcosm preparation.
Sediment samples were collected from an Atlantic Islands beach (Figueiras [FI]) in Northern Spain, which was affected by the Prestige oil spill in November 2002. Sediment cores (50 cm) were collected in May 2004 by scuba divers at a depth of 9 m with 50-cm-long cores, and an additional 30-cm core was collected at a place where a petroleum spot, intermingled with the sediment, was found by the diving team (FI-PET) (41). Cores were kept intact at 4°C until processed. Three sections were selected for analysis: the upper oxic layer (2 to 5 cm) (FI-OX), a transition layer (11 to 14 cm) (FI-TR), and a sulfidic zone (32 to 35 cm) (FI-AN). An additional core from a neighboring less-affected beach (Rodas beach [RI]) was also analyzed. Microcosm experiments were set up in June 2007 with sediments collected at a second site in Alcúdia Bay (Mallorca, Spain [M]) within a pristine seabed. Three cores (50 cm) were collected as described above, at a 12-m depth. Two of them were artificially contaminated with naphthalene (M-NAPH) or crude oil (M-OIL). Naphthalene was provided by inserting a perforated 10-ml propylene pipette filled with naphthalene crystals into the originally collected core; four lanes of 1-mm holes were distributed equidistantly along the pipette to allow for diffusion. Prestige crude oil was introduced into another core after impregnation onto a nylon stick, as described previously (43). A third core remained untreated and served as a control (M-CON). The cores were sealed with duct tape and incubated at 22°C in the dark for 4 months. The sediment section at a depth of 12 to 16 cm was used for the analysis.
Enumeration of bacteria.
Most probable number (MPN) counts of hydrocarbon-oxidizing bacteria were estimated in an artificial seawater minimal medium, as described previously (41). Anaerobic medium was amended with either acetate or an aromatic compound (toluene, benzene, naphthalene, or anthracene) to a final concentration of 1 mM or with crude oil drops (only for Mallorca samples) as the sole carbon source. Either 10 mM sulfate, 20 mM ferrihydrite, or 20 mM manganese (only for Mallorca samples) was added to the medium as an electron acceptor. All tubes were incubated in the dark at 22°C for 6 months.
Hydrocarbon analysis.
Hydrocarbons were extracted from duplicate frozen sediment aliquots (2 ml) in 4 ml of hexane-acetone (2:1). A volume of 20 μl deuterated mix 37 (manufactured by Dr. Ehrenstorfer GmbH) and 10 μl of a 1,000-ppm solution of 5a-cholestane (Aldrich) were added as internal standards before the extraction. An equal volume of salt-saturated solution (40% NaCl in water) was added to each tube. After separation of the aqueous and organic phases, the supernatant was percolated through a Bond Elut TPH column with a Si-CN-U matrix (Varian). The aliphatic fraction was first eluted by gravity with 4 ml of pure hexane, and the aromatic fraction was then eluted with 6 ml of dichloromethane. Both fractions were dried under a gentle stream of nitrogen gas and then resuspended in 0.25 ml of the corresponding solvent and stored at −20°C until analysis. Hydrocarbon analysis was performed on a Varian 450-GC instrument attached to a 240-IT mass spectrometer equipped with a programmed temperature vaporization/split-splitless injector and a CTCGCpal autosampler, as previously described (41). A static headspace gas chromatography mass spectrometer (Varian 450GC 240MS with a CTC CombiPal autosampler) was used for the determination of BTEX in contaminated sediments (44). Briefly, duplicate samples of 2 g of defrosted sediment were placed into 10-ml vials. Fluorobenzene (50 ng) was added to each vial as an internal standard. The vial was incubated in a heating block at 70°C for 10 min. A volume of 1 ml was injected at 250°C in split mode (1:20) at a flow rate of 1 ml/min. The temperature was held at 50°C for 1 min, increased to 95°C at a rate of 20°C/min and held for 2 min, and then increased to 150°C at a rate of 25°C/min and held for 0.6 min. Mass spectrometry (MS) acquisition was done with 0.5-min delay, a scan range of m/z 45 to m/z 200 until 3.4 min, and m/z 80 to m/z 200 until 8 min. Identification and quantification were performed in the single-ion monitoring (SIM) mode using the most abundant ions. The detection limit of the assays was 1 to 5 ppb for the standard analytes.
Nucleic acid extraction.
An SDS-based DNA extraction method was used with duplicate samples of frozen sediment, as described previously by Zhou and colleagues (45), with some modifications: approximately 5 g of sediment was mixed with 12.5 ml of high-salt extraction buffer (1.5 M NaCl, 100 mM Tris-HCl [pH 8.0], 100 mM sodium EDTA [pH 8.0], 100 mM sodium phosphate [pH 8.0], and 1% cetyltrimethylammonium bromide [CTAB]) containing 125 μl of proteinase K (12.5 mg/ml) and lysozyme (100 mg/ml) and incubated for 30 min at 37°C with constant horizontal shaking at 200 rpm. Next, 1.5 ml of SDS (20%, wt/vol) was added, and tubes were incubated for 2 h at 65°C. Extracts were centrifuged at 6,000 × g for 20 min, and supernatants were transferred into a new tube. Extraction was repeated once by using 7.5 ml of extraction buffer with 75 μl of proteinase K and lysozyme prepared as described above. Supernatants from both extractions were combined and treated with 1 volume of phenol-chloroform-isoamyl alcohol (25:24:1, vol/vol) and then vortexed and centrifuged at 6,000 × g for 10 min. The supernatant was extracted again with chloroform-isoamyl alcohol (24:1, vol/vol). Nucleic acids were precipitated from the aqueous phase after addition of NaCl to a final concentration of 0.2 M and isopropanol to 0.7 volumes. The pellet was washed with 70% ethanol and resuspended in 100 μl of milli-Q water.
Amplification of the benzylsuccinate synthase alpha-subunit gene (bssA).
Primers described previously (35) were used to amplify the alpha-subunit of the benzylsuccinate synthase gene (bssA). A gradient PCR was performed with annealing temperatures ranging from 45°C to 60°C for all primer combinations. After 32 cycles, primers 7772f and 8542r successfully amplified partial bssA genes of the expected size and were chosen for the gene library preparations. DNA from Thauera aromatica K172 was used as a positive control for the PCR. As unspecific bands were obtained with these primers, the amplified fragment with the expected size was separated on a 15-cm-long agarose gel (1.5%, wt/vol) over 2 h and extracted with the Qiagen gel extraction kit according to the manufacturer's instructions. Purified fragments were cloned into the pGEM-T vector (Promega), and 70 positive clones were selected for sequencing.
Sequence analysis.
DNA sequences obtained in this work were translated to perform a protein blast (blastp) search against NCBI databases (nr, refseq_protein, swissprot, pat, pdb, and env_nr) and IMG 4 databases (46) in order to select the closest matches. Some matching sequences found in the blastp search included sequences that were not annotated as BssA but showed at least 60% amino acid similarity with the query and were included in subsequent analyses. Sequences including less than 75% of the protein region analyzed in this work (region amplified with primers 7772f and 8542r) were discarded. To compile all possible BssA-like sequences, we first retrieved all published BssA, NmsA, and AssA sequences described in the literature (see Table S1 in the supplemental material). In addition, we searched all NCBI databases for the key words “benzylsuccinate synthase,” “naphthylmethylsuccinate synthase,” or “alkylsuccinate synthase.” Mega 5 software was used to align the sequences and construct phylogenetic trees. FastUniFrac was used for PCoA (47). Amino acid sequences were compiled in OTUs (operational taxonomic units) at 5% (OTU0.05) and 2% (OTU0.02) sequence dissimilarity levels for tree construction and PCoA, respectively. SPEER software (specificity prediction using amino acids' properties, entropy, and evolution rate), available at the SPEER-SERVER website, was applied to elucidate the specific conservation pattern of the amino acid residues by using the automated subgrouping tool SCI-PHY, allowing a gap of 20% per column and a weight of 1.0 for relative entropy and physicochemical (PC) property distance. This method can define protein subfamilies and predicts residues that are relevant for family protein functions by quantifying patterns of conserved sites based on their physicochemical properties and the heterogeneity of evolutionary changes between and within the protein subfamilies (48, 49). This method identifies type I sites (conserved in one subgroup and variable in the remaining groups) and type II sites, where different types of amino acids are conserved across different subfamilies. WebLogo software (50) was used to build the logo of each sequence cluster. Exportable documents were edited with Illustrator 5.0 to compare the different profiles. A computational model of the three-dimensional structure of the translated amplified bssA fragment was constructed with the Phyre2 server (Protein Homology/analogY Recognition Engine V 2.0 [http://www.sbg.bio.ic.ac.uk/phyre2/]) (51). The structures were displayed as ribbon diagrams by using Pymol software.
Nucleotide sequence accession numbers.
The partial bssA gene sequences obtained from clone libraries have been deposited in GenBank under accession numbers KC463912 to KC464320.
RESULTS AND DISCUSSION
Sample characterization.
In order to assess the diversity of the bssA gene as an indication of anaerobic hydrocarbon degradation, samples were collected from Figueiras beach (FI samples), a long-term oil-contaminated beach in northern Spain, and from sediment microcosms collected in Mallorca (Mediterranean sea) and spiked with naphthalene or crude oil (M samples). BTEX (benzene, toluene, ethylbenzene, and xylene) compounds were detected in only two samples: the oil-contaminated microcosms in Mallorca (M-OIL) and the petroleum patch in Figueiras sediment (FI-PET) (see Fig. S1a and Table S2a in the supplemental material). The crude oil contamination was composed of aliphatic and aromatic hydrocarbons, with a high proportion of naphthalene and alkylated derivatives (see Fig. S1b and Table S2b in the supplemental material). The diversity and aromatic biodegradation potential of samples collected from the Atlantic Islands were described previously (41). Delta- and Gammaproteobacteria constituted more than 60% of the retrieved 16S rRNA gene sequences, and the proportion of Deltaproteobacteria increased toward the bottom of the sediment core, while Gammaproteobacteria were predominant in FI-PET. The highest counts of aromatic-oxidizing bacteria were obtained under sulfate-reducing conditions with benzene, toluene, or naphthalene as a carbon source (see Table S3 in the supplemental material). The microbial diversity and potential for sulfate reduction-dependent hydrocarbon degradation in the sediments where M samples were collected was verified previously (43). As observed for the Atlantic Islands samples, Gamma- and Deltaproteobacteria were the most abundant communities in these sediments. After 4 months of incubation, the MPN of sulfate-reducing bacteria (SRB) determined in the three cores indicated an oil-dependent stimulation of the bacterial populations in all carbon sources assayed. The highest counts were observed with acetate, and the community able to use crude oil exhibited counts within this range (see Table S3 in the supplemental material). In contrast, the naphthalene treatment was toxic to most microbial populations, except for those that were able to oxidize naphthalene or crude oil, which increased by 4-fold and 2-fold, respectively. Thus, as observed for the Atlantic Islands sediments (41), the abundances of the bacterial communities able to use crude oil carbon sources increased in response to high pollution levels, despite a large decrease in the total cell counts.
Detection of bssA-like genes in the sediments.
We tested several previously designed primer pairs to monitor bssA genes in sediment samples (35). Only primers 7772f and 8542r consistently produced PCR amplification products with DNA extracted from sediment. The amplified sequence covered the gene region that codes for part of the active site of the enzyme, including the conserved cysteine residue, where the highly reactive thiyl radical is formed. Eight bssA gene libraries were constructed from DNA isolated from each sediment sample, and 70 clones from each library were sequenced. As previously described, non-bssA-related DNA fragments of the same size were also obtained by using these primers, which represented up to one-third of the analyzed sequences (see Table S4 in the supplemental material) (35, 52).
Sequence analysis of the PCR products revealed fragments of two different lengths: the expected 794-bp fragment and a group of 773-bp-long fragments that also showed strong similarity to the bssA gene. The amplified bssA sequences from the eight libraries were translated into amino acid sequences (BssA) and compared with available BssA sequences; a phylogenetic tree of the amplified and database sequences is provided in Fig. 1. Four main clusters that grouped sequences sharing 75% amino acid similarity were identified.
Fig 1.

Phylogeny of partial BssA-like amino acid sequences retrieved from marine sediments from Figueiras beach (FI) (blue), a petroleum patch detected in the sediment (FI-PET) (green), and Rodas beach (RI) (brown) in the Atlantic Islands and sediment microcosms from Mallorca (M) (red). The closest relatives, detected in anaerobic hydrocarbon-oxidizing strains or enrichments, and putative environmental sequences available in the databases at the beginning of this study are included. Different depths correspond to oxic (FI-OX), transition (FI-TR), and anoxic (FI-AN) zones and were analyzed in Figueiras. Mallorca sediments were treated with Prestige oil (M-OIL), naphthalene (M-NAP), and nothing (M-CON). Sequences in the databases that were retrieved from marine environments are marked with an asterisk. Numbers in parentheses represent protein GenBank accession numbers. A sequence cutoff of a 5% amino acid dissimilarity (OTU0.05) was used to select the sequences and calculate the sequence frequency (showed as horizontal bars at the right). The tree was rooted with pyruvate formate lyase (PFL2) paralogues as an outgroup.
Cluster I included the canonical, toluene-specific BssA sequences (bssA sensu stricto), present in well-characterized toluene-degrading strains that belong mostly to the Proteobacteria group, as well as environmental sequences retrieved from natural samples, including hydrocarbon-polluted aquifers and aquifer sediments, oil fields, sludge, and polluted soils. All 794-bp sequences retrieved from marine sediments (60 from Mallorca samples and 57 from Atlantic Islands samples) were located within this cluster. The 773-bp sequences were distributed into the remaining clusters. Cluster II included the previously described clusters T1 and T2 of unidentified BssA homologues, which constituted two distinct groups within this cluster (35). Sequences included in cluster II were associated with the in situ oxidation of BTEX compounds, including p-xylene, in sediment enrichments under sulfate-reducing conditions (35, 52). Only 26 of our sediment sequences clustered within this group and were retrieved primarily from the Atlantic Islands contaminated sediments.
Separation between clusters I and II could not be attributed to a distant phylogenetic origin of the sequences. Cluster I included a bssA-like sequence (GenBank accession number EHG03004; annotated as formate-C-acetyltransferase) from the nonproteobacterial Desulfotomaculum gibsoniae DSM 7213 genome (GI:355360296 and GenBank accession number AGJQ01000002.1) (Fig. 1; see Fig. S5 in the supplemental material). Sequences closely related to this hypothetical D. gibsoniae bssA gene were also found in environmental samples where methanogenesis was prevalent and where a Clostridium counterpart was suggested to be involved in toluene degradation (32, 53). Moreover, several sequences in cluster II were retrieved from environments dominated by sulfate-reducing conditions, where hydrocarbon oxidizers were associated with communities where Proteobacteria were dominant (35, 52).
Two subgroups were distinguishable in cluster III. The first one (cluster IIIa), defined as the nmsA sensu stricto cluster (38), included sequences from oil-polluted marine sites, contaminated aquifer sediments (38), and 2MN-specific nmsA gene products recently described for some SRB isolates and enrichments able to degrade naphthalene and 2MN (8, 9). The second subgroup (cluster IIIb) comprised sequences obtained from the Atlantic Islands sediments and a few additional sequences of marine origin, the putative bssA gene from the nonproteobacterial Desulfotomaculum sp. strain Ox39, and sequences retrieved from enrichments able to grow on o-xylene as the only carbon source. We could amplify nmsA-related sequences from the Prestige-contaminated FI samples, although our attempts to amplify the nmsA sequence from Deltaproteobacteria strain NaphS2 with primers 7757f and 8857r were unsuccessful, as previously described for the well-characterized 2MN-degrading consortium N47 (38).
Finally, cluster IV corresponded to sequences annotated as AssA. Two clusters could be distinguished within this group. Cluster IVa comprised several environmental sequences retrieved from a methanogenic octacosane-degrading enrichment (designated SDB) and from hydrocarbon-impacted aquifer sediments where the presence of methanogens in 16S rRNA libraries was reported as evidence of a methanogenic metabolism (30). Cluster IVa also included sequences from a methanogenic consortium growing on alkanes of different lengths, which had previously been annotated as AssA sequences in published (54) and unpublished (see Table S1 in the supplemental material) studies. None of the sequences retrieved from our sediment samples clustered within this group. It is worth noting that Archaea were not detected in the Atlantic Islands samples by using fluorescence in situ hybridization (FISH) analysis (41). Cluster IVb comprised sediment sequences derived in the current study, previously described alkane-specific AssA sequences retrieved from well-characterized alkane-degrading strains, and several environmental sequences from hydrocarbon-impacted sediments (Fig. 1). The only plausible factor that explained the separation into clusters IVa an IVb was the phylogenetic divergence between bacterial populations associated with methanogenic or nonmethanogenic conditions. Some other sequences retrieved from the methanogenic enrichment culture SDB (30) were found in cluster IVb. Thus, the methanogenic consortium in this culture probably had many bacterial counterparts responsible for hydrocarbon degradation.
Based on the literature review of the representative strains and enrichments identified in Fig. 1, the primary factor controlling the clustering of BssA-like sequences was likely the range of substrates that could be recognized by each sequence-encoded protein. Principal coordinate analysis (PCoA) based on the phylogenetic reconstruction of all BssA-like sequences grouped sequences into the same four clusters observed in the tree (Fig. 2A); coordinates P1 and P2 accounted for 30% and 21.7% of the variance, respectively. P1 separated proteins that could degrade alkane from those degrading aromatics, while P2 separated methyl-naphthalene from substituted monoaromatic or alkane-degrading enzymes. An apparent phylogeny-dependent clustering could also be observed for sequences with a known phylogenetic origin within each cluster.
Fig 2.
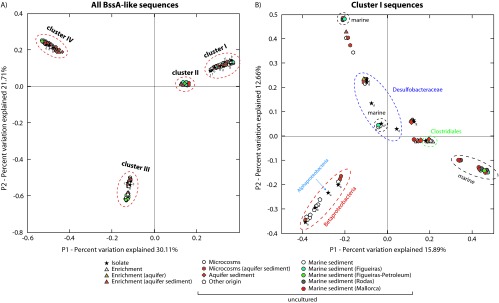
Unweighted PCoA plot based on distances between BssA-like sequences present in the sediment samples collected from marine sediments (Figueiras beach, Rodas beach, and Mallorca) and available in the databases (see Table S1 in the supplemental material). (A) PCoA of all BssA-like sequences; (B) PCoA of BssA cluster I sequences. The percentage of the variation between the samples by principal components is indicated on the axes. Spots of sequences from bacteria isolates (stars) are labeled with a letter that indicates the taxonomic affiliation: Alphaproteobacteria (a), Betaproteobacteria (b), Clostridiales (c), Geobacterales (g), and Desulfobacterales (d). Bacterial isolates from marine environments are marked with an asterisk. Colored circles indicate samples analyzed in this work. A sequence cutoff of a 2% amino acid similarity (OTU0.02) was used to select the sequences for the analysis.
The database search for bssA homologues also retrieved some entries that were not affiliated with any cluster despite their high level of homology with bssA-like sequences (see Fig. S5 in the supplemental material). These sequences include a second bssA homologue in the D. gibsoniae DSM7213 genome (GenBank accession number EHG03013), two putative PFL sequences amplified from a polluted aquifer, five metagenomic sequences derived from methane-oxidizing archaeal communities in the Santa Barbara Basin, the recently described hydroxybenzylsuccinate synthase gene hbsA from Desulfobacula toluolica, and a third bssA-like gene from this strain (55) (see Table S1 and Fig. S5 in the supplemental material). These sequences seemingly represent new phylogenetic branches of bssA homologues (see Fig. S5 in the supplemental material) (55).
Diversity of bssA-like genes in marine sediments of two different origins.
In the different libraries, the proportion of sequences present in each cluster varied according to the sample origin. The highest proportion of AssA sequences was observed in the M samples (Fig. 3). The majority (94%) of sequences in the three gene libraries obtained from Mallorca grouped into either BssA cluster I (60 sequences) or AssA cluster IVb (81 sequences), most of which were derived from the M-OIL sample (Fig. 3). The 9 remaining sequences were assigned to cluster II. In contrast to the Mallorca sediments, less than 4% of sequences in the FI samples were assigned to the AssA group. After 4 months of incubation of Mallorca sediments with crude oil, the increase in hydrocarbon levels was concomitant with a slight increase in the relative abundance of assA sequences (see Table S2b in the supplemental material). Because the Prestige oil was composed of both alkanes and aromatic hydrocarbons, the stimulation of a specific degrader population suggests that the alkane-degrading community responded more rapidly in these sediments; this conclusion was supported by a low nC18/phytane ratio (see Fig. S1b in the supplemental material) (56). Still, the presence of bssA-like sequences in the untreated sample suggests a low basal level of hydrocarbons in this area (e.g., through sporadic bilge water discharges), which would sustain such microbial populations.
Fig 3.

Frequency distribution of BssA-like sequence types in the different libraries constructed from sediment samples.
PCoA of all AssA sequences (cluster IV) identified cluster IVa at one end of the P2 coordinate (see Fig. S2 in the supplemental material). In addition, a group formed primarily by sequences retrieved from Passaic aquifer sediments (30) could be distinguished within cluster IVb. The phylogenetic tree showed that these sequences were more closely related to sequences from Desulfatibacillum alkenivorans AK-01 than to previously described environmental sequences retrieved from aquifer sediments (Fig. 1). This is consistent with the great abundance of Deltaproteobacteria in the Passaic samples and the detection of an active SRB population surviving on crude oil components. The minor differences observed between FI and M samples in cluster IVb suggest that these could belong to closely related microorganisms.
Independently of their environmental origin (geography, depth, or pollution level), the sequences retrieved from the marine sediment samples clustered together and separately from the other BssA-like sequences. In cluster I, some marine sediment sequences were associated with previously described proteobacterial and environmental BssA sequences, forming in most cases separated clusters (Fig. 1). The few marine-derived sequences available in the literature (38) formed a distinct cluster with BssA of the sole marine isolate D. toluolica Tol2 and with clone FI-TR006 retrieved in this work. In a PCoA of all sequences belonging to cluster I, both the P1 and P2 coordinates separated marine samples into two main clusters. One cluster was close to the Desulfobacterales and may represent diverging marine organisms within this group; the second cluster could not be assigned to any phylogenetic group (Fig. 2B). Betaproteobacteria formed a consistent cluster that contained no marine sediment sequence, and a group of sequences from Mallorca clustered with a central group related to Clostridiales isolates and sequences retrieved from methanogenic enrichments.
On the other hand, clusters II and III were clearly dominated by sequences from the Atlantic Islands sediments. The majority of sediment sequences clustered together with sequences of marine origin and formed a distinct group within cluster III (Fig. 1). More than 90% of the retrieved sequences in the Rodas beach sample (RI), where no AssA-type sequences were detected, and almost 90% of the sequences from the petroleum aggregate sample (FI-PET) clustered within cluster III and close to the putative Desulfotomaculum sp. Ox39 bssA sequence (Fig. 3). Incubation of this o-xylene-degrading strain with a mixture of m-xylene and o-xylene demonstrated that the degradation pathways of these two compounds were initiated by different fumarate-adding enzymes (57). It seems that the amplified sequence of this strain did not correspond to a toluene/xylene-specific bssA gene but rather to a related bssA-like enzyme. Our attempts to grow Desulfotomaculum sp. Ox39 with 2MN as the only carbon source were unsuccessful, but there were strong indications that this strain could cometabolize dimethylnaphthalenes when m-xylene was the major organic substrate (57). The main contaminants in the Prestige-affected sediments were naphthalene and alkylated derivatives (see Tables S2a and S2b in the supplemental material) (41, 58), including primarily methylated and dimethylated compounds. We thus associated the Atlantic Islands sequences in this cluster with the degradation of methylated naphthalenes. Interestingly, none of the sequences retrieved from the Mediterranean sediment microcosms were found in this group.
Rarefaction analysis of all marine sediment libraries showed that the level of diversity of the Mediterranean BssA-like sequences was low compared to that of the sequences derived from the Atlantic Islands samples, except for sequences from the oil-polluted M-OIL sample, which were mainly represented in clusters I and IVb (see Fig. S3 in the supplemental material). Comparison of BTEX compounds in the original Prestige oil and in the oil-treated microcosm indicated more rapid consumption of toluene and ethylbenzene than of xylenes in these samples (see Fig. S1a in the supplemental material), which could explain the near absence of Mallorca sequences in cluster II. This suggests that the biodegradation of hydrocarbon contaminants may require a longer exposure period. FI-PET displayed the greatest sequence diversity (see Fig. S3 in the supplemental material) and included sequences that were distributed among all clusters, as was the case for the remaining FI samples. In contrast, only two sequence types were observed in the RI sample collected from a less polluted beach in the same area: three sequences belonged to the same OTU0.02 in cluster I, and the remaining sequences formed a single OTU0.02 in cluster III. This sample displayed the lowest level of diversity and also showed a higher proportion of unspecific amplified sequences. Altogether, our data suggest that the stimulation of bacterial hydrocarbon oxidizers by the presence of pollutants resulted in a substantial increase in functional gene diversity compared to that in nonpolluted sites.
Protein sequence determinants of functional diversity.
The PCoA performed with all available BssA-like sequences clearly identified different clusters that could be related to the particular range of substrates utilized by each enzyme (Fig. 3A). If true, alignment of the protein sequences in each cluster should reveal both similarities in the active-site residues that are relevant for substrate specificity among homologues sharing similar substrates and major differences at these sites for enzymes that target different groups of substrates. The sequences in the amplified PCR fragments analyzed in this work included the active site of the enzyme, especially the conserved Cys residue directly involved in the abstraction of a hydrogen atom from the substrate (10). The binding pocket of the active site has been accurately defined by the structure of four crystallized proteins belonging to the radical enzyme family: Clostridium butyricum glycerol dehydratase (59), Archaeoglobus fulgidus PFL2 (60), Clostridium scatologenes hydroxyphenylacetate decarboxylase (61), and Escherichia coli pyruvate formate-lyase (62). The residue positions shaping this pocket have been identified in each structure and mapped in the multiple-sequence alignment. Despite the relatively low level of homology between the four proteins (41 to 53%), the position of these residues in the structure was conserved and could be superimposed between structures (61). To locate these positions in the BssA-like sequences, we built sequence logos for each cluster (see Fig. S4 in the supplemental material). Inspection of the different logos revealed a high degree of conservation between the sequences of the different clusters and identified variable regions and residues. A gap of 7 residues between positions 467 and 474 (all positions henceforth refer to T. aromatica K172 BssA numbering [13]) distinguished all sequences with respect to cluster I. In addition, cluster IV showed two single gaps at positions 508 and 528 and an insertion after position 603 with respect to the remaining sequences.
Positions 491 and 507 are two of the five main residues that shape the active-site pocket (in addition to the reactive cysteine) that were included in the amplified fragment. In addition, residues His536 and Glu637 of C. scatologenes hydroxyphenylacetate decarboxylase (corresponding to positions 509 and 611 in the alignment) were shown to interact with the substrate p-hydroxyphenylacetate (61). To map these residues in the protein active site, we built a tentative structure model of a representative protein sequence fragment from cluster III (FI-PET068) using C. butyricum glycerol dehydratase chain A (Protein Data Bank [PDB] accession number 1r9d_A) as the template (59), with which it shared 47% homology (Fig. 4). Several residues in the FI-PET068 sequence could not be modeled and are presented as a dotted black line. Glycyl radical enzymes share a global protein topology, consisting of a 10-stranded α/β-barrel composed of two antiparallel 5-stranded sheets surrounded by α-helixes. The active-site pocket is embedded in the core of this structure. The putative active-site pocket positions were occupied by a different residue in the different cluster proteins and are summarized in Fig. 4: position 491 was Ser in all clusters except cluster III, where it was Ala; positions 507 and 509, which flanked a characteristic gap in cluster IV, had a conserved Gly residue in all clusters except clusters IV, where the residues were Ser and Ala; and position 611 was conserved only in cluster I proteins (Fig. 4). To detect additional positions that could be relevant to protein specificity (the so-called specificity-determining sites [SDSs]), we used SPEER methods, which can define protein subfamilies and predict family protein functionally relevant residues (48, 49). Analysis of a multiple alignment of all available BssA-like sequences with SPEER software gave four different groups that matched the clusters previously defined in the phylogenetic tree and left 10 sequences grouped into five unclassified clusters. The SPEER software identified several residues predicted to be relevant for protein substrate specificity (Fig. 4). All predicted SDSs were located downstream of the conserved Cys in the active site. We observed stronger differences between the AssA group (cluster IV) and the remaining groups, such that all SDS residues in this group differed from the rest. Finally, position 605, which was occupied by a different amino acid residue in each cluster, could distinguish the four subfamilies. The structural consequences of all these changes are difficult to predict but could result in slight adjustments in the active-site cavity that would alter its properties (polarity, shape, and volume) and could determine differences in the range of substrates recognized by the enzyme.
Fig 4.

Mapping of the putative residues involved in substrate specificity in the FI-PET068 BssA fragment model structure obtained by using the crystal structure of Clostridium butyricum glycerol dehydratase chain A (PDB accession number 1r9d_A) as the template. Alpha-helices are represented as blue spirals, and β-strands are represented as red arrows. The active-site Cys residue is shown as a yellow sphere. The sequence fragments (residues 474, 504 to 507, and 527 to 529) that were not resolved in the model are shown as black dotted lines. The relevant active-site pocket residues and specificity-determining site (SDS) residues with the highest score according to SPEER analysis are depicted as orange spheres, except for positions 507 and 529, located in a nonmodeled sequence fragment, which are shown in white rectangles. Positions are numbered according to the T. aromatica K172 sequence (GenBank accession number CAA05052). The table shows the conserved amino acid residues occupying the predicted cluster, where X means that the position was not conserved.
Concluding remarks.
In this work, we have been able to detect different analogues of the bssA gene, which reflect the strong potential for hydrocarbon degradation in these coastal sediments. The bssA-like gene diversity was correlated with the presence of pollutants like monoaromatics and naphthalene derivatives. The long persistence of pollutants seemed to determine the presence of BssA-like variants; sequences retrieved from the Atlantic Islands sediments could be found in all clusters and were more diverse than Mallorca sequences. Mallorca samples were deliberately contaminated during a short incubation period and exhibited only small differences compared to nonpolluted sediments. The global analysis of the bssA gene suggested a broad substrate-dependent clustering and the existence of new clusters of analogous enzymes detected mainly by metagenomic analyses. Sequences of marine origin were separated from terrestrial homologues, indicating a geographical divergence within clusters. The description of new gene variants would require both enhanced probe design and the exploration of new environments, as this work has shown. Although the number of metagenomes in databases is increasing, the number of bssA sequences retrieved with this amplification-independent approach is limited. Thus, the detection of functional biomarkers will continue to be a good indicator of biodegradation potential.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by FEDER and by grants from the Spanish Ministry of Science and Technology (BIO2011-23615 and CSD2007-0005), from the BBVA Foundation (Biocon 05-094/06), and from the Junta de Andalucía (P08-CVI03591).
We thank Carlos Durán and the Centro de Investigaciones Submarinas, S.L. (CIS), for skillful help during the sampling campaigns in the Atlantic Islands; Miguel Anxo Murado at the Instituto de Investigaciones Mariñas (CSIC) in Vigo for kindly providing laboratory facilities; and Patricia Marín for excellent technical assistance. We are grateful to Mercedes Urdiain and Ana Suárez-Suárez for their help in processing the Mallorca samples and to Antonio Rosselló for his help as skipper in the sampling campaigns in Mallorca. We thank Frederick von Netzer and Tillmann Lueders for kindly providing the sequence data sets prior to public release.
Footnotes
Published ahead of print 5 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03934-12.
REFERENCES
- 1. Brune A, Frenzel P, Cypionka H. 2000. Life at the oxic-anoxic interface: microbial activities and adaptations. FEMS Microbiol. Rev. 24:691–710 [DOI] [PubMed] [Google Scholar]
- 2. Rockne K, Strand S. 1998. Biodegradation of bicyclic and polycyclic aromatic hydrocarbons in anaerobic enrichments. Environ. Sci. Technol. 32:3962–3967 [Google Scholar]
- 3. Grossi V, Cravo-Laureau C, Guyoneaud R, Ranchou-Peyruse A, Hirschler-Réa A. 2008. Metabolism of n-alkanes and n-alkenes by anaerobic bacteria: a summary. Org. Geochem. 39:1197–1203 [Google Scholar]
- 4. Chakraborty R, Coates JD. 2004. Anaerobic degradation of monoaromatic hydrocarbons. Appl. Microbiol. Biotechnol. 64:437–446 [DOI] [PubMed] [Google Scholar]
- 5. Spormann AM, Widdel F. 2000. Metabolism of alkylbenzenes, alkanes, and other hydrocarbons in anaerobic bacteria. Biodegradation 11:85–105 [DOI] [PubMed] [Google Scholar]
- 6. Heider J. 2007. Adding handles to unhandy substrates: anaerobic hydrocarbon activation mechanisms. Curr. Opin. Chem. Biol. 11:188–194 [DOI] [PubMed] [Google Scholar]
- 7. Zhang X, Young LY. 1997. Carboxylation as an initial reaction in the anaerobic metabolism of naphthalene and phenanthrene by sulfidogenic consortia. Appl. Environ. Microbiol. 63:4759–4764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DiDonato RJ, Jr, Young ND, Butler JE, Chin KJ, Hixson KK, Mouser P, Lipton MS, DeBoy R, Methe BA. 2010. Genome sequence of the deltaproteobacterial strain NaphS2 and analysis of differential gene expression during anaerobic growth on naphthalene. PLoS One 5:e14072 doi: 10.1371/journal.pone.0014072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Selesi D, Jehmlich N, von Bergen M, Schmidt F, Rattei T, Tischler P, Lueders T, Meckenstock RU. 2010. Combined genomic and proteomic approaches identify gene clusters involved in anaerobic 2-methylnaphthalene degradation in the sulfate-reducing enrichment culture N47. J. Bacteriol. 192:295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Selmer T, Pierik AJ, Heider J. 2005. New glycyl radical enzymes catalysing key metabolic steps in anaerobic bacteria. Biol. Chem. 386:981–988 [DOI] [PubMed] [Google Scholar]
- 11. Biegert T, Fuchs G, Heider J. 1996. Evidence that anaerobic oxidation of toluene in the denitrifying bacterium Thauera aromatica is initiated by formation of benzylsuccinate from toluene and fumarate. Eur. J. Biochem. 238:661–668 [DOI] [PubMed] [Google Scholar]
- 12. Beller HR, Spormann AM. 1997. Anaerobic activation of toluene and o-xylene by addition to fumarate in denitrifying strain T. J. Bacteriol. 179:670–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leuthner B, Leutwein C, Schulz H, Horth P, Haehnel W, Schiltz E, Schagger H, Heider J. 1998. Biochemical and genetic characterization of benzylsuccinate synthase from Thauera aromatica: a new glycyl radical enzyme catalysing the first step in anaerobic toluene metabolism. Mol. Microbiol. 28:615–628 [DOI] [PubMed] [Google Scholar]
- 14. Krieger CJ, Beller HR, Reinhard M, Spormann AM. 1999. Initial reactions in anaerobic oxidation of m-xylene by the denitrifying bacterium Azoarcus sp. strain T. J. Bacteriol. 181:6403–6410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Müller JA, Galushko AS, Kappler A, Schink B. 1999. Anaerobic degradation of m-cresol by Desulfobacterium cetonicum is initiated by formation of 3-hydroxybenzylsuccinate. Arch. Microbiol. 172:287–294 [DOI] [PubMed] [Google Scholar]
- 16. Müller JA, Galushko AS, Kappler A, Schink B. 2001. Initiation of anaerobic degradation of p-cresol by formation of 4-hydroxybenzylsuccinate in Desulfobacterium cetonicum. J. Bacteriol. 183:752–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kniemeyer O, Fischer T, Wilkes H, Glockner FO, Widdel F. 2003. Anaerobic degradation of ethylbenzene by a new type of marine sulfate-reducing bacterium. Appl. Environ. Microbiol. 69:760–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Musat F, Galushko A, Jacob J, Widdel F, Kube M, Reinhardt R, Wilkes H, Schink B, Rabus R. 2009. Anaerobic degradation of naphthalene and 2-methylnaphthalene by strains of marine sulfate-reducing bacteria. Environ. Microbiol. 11:209–219 [DOI] [PubMed] [Google Scholar]
- 19. Rabus R, Wilkes H, Behrends A, Armstroff A, Fischer T, Pierik AJ, Widdel F. 2001. Anaerobic initial reaction of n-alkanes in a denitrifying bacterium: evidence for (1-methylpentyl)succinate as initial product and for involvement of an organic radical in n-hexane metabolism. J. Bacteriol. 183:1707–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kropp KG, Davidova IA, Suflita JM. 2000. Anaerobic oxidation of n-dodecane by an addition reaction in a sulfate-reducing bacterial enrichment culture. Appl. Environ. Microbiol. 66:5393–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grundmann O, Behrends A, Rabus R, Amann J, Halder T, Heider J, Widdel F. 2008. Genes encoding the candidate enzyme for anaerobic activation of n-alkanes in the denitrifying bacterium, strain HxN1. Environ. Microbiol. 10:376–385 [DOI] [PubMed] [Google Scholar]
- 22. Callaghan AV, Wawrik B, Ni Chadhain SM, Young LY, Zylstra GJ. 2008. Anaerobic alkane-degrading strain AK-01 contains two alkylsuccinate synthase genes. Biochem. Biophys. Res. Commun. 366:142–148 [DOI] [PubMed] [Google Scholar]
- 23. Beller HR, Kane SR, Legler TC, McKelvie JR, Lollar BS, Pearson F, Balser L, Mackay DM. 2008. Comparative assessments of benzene, toluene, and xylene natural attenuation by quantitative polymerase chain reaction analysis of a catabolic gene, signature metabolites, and compound-specific isotope analysis. Environ. Sci. Technol. 42:6065–6072 [DOI] [PubMed] [Google Scholar]
- 24. Achong GR, Rodriguez AM, Spormann AM. 2001. Benzylsuccinate synthase of Azoarcus sp. strain T: cloning, sequencing, transcriptional organization, and its role in anaerobic toluene and m-xylene mineralization. J. Bacteriol. 183:6763–6770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Coschigano PW, Wehrman TS, Young LY. 1998. Identification and analysis of genes involved in anaerobic toluene metabolism by strain T1: putative role of a glycine free radical. Appl. Environ. Microbiol. 64:1650–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kane SR, Beller HR, Legler TC, Anderson RT. 2002. Biochemical and genetic evidence of benzylsuccinate synthase in toluene-degrading, ferric iron-reducing Geobacter metallireducens. Biodegradation 13:149–154 [DOI] [PubMed] [Google Scholar]
- 27. Kube M, Heider J, Amann J, Hufnagel P, Kuhner S, Beck A, Reinhardt R, Rabus R. 2004. Genes involved in the anaerobic degradation of toluene in a denitrifying bacterium, strain EbN1. Arch. Microbiol. 181:182–194 [DOI] [PubMed] [Google Scholar]
- 28. Shinoda Y, Sakai Y, Uenishi H, Uchihashi Y, Hiraishi A, Yukawa H, Yurimoto H, Kato N. 2004. Aerobic and anaerobic toluene degradation by a newly isolated denitrifying bacterium, Thauera sp. strain DNT-1. Appl. Environ. Microbiol. 70:1385–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shinoda Y, Akagi J, Uchihashi Y, Hiraishi A, Yukawa H, Yurimoto H, Sakai Y, Kato N. 2005. Anaerobic degradation of aromatic compounds by Magnetospirillum strains: isolation and degradation genes. Biosci. Biotechnol. Biochem. 69:1483–1491 [DOI] [PubMed] [Google Scholar]
- 30. Callaghan AV, Davidova IA, Savage-Ashlock K, Parisi VA, Gieg LM, Suflita JM, Kukor JJ, Wawrik B. 2010. Diversity of benzyl- and alkylsuccinate synthase genes in hydrocarbon-impacted environments and enrichment cultures. Environ. Sci. Technol. 44:7287–7294 [DOI] [PubMed] [Google Scholar]
- 31. Botton S, van Harmelen M, Braster M, Parsons JR, Roling WF. 2007. Dominance of Geobacteraceae in BTX-degrading enrichments from an iron-reducing aquifer. FEMS Microbiol. Ecol. 62:118–130 [DOI] [PubMed] [Google Scholar]
- 32. Washer CE, Edwards EA. 2007. Identification and expression of benzylsuccinate synthase genes in a toluene-degrading methanogenic consortium. Appl. Environ. Microbiol. 73:1367–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beller HR, Kane SR, Legler TC, Alvarez PJ. 2002. A real-time polymerase chain reaction method for monitoring anaerobic, hydrocarbon-degrading bacteria based on a catabolic gene. Environ. Sci. Technol. 36:3977–3984 [DOI] [PubMed] [Google Scholar]
- 34. Oka AR, Phelps CD, Zhu X, Saber DL, Young LY. 2011. Dual biomarkers of anaerobic hydrocarbon degradation in historically contaminated groundwater. Environ. Sci. Technol. 45:3407–3414 [DOI] [PubMed] [Google Scholar]
- 35. Winderl C, Schaefer S, Lueders T. 2007. Detection of anaerobic toluene and hydrocarbon degraders in contaminated aquifers using benzylsuccinate synthase (bssA) genes as a functional marker. Environ. Microbiol. 9:1035–1046 [DOI] [PubMed] [Google Scholar]
- 36. Pilloni G, von Netzer F, Engel M, Lueders T. 2011. Electron acceptor-dependent identification of key anaerobic toluene degraders at a tar-oil-contaminated aquifer by Pyro-SIP. FEMS Microbiol. Ecol. 78:165–175 [DOI] [PubMed] [Google Scholar]
- 37. Staats M, Braster M, Roling WF. 2011. Molecular diversity and distribution of aromatic hydrocarbon-degrading anaerobes across a landfill leachate plume. Environ. Microbiol. 13:1216–1227 [DOI] [PubMed] [Google Scholar]
- 38. von Netzer F, Pilloni G, Kleindienst S, Krüger M, Knittel K, Gründger F, Lueders T. 2013. Enhanced gene detection assays for fumarate-adding enzymes allow uncovering anaerobic hydrocarbon degraders in terrestrial and marine systems. Appl. Environ. Microbiol. 79:543–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zedelius J, Rabus R, Grundmann O, Werner I, Brodkorb D, Schreiber F, Ehrenreich P, Behrends A, Wilkes H, Kube M, Reinhardt R, Widdel F. 2011. Alkane degradation under anoxic conditions by a nitrate-reducing bacterium with possible involvement of the electron acceptor in substrate activation. Environ. Microbiol. Rep. 3:125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mbadinga SM, Li KP, Zhou L, Wang LY, Yang SZ, Liu JF, Gu JD, Mu BZ. 2012. Analysis of alkane-dependent methanogenic community derived from production water of a high-temperature petroleum reservoir. Appl. Microbiol. Biotechnol. 96:531–542 [DOI] [PubMed] [Google Scholar]
- 41. Acosta-González A, Rosselló-Mora R, Marqués S. 2013. Characterization of the anaerobic microbial community in oil-polluted subtidal sediments: aromatic biodegradation potential after the Prestige oil spill. Environ. Microbiol. 15:77–92 [DOI] [PubMed] [Google Scholar]
- 42. Alonso-Gutiérrez J, Costa MM, Figueras A, Albaigés J, Viñas L, Solanas AM, Novoa B. 2008. Alcanivorax strain detected among the cultured bacterial community from sediments affected by the Prestige oil spill. Mar. Ecol. Prog. Ser. 362:25–36 [Google Scholar]
- 43. Suárez-Suárez A, López-López A, Tovar-Sánchez A, Yarza P, Orfila A, Terrados J, Arnds J, Marqués S, Niemann H, Schmitt-Kopplin P, Amann R, Rossello-Mora R. 2011. Response of sulfate-reducing bacteria to an artificial oil-spill in a coastal marine sediment. Environ. Microbiol. 13:1488–1499 [DOI] [PubMed] [Google Scholar]
- 44. Shin H-S. 2012. Determination of MTBE, TBA and BTEX in soil by headspace gas chromatography-mass spectrometry. Bull. Korean Chem. Soc. 33:1693–1698 [Google Scholar]
- 45. Zhou J, Bruns MA, Tiedje JM. 1996. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62:316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Markowitz VM, Chen IM, Chu K, Szeto E, Palaniappan K, Grechkin Y, Ratner A, Jacob B, Pati A, Huntemann M, Liolios K, Pagani I, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. 2012. IMG/M: the integrated metagenome data management and comparative analysis system. Nucleic Acids Res. 40:D123–D129 doi: 10.1093/nar/gkr975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hamady M, Lozupone C, Knight R. 2010. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 4:17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chakrabarti S, Bryant SH, Panchenko AR. 2007. Functional specificity lies within the properties and evolutionary changes of amino acids. J. Mol. Biol. 373:801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chakraborty A, Mandloi S, Lanczycki CJ, Panchenko AR, Chakrabarti S. 2012. SPEER-SERVER: a Web server for prediction of protein specificity determining sites. Nucleic Acids Res. 40:W242–W248 doi: 10.1093/nar/gks559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kelley LA, Sternberg MJE. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371 [DOI] [PubMed] [Google Scholar]
- 52. Herrmann S, Vogt C, Fischer A, Kuppardt A, Richnow H-H. 2009. Characterization of anaerobic xylene biodegradation by two-dimensional isotope fractionation analysis. Environ. Microbiol. Rep. 1:535–544 [DOI] [PubMed] [Google Scholar]
- 53. Fowler SJ, Dong X, Sensen CW, Suflita JM, Gieg LM. 2012. Methanogenic toluene metabolism: community structure and intermediates. Environ. Microbiol. 14:754–764 [DOI] [PubMed] [Google Scholar]
- 54. Wang L-Y, Li W, Mbadinga SM, Liu J-F, Gu J-D, Mu B-Z. 2012. Methanogenic microbial community composition of oily sludge and its enrichment amended with alkanes incubated for over 500 days. Geomicrobiol. J. 29:716–726 [Google Scholar]
- 55. Wöhlbrand L, Kube M, Mussmann M, Jarling R, Beck A, Amann R, Wilkes H, Reinhardt R, Rabus R. 4 September 2012. Complete genome, catabolic sub-proteomes and key-metabolites of Desulfobacula toluolica Tol2, a marine, aromatic compound-degrading, sulfate-reducing bacterium. Environ. Microbiol. [Epub ahead of print.] doi: 10.1111/j.1462-2920.2012.02885.x [DOI] [PubMed] [Google Scholar]
- 56. Díez S, Sabaté J, Viñas M, Bayona JM, Solanas AM, Albaigés J. 2005. The Prestige oil spill. I. Biodegradation of a heavy fuel oil under simulated conditions. Environ. Toxicol. Chem. 24:2203–2217 [DOI] [PubMed] [Google Scholar]
- 57. Morasch B, Schink B, Tebbe C, Meckenstock R. 2004. Degradation of o-xylene and m-xylene by a novel sulfate-reducer belonging to the genus Desulfotomaculum. Arch. Microbiol. 181:407–417 [DOI] [PubMed] [Google Scholar]
- 58. Alzaga R, Montuori P, Ortíz L, Bayona JM, Albaigés J. 2004. Fast solid-phase extraction-gas chromatography-mass spectrometry procedure for oil fingerprinting: application to the Prestige oil spill. J. Chromatogr. A 1025:133–138 [DOI] [PubMed] [Google Scholar]
- 59. O'Brien JR, Raynaud C, Croux C, Girbal L, Soucaille P, Lanzilotta WN. 2004. Insight into the mechanism of the B12-independent glycerol dehydratase from Clostridium butyricum: preliminary biochemical and structural characterization. Biochemistry 43:4635–4645 [DOI] [PubMed] [Google Scholar]
- 60. Lehtiö L, Grossmann JG, Kokona B, Fairman R, Goldman A. 2006. Crystal structure of a glycyl radical enzyme from Archaeoglobus fulgidus. J. Mol. Biol. 357:221–235 [DOI] [PubMed] [Google Scholar]
- 61. Martins BM, Blaser M, Feliks M, Ullmann GM, Buckel W, Selmer T. 2011. Structural basis for a Kolbe-type decarboxylation catalyzed by a glycyl radical enzyme. J. Am. Chem. Soc. 133:14666–14674 [DOI] [PubMed] [Google Scholar]
- 62. Becker A, Fritz-Wolf K, Kabsch W, Knappe J, Schultz S, Volker Wagner AF. 1999. Structure and mechanism of the glycyl radical enzyme pyruvate formate-lyase. Nat. Struct. Biol. 6:969–975 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.