

Abstract
Saturation point mutagenesis was carried out at position 479 in the polyhydroxyalkanoate (PHA) synthase from Chromobacterium sp. strain USM2 (PhaCCs) with specificities for short-chain-length (SCL) [(R)-3-hydroxybutyrate (3HB) and (R)-3-hydroxyvalerate (3HV)] and medium-chain-length (MCL) [(R)-3-hydroxyhexanoate (3HHx)] monomers in an effort to enhance the specificity of the enzyme for 3HHx. A maximum 4-fold increase in 3HHx incorporation and a 1.6-fold increase in PHA biosynthesis, more than the wild-type synthase, was achieved using selected mutant synthases. These increases were subsequently correlated with improved synthase activity and increased preference of PhaCCs for 3HHx monomers. We found that substitutions with uncharged residues were beneficial, as they resulted in enhanced PHA production and/or 3HHx incorporation. Further analysis led to postulations that the size and geometry of the substrate-binding pocket are determinants of PHA accumulation, 3HHx fraction, and chain length specificity. In vitro activities for polymerization of 3HV and 3HHx monomers were consistent with in vivo substrate specificities. Ultimately, the preference shown by wild-type and mutant synthases for either SCL (C4 and C5) or MCL (C6) substrates substantiates the fundamental classification of PHA synthases.
INTRODUCTION
Polyhydroxyalkanoates (PHAs) are polyoxoesters synthesized by a wide range of bacteria as intracellular storage materials (1, 2). The unique properties of PHA, such as its thermoplastic capabilities and inherent degradability, have made it a worthwhile alternative to conventional petrochemical plastics (3–5). These biobased polymers vary substantially in their compositions, resulting in a huge diversity of material properties (6), ranging from the strong but brittle 3-hydroxybutyrate (3HB) (C4) homopolymer to the flexible copolymer of 3HB and 3-hydroxyhexanoate (3HHx) (C6).
As the central enzyme for PHA production, the PHA synthase, or PhaC, determines the compositional variation of the resultant polymer to a certain extent, and hence, enzyme evolution has been employed for the generation of mutant enzymes with greatly enhanced properties. Extensive efforts in this direction have yielded various mutant PHA synthases with dramatic improvements in their activities for polymerization, as well as novel substrate-binding properties (7, 8). Engineered synthases with up to 5-fold enhancement in activity, increased efficiency of substrate incorporation, and extended substrate range have been documented in several studies (9–13). These effects, particularly those related to substrate specificity, are of industrial importance, as they are feasible strategies for efficient production of PHAs with desirable properties.
PHA synthases are generally divided into four classes based on their primary amino acid sequences and substrate specificities (preference for short-chain-length [SCL] [C3 to C5] or medium-chain-length [MCL] [C6 to C14] monomers) (14, 15). Some synthases are difficult to classify with this system as, intriguingly, they are able to polymerize both SCL and MCL monomers, such as the PHA synthase from Chromobacterium sp. strain USM2 (PhaCCs) (16). While a few other bacteria, such as Aeromonas caviae (17, 18), Aeromonas hydrophila (19, 20), and Rhodococcus ruber (21), have been reported to also possess synthases that exhibit specificity toward both SCL and MCL monomers, PhaCCs exhibited superior polymerization activity (22) that provides the added advantage of increased PHA production efficiency. Mutations to further enhance the preference of this highly active synthase with broad substrate specificity for comonomer units can greatly improve its versatility, making PhaCCs the chosen subject for this study.
Multiple-sequence alignment of PhaCCs with other comprehensively studied synthases in this area (belonging to class I types, Ralstonia eutropha and A. caviae, as well as class II PHA synthases [PhaC1] from Pseudomonas sp. strain 61-3, Pseudomonas aeruginosa, and Pseudomonas oleovorans) revealed a highly conserved alanine (class I) or glutamine (class II) residue (Fig. 1) located at position 479 in PhaCCs. Functional implications of amino acid substitutions at the position based on previous studies include alterations in substrate specificity, and in certain cases, variations in the molecular weight of the resultant polymer were observed (10, 12, 23). Position 479 in PhaCCs, therefore, appears to be a suitable candidate for the mutagenesis approach in order to achieve similar positive effects.
Fig 1.

Partial amino acid sequence alignment of the PHA synthase from Chromobacterium sp. strain USM2 (PhaCCs) with those from R. eutropha (PhaCRe), A. caviae (PhaCAc), Pseudomonas sp. strain 61-3 (PhaC1Ps), P. aeruginosa (PhaC1Pa), and P. oleovorans (PhaC1Po). Classes I and II indicate the classification of PHA synthases. Putative active-site residues, aspartic acid (D) and histidine (H), are shaded in gray. The asterisks mark highly conserved amino acid residues, which are present in all PHA synthases. Amino acid residues corresponding to A479 in PhaCCs are boxed.
In the present study, saturation mutagenesis of the PhaCCs-encoding gene (phaCCs) was carried out, and the mutagenized fragments were introduced into Escherichia coli LS5218, individually, for evaluation of poly[(R)-3-hydroxybutyrate-co-(R)-3-hydroxyhexanoate] [P(3HB-co-3HHx)] production from dodecanoic acid. With the lack of structural data for PHA synthases, analysis of mutant enzymes generated in this study will contribute to a deeper understanding of PhaC enzymology. Additionally, increased understanding of reaction mechanisms and structure-function relationships of PhaCCs will enable unrestrained application of this highly useful enzyme. While numerous PHA synthases have been genetically engineered and characterized to date, a majority of these studies have been conducted using (R)-3-hydroxybutyryl-coenzyme A (CoA) (3HB-CoA) as the substrate for PhaC. Little is known for most PHA synthases about the substrate specificity in relation to the formation of PHA copolymers. With information on substrate specificity for in vitro polymerization of secondary monomers, (R)-3-hydroxyvaleryl-CoA (3HV-CoA) and (R)-3-hydroxyhexanoyl-CoA (3HHx-CoA), PhaCCs can serve as a benchmark for comparison with other synthases in similar studies. Alteration in the substrate specificity of PhaCCs is also of interest as a means of incorporating novel substrate analogues into the polymer chain by a PhaCCs-catalyzed polymerization reaction.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
E. coli JM109 (Stratagene, La Jolla, CA) was used for all standard cloning procedures and as the host strain for expression of pGEM″AB(phaCCs) (22) to obtain crude PhaCCs enzyme. E. coli BL21(DE3) (Novagen, Madison, WI) was used as the host strain for expression of pET-phaCCs (22) to obtain purified PhaCCs enzyme. To examine the effects of amino acid substitutions, E. coli LS5218 [fadR601 atoC(Con)] (24) was cotransformed with pGEM″AB(phaCCs) and pBPP-phaJAc (kindly provided by Toshiaki Fukui, Tokyo Institute of Technology, Tokyo, Japan) for expression of (R)-specific enoyl-CoA hydratase (PhaJ) to enable biosynthesis of P(3HB-co-3HHx). The transformants were cultivated for 72 h at 30°C in M9 medium (25) supplemented with 0.25% (wt/vol) dodecanoic acid as the sole carbon source, solubilized with 0.4% (vol/vol) Brij 35. When needed, 100 μg/ml of ampicillin and/or 50 μg/ml of kanamycin was added to maintain plasmid stability.
DNA manipulation and sequencing.
Standard recombinant DNA manipulations (25) were used for plasmid isolation, restriction enzyme digestion, ligation, and bacterial transformation. All of the enzymes (Toyobo, Osaka, Japan) were used according to the manufacturers' protocols. All other chemicals used were of analytical grade. DNA sequencing for analysis of the mutation points and confirmation of plasmid constructs was carried out by the dideoxy chain termination method with a Prism 310 genetic analyzer DNA sequencer using a BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, Foster City, CA).
Site-specific saturation mutagenesis of A479.
Site-specific saturation mutagenesis at the A479 position was carried out via PCR using tail-to-tail forward and reverse primers. The pGEM″AB(phaCCs) and pET-phaCCs plasmids harboring phaCCs served as the templates for amplification. The primers used were the forward primer A479X (for amino acid substitution) and the reverse primer A479rev (see Table S1 in the supplemental material), with PCR cycle conditions as follows: an initial activation step of 94°C for 2 min and 20 cycles of 98°C for 10 s, 61°C for 30 s, and 68°C for 7.5 min. The amplified products were self-ligated, and the phaCCs genes were sequenced to verify the correctness of each substituted codon, as well as the overall gene sequence.
Analysis of PHA.
To determine the PHA content and monomer composition, the freeze-dried cells were subjected to methanolysis (26), followed by gas chromatography analysis on a Shimadzu (Kyoto, Japan) GC-2010 system equipped with a Neutra Bond-1 capillary column (30 m by 0.25 mm) and a flame ionization detector. SPSS 17.0 for Windows was employed for statistical analysis. Tukey's honestly significant difference (HSD) test was used, in conjunction with analysis of variance (ANOVA) for single-step multiple comparisons. Significant differences in PHA content and 3HHx content were determined.
PHA was extracted by refluxing freeze-dried cells with chloroform for 48 h at 60°C and purified by precipitation with hexane and methanol, consecutively. Molecular weights (weight-average molecular weight [Mw] and number-average molecular weight [Mn]) of the polymers were obtained using the Shimadzu (Kyoto, Japan) LC-10A gel permeation chromatography system with a Shodex K-806 column (Showa Denko, Tokyo, Japan) at 40°C. Chloroform was used as the eluent at a flow rate of 0.8 ml/min, and the concentration of samples used was 1.0 mg/ml. The calibration curve used for estimation of molecular weight was generated using polystyrene standards with low polydispersity (Showa Denko, Tokyo, Japan).
The differential scanning calorimetry (DSC) data were recorded at a temperature ranging from −90 to 210°C on a Perkin-Elmer (Waltham, MA) DSC 8500 instrument equipped with a cooling accessory under a nitrogen flow rate of 20 ml/min. The polymers (approximately 3 mg) were encapsulated in aluminum pans and heated from 100 to 210°C at 20°C/min. The melting temperature (Tm) and enthalpy of fusion (ΔHm) were determined from the DSC endotherms. The glass transition temperature (Tg), which was analyzed based on the DSC curves from the second heating, was taken as the midpoint of the heat capacity change.
The composition and sequence distribution of the polymers were determined by 1H and 13C nuclear magnetic resonance (NMR) spectra (Varian NMR System 500). The 500-MHz 1H NMR spectrum was recorded at 27°C in a CDCl3 solution of PHA (10 mg/ml), with 5-ms pulse width, 32,000 data points, and 32 accumulations. The 125-MHz 13C NMR spectrum was recorded at 27°C in a CDCl3 solution of PHA (20 mg/ml) with 5-ms pulse width (pulse angle, 45°), 0.7-s pulse repetition, 23,000-Hz spectral width, 32,000 data points, and 10,000 accumulations. Tetramethylsilane was used as an internal chemical-shift standard.
Western blot analysis.
Extraction of crude PhaCCs and determination of the total protein concentration were performed as described previously (22). A total of 40 μg of protein fractions prepared from the supernatant of the disrupted cells was separated on 10% SDS-PAGE using the Mini-Protean 3 electrophoresis system (Bio-Rad, Hercules, CA). The separated proteins were then transferred to an Invitrolon polyvinylidene difluoride (PVDF) membrane (Invitrogen, Carlsbad, CA) using a Mini Trans-Blot SD Semi-Dry Electrophoretic Transfer Cell (Bio-Rad, Hercules, CA). Western blot analysis of PhaC was carried out using specific rabbit antiserum raised against the C-terminal peptides of PhaC as described previously (27). PhaC was detected using goat anti-rabbit IgG conjugated with alkaline phosphatase as a secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
In vitro enzymatic assay of crude and purified PhaCCs.
Characterization of the wild-type and mutant synthases in terms of activity and substrate specificity was carried out by an in vitro enzymatic assay. Crude and purified synthase extracts were prepared as described previously (22). The assay mixture contained either 40 μg of protein from the soluble fraction of the crude synthase extract or a 7.5 nM to 0.3 μM concentration of purified synthase extract. Substrates used in the synthase activity assays, (R)-3HB-CoA, (R)-3HV-CoA, and (R)-3HHx-CoA, were prepared based on a mixed-anhydride method (28). (R)-3HB-CoA was used as the substrate in reaction mixtures containing crude or purified synthase, while (R)-3HV-CoA and (R)-3HHx-CoA were employed only for reactions using purified synthase. All substrates were used at a final concentration of 600 μM. The concentration of CoA was determined spectrophotometrically using a molar absorption coefficient of 13,600 M−1 cm−1 at 412 nm. One unit of enzyme activity (U) is defined as the amount of enzyme that catalyzed the release of 1 μmol CoA/min. Enzyme assays were performed in triplicate.
RESULTS
Analysis of PHA production and in vivo level of PhaCCs.
To determine the mutational effects with respect to PHA accumulation and monomer composition, the content and monomer composition of the produced PHA were analyzed by gas chromatography. As shown in Fig. 2A and Table S2 in the supplemental material, all strains were able to accumulate considerable amounts of PHA, with the exception of those harboring A479F/K/L/N/R mutations. The possibility that the amino acid substitution at this position affected the primary polymerization function of the enzyme cannot be dismissed, although these strains were still able to accumulate trace amounts (less than 1 weight percent [wt%]) of PHA. Overall, the PHA content varied from trace amounts up to approximately 60 wt% for the A479G mutant, which was approximately 1.6-fold higher than that produced using the wild-type synthase. Higher elevations in PHA production could be achieved with (in increasing order of PHA content) valine, methionine, tryptophan, and glycine substitutions.
Fig 2.

Contents (A) and 3HHx monomer fractions (B) of PHA accumulated by E. coli LS5218 transformants harboring wild-type and individual A479X mutant PhaCCs. E. coli LS5218 transformants were cultivated in 100 ml of M9 medium containing 0.25% (wt/vol) dodecanoic acid and 0.4% (vol/vol) Brij 35 for 72 h at 30°C. Ampicillin and kanamycin were supplemented at final concentrations of 100 μg/ml and 50 μg/ml, respectively, for plasmid maintenance. The data shown are means of triplicate experiments. Mean values indicated by different letters are significantly different (Tukey's HSD test; P < 0.05). The error bars indicate standard deviations. (C) Comparison of the expression levels of wild-type and A479X mutants of PhaCCs in E. coli LS5218 transformants by Western blotting. A total of 40 μg of protein was used for the analysis.
The A479X mutation also affected the monomeric composition of the resultant polymer (Fig. 2B), in fact, to a greater extent than it did the PHA content. Ranging from as low as no 3HHx content up to 6.6 mol%, a maximum of 4-fold-higher 3HHx content than that with the wild-type synthase was achieved with the A479S mutant. Some amino acid substitutions, on the other hand, diminished 3HHx fractions completely, thereby resulting in mutant synthases capable of poly[(R)-3-hydroxybutyrate] [P(3HB)] synthesis only. All of these mutants, coincidentally, were also found to be incapable of PHA accumulation beyond 1 wt%, except for one (A479E) that could facilitate PHA accumulation of 3.5 wt%. A noteworthy observation was that certain amino acid substitutions resulted in mutant synthases with simultaneous enhancements in both aspects (PHA content and 3HHx fraction), namely, A479G/M/Q/T/V.
Subsequently, Western blot analysis was carried out to investigate the effect of the A479 mutation on expression of the synthase. From the profile shown in Fig. 2C, no distinguishable variation could be seen in the expression level of each mutant synthase compared to that of the wild-type synthase.
Determination of PhaCCs activity in E. coli.
Different enzyme-engineering approaches for the creation of various mutant PHA synthases have claimed success in the improvement of synthase activities (7). To verify if similar effects were also accomplished by the A479X mutation in PhaCCs, wild-type PhaCCs and four mutants (A479C/E/G/N) were selected based on distinct variations in PHA content, and the crude enzymes obtained from cultures were subjected to an in vitro assay of PHA synthase activity. The activity values presented in Table 1 confirmed the effects of the selected mutations on the catalytic activity of PhaCCs. While the strain harboring the wild-type synthase exhibited 121 ± 9 U/g of activity, those harboring mutant synthases (A479C/E/N) showed lower activity, in the approximate range of 24 to 83 U/g, and the strain harboring A479G recorded an activity of 180 ± 20 U/g. An apparent observation was that the increase or decrease in synthase activity corresponded to PHA accumulation by strains harboring each of these mutant synthases, although not in a proportional manner. To validate this observation, the PHA synthase activities of the remaining PhaCCs mutants were determined and a correlation was made between the PHA content and the activity values obtained. From the plot shown in Fig. 3, consistency could be seen in the in vitro and in vivo catalytic activities of these mutant synthases.
Table 1.
In vitro and in vivo activities of wild-type and A479X mutants of PhaCCsa
| PHA synthaseb | PHA content (wt%)c | Relative PHA content (%)d | PHA synthase activity (U/g)e | Relative PHA synthase activity (%)f |
|---|---|---|---|---|
| Wild type | 35.6 ± 2.9 | 100 ± 8 | 121 ± 9 | 100 ± 7 |
| A479C | 19.0 ± 6.9 | 53 ± 19 | 83 ± 2 | 69 ± 2 |
| A479E | 3.5 ± 1.8 | 10 ± 5 | 31 ± 6 | 26 ± 5 |
| A479G | 58.2 ± 2.8 | 163 ± 8 | 180 ± 20 | 149 ± 17 |
| A479N | 0.9 ± 0.1 | 2 ± 0.2 | 24 ± 4 | 20 ± 3 |
The data shown are means of triplicate experiments.
Crude extract from E. coli LS5218 transformants harboring wild-type and individual A479 mutant PhaCCs grown on LB medium containing 100 μg/ml of ampicillin and 50 μg/ml of kanamycin for 9 h at 30°C.
Determined from freeze-dried cells via gas chromatography.
Percentage relative to PHA accumulation by wild-type PhaCCs in the E. coli transformant.
One unit of enzyme activity is defined as the amount of enzyme that catalyzed the release of 1 μmol CoA per minute.
Percentage relative to the activity of wild-type PhaCCs for the polymerization of (R)-3HB-CoA.
Fig 3.
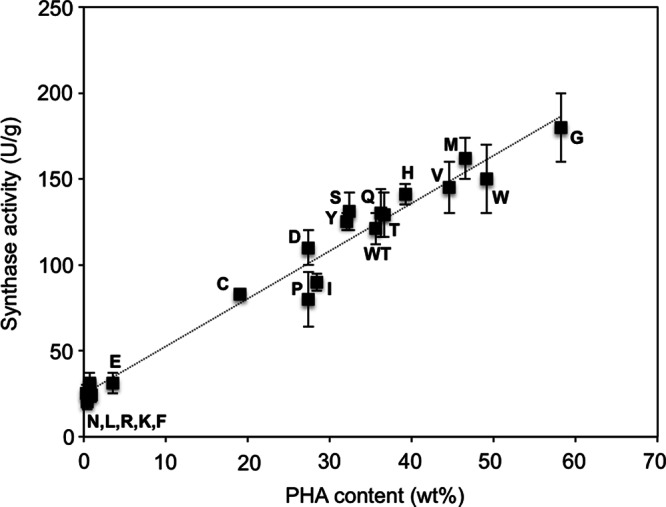
Correlation between in vitro and in vivo activities of wild-type and A479X mutants of PhaCCs. Each letter of the alphabet represents 1 of the 19 amino acid residues replacing the original one (WT) at position 479 in phaCCs. The data points show a linear correlation (dotted line). In vitro activity was determined by an enzymatic assay of crude synthase extracts (40 μg) using (R)-3HB-CoA as the substrate (600 μM). One unit of enzyme activity is defined as the amount of enzyme that catalyzed the release of 1 μmol CoA/min. In vivo activity was determined from freeze-dried cells via gas chromatography. The data shown are means of triplicate experiments. The error bars indicate standard deviations.
Characterization of P(3HB) and P(3HB-co-3HHx).
P(3HB-co-6.6 mol% 3HHx), with the largest 3HHx fraction, was isolated from the transformant strain harboring the A479S mutant PhaCCs as a representative sample for NMR analysis in order to obtain compositional sequence information for the copolymers produced. The 1H NMR spectrum revealed that the copolymer had typical resonances of methyl, methylene, and methine protons that could be assigned to 3HB and 3HHx units, thus verifying the presence of these monomers in the copolymer (see Fig. S1A in the supplemental material). 13C NMR analysis of the same sample, as shown in Fig. S1B in the supplemental material, provided supporting structural evidence for the 3HB and 3HHx units, in addition to information on the sequence structure of the copolymer. The four evidently strong signals were unambiguously assigned to the carbon resonances of the 3HB unit (29). Meanwhile, the carbonyl resonance at 169.1 to 169.4 ppm exhibited two groups of peaks arising from the different dyad sequences of connecting 3HB and 3HHx units (3HB*-3HB and 3HB*3HA/3HA*-3HB). Some peaks, such as the 3HA*-3HA dyad carbonyl resonance, however, did not display a well-resolved structure due to low 3HHx fraction in the copolymer. Overall, the observations indicated that the 3HB units were connected to the 3HHx units, implying that the PHA copolymer produced in this study was a random copolymer comprising 3HB and 3HHx units rather than a blend of homopolymers with different units.
To further support this implication, the thermal properties of the copolymers were investigated. The glass transition temperatures of P(3HB-co-3HHx) copolymers with various 3HHx fractions were similar to or lower than that of P(3HB). A single peak corresponding to the glass transition temperature, observed in the DSC thermographs of all analyzed copolymer samples, was characteristic of a random copolymer (17). Additionally, the incorporation of 3HHx into the polymer chain had a noticeable effect on the thermal properties of the resultant polymer (see Table S3 in the supplemental material). The melting temperature and enthalpy of fusion of the polymers exhibited a decrease with increasing 3HHx fractions. The Tm values were found to be in the range of 154 to 165°C when the 3HHx monomer fraction ranged from 0.8 to 6.6 mol%. This was lower than that of P(3HB) produced using the A479E mutant synthase, with a Tm of 172°C. Similarly, the ΔHm values (40 to 68 J/g) were lower than that of the P(3HB) homopolymer (80 J/g). It was evident that the presence of 3HHx monomer in the polymer chain, even in small amounts of 0.8 mol%, was sufficient to cause a change in the thermal property of the polymer. As expected, P(3HB-co-6.6 mol% 3HHx), with the largest 3HHx fraction, had the lowest Tm and ΔHm values.
Based on the data shown in Table 2, slight variation in the molecular weight of the synthesized polymers could be observed, ranging from 113 × 103 to 272 × 103 (Mw) and from 40 × 103 to 79 × 103 (Mn). The homopolymer with the highest molecular weight had a molecular weight that was 1.5-fold in excess of that of P(3HB-co-1.6 mol% 3HHx) synthesized using the wild-type synthase, while the copolymer with the lowest molecular weight had a molecular weight that was 0.8-fold lower. The polydispersity indexes were relatively consistent for polymers synthesized using the wild-type and various mutant synthases, in a range of 2.7 to 3.8. Here, it could be seen that the polymer with the highest molecular weight was P(3HB), synthesized using the A479E mutant synthase. In relation to this observation, mutant synthases capable of polymerizing P(3HB-co-3HHx) but with 3HHx fractions (0 to 1.2 mol%) lower than that synthesized by the wild-type synthase (1.6 mol%), in comparison, also tended to have higher molecular weights.
Table 2.
Molecular weights of P(3HB) and P(3HB-co-3HHx) synthesized by E. coli LS5218 transformants harboring wild-type and various A479X mutant PhaCCsa
| PHA synthase | 3HHx fraction (mol%) | Mw (103) | Mn (103) | Mw/Mnb |
|---|---|---|---|---|
| Wild type | 1.6 | 168 | 52 | 3.2 |
| A479C | 0.7 | 223 | 73 | 3.1 |
| A479D | 1.2 | 240 | 66 | 3.6 |
| A479E | 0 | 272 | 79 | 3.4 |
| A479F | 0 | NDc | ND | ND |
| A479G | 2.6 | 113 | 40 | 2.8 |
| A479H | 1.9 | 261 | 74 | 3.5 |
| A479I | 0.8 | 190 | 71 | 2.7 |
| A479K | 0 | ND | ND | ND |
| A479L | 0 | 242 | 67 | 3.6 |
| A479 M | 5.4 | 166 | 46 | 3.6 |
| A479N | 0 | ND | ND | ND |
| A479P | 1.0 | 196 | 56 | 3.5 |
| A479Q | 2.5 | 175 | 62 | 2.8 |
| A479R | 0 | ND | ND | ND |
| A479S | 6.6 | 196 | 54 | 3.6 |
| A479T | 4.4 | 187 | 49 | 3.8 |
| A479V | 2.0 | 170 | 62 | 2.7 |
| A479W | 0.8 | 195 | 56 | 3.5 |
| A479Y | 1.1 | 215 | 59 | 3.6 |
Cells were cultivated in 100 ml of M9 medium containing 0.25% (wt/vol) dodecanoic acid, 0.4% (vol/vol) Brij 35, 100 μg/ml of ampicillin, and 50 μg/ml of kanamycin for 72 h at 30°C.
Polydispersity index.
ND, not determined.
Evaluation of PhaCCs substrate specificity.
To evaluate the mutational effect of amino acid substitution at position A479 on the substrate specificity of PhaCCs, the wild type and a few A479X mutants of PhaCCs, selected on the basis of their distinct 3HHx contents, were purified for an in vitro enzymatic assay. The resultant enzymes were assayed to determine their specificities for the polymerization of (R)-3HHx-CoA in an effort to understand the variation in the 3HHx fractions of polymers produced by transformant strains harboring these different synthases. Given that PhaCCs is able to polymerize (R)-3HB-CoA and (R)-3HV-CoA in vivo (30), in addition to (R)-3HHx-CoA, it is of interest to investigate possible changes in in vitro substrate specificities, due to mutation, for polymerization of these substrates, as well. Based on substrate specificity studies conducted using the wild-type synthase, PhaCCs was found to have a 2-fold-higher activity for (R)-3HV-CoA polymerization (442 ± 81 U/mg) and a 10-fold-lower activity for (R)-3HHx-CoA polymerization (11 ± 1 U/mg) than that for (R)-3HB-CoA polymerization (253 ± 13 U/mg) (Table 3). It was apparent that mutations at this position impacted the substrate specificities of PhaCCs, although the effect was different, and to a different extent, for each substrate. The specific activities for polymerization of (R)-3HB-CoA did not differ significantly and were almost constant for all mutant synthases, ranging from 228 to 338 U/mg in comparison with the original 253-U/mg activity exhibited by the wild-type synthase. In contrast, a larger difference in substrate specificity alteration was observed with longer-chain-length (C5 and C6) substrates, namely, (R)-3HV-CoA and (R)-3HHx-CoA. The A479E mutant exhibited the highest preference for the C5 substrate, with polymerization activities 1.3-fold in excess of that of the wild-type synthase. The A479S mutant, which exhibited the lowest preference for C5 substrate polymerization (296 ± 25 U/mg), coincidentally, polymerized the C6 substrate with the highest efficiency (20 ± 3 U/mg), almost twice as fast as the wild-type synthase. In contrast, the A479E mutant synthase had the lowest efficiency for the polymerization of the C6 substrate. Overall, the activities for polymerization of substrates by all PhaCCs mutants were similar to that by the wild-type synthase, that is, in increasing order of C6 < C4 < C5.
Table 3.
In vitro substrate specificities of wild-type and A479X mutants of PhaCCs
| PHA synthasea | Value for substrateb |
|||||
|---|---|---|---|---|---|---|
| (R)-3HB-CoAc |
(R)-3HV-CoAc |
(R)-3HHx-CoAd |
||||
| Sp act (U/mg)e | Relative sp act (%)f | Sp act (U/mg) | Relative sp act (%) | Sp act (U/mg) | Relative sp act (%) | |
| Wild type | 253 ± 13 | 100 ± 5 | 442 ± 81 | 100 ± 18 | 11 ± 1 | 100 ± 9 |
| A479E | 338 ± 38 | 134 ± 15 | 589 ± 55 | 133 ± 13 | 9 ± 2 | 82 ± 18 |
| A479T | 236 ± 8 | 93 ± 3 | 380 ± 20 | 86 ± 5 | 14 ± 1 | 127 ± 9 |
| A479S | 228 ± 19 | 90 ± 8 | 296 ± 25 | 67 ± 6 | 20 ± 3 | 182 ± 27 |
Isolated from E. coli BL21(DE3) and purified by affinity chromatography using a Strep-Tactin column.
A final concentration of 600 μM was used in each assay. The data shown are means of triplicate experiments.
Polymerization catalyzed by 7.5 nM, 15 nM, and 30 nM enzyme, respectively, in each of three assays performed.
Polymerization catalyzed by 100 nM, 200 nM, and 300 nM enzyme, respectively, in each of three assays performed.
One unit of enzyme activity is defined as the amount of enzyme that catalyzed the release of 1 μmol CoA per minute.
Percentage relative to the activity of wild-type PhaCCs for the polymerization of each substrate.
DISCUSSION
Effects of the A479X mutation on PHA production.
Variation in the contents of PHA produced by strains harboring various mutant synthases in this study allowed the analysis of amino acid substitutions and the corresponding effects. Of the 19 amino acid substitutions, 4 resulted in an obvious increase in PHA content (44.6 to 58.2 wt%) compared to that with the wild-type synthase (35.6 wt%). Another five had modest effects on the PHA synthase (32.1 to 39.2 wt% accumulation), while the remaining 10 decreased PHA accumulation to as low as 0.3 wt%. A classification could be made as follows: nonpolar amino acid substitutions increased PHA accumulation (in increasing order of V < M < W < G), polar but neutral/uncharged amino acids (H/T/Q/S/Y) did not interfere with the ability of the synthase in this respect, while charged residues (D/E/K/R) negatively impacted PHA accumulation. Although histidine has a charged imidazole functional group, at the cultivation pH of 7 used in this study, the amino acid maintained a neutral charge. As position 479 is a constituent of the substrate-binding pocket and the original residue at this position is the nonpolar alanine, it could be inferred that substitutions with polar amino acid residues resulted in interactions with other neighboring atoms, thus changing the geometry of the substrate-binding pocket. This, in turn, possibly reduced the number of monomer substrates able to interact with PhaCCs for polymerization into polymer chains, as reflected in the decreased PHA accumulation. The most prominent increase in PHA accumulation was obtained by substitution with glycine, which has the lowest van der Waals volume, as this may have enlarged the substrate-binding pocket of PhaCCs and/or improved the accessibility of the substrate to crucial neighboring atoms, thereby increasing its catalytic efficiency.
In vivo and in vitro synthase activities, 3HHx content, and molecular weight.
The linear relationship between in vitro and in vivo synthase activities of the mutant synthases (Fig. 3) provided further evidence in support of the notion of a linear correlation between synthase activity and PHA accumulation proposed in several earlier studies (31–33). An obvious advantage of this correlation is that it allows the estimation of synthase activity based on the in vivo PHA production level. It is also apparent here that mutation at position 479 was effective for controlling the catalytic activity of PhaCCs, although all introduced mutations did not appear to have affected synthase production, nor did they result in misfolding or degradation of the enzyme (Fig. 2C).
Additionally, correlations could be made between the synthase activities, 3HHx fractions, and molecular weights of the polymers produced in this study (Fig. 4). The dependence of the polymer molecular weight on the synthase concentration and activity has been demonstrated both in vitro (34) and in vivo (35). In both of these different environments, the molecular weight of the polymer decreased with increased concentrations of PHA synthase. Incorporation of a second monomer unit was also found to reduce the molecular weight of the resultant polymer, and the molecular weight varied with the degree of incorporation of the comonomer unit (2). Similar to previous observations, the molecular weights of the produced polymers tended to increase with a reduction in 3HHx content and a decrease in synthase activity. As the wild-type and mutant synthases generated in this study were found to differ in catalytic activities and produced polymers that varied in 3HHx contents, it can be seen that both factors, in combination, influenced the molecular weight of the polymers. In fact, based on this correlation, a linear relationship could be seen between synthase activity and 3HHx fraction, suggesting that the activity of the synthase, and not just its substrate preference, determines the level of incorporation of the 3HHx monomer. The dependence of the 3HHx fraction on the synthase activity was also found to be true for copolymers synthesized using PHA synthase from A. caviae (PhaCAc) (23, 36, 37).
Fig 4.
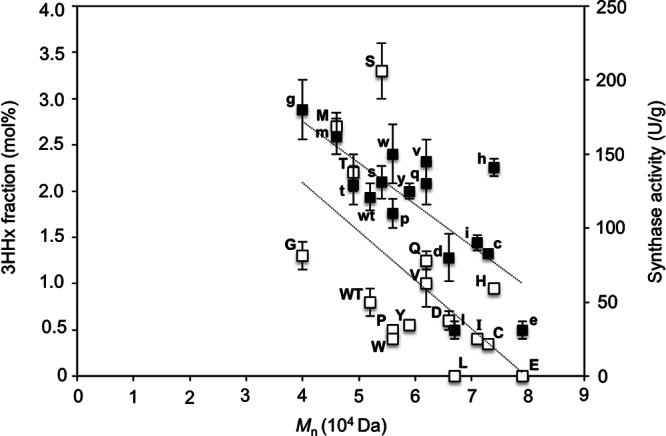
Correlation between molecular weight, 3HHx fraction (white boxes), and synthase activity (black boxes) of wild-type and A479X mutants of PhaCCs. Each letter of the alphabet represents 1 of the 19 amino acid residues replacing the original one (WT) at position 479 in phaCCs. The data points show linear correlations (dotted lines). Molecular weight was determined using PHA extracted from freeze-dried cells via gel permeation chromatography. The 3HHx fraction was determined from freeze-dried cells via gas chromatography. Synthase activity was determined by an enzymatic assay of crude synthase extracts (40 μg) using (R)-3HB-CoA as the substrate (600 μM). One unit of enzyme activity is defined as the amount of enzyme that catalyzed the release of 1 μmol CoA/min. The data shown are means of triplicate experiments. The error bars indicate standard deviations.
Substrate specificity of PhaCCs.
The high affinity of PhaCCs for 3-hydroxyvalerate (3HV) was supported by findings in preceding studies: (i) a copolymer of P(3HB-co-3HV) with a high 3HV monomer composition of 98 mol% was produced by the native strain grown on sodium valerate (16) and (ii) addition of a 3HV precursor induced synthesis of a terpolymer with 85 mol% 3HV by an R. eutropha transformant expressing phaCCs (30). This showed that the high affinity for 3HV-CoA was not strain dependent, is a property of the synthase, and was consistent both in vivo and in vitro.
The consistency of in vivo results in correlation with in vitro activities was similarly observed for polymerization of the 3HHx monomer. Polymerization of (R)-3HHx-CoA in vitro using purified PhaCCs was congruous with in vivo PHA production from crude palm kernel oil by R. eutropha harboring PhaCCs (16). In terms of the polymerization rate, an approximate ratio of 1 to 20 for monomers of 3HHx to 3HB could be observed from the specific-activity data (11 U/mg for 3HHx to 253 U/mg for 3HB) and monomeric composition (4 mol% for 3HHx to 96 mol% for 3HB) of copolymers produced in the study mentioned above. The results here provide solid evidence of a linear relationship between in vivo and in vitro substrate specificities of synthases, despite conflicting observations in previous studies (38, 39). Overall, the affinity of PhaCCs for the polymerization of (R)-3HV-CoA was found to be superior to those of other class I synthases characterized so far (38, 39), and its affinity was comparable to that of PhaCAc for the polymerization of (R)-3HHx-CoA (40).
It is interesting that the chain length dependence on the rate of polymerization by PhaCCs (in the order of C5 > C4 > C6) differed from that of other synthases belonging to R. eutropha and Chromatium vinosum (39), although all the synthases mentioned preferentially polymerize short-chain-length (C3 to C5) substrates. It was postulated that these synthases with a preference for short-chain-length substrates either fail or are only able to feebly polymerize large monomers (such as the C6 substrate) due to a spatial limitation for fitting a second, larger monomer into the binding pocket of the synthase (41). These synthases optimally polymerize C4 substrates, with the ability to also polymerize C5 substrates, albeit with reduced efficiency, and C6 substrates, if at all, with limited activity. On the other hand, PhaCCs, by nature, exhibited very high affinity for polymerization of a C5 substrate, which is larger than the C4 substrate. This may well be the reason behind the ability of PhaCCs to accept the C6 substrate with greater ease than the other short-chain synthases.
Effect of the A479 mutation on the substrate specificity of PhaCCs.
The A479 mutation point had a greater influence on the alteration of substrate preference than the PHA polymerization activity of PhaCCs. Substitutions with charged residues produced adverse effects, in this case, reduced or completely diminished affinity for 3HHx. Meanwhile, substitutions with nonpolar (G/M/V) or uncharged polar (S/T/Q) residues caused a shift in specificity toward 3HHx incorporation. This datum not only provides support for the previous inference on reduced efficiency of PhaCCs due to changes in the topography of its substrate-binding pocket resulting from substitution with charged residues, it further suggests that the shape and size of the binding pocket are determinants of chain length specificity. Substitution with uncharged residues (G/M/S/T/Q) was concluded to be best for enhancement of both PHA accumulation and 3HHx fraction. Interestingly, it was found that A510M/Q in PhaCRe also improved PHA accumulation and 3HHx fraction in the resultant copolymer (12).
For in vitro substrate specificity evaluation, the amino acid residues at position 479 of selected wild-type and mutant synthases differed in their properties: nonpolar (wild type), polar but uncharged (A479S/T), and charged (A479E). For the longer-chain substrate (C6), an enhanced affinity was observed with substitutions of serine and threonine, which have in common a hydroxyl group in their side chains. As the side chain hydroxyl atom has a tendency to form a hydrogen bond with the protein backbone, this may have facilitated conformational changes in the substrate pocket to adapt to a larger substrate molecule. On the other hand, substitution by a charged residue (glutamate) with a large van der Waals volume negatively affected insertion of the C6 substrate into the substrate-binding pocket of PhaCCs. The van der Waals volume of an amino acid residue has been demonstrated to affect incorporation of 3HHx monomer by PhaCAc, where an increase in the former resulted in reduced amounts of the latter (23). In other studies where selection of residues for substitution was aided by homology-modeled protein structures, replacement of large amino acids in the substrate-binding pocket by smaller ones allowed larger substrates to bind while preserving activity toward a naturally preferred substrate (42, 43). This was true for PhaCCs if, based on substrate specificities exhibited by the synthase, we consider 3HB and 3HV monomers to be the substrates preferred over 3HHx. In this case, the presence of smaller amino acid residues (alanine, serine, and threonine) in position 479, in comparison to glutamine, allowed improved incorporation of a longer carbon chain (C6) substrate.
Amino acid replacements at position 479 shifted the preference of the synthase toward or away from 3HV or 3HHx, but affinity for 3HB remained relatively constant. From the ratios of synthase activities toward C4, C5, and C6 substrates (Table 4), it was clear that the mutation successfully yielded the desired effect of increasing the tendency for polymerization of the C6 substrate. This confirmed that variation in the 3HHx fraction of the polymers produced in this study could be attributed to changes in activity, as well as the substrate specificity of the synthase, as proposed previously based on the linear correlation between synthase activity and 3HHx content shown in Fig. 4. The highest 3HHx monomer-incorporating mutant, A479S, showed an increased affinity 2.3-fold higher than that of the wild-type synthase, corresponding to an increase in 3HHx monomer fraction in vivo from 1.6 mol% (wild type) to 6.6 mol% (A479S). This provides a rough estimate that a 2-fold increase in the specific activity of the synthase leads to a 4-fold increase in the 3HHx monomer fraction. Likewise, the A479T mutant, which was able to incorporate approximately 3 times more 3HHx monomer (4.4 mol%) than the wild-type synthase, showed about a 1.3-fold increase in specific activity. The data suggest a linear relationship between specific activity and 3HHx monomer fraction, which is shown clearly in Fig. 5A.
Table 4.
Ratios of in vitro synthase activities for the polymerization of different substrates
| PHA synthase | Ratio of activity for substrate polymerization |
|
|---|---|---|
| C4/C5 | C4/C6 | |
| Wild type | 1:1.7 | 1:0.04 |
| A479E | 1:1.7 | 1:0.03 |
| A479T | 1:1.6 | 1:0.06 |
| A479S | 1:1.3 | 1:0.09 |
Fig 5.

Correlation between 3HHx fraction and specific synthase activity for polymerization of (R)-3HHx-CoA (A) and (R)-3HB-CoA (white boxes) and (R)-3HV-CoA (black boxes) (B) by wild-type and selected A479X mutants of PhaCCs. The data points show linear correlations (dotted lines). The 3HHx fraction was determined from freeze-dried cells via gas chromatography. In vitro activity was determined by an enzymatic assay of purified synthase extracts (7.5 nM to 0.3 μM) using (R)-3HB-CoA, (R)-3HV-CoA, and (R)-3HHx-CoA as substrates (600 μM). One unit of enzyme activity is defined as the amount of enzyme that catalyzed the release of 1 μmol CoA/min. The data shown are means of triplicate experiments. The error bars indicate standard deviations.
A slight decrease in affinity for the C5 substrate for mutants with increased tendencies toward the polymerization of the C6 substrate indirectly indicates that 3HHx monomer incorporation improved with reduced specificity of the synthase for short-chain-length C4 and C5 substrates (Fig. 5B). The consistency observed in the preference of synthases, wild type and mutants alike, for either C4 and C5 substrates or C6 substrate clearly emphasizes the boundary between short-chain-length (C3 to C5) and medium-chain-length (C6 to C14) PHA synthases, which was the basis for classification of PHA synthases (15, 44). The data obtained here substantiate this fundamental classification.
In conclusion, mutation at position 479 in PhaCCs was not only found to be beneficial for enhancement of synthase activity, but also successfully altered the substrate preference of the synthase, leading to accumulation of copolymers that varied in monomer composition. Increases in 3HHx content, though modest, affected the molecular weight, as well as the thermal properties, of the resultant copolymer. Elevated synthase activity and enhanced preference of the synthase for 3HHx-CoA were found to have synergistic effects in promoting the incorporation of more 3HHx units into the polymer chain. Analysis of mutational effects revealed that replacements with nonpolar or neutral amino acid residues constituting the substrate-binding pocket yielded mutant synthases exhibiting simultaneous improvements in PHA production and 3HHx incorporation. In vitro characterization of PhaCCs for polymerization of comonomers revealed two times greater activity for 3HV-CoA polymerization than for 3HB-CoA, while corresponding activity for 3HHx-CoA polymerization rivaled that of another synthase, from A. caviae, also belonging to the rare group of SCL-MCL synthases. Additionally, in vivo PhaCCs activity and specificity for 3HB, 3HV, and 3HHx monomers were in linear correlation with in vitro results. On the whole, the preference exhibited by wild-type and mutant synthases for either SCL or MCL monomers provided insights into the classification of PHA synthases, which is based on substrate preference.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the RIKEN Biomass Engineering Program and USM APEX DE. J.-A.C. acknowledges Universiti Sains Malaysia's Fellowship Scheme and RIKEN′s International Program Associate Fellowship for financial support.
We are very grateful to Toshiaki Fukui (Tokyo Institute of Technology, Tokyo, Japan) for his kindness in providing the pBBR-PPJAc plasmid.
Footnotes
Published ahead of print 12 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00564-13.
REFERENCES
- 1. Anderson AJ, Dawes EA. 1990. Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol. Rev. 54:450–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doi Y. 1990. Microbial polyesters. VCH, New York, NY [Google Scholar]
- 3. Mergaert J, Webb A, Anderson C, Wouters A, Swings J. 1993. Microbial degradation of poly(3-hydroxybutyrate) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) in soils. Appl. Environ. Microbiol. 59:3233–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steinbüchel A. 2001. Perspectives for biotechnological production and utilization of biopolymers: metabolic engineering of polyhydroxyalkanoate biosynthesis pathways as a successful example. Macromol. Biosci. 1:1–24 [Google Scholar]
- 5. Sudesh K, Iwata T. 2008. Sustainability of biobased and biodegradable plastics. Clean 36:433–442 [Google Scholar]
- 6. Steinbüchel A, Lütke-Eversloh T. 2003. Metabolic engineering and pathway construction for biotechnological production of relevant polyhydroxyalkanoates in microorganisms. Biochem. Eng. J. 16:81–96 [Google Scholar]
- 7. Nomura CT, Taguchi S. 2007. PHA synthase engineering toward superbiocatalysts for custom-made biopolymers. Appl. Microbiol. Biotechnol. 73:969–979 [DOI] [PubMed] [Google Scholar]
- 8. Taguchi S, Doi Y. 2004. Evolution of polyhydroxyalkanoate (PHA) production system by “enzyme evolution”: successful case studies of directed evolution. Macromol. Biosci. 4:145–156 [DOI] [PubMed] [Google Scholar]
- 9. Amara AA, Steinbüchel A, Rehm BH. 2002. In vivo evolution of the Aeromonas punctata polyhydroxyalkanoate (PHA) synthase: isolation and characterization of modified PHA synthases with enhanced activity. Appl. Microbiol. Biotechnol. 59:477–482 [DOI] [PubMed] [Google Scholar]
- 10. Takase K, Taguchi S, Doi Y. 2003. Enhanced synthesis of poly(3-hydroxybutyrate) in recombinant Escherichia coli by means of error-prone PCR mutagenesis, saturation mutagenesis and in vitro recombination of the type II polyhydroxyalkanoate synthase gene. J. Biochem. 133:139–145 [DOI] [PubMed] [Google Scholar]
- 11. Takase K, Matsumoto K, Taguchi S, Doi Y. 2004. Alteration of substrate chain-length specificity of type II synthase for polyhydroxyalkanoate biosynthesis by in vitro evolution: in vivo and in vitro enzyme assays. Biomacromolecules 5:480–485 [DOI] [PubMed] [Google Scholar]
- 12. Tsuge T, Saito Y, Narike M, Muneta K, Normi YM, Kikkawa Y, Hiraishi T, Doi Y. 2004. Mutation effects of a conserved alanine (Ala510) in type I polyhydroxyalkanoate synthase from Ralstonia eutropha on polyester biosynthesis. Macromol. Biosci. 4:963–970 [DOI] [PubMed] [Google Scholar]
- 13. Tsuge T, Watanabe S, Shimada D, Abe H, Doi Y, Taguchi S. 2007. Combination of N149S and D171G mutations in Aeromonas caviae polyhydroxyalkanoate synthase and impact on polyhydroxyalkanoate biosynthesis. FEMS Microbiol. Lett. 277:217–222 [DOI] [PubMed] [Google Scholar]
- 14. Potter M, Steinbüchel A. 2005. Poly(3-hydroxybutyrate) granule-associated proteins: impacts on poly(3-hydroxybutyrate) synthesis and degradation. Biomacromolecules 6:552–560 [DOI] [PubMed] [Google Scholar]
- 15. Rehm BHA. 2003. Polyester synthases: natural catalysts for plastics. Biochem. J. 376:15–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhubalan K, Yong KH, Kam YC, Sudesh K. 2010. Cloning and expression of the PHA synthase gene from a locally isolated Chromobacterium sp. USM2. Mal. J. Microbiol. 6:81–90 [Google Scholar]
- 17. Doi Y, Kitamura S, Abe H. 1995. Microbial synthesis and characterization of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate). Macromolecules 28:4822–4828 [Google Scholar]
- 18. Kobayashi G, Shiotani T, Shima Y, Doi Y. 1994. Biosynthesis and characterization of poly(3-hydroxybutyrate-co-3-hydroxyalkanoate) from oils and fats by Aeromonas sp. OL-338 and Aeromonas sp. FA440, p 410–416 In Doi Y, Fukuda K. (ed), Biodegradable plastics and polymers. Elsevier, Amsterdam, The Netherlands [Google Scholar]
- 19. Chen G, Zhang G, Park S, Lee S. 2001. Industrial scale production of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate). Appl. Microbiol. Biotechnol. 57:50–55 [DOI] [PubMed] [Google Scholar]
- 20. Lee Y, Lee SH, Lee SY. 1999. Fed-batch culture of Aeromonas hydrophila for the production of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) using two carbon sources. Biotechnol. Bioproc. Eng. 4:195–198 [Google Scholar]
- 21. Haywood GW, Anderson AJ, Williams DR, Dawes EA, Ewing DF. 1991. Accumulation of poly(hydroxyalkanoate) copolymer containing primarily 3-hydroxyvalerate from simple carbohydrate substrates by Rhodococcus sp. NCIMB 40126. Int. J. Biol. Macromol. 13:83–88 [DOI] [PubMed] [Google Scholar]
- 22. Bhubalan K, Chuah J, Shozui F, Brigham CJ, Taguchi S, Sinskey AJ, Rha C, Sudesh K. 2011. Characterization of the highly active polyhydroxyalkanoate synthase of Chromobacterium sp. strain USM2. Appl. Environ. Microbiol. 77:2926–2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsuge T, Watanabe S, Sato S, Hiraishi T, Abe H, Doi Y, Taguchi S. 2007. Variation in copolymer composition and molecular weight of polyhydroxyalkanoate generated by saturation mutagenesis of Aeromonas caviae PHA synthase. Macromol. Biosci. 7:846–854 [DOI] [PubMed] [Google Scholar]
- 24. Spratt SK, Ginsburgh CL, Nunn WD. 1981. Isolation and genetic characterization of Escherichia coli mutants defective in propionate metabolism. J. Bacteriol. 146:1166–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 26. Braunegg G, Sonnleitner B, Lafferty RM. 1978. A rapid gas chromatographic method for the determination of poly-β-hydroxybutyric acid in microbial biomass. Eur. J. Appl. Microbiol. Biotechnol. 6:29–37 [Google Scholar]
- 27. Murata T, Takase K, Yamato I, Igarashi K, Kakinuma Y. 1997. Purification and reconstitution of Na+-translocating vacuolar ATPase from Enterococcus hirae. J. Biol. Chem. 272:24885–24890 [DOI] [PubMed] [Google Scholar]
- 28. Simon EJ, Shemin D. 1953. The preparation of S-succinyl coenzyme A. J. Am. Chem. Soc. 75:2520 [Google Scholar]
- 29. Abe H, Doi Y, Fukushima T, Eya H. 1994. Biosynthesis from gluconate of a random copolyester consisting of 3-hydroxybutyrate and medium-chain-length 3-hydroxyalkanoates by Pseudomonas sp. 61-3. Int. J. Biol. Macromol. 16:115–119 [DOI] [PubMed] [Google Scholar]
- 30. Bhubalan K, Rathi DN, Abe H, Iwata T, Sudesh K. 2010. Improved synthesis of P(3HB-co-3HV-co-3HHx) terpolymers by mutant Cupriavidus necator using the PHA synthase gene of Chromobacterium sp. USM2 with high affinity towards 3HV. Polym. Degrad. Stab. 95:1436–1442 [Google Scholar]
- 31. Taguchi S, Maehara A, Takase K, Nakahara M, Nakamura H, Doi Y. 2001. Analysis of mutational effects of a polyhydroxybutyrate (PHB) polymerase on bacterial PHB accumulation using an in vivo assay system. FEMS Microbiol. Lett. 198:65–71 [DOI] [PubMed] [Google Scholar]
- 32. Taguchi S, Nakamura H, Hiraishi T, Yamato I, Doi Y. 2002. In vitro evolution of a polyhydroxybutyrate synthase by intragenic suppression-type mutagenesis. J. Biochem. 131:801–806 [DOI] [PubMed] [Google Scholar]
- 33. Kichise T, Taguchi S, Doi Y. 2002. Enhanced accumulation and changed monomer composition in polyhydroxyalkanoate (PHA) copolyester by in vitro evolution of Aeromonas caviae PHA synthase. Appl. Environ. Microbiol. 68:2411–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gerngross TU, Martin DP. 1995. Enzyme-catalyzed synthesis of poly[(R)-(−)-3-hydroxybutyrate]: formation of macroscopic granules in vitro. Proc. Natl. Acad. Sci. U. S. A. 92:6279–6283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sim SJ, Snell KD, Hogan SA, Stubbe J, Rha CK, Sinskey AJ. 1997. PHA synthase activity controls the molecular weight and polydispersity of polyhydroxybutyrate in vivo. Nat. Biotechnol. 15:63–67 [DOI] [PubMed] [Google Scholar]
- 36. Fukui T, Kichise T, Iwata T, Doi Y. 2001. Characterization of 13 kDa granule-associated protein in Aeromonas caviae and biosynthesis of polyhydroxyalkanoates with altered molar composition by recombinant bacteria. Biomacromolecules 2:148–153 [DOI] [PubMed] [Google Scholar]
- 37. Kichise T, Fukui T, Yoshida Y, Doi Y. 1999. Biosynthesis of polyhydroxyalkanoates (PHA) by recombinant Ralstonia eutropha and effects of PHA synthase activity on in vivo PHA biosynthesis. Int. J. Biol. Macromol. 25:69–77 [DOI] [PubMed] [Google Scholar]
- 38. Haywood GW, Anderson AJ, Dawes EA. 1989. The importance of PHB-synthase substrate specificity in polyhydroxyalkanoate synthesis by Alcaligenes eutrophus. FEMS Microbiol. Lett. 57:1–6 [Google Scholar]
- 39. Yuan W, Jia Y, Tian J, Snell KD, Müh U, Sinskey AJ, Lambalot RH, Walsh CT, Stubbe J. 2001. Class I and III polyhydroxyalkanoate synthases from Ralstonia eutropha and Allochromatium vinosum: characterization and substrate specificity studies. Arch. Biochem. Biophys. 394:87–98 [DOI] [PubMed] [Google Scholar]
- 40. Numata K, Motoda Y, Watanabe S, Tochio N, Kigawa T, Doi Y. 2012. Active intermediates of polyhydroxyalkanoate synthase from Aeromonas caviae in polymerization reaction. Biomacromolecules 13:3450–3455 [DOI] [PubMed] [Google Scholar]
- 41. Zhang S, Kamachi M, Takagi Y, Lenz RW, Goodwin S. 2001. Comparative study of the relationship between monomer structure and reactivity for two polyhydroxyalkanoate synthases. Appl. Microbiol. Biotechnol. 56:131–136 [DOI] [PubMed] [Google Scholar]
- 42. Creaser EH, Murali C, Britt KA. 1990. Protein engineering of alcohol dehydrogenases: effects of amino acid changes at positions 93 and 48 of yeast ADH1. Protein Eng. 3:523–526 [DOI] [PubMed] [Google Scholar]
- 43. Wilks HM, Halsall DJ, Atkinson T, Chia WN, Clarke AR, Holbrook JJ. 1990. Designs for a broad substrate specificity keto acid dehydrogenase. Biochemistry 29:8587–8591 [DOI] [PubMed] [Google Scholar]
- 44. Liebergesell M, Sonomoto K, Madkour M, Mayer F, Steinbüchel A. 1994. Purification and characterization of the poly(hydroxyalkanoic acid) synthase from Chromatium vinosum and localization of the enzyme at the surface of poly(hydroxyalkanoic acid) granules. Eur. J. Biochem. 226:71–80 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.