

Abstract
Previous studies indicated that the Lyme disease spirochete Borrelia burgdorferi expresses the RevA outer surface protein during mammalian infection. As an adhesin that promotes bacterial interaction with fibronectin, RevA appears to be a good target for preventive therapies. RevA proteins are highly conserved across all Lyme borreliae, and antibodies against RevA protein are cross-reactive among RevA proteins from diverse strains. Mice infected with B. burgdorferi mounted a rapid IgM response to RevA, followed by a strong IgG response that generally remained elevated for more than 12 months, suggesting continued exposure of RevA protein to the immune system. RevA antibodies were bactericidal in vitro. To evaluate the RevA antigen as a potential vaccine, mice were vaccinated with recombinant RevA and challenged with B. burgdorferi by inoculation with a needle or by a tick bite. Cultured tissues from all treatment groups were positive for B. burgdorferi. Vaccinated animals also appeared to have similar levels of B. burgdorferi DNA compared to nonvaccinated controls. Despite its antigenicity, surface expression, and the production of bactericidal antibodies against it, RevA does not protect against Borrelia burgdorferi infection in a mouse model. However, passive immunization with anti-RevA antibodies did prevent infection, suggesting the possible utility of RevA-based immunotherapeutics or vaccine.
INTRODUCTION
Borrelia burgdorferi is the causative agent of Lyme disease, the most common arthropod-borne infection in the United States (1). Early diagnosis and treatment are key to preventing the debilitating long-term sequelae such as musculoskeletal, cardiovascular, and neurological damage (2). A preventative vaccine was approved for human use in 1998, but production was discontinued in early 2002 (3). The incidence of this disease has been steadily increasing since it was first described in the late 1970s, and all evidence indicates that Lyme disease will continue to be a widespread public health problem.
B. burgdorferi can infect immunocompetent humans and other vertebrates for extensive periods of time, even for the animal's lifetime (4, 5, 6). The Lyme disease spirochete is an extracellular organism, but a complete picture of how it manages to avoid clearance from its hosts is lacking. Antigenic variation at the vls locus, which occurs only in vivo, is continuous throughout infection (7, 8). The outer surface protein VslE appears to be crucial for persistence in the mammalian host, as bacteria lacking VslE are completely cleared (9, 10). Antibody appears to be important for clearance of B. burgdorferi, as the variable regions of VslE are accessible to antibodies (11). Other aspects of B. burgdorferi, including its tropism for immunologically isolated sites, may also contribute to its persistence in vivo.
Extracellular matrix (ECM) has been suggested to provide a protective niche for the spirochete (12). B. burgdorferi is frequently found associated with connective tissues (12, 13, 14, 15) and is often detected in and isolated from infected cartilaginous or membranous tissues, such as skin and joints. This suggests specific interactions between the pathogen and host skin tissues (5, 16, 17, 18). In vitro, B. burgdorferi shows affinity for host extracellular matrix components, such as fibronectin (12, 19, 20, 21). Bacteria deficient in one of the fibronectin-binding proteins, BBK32, exhibit reduced virulence in vivo (22, 23). Together, these data indicate that B. burgdorferi interacts with its host's ECM and suggest that those interactions are critical in both B. burgdorferi pathogenesis and persistence in mammals. Recently, we discovered that an antigenic 17-kDa outer surface lipoprotein, RevA, binds to fibronectin (19). We hypothesize that borrelia-ECM interactions, especially those mediated by RevA fibronectin-binding protein, are crucial for mammalian infection and persistence in the host.
The gene encoding RevA (so named because it is transcribed in the reverse direction from its neighboring genes) is located on a circular prophage (cp32). RevA has no significant homology to any proteins outside Borrelia species, yet it is highly conserved within the Lyme disease borrelial genospecies. The revA genes are widely distributed among Lyme disease spirochetes, and the predicted amino acid sequences of RevA proteins are highly conserved (19). Many strains of B. burgdorferi carry two copies of the revA gene; for example, the type strain B31 has two copies, and the well-characterized isolate 297 also has two copies of revA. In contrast, B. burgdorferi strain N40 and Borrelia garinii strain PBi each carry only one revA locus (19).
Serological studies indicate that humans and laboratory animals are frequently exposed to RevA during B. burgdorferi infection (24, 25). Using quantitative real-time PCR, it was confirmed that revA is indeed transcribed during mammalian infection, but not during colonization of vector ticks (19). Sera from patients in the initial stages of Lyme disease contained antibodies against RevA, demonstrating that this protein is expressed early in human infection (26).
In the current study, we propose that RevA is the target of protective antibodies and that RevA expression remains elevated throughout mammalian infection. To test our hypotheses, we examined mammalian response to RevA expression throughout the natural course of infection. In addition, we vaccinated mice with recombinant RevA antigen and challenged them with B. burgdorferi.
MATERIALS AND METHODS
Bacteria.
B. burgdorferi strain B31 MI-16 is an infectious clone of the sequenced type strain (27, 28) which contains all parental plasmids (29). Bacteria were grown at 34°C to cell densities of approximately 1 × 107 bacteria/ml in modified Barbour-Stoenner-Kelly (BSK-II) medium supplemented with 6% rabbit serum (30). Total DNA (genomic and plasmids) was isolated using a DNeasy blood and tissue kit (Qiagen, Valencia, CA). Plasmid content was monitored by multiplex PCR by the method of Bunikis et al. (31).
Recombinant proteins.
Recombinant proteins contained amino-terminal polyhistidine tags, with the RevA segment beginning with that protein's first amino acid following the cysteine lipidation site. The revA gene was PCR amplified from total genomic DNA of B. burgdorferi strain B31 MI-16 using oligonucleotides 5′-TGTAAAGCATATGTAGAAGAAAAG-3′ and 5′-TTAATTAGTGCCCTCTTCGAGGAA-3′. Amplicons were cloned into pET200 (Invitrogen, Carlsbad, CA). The resultant plasmid inserts were entirely sequenced on both strands to ensure that no undesired mutations had occurred during PCR or cloning procedures. Recombinant proteins were expressed in Escherichia coli strain Rosetta (DE3) pLysS (Novagen, Madison, WI) upon induction with isopropyl thiogalactopyranoside. Induced E. coli cultures were harvested and lysed by sonication or treatment with a French press, and debris was cleared by centrifugation. Recombinant proteins were purified from cleared lysates by using MagneHis nickel-conjugated magnetic beads (Promega, Madison, WI). All recombinant proteins were dialyzed at 4°C overnight against phosphate-buffered saline (PBS) using 3,500-molecular-weight-cutoff (MWCO) Slide-A-Lyzer cassettes (Pierce, Rockford, IL). Protein purity was assessed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis followed by staining with Coomassie brilliant blue (see Fig. 1A; also data not shown). Protein concentrations were determined by bicinchoninic acid protein assays (Pierce). Synthetic 20-amino-acid RevA peptides were produced commercially from GenScript (Piscataway, NJ); sequences are detailed in Fig. S2 in the supplemental material.
Fig 1.
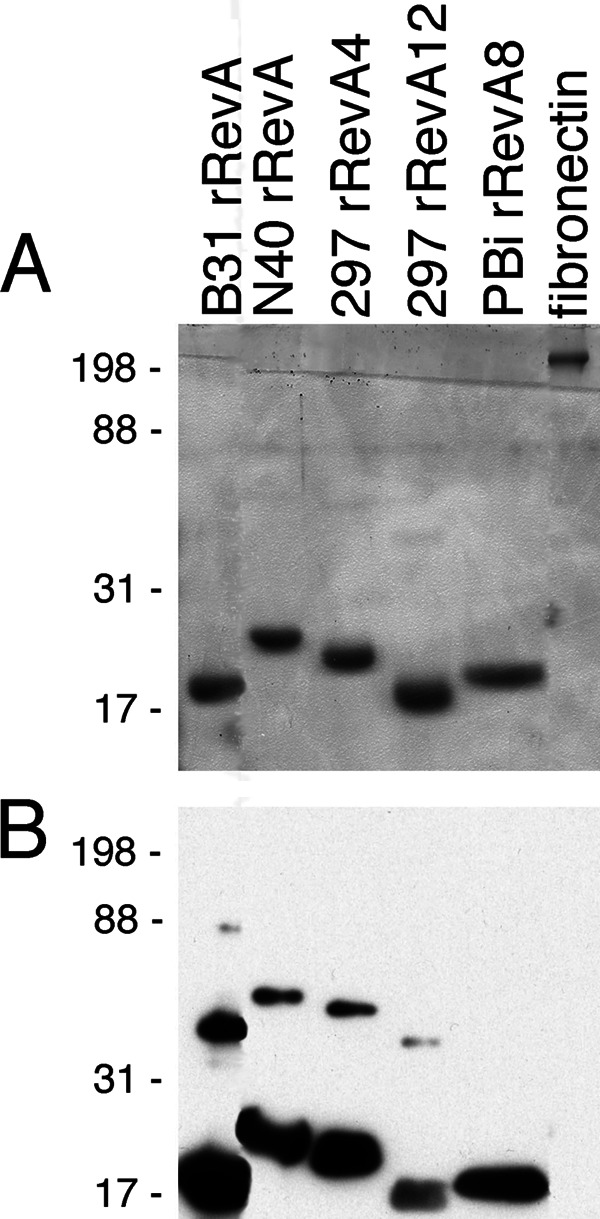
Antisera against B. burgdorferi B31 RevA recognizes Rev proteins across the Lyme borreliae. (A) Coomassie brilliant blue-stained 12.5% acrylamide gel of recombinant Rev (rRev) proteins. The positions of molecular weight markers are indicated to the left of the gel. B31 is the type strain of B. burgdorferi (28). N40 is an B. burgdorferi strain isolated from a tick (47). Strain 297, originally isolated from cerebrospinal fluid from a Lyme disease patient with meningitis (48), has two separate copies of RevA (19). PBi is a European isolate of B. garinii. (B) Western blot of the gel in panel A with affinity-purified antibody to RevA from strain B31. Mass spectrometric analysis (University of Kentucky Center for Structural Biology Protein Core Facility) indicated that the higher-molecular-weight bands present in most lanes are also RevA, suggesting that RevA may form homomultimers. Note that the affinity-purified antibodies to RevA from strain B31 do not cross-react with human plasma fibronectin.
Immunoblot.
Polyclonal antiserum directed against RevA was produced by inoculation of purified recombinant protein into a New Zealand White rabbit at AnimalPharm (Healdsburg, CA), using one round of their standard protocol. Antiserum was adsorbed against sonicated Escherichia coli Rosetta (DE3) pLysS (Novagen) and then affinity purified using HiTrap protein A columns (GE Healthcare) according to the manufacturer's instructions. The specificity of the purified antibody for RevA was tested by immunoblotting against recombinant RevA proteins, B. burgdorferi lysates, and control proteins (bovine serum albumin [BSA] and human plasma fibronectin). Briefly, proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose. Membranes were blocked overnight at 4°C with 5% (wt/vol) BSA in Tris-buffered saline-Tween 20 (TBS-T) (20 mM Tris [pH 7.5], 150 mM NaCl, 0.05% [vol/vol] Tween 20). The membranes were next washed with TBS-T and incubated for 2 h at room temperature with purified anti-RevA antibody diluted 1:500 in TBS-T. After the membranes were washed extensively with TBS-T, they were incubated for 1 h at room temperature with horseradish peroxidase-conjugated donkey anti-rabbit immunoglobulin G (IgG) antibody (GE Healthcare) diluted 1:5,000 in TBS-T. After a final series of washes with TBS-T, bound antibodies were detected by using SuperSignal West Pico enhanced chemiluminescence substrate (Pierce).
Infection of mice and ticks.
Female C3H/HEN or BALB/c mice (4 to 6 weeks old) were infected by subcutaneous injection of 1 × 106 B. burgdorferi B31 MI-16 bacteria from a mid-exponential-phase culture grown at 34°C. These mice then served to infect Ixodes scapularis larvae as follows. Egg masses laid by pathogen-free I. scapularis ticks were obtained from the Department of Entomology, Oklahoma State University—Stillwater and held in a humidified chamber until they hatched. For B. burgdorferi acquisition studies, approximately 200 naive larvae were placed on each of the above-described B. burgdorferi-infected mice. After 96 h, the ticks had fully engorged and naturally dropped off the mice. These ticks were returned to the humidified chamber and were allowed to molt to the nymphal stage. Approximately 3 weeks after ecdysis, the ticks were fed upon vaccinated female C3H/HEN or BALB/c mice. Infection of mice was confirmed by analysis of serum samples by enzyme-linked immunosorbent assay (ELISA) for antibodies directed against B. burgdorferi whole-cell lysate (see below). Mice infected through feeding by infected nymphs were killed 2 weeks after completion of tick feeding, and their ear pinnae, hearts, bladders, spleens, and tibiotarsal joints were collected and either frozen for DNA extraction and quantitative PCR (qPCR) or cultured in BSK-II medium plus 6% rabbit serum and 50 μg/ml rifampin.
Immunization.
For vaccination protocol 1, C3H/HEN mice were injected with 12.5 μg recombinant RevA in PBS 1:1 with adjuvant (Alhydrogel; Invivogen, San Diego, CA). Mice received 2 boosts at 3-week intervals. Three weeks after the final boost, mice were infected with 1 × 105 B. burgdorferi B31 MI-16 via subcutaneous injection or infected via tick bite (20 infected nymphs per mouse). For vaccination protocol 2, C3H/HEN mice were injected with 12.5 μg recombinant RevA in PBS (1:1) with adjuvant (complete Freund's adjuvant; Sigma). Mice received 2 boosts (incomplete Freund's adjuvant) at 10-day intervals. Ten days after the final boost, mice were infected with 1 × 105 B. burgdorferi B31 MI-16 via subcutaneous injection. For passive immunization, C3H/HEN mice were injected with approximately 200 μg anti-RevA IgG rabbit sera or rabbit preimmune sera (AnimalPharm). Twenty-four hours after injection, mice were infected with 1 × 104 B. burgdorferi B31 MI-16 via subcutaneous injection. Two weeks postinfection, mice were sacrificed and exsanguinated. Joints, ears, bladders, and hearts were cultured for 2 weeks in BSK-II medium plus 6% rabbit serum. The presence or absence of B. burgdorferi was confirmed via dark-field microscopy in 10 random fields per culture.
Enzyme-linked immunosorbent assay (ELISA).
Mouse blood was drawn from the saphenous vein and collected in heparin-coated tubes. Blood samples were centrifuged (6,000 × g) to remove red blood cells, and serum samples were stored at −20°C. To measure mouse IgM or IgG against B. burgdorferi, the wells on 96-well plates were coated overnight with 100 μl/well of 10-μg/ml B. burgdorferi lysate (mid-log-phase B. burgdorferi pelleted and washed three times in PBS) in carbonate coating buffer (0.32 g Na2CO3 and 0.586 g NaHCO3 [both per 200 ml] [pH 9.6]) at 4°C. To measure mouse antibody response against RevA, the wells were coated with 10 μg/ml recombinant RevA in carbonate coating buffer. Room temperature plates were washed three times with PBS containing 0.05% Tween 20 (by volume) (PBS-T). The wells were blocked for 2 h at room temperature with PBS containing 10% fetal bovine serum and then washed three times with PBS-T. At the time of the assay, a 1:100 dilution of serum was placed on the plate and incubated for 2 h at 37°C. The wells were washed three times with PBS-T and then incubated for 1 h at room temperature with horseradish peroxidase (HRP)-conjugated goat antiserum against mouse IgM (Pierce) or IgG (GE Healthcare, Piscataway, NJ) diluted 1:5,000 in PBS. Color development was performed using a tetramethylbenzidine (TMB) substrate (Thermo Fisher Scientific, Waltham, MA) for 15 min and stopped with the addition of an equal volume of 2 N sulfuric acid. The plates were read on an Epoch plate reader at 450 nm (BioTek, Winooski, VT). For epitope mapping, recombinant RevA or RevA peptides were solubilized according to the manufacturer's instructions and were coated on a 96-well plate overnight (10 μg/ml in carbonate coating buffer). After the wells were blocked and washed three times with PBS-T, pooled 2- to 4-week-infected mouse serum at 1:200 dilution was added for 1 h at 37°C, followed by washes, incubation with HRP-conjugated anti-mouse IgG, and detection as described above.
Bactericidal assay.
B. burgdorferi (5 × 106/ml) in BSK-II medium was treated with 1:25 dilution of rabbit anti-RevA antiserum (produced commercially by AnimalPharm [19]), sera from vaccinated mice, or preimmune sera for 24 h. Fifty microliters from each tube was transferred to 450 μl fresh BSK-II medium to examine the ability of B. burgdorferi to replicate after antiserum exposure. One hundred twenty hours after transfer, the bacteria were enumerated by dark-field microscopy; the number of motile bacteria in 10 random fields was determined. Cultures were then examined after an additional week in culture by dark-field microscopy.
Analysis of B. burgdorferi DNA levels.
Total DNA was extracted from tissue samples by using a DNeasy kit according to the manufacturer's instructions (Qiagen). Frozen mouse tissue samples (20 mg) were first minced with sterile single-use razor blades on a DNA/DNase-free glass surface and resuspended in buffer ATL (Qiagen) with proteinase K for overnight digestion at 56°C as recommended by the manufacturer. qPCR was performed by using a Bio-Rad MyIQ2 thermal cycler and Bio-Rad SYBR green supermix. All DNA samples were analyzed in triplicate. Each run included a sample that lacked template to test for DNA contamination of reagents. Oligonucleotide primers used for amplification are B. burgdorferi recA nTM17F (F stands for forward) (5′-GTGGATCTATTGTATTAGATGAGGCTCTCG-3′), B. burgdorferi recA nTM17R (R stands for reverse) (5′-GCCAAAGTTCTGCAACATTAACACCTAAAG-3′) (32), mouse nidogen F 5′-CCAGCCACAGAATACCATCC-3′, and mouse nidogen R 5′-GGACATACTCTGCTGCCATC-3′. The reaction conditions were as follows: (i) a 10-min initial denaturation step at 95°C; (ii) 40 cycles, with 1 cycle consisting of 15 s at 95°C and 1 min at 55°C (for recA) or 60°C (for nidogen); (iii) 1 min at 95°C and 1 min at 60°C for 1 min; and (iv) melting-curve analysis starting at 60°C plus 0.5°C with a hold at each temperature for 10 s. Tenfold serial dilutions of B. burgdorferi genomic DNA or mouse genomic DNA were included in every assay for each primer set. This enabled the generation of standard curves from which the amount of DNA present in each sample could be calculated, which was done using the Bio-Rad MyIQ2 software. The same software package was also used for melting-curve analyses. To verify amplicon sizes and purities, all products were separated by agarose gel electrophoresis, and DNA was visualized with ethidium bromide. Average values obtained from triplicate runs of each DNA sample for B. burgdorferi recA copies were calculated relative to the average triplicate value for the mouse nidogen housekeeping gene from the same DNA preparation. Statistical analyses of data were performed using Student's t test and assuming unequal variances.
RESULTS
Antiserum against Borrelia burgdorferi B31 RevA recognizes RevA proteins from other strains.
Previously, we tested 7 recombinant RevA proteins representing 7 distinct revA alleles from 3 different strains and 3 distinct species of Lyme disease borrelia (B. burgdorferi, Borrelia garinii, and Borrelia spielmanii), and all bound fibronectin (19; our unpublished results). Some outer surface proteins of B. burgdorferi are poor vaccine candidates due to their sequence variability or strain-to-strain heterogeneity. An alignment of known RevA sequences demonstrates extensive amino acid identity (see Fig. S1 in the supplemental material). Therefore, we examined whether antiserum against the B. burgdorferi type strain B31 RevA would recognize other RevA proteins. Antiserum directed against the B. burgdorferi type strain B31 RevA allele is cross-reactive against all tested RevA proteins (Fig. 1). These data suggest that antibodies against the B31 RevA protein will recognize RevA proteins across Lyme borreliae.
RevA antibody production during long-term infection.
Preliminary serological studies from infected humans and mice indicate the frequent presence of anti-RevA antibodies. To determine the characteristics of the antibody response over time, female BALB/c mice were infected with B. burgdorferi B31 via tick bite and monitored over 1 year. Serum samples were collected at 4- to 6-week intervals and tested for the presence of antibodies against RevA. IgM levels increased upon infection and remained steady throughout the course of infection, never reaching a titer higher than 100 (Fig. 2A). IgG levels varied from animal to animal but once elevated tended to remain high (Fig. 2B and C).
Fig 2.

Antibodies to RevA in infected mouse serum. BALB/c mice were infected via tick bite with B. burgdorferi. RevA-specific IgG and IgM from three individual mice were measured by ELISA. (A) IgM diluted 1:100. Data represent the mean absorbance and standard errors from 3 replicates per time point. (B and C) IgG diluted 1:100 (B) and 1:1,000 (C). Data in all three panels represent the mean absorbance ± standard errors (error bars) from 3 replicates per time point.
RevA antibodies are bactericidal.
Lyme disease borreliae are relatively resistant to killing by complement present in mammalian serum in the absence of specific antiborrelia antibodies (33, 34, 35, 36, 37). To determine whether anti-RevA antibodies were bactericidal, B. burgdorferi bacteria were incubated in the standard growth medium (BSK-II medium plus 6% non-heat-inactivated rabbit serum) for 24 h in the presence of rabbit polyclonal anti-RevA antiserum (19), preimmune serum from the same animal, or an equivalent volume of BSK-II medium. Bacteria were subcultured into fresh medium to determine whether B. burgdorferi could replicate after antibody exposure. As shown in Fig. 3, treatment with anti-RevA antibodies significantly impaired the ability of B. burgdorferi to replicate, while treatment with preimmune serum had no effect.
Fig 3.
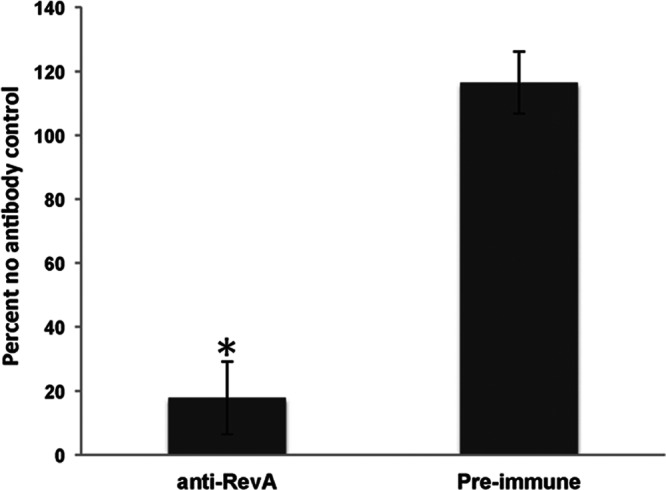
RevA antibodies are bactericidal. B. burgdorferi (5 × 106/ml) in BSK-II medium was treated with a 1:25 dilution of anti-RevA antiserum (19) or preimmune sera for 24 h. Fifty microliters from each tube was transferred to 450 μl fresh BSK-II medium plus 6% rabbit serum. Motile bacteria were enumerated in 10 random fields after 5 days by dark-field microscopy. Data are normalized to the percentage for the no-antibody control and represent the means plus standard errors from 3 independent counts for each condition. ∗, P < 0.001 by Student's t test assuming unequal variances.
Assessment of the efficacy of RevA as a vaccine.
Female C3H/HEN mice were vaccinated with recombinant RevA protein and challenged 3 weeks after the final boost with B. burgdorferi by inoculation with a needle. Serum samples from all mice were examined for the presence of anti-RevA and anti-B. burgdorferi antibodies prior to vaccination to ensure that the animals had no prior exposure to the pathogen (data not shown). After vaccination, serum was tested for the presence of RevA-specific IgG or IgM antibodies (Fig. 4). Both mice vaccinated with RevA alone and mice vaccinated with RevA plus adjuvant showed a robust IgG response to RevA, but no measurable difference among groups in the levels of RevA-specific IgM.
Fig 4.
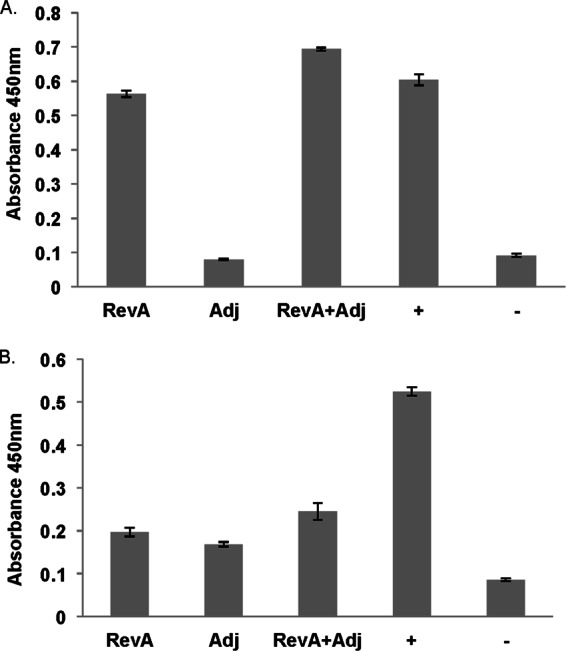
RevA antibody prior to B. burgdorferi challenge. After RevA immunization and prior to B. burgdorferi challenge, IgG (A) and IgM antibodies against RevA (B) in serum samples was measured by ELISA. Data represent the means ± standard errors (error bars) from 1 experiment with eight mice. RevA, RevA only; Adj, adjuvant alone; +, positive control (confirmed infected mouse); −, negative control (naive mouse).
Next we tested the ability of RevA vaccination to protect mice against B. burgdorferi infection. C3H/HEN mice were inoculated subcutaneously with B. burgdorferi. Two weeks later, the mice were sacrificed, and their organs were cultured for the presence of live spirochetes. As shown in Table 1 (inoculation with a needle), mice vaccinated with RevA in the presence of adjuvant were not protected from subsequent infection with B. burgdorferi.
Table 1.
Immunization with RevA in Alhydrogel
| Method of inoculation | Treatment group | Culture positivea |
|---|---|---|
| Needle | Adjuvant only | 9/9 |
| RevA | 8/8 | |
| RevA + adjuvant | 8/8 | |
| Tick | Adjuvant only | 7/7 |
| RevA | 7/7 | |
| RevA + adjuvant | 7/7 |
At least one tissue type cultured was positive for the presence of live B. burgdorferi. For mice inoculated with a needle, the tissues cultured included heart, ear, bladder, and tibiotarsal joint. For mice inoculated by tick bites, the tissues cultured included ear and tibiotarsal joint.
We also examined whether there was a difference in tissue load or dissemination between vaccinated and control groups by qPCR. We observed no differences in the number of positive samples or amount of B. burgdorferi DNA in spleen, heart, or joint samples (Table 2). We were unable to detect any B. burgdorferi DNA in the ears of challenged mice (data not shown).
Table 2.
B. burgdorferi tissue loads measured by qPCRa
| Parameter | Spleen |
Heart |
Joint |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Adj | RevA | RevA + Adj | Adj | RevA | RevA + Adj | Adj | RevA | RevA + Adj | |
| Copy no.b | ND | 4 × 10−6 ± 1 × 10−6 | 2.5 × 10−7 | 3 × 10−11 ± 2 × 10−11 | 10 × 10−11 ± 3 × 10−11 | 5 ×10−11 ± 2 × 10−11 | ND | 1 × 10−5 ± 7 × 10−11 | 4 × 10−5 ± 3 × 10−12 |
| No. of positive samples/no. of tissues sampled | 0/8 | 4/8 | 1/8 | 4/8 | 4/8 | 7/8 | 0/8 | 3/8 | 3/8 |
For each tissue type, the value is shown for mice treated with adjuvant only (Adj), RevA alone, and RevA plus Adj. ND, not detected.
Average B. burgdorferi copy number per pg of mouse DNA ± standard error of the mean for B. burgdorferi-positive specimens only.
We challenged mice with a large bolus of B. burgdorferi by inoculation with a needle, a situation that does not accurately reflect natural infection (transfer of a few organisms by tick bite). A second group of vaccinated mice was challenged by tick bite. Mice were infected via tick bite from B. burgdorferi-carrying I. scalpularis nymphs. Three weeks postchallenge, the animals were sacrificed and their tissues were cultured for the presence of B. burgdorferi. All mice produced anti-RevA antibodies postchallenge, regardless of whether they were vaccinated with RevA or adjuvant alone, suggesting that all mice were indeed infected (Fig. 5). Tissue samples were cultured, and as shown in Table 1 (tick bites), all mice were culture positive regardless of vaccination status.
Fig 5.
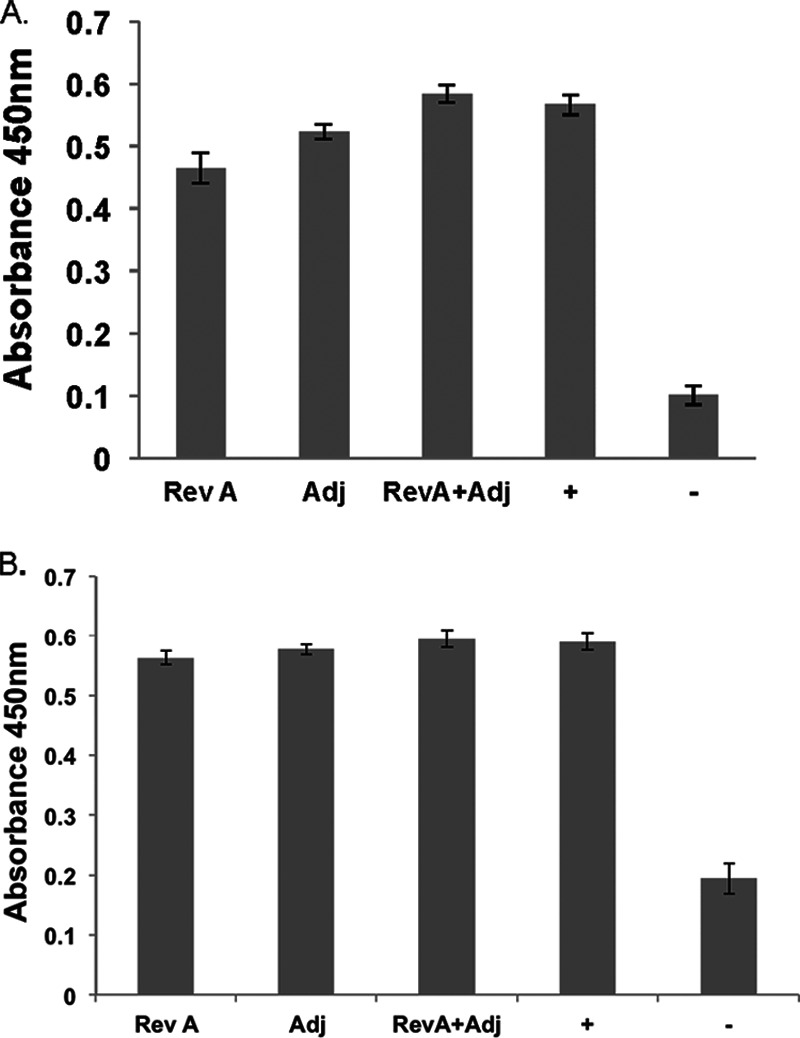
RevA antibodies in mouse serum postinfection. C3H/HEN mice were infected via tick bite with B. burgdorferi. Three weeks after the final boost, mice were bled from the saphenous vein, serum samples were collected, and RevA-specific IgG (A) and IgM (B) were measured by ELISA. Data represent the means ± standard errors from 6 wells per mouse (n = 7) per condition. RevA, RevA only; Adj, adjuvant alone; +, positive control (confirmed infected mouse); −, negative control (naive mouse).
Alum is a relatively weak adjuvant, so to better assess the immunoprotective activity of RevA, we also performed immunization with a stronger adjuvant. Mice were immunized with RevA in complete Freund's adjuvant, followed by two boosts in incomplete Freund's adjuvant. As shown in Table 3, mice immunized with RevA by this protocol were also not protected from subsequent infection with the Lyme disease spirochete.
Table 3.
Immunization with RevA in Freund's adjuvanta
| Immunization | No. of positive samples/total no. of samples cultured |
ELISA IgGb | |||
|---|---|---|---|---|---|
| Heart | Bladder | Ear | Joint | ||
| RevA | 4/6 | 6/6 | 6/6 | 6/6 | 6/6 |
| RevA + Adj | 5/6 | 5/6 | 6/6 | 6/6 | 6/6 |
| Adj | 6/6 | 5/5 | 6/6 | 6/6 | 6/6 |
Mice were immunized with RevA alone (control), RevA plus adjuvant (Adj), and Adj alone (control).
ELISA for B. burgdorferi-positive IgG. The numbers of positive serum samples/total numbers of samples tested are shown.
Next, we revisited the question of the bactericidal nature of anti-RevA antibodies. To determine whether anti-RevA antibodies produced in our vaccinated mice were bactericidal, B. burgdorferi bacteria were incubated in the standard growth medium (BSK-II medium plus 6% non-heat-inactivated rabbit serum) for 24 h in the presence of serum from the vaccinated mice, preimmune serum from the same animal, or an equivalent volume of BSK-II medium. Bacteria were subcultured into fresh medium to determine whether B. burgdorferi could replicate after antibody exposure. At 5 days posttransfer, there was no difference between B. burgdorferi growth between bacteria incubated in the presence of preimmune serum or serum from vaccinated mice (Fig. 6).
Fig 6.
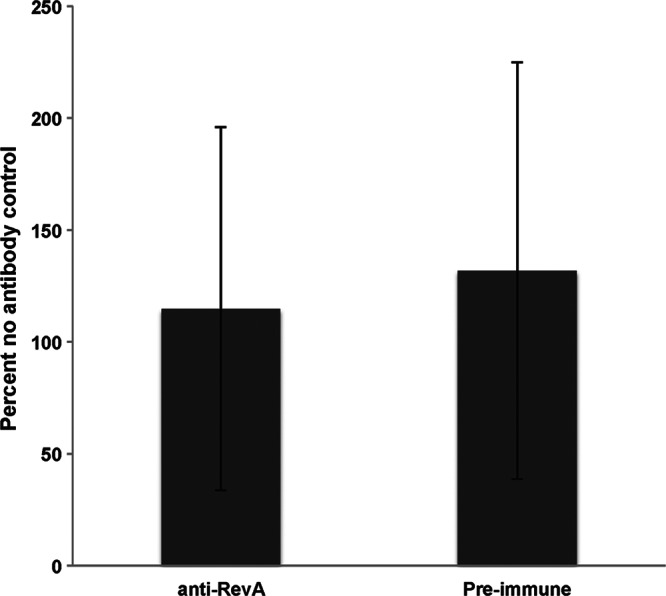
Serum from vaccinated mice is not bactericidal. B. burgdorferi (5 × 106/ml) in BSK-II medium was treated with a 1:25 dilution of antiserum from vaccinated mice or preimmune sera for 24 h. Fifty microliters from each tube was transferred to 450 μl fresh BSK-II medium plus 6% rabbit serum. Bacteria were enumerated after 5 days by dark-field microscopy with a Petroff-Hausser chamber. Data are normalized to the percentage for the no-antibody control and represent the means and standard errors from 3 independent counts for each condition.
Finally, we assessed passive immunization with the anti-RevA polyclonal serum produced in rabbits, which proved bactericidal in vitro (Fig. 3). Groups of six mice were immunized with either anti-RevA antiserum or rabbit preimmune serum. Two weeks after challenge, the ears, hearts, and tibiotarsal joints of the mice were cultured and examined for B. burgdorferi. Of the six mice immunized with preimmune serum, five were positive for infection. In contrast, of the six mice receiving anti-RevA antibodies, only one mouse became infected (Table 4). These results suggest that in vivo, anti-RevA antibodies are protective.
Table 4.
Passive immunization with anti-RevA antibodiesa
| Immunization | No. of positive samples (n = 6) |
ELISA IgG (n = 6)b | ||
|---|---|---|---|---|
| Heart | Joint | Ear | ||
| Anti-RevA | 1 | 1 | 1 | 1 |
| Preimmune serum | 5 | 5 | 5 | 5 |
Mice were immunized with anti-RevA polyclonal rabbit bactericidal antibody or with preimmune serum from a rabbit.
ELISA for B. burgdorferi-positive IgG. The numbers of positive serum samples are shown.
DISCUSSION
RevA is a surface-exposed protein of B. burgdorferi and is an early antigen of Lyme disease. Its expression is upregulated upon mammalian infection, and RevA-specific antibodies are frequently detected in experimentally infected animals and Lyme disease patients (19, 26, 38, 39, 40). RevA has also been shown to bind host fibronectin; the ability to adhere to host extracellular matrix and cells is a critical virulence factor for many bacteria (41). As a surface-exposed, highly antigenic adhesin, RevA is a prime target for the host immune system. The current study demonstrates that antibodies against RevA are made continuously throughout long-term, natural B. burgdorferi infection. Infection with B. burgdorferi results in a rapid IgM response, followed by a variable IgG response. IgM levels appeared to remain low, but steady, throughout infection (Fig. 2A). However, when the levels were examined individually, serum IgM levels often spiked, a pattern suggestive of restimulation of the immune system. A similar pattern has been seen for the Erp and OspC outer surface proteins of B. burgdorferi (29). The episodic nature of many Lyme disease symptoms, such as arthritis, may be linked to the reemergence of B. burgdorferi from tissue and the subsequent reactivation of inflammatory responses by the host immune system (6).
Specific antibodies produced against RevA in rabbits are bactericidal in vitro, while RevA antibodies from vaccinated mice had no effect on B. burgdorferi growth in culture. This could reflect species specificity of complement—B. burgdorferi is cultured in rabbit serum—but this idea was not borne out, as passive immunization of rabbit anti-RevA antibodies was protective in vivo. Immunization with RevA in Alhydrogel or Freund's adjuvant, however, was not protective in vivo, as vaccinated mice were infected despite a robust anti-RevA response. There are numerous differences between cultured B. burgdorferi and bacteria growing in vivo (42, 43) that may account for our results. The amount and accessibility of the RevA protein on the surfaces of organisms in vivo, for instance, may be altered compared to those of cultured bacteria. The interaction of antibodies against RevA with B. burgdorferi organisms in vivo may also be limited by both the paucibacilliary nature of B. burgdorferi infection and the ability of the organism to disseminate widely throughout its host. Another factor is suggested by a recent study by Hastey et al. (44); B. burgdorferi may evade B cell immunity by interfering with the quality of the antibody response. B. burgdorferi infection results in a strong but ineffective serum antibody response due to a lack of accumulation of long-lived plasma cells (44).
Animals injected with RevA or RevA plus Alhydrogel adjuvant had roughly equivalent anti-RevA IgG responses (Fig. 5). We expected a more vigorous response to RevA in the presence of this adjuvant. The purified recombinant RevA was free of protein contaminants (Fig. 1A). Similar results were seen when we immunized animals with a more potent adjuvant (complete Freund's adjuvant; data not shown). Despite the ability of the host to mount a strong antibody response to RevA, in the presence or absence of adjuvant, we were unable to stimulate a protective response in vivo.
Our results suggest that while rabbit anti-RevA antibodies are bactericidal in vitro, vaccination with RevA fails to protect mice from subsequent infection by B. burgdorferi, either through inoculation with a needle or the natural mode of infection, tick bite. In contrast, passive immunization with bactericidal anti-RevA antibodies prevented infection, suggesting that RevA is indeed highly expressed in the early stages of infection. Coupled with the fact that RevA is a highly expressed, surface-exposed protein that elicits a long-lasting antibody response, our data suggest that RevA may still be a useful target for rational vaccine development. For example, quantitative response to the lipoprotein OspA is not indicative of protection; instead, protective immunity correlates with a specific epitope (45). Using linear, overlapping peptides, we were unable to detect an immunodominant epitope (see Fig. S2 in the supplemental material). This suggests that for RevA, conformational, rather than linear, epitopes are important. Further research to identify protective epitopes, combined with a more exhaustive survey of RevA sequence variation, may aid in the design of an effective vaccine (46).
Finally, our studies emphasize the fact that B. burgdorferi, like many spirochetes, causes persistent, life-long infections in immunocompetent hosts. How spirochetes continually evade the host immune response and resist clearance is a conundrum that warrants further investigation.
Supplementary Material
ACKNOWLEDGMENTS
We thank John Watt, Brian Stevenson, and Ann Flower for helpful discussion, and Carol Beach of the University of Kentucky Center for Structural Biology Protein Core Facility for mass spectrometry.
This work was supported by grants from NovaDigm Therapeutics and NIH/NIAID 1K22AI093671-01 to C.A.B.
Footnotes
Published ahead of print 17 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/CVI.00758-12.
REFERENCES
- 1. Centers for Disease Control and Prevention 2011. Summary of notifiable diseases—United States, 2009. Morb. Mortal. Wkly. Rep. 58:1–100 [PubMed] [Google Scholar]
- 2. Stanek G, Wormser GP, Gray J, Strle F. 2012. Lyme borreliosis. Lancet 379:461–473 [DOI] [PubMed] [Google Scholar]
- 3. Nigrovic LE, Thompson KM. 2007. The Lyme vaccine: a cautionary tale. Epidemiol. Infections 135:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Souza MS, Smith AL, Beck DS, Terwilliger GA, Fikrig E, Barthold SW. 1993. Long-term study of cell-mediated responses to Borrelia burgdorferi in the laboratory mouse. Infect. Immun. 61:1814–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stanek G, Strle F. 2003. Lyme borreliosis. Lancet 362:1639–1647 [DOI] [PubMed] [Google Scholar]
- 6. Steere AC. 2001. Lyme disease. N. Engl. J. Med. 345:115–125 [DOI] [PubMed] [Google Scholar]
- 7. Zhang J-R, Norris SJ. 1998. Genetic variation of the Borrelia burgdorferi gene vlsE involves cassette-specific, segmental gene conversion. Infect. Immun. 66:3698–3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang J-R, Norris SJ. 1998. Kinetics and in vivo induction of genetic variation of vlsE in Borrelia burgdorferi. Infect. Immun. 66:3689–3697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bankhead T, Chaconas G. 2007. The role of VlsE antigenic variation in the Lyme disease spirochete: persistence through a mechanism that differs from other pathogens. Mol. Microbiol. 65:1547–1558 [DOI] [PubMed] [Google Scholar]
- 10. Palmer GH, Bankhead T, Lukehart SA. 2009. ‘Nothing is permanent but change’- antigenic variation in persistent bacterial pathogens. Cell. Microbiol. 11:1697–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eicken C, Sharma V, Klabunde T, Lawrenz MB, Hardham JM, Norris SJ, Sacchettini JC. 2002. Crystal structure of Lyme disease variable surface antigen VlsE of Borrelia burgdorferi. J. Biol. Chem. 277:21691–21696 [DOI] [PubMed] [Google Scholar]
- 12. Cabello FC, Godfrey HP, Newman SA. 2007. Hidden in plain sight: Borrelia burgdorferi and the extracellular matrix. Trends Microbiol. 15:350–354 [DOI] [PubMed] [Google Scholar]
- 13. Coburn J, Medrano M, Cugini C. 2002. Borrelia burgdorferi and its tropisms for adhesion molecules in the joint. Curr. Opin. Rheumatol. 14:394–398 [DOI] [PubMed] [Google Scholar]
- 14. Franz JK, Fritze O, Rittig M, Keysser G, Priem S, Zacher J, Burmester GR, Krause A. 2001. Insights from a novel three-dimensional in vitro model of Lyme arthritis: standardized analysis of cellular and molecular interactions between Borrelia burgdorferi and synovial explants and fibroblasts. Arthritis Rheum. 44:151–162 [DOI] [PubMed] [Google Scholar]
- 15. Häupl T, Hahn G, Rittig M, Krause A, Schoerner C, Schonherr U, Kalden JR, Burmester GR. 1993. Persistence of Borrelia burgdorferi in ligamentous tissue from a patient with chronic Lyme borreliosis. Arthritis Rheum. 36:1621–1626 [DOI] [PubMed] [Google Scholar]
- 16. Asbrink E, Hovmark A. 1988. Early and late cutaneous manifestations in Ixodes-borne borreliosis (erythema migrans borreliosis, Lyme borreliosis), p 4–15 In Benach JL, Bosler EM. (ed), Lyme disease and related disorders. New York Academy of Sciences, New York, NY: [DOI] [PubMed] [Google Scholar]
- 17. Berger BW. 1989. Dermatologic manifestations of Lyme disease. Rev. Infect. Dis. 11(Suppl 6):S1475–S1481 [DOI] [PubMed] [Google Scholar]
- 18. Ohlenbusch A, Matuschka FR, Richter D, Christen HJ, Thomssen R, Spielman A, Eiffert H. 1996. Etiology of the acrodermatitis chronica atrophicans lesion in Lyme disease. J. Infect. Dis. 174:421–423 [DOI] [PubMed] [Google Scholar]
- 19. Brissette CA, Bykowski T, Cooley AE, Bowman A, Stevenson B. 2009. Borrelia burgdorferi RevA antigen binds host fibronectin. Infect. Immun. 77:2802–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Coburn J. 2001. Adhesion mechanisms of the Lyme disease spirochete, Borrelia burgdorferi. Curr. Drug Targets Infect. Disord. 1:171–179 [DOI] [PubMed] [Google Scholar]
- 21. Grab DJ, Givens C, Kennedy R. 1998. Fibronectin-binding activity in Borrelia burgdorferi. Biochim. Biophys. Acta 1407:135–145 [DOI] [PubMed] [Google Scholar]
- 22. Seshu J, Esteve-Gassent MD, Labandeira-Rey M, Kim JH, Trzeciakowski JP, Hook M, Skare JT. 2006. Inactivation of the fibronectin-binding adhesin gene bbk32 significantly attenuates the infectivity potential of Borrelia burgdorferi. Mol. Microbiol. 59:1591–1601 [DOI] [PubMed] [Google Scholar]
- 23. Li X, Liu X, Beck DS, Kantor FS, Fikrig E. 2006. Borrelia burgdorferi lacking BBK32, a fibronectin-binding protein, retains full pathogenicity. Infect. Immun. 74:3305–3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nowalk AJ, Gilmore RD, Jr, Carroll JA. 2006. Serologic proteome analysis of Borrelia burgdorferi membrane-associated proteins. Infect. Immun. 74:3864–3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gilmore RD, Jr, Mbow ML, Stevenson B. 2001. Analysis of Borrelia burgdorferi gene expression during life cycle phases of the tick vector Ixodes scapularis. Microbes Infect. 3:799–808 [DOI] [PubMed] [Google Scholar]
- 26. Brissette CA, Rossmann E, Bowman A, Cooley AE, Riley SP, Hunfeld KP, Bechtel M, Kraiczy P, Stevenson B. 2010. The borrelial fibronectin-binding protein RevA is an early antigen of human Lyme disease. Clin. Vaccine Immunol. 17:274–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Casjens S, Palmer N, van Vugt R, Huang WM, Stevenson B, Rosa P, Lathigra R, Sutton G, Peterson J, Dodson RJ, Haft D, Hickey E, Gwinn M, White O, Fraser C. 2000. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs of an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol. 35:490–516 [DOI] [PubMed] [Google Scholar]
- 28. Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, Gwinn M, Dougherty B, Tomb J-F, Fleischmann RD, Richardson D, Peterson J, Kerlavage AR, Quackenbush J, Salzberg S, Hanson M, van Vugt R, Palmer N, Adams MD, Gocayne J, Weidmann J, Utterback T, Watthey L, McDonald L, Artiach P, Bowman C, Garland S, Fujii C, Cotton MD, Horst K, Roberts K, Hatch B, Smith HO, Venter JC. 1997. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390:580–586 [DOI] [PubMed] [Google Scholar]
- 29. Miller JC, von Lackum K, Babb K, McAlister JD, Stevenson B. 2003. Temporal analysis of Borrelia burgdorferi Erp protein expression throughout the mammal-tick infectious cycle. Infect. Immun. 71:6943–6952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zückert WR. 2007. Laboratory maintenance of Borrelia burgdorferi. Curr. Protoc. Microbiol. Chapter 12:Unit 12C.1 doi: 10.1002/9780471729259.mc12c01s4 [DOI] [PubMed] [Google Scholar]
- 31. Bunikis I, Kutschan-Bunikis S, Bonde M, Bergstrom S. 2011. Multiplex PCR as a tool for validating plasmid content of Borrelia burgdorferi. J. Microbiol. Methods 86:243–247 [DOI] [PubMed] [Google Scholar]
- 32. Ornstein K, Barbour AG. 2006. A reverse transcriptase-polymerase chain reaction assay of Borrelia burgdorferi 16S rRNA for highly sensitive quantification of pathogen load in a vector. Vector Borne Zoonotic Dis. 6:103–112 [DOI] [PubMed] [Google Scholar]
- 33. Breitner-Ruddock S, Würzner R, Schulze J, Brade V. 1997. Heterogeneity in the complement-dependent bacteriolysis within the species of Borrelia burgdorferi. Med. Microbiol. Immunol. 185:253–260 [DOI] [PubMed] [Google Scholar]
- 34. Brissette CA, Cooley AE, Burns LH, Riley SP, Verma A, Woodman ME, Bykowski T, Stevenson B. 2008. Lyme borreliosis spirochete Erp proteins, their known host ligands, and potential roles in mammalian infection. Int. J. Med. Microbiol. 298(Suppl 1):257–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Herzberger P, Siegel C, Skerka C, Fingerle V, Schulte-Spechtel U, van Dam A, Wilske B, Brade V, Zipfel PF, Wallich R, Kraiczy P. 2007. Human pathogenic Borrelia spielmanii sp. nov. resists complement-mediated killing by direct binding of immune regulators factor H and factor H-like protein 1. Infect. Immun. 75:4817–4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kurtenbach K, Sewell H-S, Ogden NH, Randolph SE, Nuttall PA. 1998. Serum complement sensitivity as a key factor in Lyme disease ecology. Infect. Immun. 66:1248–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Dam AP, Oei A, Jaspars R, Fijen C, Wilske B, Spanjaard L, Dankert J. 1997. Complement-mediated serum sensitivity among spirochetes that cause Lyme disease. Infect. Immun. 65:1228–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carroll JA, El-Hage N, Miller JC, Babb K, Stevenson B. 2001. Borrelia burgdorferi RevA antigen is a surface-exposed outer membrane protein whose expression is regulated in response to environmental temperature and pH. Infect. Immun. 69:5286–5293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gilmore RD, Jr, Mbow ML. 1998. A monoclonal antibody generated by antigen inoculation via tick bite is reactive to the Borrelia burgdorferi Rev protein, a member of the 2.9 gene family locus. Infect. Immun. 66:980–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mbow ML, Gilmore RD, Jr, Stevenson B, Golde WT, Piesman J, Johnson BJB. 2002. Borrelia burgdorferi-specific monoclonal antibodies derived from mice primed with Lyme disease spirochete-infected Ixodes scapularis ticks. Hybrid Hybridomics 21:179–182 [DOI] [PubMed] [Google Scholar]
- 41. Henderson B, Nair S, Pallas J, Williams MA. 2011. Fibronectin: a multidomain host adhesin targeted by bacterial fibronectin-binding proteins. FEMS Microbiol. Rev. 35:147–200 [DOI] [PubMed] [Google Scholar]
- 42. Brooks CS, Hefty PS, Jolliff SE, Akins DR. 2003. Global analysis of Borrelia burgdorferi genes regulated by mammalian host-specific signals. Infect. Immun. 71:3371–3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Caimano MJ. 2005. Cultivation of Borrelia burgdorferi in dialysis membrane chambers in rat peritonea. Curr. Protoc. Microbiol. Chapter 12:Unit 12C.3 doi: 10.1002/9780471729259.mc12c03s00 [DOI] [PubMed] [Google Scholar]
- 44. Hastey CJ, Elsner RA, Barthold SW, Baumgarth N. 2012. Delays and diversions mark the development of B cell responses to Borrelia burgdorferi infection. J. Immunol. 188:5612–5622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kramer MD, Schaible UE, Wallich R, Moter SE, Petzoldt D, Simon MM. 1990. Characterization of Borrelia burgdorferi associated antigens by monoclonal antibodies. Immunobiology 181:357–366 [DOI] [PubMed] [Google Scholar]
- 46. Luft BJ, Dunn JJ, Lawson CL. 2002. Approaches toward the directed design of a vaccine against Borrelia burgdorferi. J. Infect. Dis. 185(Suppl 1):S46–S51 [DOI] [PubMed] [Google Scholar]
- 47. Barthold SW, Moody KD, Terwilliger GA, Duray PH, Jacoby RO, Steere AC. 1988. Experimental Lyme arthritis in rats infected with Borrelia burgdorferi. J. Infect. Dis. 157:842–846 [DOI] [PubMed] [Google Scholar]
- 48. Steere AC, Grodzicki RL, Kornblatt AN, Craft JE, Barbour AG, Burgdorfer W, Schmid GP, Johnson E, Malawista SE. 1983. The spirochetal etiology of Lyme disease. N. Engl. J. Med. 308:733–740 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.