

Abstract
The choline oxidase (CHOA) and betaine aldehyde dehydrogenase (BADH) genes identified in Aspergillus fumigatus are present as a cluster specific for fungal genomes. Biochemical and molecular analyses of this cluster showed that it has very specific biochemical and functional features that make it unique and different from its plant and bacterial homologs. A. fumigatus ChoAp catalyzed the oxidation of choline to glycine betaine with betaine aldehyde as an intermediate and reduced molecular oxygen to hydrogen peroxide using FAD as a cofactor. A. fumigatus Badhp oxidized betaine aldehyde to glycine betaine with reduction of NAD+ to NADH. Analysis of the AfchoAΔ::HPH and AfbadAΔ::HPH single mutants and the AfchoAΔAfbadAΔ::HPH double mutant showed that AfChoAp is essential for the use of choline as the sole nitrogen, carbon, or carbon and nitrogen source during the germination process. AfChoAp and AfBadAp were localized in the cytosol of germinating conidia and mycelia but were absent from resting conidia. Characterization of the mutant phenotypes showed that glycine betaine in A. fumigatus functions exclusively as a metabolic intermediate in the catabolism of choline and not as a stress protectant. This study in A. fumigatus is the first molecular, cellular, and biochemical characterization of the glycine betaine biosynthetic pathway in the fungal kingdom.
INTRODUCTION
Glycine betaine (GB) is a small, water-soluble organic molecule that is essential to protect plants, animals, and bacteria against abiotic stress. Two main mechanisms have been proposed for GB's responsibility for enhanced stress tolerance: (i) osmotic adjustment controlling the absorption of water from the surroundings and (ii) reactive oxygen species (ROS) scavenging (1). In plants, this molecule is essential to fight salt, cold, heat, and drought stresses. The antistress properties of GB have led to the production of transgenic plants that are able to resist abiotic stress and especially to become tolerant to salt and drought (2). It has also been suggested that the same mechanisms would be responsible for the capacity of many human-pathogenic bacteria, including Pseudomonas aeruginosa, Escherichia coli, Staphylococcus aureus, Listeria monocytogenes, and Mycobacterium tuberculosis, to survive and grow in their human host (3–7). In the majority of biological systems, GB is synthesized by a two-step oxidation reaction: choline is first oxidized to betaine aldehyde (BA), which is later oxidized to yield GB. The first oxidation is catalyzed by a choline monooxygenase in plants and by a choline dehydrogenase (CDH) in bacteria, whereas the second reaction is catalyzed by an NADP+-dependent betaine aldehyde dehydrogenase (BADH) (1).
Acquisition of osmoprotectants is important for the survival of the conidia and for mycelial growth of filamentous fungal pathogens such as Aspergillus fumigatus in their human host (8, 9). However, the mechanisms controlling the osmotic pressure changes during the biological cycle of fungal species remain insufficiently understood. Although trehalose has been repeatedly cited as the major metabolite involved in the osmoprotection of A. fumigatus, several arguments suggest that it is not the only one: (i) trehalose is rapidly degraded upon induction of conidial germination and increases again later during mycelial growth; (ii) two metabolic pathways for the biosynthesis of trehalose have been identified, but their respective roles have not been evaluated; and (iii) other polyols have been also suggested to be associated with changes in osmotic pressure (8, 9). This also raised the question of the existence of other molecules that could be associated with osmoprotection during the fungal biological cycle. GB was an obvious candidate to study since (i) it is an osmoprotectant in plants and bacteria and (ii) a transcriptomic study of the conidial germination of A. fumigatus showed that a gene orthologous to the bacterial choline oxidase gene was one of the most upregulated genes in the early steps of conidial germination (10).
The GB pathway in A. fumigatus was analyzed here to investigate the role of this molecule in the resistance of fungi to abiotic stress. This is the first time that the GB pathway in the fungal kingdom has been analyzed. Biochemical studies have shown that (i) the choline oxidase AfChoAp catalyzes the flavin-linked oxidation of choline or betaine aldehyde (BA) to GB, with reduction of molecular oxygen to hydrogen peroxide, and (ii) the betaine aldehyde dehydrogenase AfBadAp catalyzes the oxidation of BA to GB with reduction of NAD+ to NADH. Studies on single and double deletion mutants showed that GB is not involved in osmotic regulation but is used as a nutrient by this fungal species.
MATERIALS AND METHODS
Strains, media, and culture conditions.
The A. fumigatus parental strains CBS 144.89 (= Dal = Af1163) (10) and its akuBku80 derivative (11) and the mutant strains used in this study are shown in Table S1 in the supplemental material. A. fumigatus strains were maintained on 2% malt agar tubes. Conidial germination and mycelial growth were studied in different 2% agar or shake liquid media at 37°C. Complex media were Sabouraud (SAB) medium, 2% malt medium, yeast extract-peptone-dextrose (YPD) medium, 1% yeast extract (YE), or RPMI synthetic medium. Mutant and parental strains of A. fumigatus were also grown in MM medium containing 10 g/liter glucose, 0.92 g/liter ammonium tartrate, 0.5 g KCl, 0.5 g MgSO4 · 7H2O, 2 g KH2PO4, and 1 ml of trace element solution at pH 6.5. The various media and stresses tested in this study are summarized in Table S2 in the supplemental material. Briefly, media were supplemented with 1.2 M mannitol, 1.2 M sorbitol, or 1 to 2.5 M NaCl to test the effect of osmotic pressure. Growth was also tested at pH 5.0 to 9.0, cold (4 to 10°C) or hot (37 to 50°C) temperatures, and in the presence of 1 to 5 mM hydrogen peroxide or 5 to 80 μM menadione in complex media or in MM without a nitrogen source but containing 20 mM GB. Glucose or/and ammonium tartrate was replaced in the MM medium with 20 mM choline chloride, 20 mM GB, or 6.6 mM L-α-phosphatidylcholine as a sole carbon, nitrogen, or carbon and nitrogen source. Curosurf (Chiesi Farmaceutici, Parma, Italy) was tested at 1 to 20 mg/ml on a 2% agar medium.
DNA and RNA analyses.
For DNA extraction, mycelium was grown for 16 h at 37°C in a liquid medium containing 3% glucose and 1% yeast extract. For the RNA extraction, the fungus was grown in YPD liquid medium at 37°C. Total DNA and RNA were isolated as previously described (12, 13). Reverse transcriptase PCRs (RT-PCRs) were performed with 2 μg total RNA, using a Thermoscript RT-PCR system kit (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions with gene-specific primers for AfCHOA and AfBADA (see Table S3 in the supplemental material).
Production of recombinant AfChoAp and AfBadAp.
The gene AfCHOA was subcloned into pET20b(+), which allows the expression of the enzyme without any tag, whereas the gene AfBADA was expressed in the pET16 vector, which results in the addition of His6 at the N-terminal region of the protein. The AfCHOA and AfBADA cDNAs were obtained by PCR amplification using, respectively, the primer pairs NdeI-ATG-AfCHOA/AfCHOA-stop-BamHI and NdeI-ATG-AfBADA/AfBADA-stop-BamHI and cDNA from A. fumigatus mycelium as the template (see Table S3 in the supplemental material). The obtained PCR products were gel purified and digested by NdeI and BamHI. These fragments were introduced into pET16b (NdeI/BamHI). The vector pET20b was used for the final expression of the AfCHOA cDNA. For cloning into the pET20b vector, the AfCHOA cDNA was excised with NdeI (5′ site) and BamHI (3′ site) from the pET16b plasmid and inserted with NdeI/BamHI sites of pET20b. After confirming the absence of mutations by DNA sequencing at the DNA Core Facility at Georgia State University, the enzyme was expressed in E. coli Rosetta(DE3)pLysS. After transformation, E. coli Rosetta(DE3)pLysS (Novagen) cells were grown at 37°C in Luria-Bertani medium supplemented with 50 μg/ml ampicillin and 34 μg/ml chloramphenicol, and protein production was induced for 16 h at 20 or 25°C in the presence of 0.2 or 0.4 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for AfChoAp or AfBadAp, respectively. AfChoAp was recovered by sonication in 50 mM potassium phosphate (pH 7.0) containing 10% glycerol, 10 mM MgCl2, 1 mM 2-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride (PMSF), 5 μg/ml DNase I, 5 μg/ml RNase, and 0.2 mg/ml lysozyme and purified by sequential 30 and 65% saturation ammonium sulfate precipitation before loading onto a DEAE-Sepharose Fast Flow column eluted with a linear gradient from 0 to 500 mM NaCl. For best preservation of the activity during storage at −20°C, the enzyme was dialyzed and stored in 100 mM imidazole (pH 7.0) with 10% glycerol. E. coli cells expressing AfBadAp were resuspended in 20 mM sodium phosphate (pH 7.4) containing 500 mM NaCl, 20 mM imidazole, 10% glycerol, 0.2 mg/ml lysozyme, and 1 mM PMSF and sonicated for 20 min. The soluble fraction was loaded onto a nickel-Sepharose column, and elution was with a linear gradient from 20 to 500 mM imidazole. It was verified by SDS-PAGE that these proteins were purified to homogeneity (see Fig. S1A in the supplemental material).
Spectroscopic analysis.
All UV-visible absorbance spectra were recorded using an Agilent Technologies diode array spectrophotometer (model HP 8453). The extinction coefficient of AfChoAp was determined in 20 mM Tris-Cl (pH 8.0) and 10% glycerol after denaturation of the enzyme by treatment with 4 M urea at 40°C for 30 min, based upon the ε450 value of 11.3 mM−1 cm−1 for free FAD (14). To determine the amount of covalently bound flavin, the purified enzyme was incubated on ice for 30 min after addition of 10% trichloroacetic acid, followed by removal of precipitated protein by centrifugation. The UV-visible absorbance spectrum of the supernatant was recorded again to check for the presence of unbound FAD. AfChoAp reduction was achieved by anaerobic substrate reduction carried out with an anaerobic solution of 24 μM enzyme through successive (20 times) evacuations and flushings with O2-free argon in an anaerobic cuvette (see Fig. S1B in the supplemental material).
Biochemical analysis.
AfChoAp (24 μM) reduction was achieved by anaerobic incubation with 0.5 mM choline. The enzymatic activity of AfChoAp was measured by the method of initial rates as described for the Arthrobacter globiformis wild-type choline oxidase (15). The steady-state kinetic parameters of the enzyme were determined at various concentrations of oxygen (0.1 to 1.1 mM) and choline (0.5 to 10 mM) or BA (0.5 to 10 mM) in 10 mM Tris-Cl (pH 8.0) at 25°C. AfBadAp activity was assayed spectrophotometrically by measuring NAD(P)+ reduction by the increase in extinction at 340 nm (16). The steady-state kinetic parameters of the enzyme were determined at various concentration of BA (10 to 400 μM) and NAD(P)+ (0.01 to 10 mM) in 100 mM HEPES-KOH (pH 8.0) at 25°C. In the assay coupling AfChoAp and AfBadAp, the activity of AfBadAp was measured spectrophotometrically as described above at various concentrations of NAD+ (20 to 740 μM) and fixed concentrations of choline (1 mM) and oxygen (0.25 mM) in 100 mM HEPES-KOH (pH 8.0) at 25°C. The amount of BA produced during catalytic turnover of the enzyme with choline as the substrate was determined spectrophotometrically using 2,4-dinitrophenylhydrazine as described previously (17) (see Fig. S1B in the supplemental material).
Mutant constructions.
The cassettes to produce the AfchoAΔ::HPH and AfbadAΔ::HPH single mutants and the AfchoAΔAfbadAΔ::HPH double mutant were constructed by PCR fusion as described earlier (12) using primers listed in Table S3 in the supplemental material. The resistance marker used to replace AfCHOA, AfBADA, or both was the HPH gene of E. coli, coding for hygromycin B phosphotransferase. An example of a deletion cassette constructed by PCR fusion of three PCR products (PCR1, PCR2, and PCR3) is shown in Fig. S2 in the supplemental material (deletion of AfCHOA). In a first PCR round (see Fig. S2 in the supplemental material), flanking region 1 (PCR1, primers 55-CHOA and 53-CHOA), flanking region 2 (PCR2, primers 35-CHOA and 33-CHOA), and the HPH gene (PCR3, primers 5-CHOA-HPH and 3-CHOA-HPH) were amplified from wild-type DNA template and pAN7.1 with the primers listed in Table S3 in the supplemental material. Phusion DNA Polymerase (New England BioLabs) was used, and the PCR conditions were as follows: 98°C for 30 s, 25 cycles of 98°C for 10 s, 65°C for 20 s, and 72°C for 30 s, and 72°C 10 min. Primers 53-CHOA, 5-CHOA-HPH, 35-CHOA, and 3-CHOA-HPH were 60-bp chimeric oligonucleotides containing at the 5′ end a reverse complement sequence (53-CHOA with 5-CHOA-HPH and 35-CHOA with 3-CHOA-HPH) for fusion PCR. The obtained PCR products were gel purified and used for a second PCR step that allowed fusion of these three separate fragments by using the 55-CHOA and 33-CHOA primers. The PCR conditions were the same as described above. The resulting PCR product was purified and used to transform A. fumigatus conidia. The AfchoAΔ::HPH and AfbadAΔ::HPH single mutants and the AfchoAΔAfbadAΔ::HPH double mutant were constructed in the akuBku80 strain (11). The fusion PCR product (1 to 2 μg) was used to transform A. fumigatus akuBku80 swollen conidia by the electroporation method described previously (18). For transformation experiments, minimal medium MM (1% glucose, 0.092% ammonium tartrate, 0.052% KCl, 0.052% MgSO4 · 7H2O, 0.152% KH2PO4, 1 ml trace element solution, pH 6.8) was used (19). MM containing 250 μg · ml−1 hygromycin (Sigma) was used for the screening of the mutants (AfchoAΔ::HPH, AfbadAΔ::HPH, and AfchoAΔAfbadAΔ::HPH), and MM containing 30 μg · ml−1 phleomycin (InvivoGen) was used for the selection of the AfchoAΔAfbadAΔ::HPH double deletion mutant strain transformed with pAN8.1-eGFP-AfCHOA::promoter::AfBADA-RFP-T plasmid. To check integration of the HPH cassette at the locus, the DNAs of the akuBku80 and the mutant strains were digested with restriction enzyme (Roche Applied Science) and verified by Southern blotting with probes corresponding to the first flanking region (see Fig. S2A [deletion of AfCHOA], B [deletion of AfBADA], and C [double deletion of AfCHOA and AfBADA] in the supplemental material).
Transcriptional and translational fusion of AfCHOA with eGFP and of AfBADA with RFP-T.
The transcriptional fusion (eGFP::promoter::RFP-T) of the AfCHOA and AfBADA promoter (1,650 bp) with enhanced green fluorescent protein (eGFP) and RFP-T was constructed by PCR fusion (Fig. 1C). The eGFP fragment (PCR1) was obtained by PCR amplification using p123 (pOTEF eGFP)-HPH plasmid as the template and the primer pairs NdeI-PCR1-transcript-F and PCR1-transcript-R. The AfCHOA and AfBADA promoter (PCR2) was obtained by PCR amplification using akuBku80 genomic DNA as the template and the primer pairs PCR2-transcript-F and PCR2-transcript-R. The RFP-T fragment (PCR3) was obtained by PCR amplification using pAL5-lifeact plasmid as the template and the primer pairs PCR3-transcript-F and PCR3-transcript-NdeI-R. The obtained PCR products were gel purified and used for a second PCR step that allowed fusion of these two separate fragments by using NdeI-PCR1-transcript-F and PCR3-transcript-NdeI-R primers. The resulting PCR product was purified and digested NdeI, whose restriction site is present in the NdeI-PCR1-transcript-F and PCR2-transcript-R-NdeI primers. This fragment was introduced into pAN8.1 plasmid (20) to obtain the pAN8.1-eGFP::promoter::RFP-T plasmid (NdeI). One to 2 μg of linear pAN8.1-eGFP::promoter::RFP-T (BglII) was used to transform the swollen conidia of the wild-type Dal strain. The revertant cassette corresponding to the translational fusion (eGFP::AfCHOA::promoter::AfBADA::RFP-T) of the AfCHOA open reading frame (ORF) (1,720 bp) with eGFP (720 bp) and the AfBADA ORF (1,602 bp) with RFP-T (735 bp) under the control of the AfCHOA and AfBADA promoter (1,650 bp) was obtained by PCR fusion (see Fig. S2C in the supplemental material). In a first PCR round (see Fig. S2C in the supplemental material), the eGFP (PCR1, primers NdeI-PCR1-translational-F and PCR1-translational-R), the fragment corresponding to the AfCHOA ORF and AfCHOA and AfBADA promoter and the AfBADA ORF (PCR2, primers PCR2-translational-F and PCR2-translational-R), and the RFP-T (PCR3, primers PCR3-translational-F and PCR3-translational-NdeI-R) were amplified, respectively, from the p123 (pOTEF eGFP)-HPH plasmid, wild-type DNA template, and the pAL5-lifeact plasmid. The primers are listed in Table S3 in the supplemental material. The obtained PCR products were gel purified and used for a second PCR step that allowed fusion of these four separate fragments by using the NdeI-PCR1-translational-F and PCR3-translational-NdeI-R primers. The resulting PCR product was purified and digested by NdeI, whose restriction site is present in the NdeI-PCR1-translational-F and PCR3-translational-NdeI-R primers. This fragment was introduced into pAN8.1 plasmid to obtain the pAN8.1-eGFP::AfCHOA::promoter::AfBADA::RFP-T plasmid (NdeI). One to 2 μg of linear pAN8.1-eGFP::AfCHOA::promoter::AfBADA::RFP-T (BglII) was used to transform the swollen conidia of the AfchoAΔAfbadAΔ::HPH double mutant. Fluorescence light microscopy observations were performed after the fungus was grown in MM medium supplemented with choline to boost the fluorescence.
Fig 1.

Organization and expression of BADA and CHOA gene cluster in A. fumigatus. (A) Organization of BADA and CHOA gene cluster in A. fumigatus. The BADA and CHOA tail-to-tail cluster is found in most ascomycetes (panel a). In 3 species, two (for Blastomycesdermatitidis and Paracoccidioides brasiliensis) (panel c) or three (for Histoplasma capsulatum) (panel b) genes are inserted between the CHOA and BADA genes (dotted lines). (B) Gene expression during growth was determined by RT-PCR in akuBku80 resting conidia (0 h) and in conidia incubated in YPD broth at 37°C for 0.5, 2, or 4 h (swollen conidia), for 8 h (germinated conidia), and for 20 and 30 h (mycelia). Specific primer pairs designed to include an intron in the PCR product to verify the lack of genomic DNA amplification (see Table S3 in the supplemental material) were used for cDNA amplification. The sizes of amplified products are indicated. The EF1α gene was constitutively expressed and used as a control of constitutive expression. H2O, negative control; G, A. fumigatus genomic DNA. (C) Transcriptional fusion was performed by fusion of three PCR fragments: the PCR1 fragment encoding eGFP, the PCR2 fragment corresponding to the promoter of the AfCHOA and AfBADA genes (1.650 kb), and the PCR3 fragment encoding RFP-T. (D and E) eGFP (D) and RFP-T (E) fluorescences were observed in cytosol of swollen conidia, germ tubes, and mycelium. Cultures were grown in liquid MM with choline chloride (20 mM) as the sole nitrogen source. (F) Bright-field image of the same hyphae. Note the resting conidia (small conidia, inset F) are not fluorescent in panels D and E, confirming that the AfCHOA and AfBADA genes are not expressed in resting conidia.
Nuclear magnetic resonance (NMR) analysis.
Mycelium grown for 16 h on MM at 37°C under shaking conditions was disrupted with 1-mm diameter glass beads for 2 min in a Fast-prep cell breaker (MP Biomedical). After removal of the polysaccharides and proteins by the addition of 2 volumes of ethanol, the supernatants were submitted to one-dimensional (1D) 1H- and 2D 13C-edited heteronuclear single quantum correlation (HSQC), 2D 1H,13C-heteronuclear multiple bond correlation (HMBC), and 2D 1H,14N-HSQC spectroscopies (21). As described below, the supernatants were dried in vacuo and then reconstituted with the minimum amount of water. After removing the residual insoluble materials by centrifugation, these obtained supernatants were lyophilized to obtain the intracellular fractions. Exchangeable protons were removed by dissolving the intracellular fractions in D2O and subsequent lyophilization. This exchange process was repeated thrice. All spectra were recorded in D2O at 300 K on a Bruker AVANCE-III 400-MHz Nanobay spectrometer operating at a proton frequency of 400 MHz and equipped with a 5-mm BBFO Plus Z gradient probe. 1H chemical shifts were referenced to external 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) (its methyl resonance was set to 0 ppm). 13C chemical shifts were then calculated from the 1H chemical shift and gamma ratio relative to DSS. The 13C/1H gamma ratio of 0.251449530 was used (22) based on 1D 1H- and 2D 13C-edited HSQC and 2D 1H,13C-HMBC data. For the further specific detection of choline derivatives, including glycine betaine, we used 1D-15N- and 2D 1H,14N-HSQC spectra (21). For 1H,14N-HSQC spectra, the INEPT (insensitive nuclei enhanced by polarization transfer) evolution time τ = 18 ms was used.
Follow-up of osmolyte molecules and enzymes in A. fumigatus.
The concentrations of trehalose, mannitol, arabitol, and glycerol in the resting and germinating conidia were analyzed as described previously (23). Briefly, 1-week-old conidia (produced in 2% malt extract agar medium) were incubated in Sabouraud (SAB) liquid medium for 0 h, 3 h, and 6 h at 37°C in shake flasks at 300 rpm. Resting and germinated conidia were resuspended in distilled water (dH2O), mixed with 2.5% 0.5-mm glass beads, and disrupted using a FastPred-24 instrument (MP-Biomedical) for 120 s at the speed of 6 m/s. Centrifugation at 10,000 × g for 10 min at 4°C was carried out to eliminate cells debris. Supernatant was taken and boiled for 10 min. A new centrifugation at 10,000 × g for 5 min was performed, and the supernatant was used in the following steps. Sugars and polyols were separated by liquid chromatography using an HPX-87 H column (300 by 7.8 mm; Bio-Rad) with the following conditions: temperature, 60°C; eluent, 0.008 N H2SO4; and flow, 0.6 ml/min. Compounds were detected by refractometry with trehalose, mannitol, arabitol, and glycerol standards.
The expression of enzymes catalyzing trehalose and mannitol biosynthesis was checked by RT-PCR using the same RNA preparations as described above for the analysis of CodA and BadH expression in vitro with pairs of primers presented in Table S3 in the supplemental material. The enzymes that are part of the trehalose phosphate synthase/phosphatase complex are orthologs of yeast TPS1 (AFUB_001790), TPS2 (AFUB_ 043350), TPS3 (AFUB_089470), TSL1 (AFUB_021090), and CLOCK9 (AFUA_6G03420) (10, 24, 25). The two key enzymes of the mannitol biosynthetic pathway are the mannitol 1 phosphate 5 dehydrogenase (AFUB_026440) and the mannitol 2 dehydrogenase (AFUB_071700) (M1PDH and M2DH) (26).
In vivo experiments.
Immunosuppressed mice were generated as described previously (13). Each mouse was intranasally inoculated with 6 × 107 conidia. Two days after inoculation, infected mice were euthanized with CO2. Lungs were taken from each mouse and frozen in liquid nitrogen. Three lungs pairs were mix together for each biological sample. RNA extraction was performed according to the mirVana kit (Ambion) protocol, followed by reverse transcriptase reactions performed with the iScript cDNA synthesis kit (Bio-Rad). The expression of AfBadAp and AfChoAp in lungs of mice infected with A. fumigatus was determined by PCR using the primers listed in Table S3 in the supplemental material for expression in vivo.
For assays of survival of parental and mutant strains in the lungs of infected animals, 6 × 107 fluorescein isothiocyanate (FITC)-labeled conidia in 30 μl of 0.05% Tween 20 were inoculated intranasally in 7-week-old OF1 male mice (Charles River Laboratory, L'Arbresle, France) as described previously (13). After an infection period of 36 h, bronchoalveolar washes were performed (4 mice per fungal strain). The percentage of germination was quantified after incubation of the bronchoalveolar lavage (BAL) fluid in an equal volume of 2× Sabouraud (4% glucose–2% mycopeptone) culture medium at 37°C for 7 h.
RESULTS
A gene cluster encodes the choline oxidase and betaine aldehyde dehydrogenase of A. fumigatus.
Two genes involved in GB metabolism in A. fumigatus were identified by BLAST analysis. They were located nearby on chromosome 8 and arranged in a head-to-head configuration at a distance of 1.7 kb between their ORFs. The cDNAs corresponding to the AfCHOA and AfBADA genes were sequenced in order to determine the structures (intron/exon) of these genes. The first one coded for a choline oxidase, AfChoAp (AFUB_083470). It was a 1,723-bp gene that contained two exons of 247 and 1,382 bp separated by an intron of 94 bp coding for a 542-amino-acid protein. The second one encoded a betaine dehydrogenase (AfBadAp; AFUB_083480); it was 1,605 bp long and contained two exons of 485 and 1,015 bp separated by an intron of 105 bp coding for a 499-amino-acid protein. AfChoAp displays 37% identity and 54% similarity with ChoAp of Arthrobacter globiformis (AAP68832.1), which is the only bacterial species that has a unique CHOA gene. All relevant amino acid genes of the active site previously identified in A. globiformis CHOA were present in all fungal CHOA genes (data not shown). The percentages of identity and similarity between the BadAp of A. fumigatus and the best-characterized BadAp bacterial protein, from Pseudomonas aeruginosa (NP_254060.1), were 57% and 70%, respectively, and those between AfBadAp and BadAp of Arabidopsis thaliana (AEE35649.1) were 35% and 54%, respectively.
Interestingly, BADA clustered in the opposite orientation with respect to the CHOA gene in A. fumigatus and other ascomycetous fungi, suggesting the presence of a common promoter (Fig. 1). No N-terminal secretion signal (N-terminal peptide signal) or transmembrane domains were predicted in either protein. Both AfCHOA and AfBADA mRNAs were expressed in germinating conidia (immediately after their incubation in a nutritive medium) and growing mycelia but not in resting conidia (Fig. 1). The transcriptional fusion of the AfCHOA promoter with eGFP and of the AfBADA promoter with RFP-T also showed that fluorescence of both eGFP and RFP-T was seen in swollen and germinating conidia and mycelia but not in the dormant conidia (Fig. 1).
Biochemical characterization of recombinant AfChoAp and AfBadAp.
Both recombinant AfChoAp and AfBadAp were produced in E. coli, with a molecular masses of 60 and 52 kDa, respectively (see Fig S1A in the supplemental material). The UV-visible absorbance spectra of the oxidized and reduced forms of the AfChoAp are shown in Fig. S1B in the supplemental material. With AfChoAp, the absorbance in the visible region of the oxidized species is typical of that of a flavoprotein, with bands at 361 and 448 nm and shoulders at 420 and 460 nm. The UV-visible absorbance spectrum of the reduced enzyme obtained through anaerobic reduction with choline showed a well-resolved maximum at ∼360 nm, consistent with the enzyme-bound flavin being present in the anionic form (27). The flavin was shown to be covalently bound to the enzyme as observed after protein precipitation with 10% trichloroacetic acid and removal of the denatured protein through centrifugation. The extinction coefficient of the enzyme at 448 nm was calculated to be 11,500 M−1 cm−1 from the absorbance of FAD after protein denaturation by urea. The absorbance spectrum of AfBadAp from 200 to 600 nm showed a single peak at 280 nm with no absorption in the visible region, consistent with the absence of a bound chromophore.
AfChoAp was able to catalyze the oxidation of choline to GB with BA as an intermediate and to reduce molecular oxygen to hydrogen peroxide. Under the experimental conditions tested, recombinant AfChoAp had an average specific activity of 9 μM O2 · min−1 · mg E−1, using 10 mM choline and atmospheric oxygen at pH 7.0 and 25°C. The steady-state kinetic mechanism and the associated kinetic parameters were determined by measuring the initial rates of oxygen consumption at various concentrations of both oxygen and choline or BA as the substrate. Initial velocity data yielded a series of lines converging to the left of the y axis in double-reciprocal plots for both choline and BA, indicating a mechanism in which a ternary complex must be formed before release of the first product of the reaction. In accordance with the observed kinetic pattern, the data described a sequential steady-state kinetic mechanism (Table 1). To test if BA was released from the active site of the enzyme, the amount of BA accumulated during catalytic turnover of AfChoAp with saturating choline and 1.1 mM oxygen dissolved in aqueous buffered solution was determined using 2,4-dinitrophenylhydrazine (see Fig. S1C in the supplemental material). The amount of BA released and the amount of BA expected to be released based on the kinetic parameters were similar, showing that BA was predominantly released from the active site during turnover of the enzyme with choline as the substrate.
Table 1.
Steady-state kinetic parameters for AfChoAp and AfBadApa
| Protein | Substrate | Ligand | kcat, s−1 | KA, mM | kcat/KA, M−1 s−1 | KO2, mM | kcat/KO2, M−1 s−1 | KNAD(P)+, mM | kcat/KNAD(P)+, M−1 s−1 | Kia, mM |
|---|---|---|---|---|---|---|---|---|---|---|
| AfChoAp | Choline | 40.0 ± 0.2 | 1.8 ± 0.1 | 22,200 ± 700 | 0.69 ± 0.01 | 58,000 ± 1,000 | 1.4 ± 0.1 | |||
| Betaine-aldehyde | 18.2 ± 0.4 | 1.6 ± 0.1 | 11,400 ± 800 | 0.36 ± 0.02 | 52,000 ± 3,200 | 3.8 ± 0.3 | ||||
| Choline | NAD+ | 23.0 ± 1.0 | 0.62 ± 0.03 | 37,400 ± 2,400 | 2.4 ± 0.1 | |||||
| AfBadAp | NAD+ | 10.2 ± 0.4 | 0.025 ± 0.001 | 408,000 ± 20,000 | 0.037 ± 0.004 | 276,000 ± 11,000 | 50 ± 1 | |||
| NADP+ | 1.4 ± 0.1 | 0.11 ± 0.01 | 13,000 ± 1,300 | 1.7 ± 0.1 | 820 ± 70 | 150 ± 13 |
Parameters are for AfChoAp with oxygen and choline or betaine aldehyde as substrates and for AfBadAp with betaine aldehyde and NAD(P)+ as substrates. Values are sample means and standard errors. KA is the Km for either choline or betaine aldehyde. Data for reactions with no ligand were fitted to the equation v/e = kcatAB/(KaB + KbA + AB + KiaKb) (Equation 1) and those for AfChoAp with choline and NAD+ ligand were fitted to the equation v/e = kcatAB/(KbA + AB + Kia) (Equation 2), where Ka and Kb are the Michaelis constants for choline and betaine aldehyde (Ka) and oxygen or NAD(P)+ (Kb), respectively, and kcat is the turnover enzyme (e) saturated with both substrates. The choline oxidase activity was measured in 20 mM Tris-HCl (pH 8.0) at 25°C and the betaine aldehyde dehydrogenase activity in 100 mM HEPES-KOH (pH 8.0) at 25°C.
AfBadAp activity was measured with the method of initial rates by measuring the rate of NAD(P)+ reduction at various concentrations of both BA and NAD(P)+. No enzyme inhibition by BA or NAD(P)+ was observed with concentrations as high as 200 μM NAD+, 5 mM NADP+, or 400 μM BA. The lines in the double-reciprocal plots converged to the left of the y axis, indicating a sequential mechanism. In accordance, the best fit of the data was achieved with equation 1 (Table 1). The enzyme could use either NAD+ or NADP+ as the oxidizing substrate for the reaction, but NAD+ was clearly preferred, as indicated by the ratio of the second-order rate constants for substrate capture (kcat/KNAD+)/(kcat/KNAD(P)+) being larger than 300 (Table 1). The recombinant AfBadAp had a specific activity of 33 μM NAD+ · min−1 · mg E−1, using 1 mM NAD+ and 0.5 mM BA at pH 8.0 and 25°C.
To evaluate whether NAD+ had an effect on the oxidation of choline by AfChoAp, the steady-state kinetics of AfChoAp with choline as the substrate was determined in the presence of 300 μM NAD+. With choline as the substrate, the best fit of the data was obtained with equation 2 (Table 1), which describes a steady-state mechanism with the formation of a ternary complex where the Km value for choline is significantly smaller than the term KiaKoxygen that is present in equation 1 (28). If one considers a Kcholine/KiaKoxygen ratio of at least 0.05 as being required to simplify equation 1 to equation 2, then a Km value for choline of 26 μM can be estimated from the data in Table 1. Consequently, the kcat/Km ratio for choline can be estimated to be ≥9 × 106 M−1 s−1. Thus, in the presence of NAD+, the Km value for choline decreased by at least 70 times.
AfChoAp and AfBadAp both catalyzed the oxidation of BA to GB, but only AfChoAp catalyzes the oxidation of choline to BA. Thus, to test if AfBadA was able to oxidize the aldehyde released from the active site of AfChoAp in turnover with choline, the production of NADH was measured in a reaction mixture of the two enzymes, choline, oxygen, and NAD+. Reduction of NAD+ by AfBadAp was observed, suggesting that the BA released from AfChoAp could be utilized by AfBadAp. The limiting rate for NADH production at saturating NAD+ with 1 mM choline, 0.25 mM oxygen, and 87 nM AfChoAp was 10.7 s−1. This value is comparable to the turnover number of 10.4 s−1 for AfBadAp at saturating BA and NAD+ concentrations.
This study in A. fumigatus is the first biochemical characterization of a GB biosynthetic pathway in the fungal kingdom. AfChoAp and AfBadAp function in a coordinated fashion to synthesize GB from choline, as illustrated in Fig. 2. The first step in the biosynthetic pathway is the conversion of choline to BA catalyzed by AfChoAp. This reaction requires the presence of molecular oxygen, which is used to oxidize the enzyme-bound flavin cofactor with concomitant production of hydrogen peroxide. BA is then released from the active site of AfChoAp and undergoes a fast hydration reaction in aqueous solution that results in the reversible formation of gem-diol-choline (boxed step in Fig. 2). The gem-diol-choline is further oxidized to GB in a reaction catalyzed by AfChoAp. The aldehyde is instead further oxidized to GB by AfBadAp in a reaction that requires NAD+ as the oxidizing substrate for the enzyme.
Fig 2.
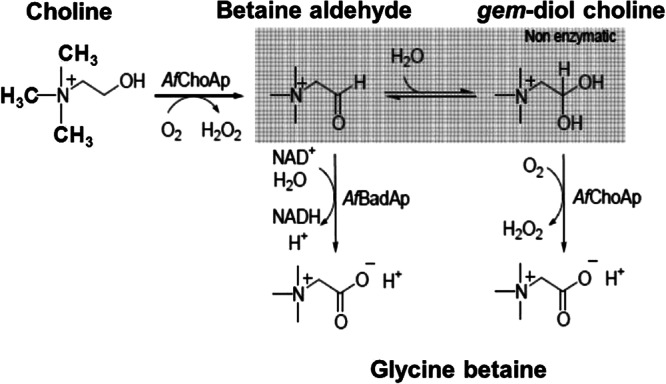
Schematic representation of the GB biosynthetic pathway in A. fumigatus. The first step in the biosynthetic pathway is the conversion of choline to BA catalyzed by AfChoAp in the presence of the flavin cofactor. BA is then released from the active site of AfChoAp and undergoes a fast hydration reaction in aqueous solution that results in the formation of gem-diol-choline (box). The gem-diol-choline is further oxidized to GB in a reaction catalyzed by AfChoAp. The aldehyde can also be further oxidized to GB by AfBadAp in a reaction that requires NAD+ as the oxidizing substrate for the enzyme. Note that the methyl groups have been represented as lines in the intermediate steps of the reaction.
Functional characterization of the AfCHOA/AfBADA cluster.
In order to understand the functional roles of these proteins, the AfchoAΔ::HPH and AfbadAΔ::HPH single mutants and the AfchoAΔAfbadAΔ::HPH double mutant were constructed. The replacement of the AfCHOA gene, the AfBADA gene, and both the AfCHOA and AfBADA genes by the HPH gene at the right locus is shown in Fig. S2 in the supplemental material. Mycelial growth and conidial germination were analyzed in agar (Fig. 3A) or liquid MM (Fig. 3B) supplemented with 20 mM choline chloride, BA, or GB or 6.6 mM phosphatidylcholine in the absence of glucose, ammonium tartrate, or both as the sole carbon and nitrogen source. In contrast to the case for the akuBku80 parental strain and the AfbadAΔ::HPH mutant, the growth and germination of the AfchoAΔ::HPH and AfchoAΔAfbadAΔ::HPH mutants were severely impaired at 37°C when choline chloride, BA, or phosphatidylcholine was used as the sole nitrogen source (Fig. 4 and data not shown). The addition of 20 mM GB restored the growth defect observed in the AfchoAΔ::HPH mutant and in the AfchoAΔAfbadAΔ::HPH double mutant (Fig. 4). Similar growth defects of the mutants were observed when choline was used as the carbon source or as carbon and nitrogen sources (see Fig. S3 in the supplemental material). However, the growth was much higher when choline was used as a nitrogen source. These results showed that GB biosynthesis in A. fumigatus is more important in the case of nitrogen starvation than in carbon starvation. It also suggested that the catabolic pathway from GB to ammonium could be a salvage pathway in the case of nitrogen starvation.
Fig 3.

Growth of AfchoAΔ::HPH, AfbadAΔ::HPH, and AfchoAΔAfbadAΔ::HPH mutants of A. fumigatus at 37°C on MM agar with choline or BA as the sole nitrogen source. (A) The akuBku80 parental and the AfchoAΔ::HPH, AfbadAΔ::HPH, and AfchoAΔAfbadAΔ::HPH mutants and revertant AfchoAΔAfbadAΔ::AfCHO-eGFP+AfBADA-RFP were grown on agar plates with MM with glucose and ammonium tartrate, MM with glucose and without a nitrogen source, or MM with glucose and without ammonium tartrate, which is replaced by choline chloride (20 mM), BA (20 mM), or GB (20 mM). (B) Microscopic observation of the akuBku80 parental and the AfchoAΔ::HPH, AfbadAΔ::HPH, and AfchoAΔAfbadAΔ::HPH mutant cultures in liquid MM without a nitrogen source and in liquid MM with choline chloride (20 mM) or BA (20 mM) as the sole nitrogen source. Experiments were repeated three times with similar results.
Fig 4.

NMR analysis. 1H,14N-HSQC spectroscopy of the intracellular fraction from the A. fumigatus akuBku80 strain (middle panel) and the A. fumigatus AfChoAΔ::HPH strain (lower panel) cultured in MM is shown. The chemical shifts of GB signals are circled in green. Upper panel, control GB.
NMR studies confirmed that in MM medium no GB was present in the intracellular fraction of the AfchoAΔ::HPH mutant, whereas it was identified in the parental strain. The NMR analysis was originally based on 1H,13C-HSQC and 1H,13C-HMBC experiments (see Fig. S4 in the supplemental material) and was further confirmed by use of a 1H,14N-HSQC method that was appropriate to detect choline derivatives NCH3 and CH2 (Fig. 4) (21). The mutants were complemented by the ectopic integration of the translational fusion construct eGFP::AfCHOA::promoter::AfBADA::RFP (see Fig. S2 in the supplemental material).
The GFP-tagged AfChoAp and RFP-tagged AfBadAp restored the growth and germination defects observed in the AfchoAΔAfbadAΔ::HPH double mutant (Fig. 3). This result indicated that these fusion proteins complement the GB biosynthesis pathway. The fluorescence of the AfChoAp-eGFP and AfBadAp-RFP-T fusion proteins was seen in the cytoplasm of mycelium and germinating conidia, demonstrating that AfChoAp and AfBadAp were cytoplasmic proteins (Fig. 5). In contrast, no fluorescence was seen in resting conidia, which, like for the transcriptional fusion, became positive as soon as they were plunged into a nutritive medium (data not shown).
Fig 5.

Localization of AfChoAp-eGFP and AfBadAp-RFP-T fusion proteins. (A and B) The AfchoAΔAfbadAΔ::HPH mutant transformed with pAN8.1 containing the AfCHOA::eGFP (A) and AfBADA::RFP-T (B) fusions under the control of the AfCHOA and AfBADA gene promoter. (C) Bright-field image of the same germinating conidia (upper panel) and hyphae (lower panel). The fungus was grown in liquid MM with choline chloride (20 mM) as the sole nitrogen source.
In contrast to the role of GB in bacteria, the GB mutants were not affected by stress and especially osmotic stress: no difference in growth was seen when the parental and mutant strains were grown on solid complex rich or solid MM in the presence of high concentrations of osmolytes (1.2 M mannitol, 1.2 M sorbitol, or 1 to 2.5 M NaCl). In addition, the addition of 0.6 M NaCl did not change the expression of the CODA and BADA genes (see Fig. S5 in the supplemental material). The degradation of the nonreducing sugar trehalose and the polyols mannitol and arabinitol of the resting conidia was similar during the conidial germination of the wild type and the AfchoAΔAfbadAΔ::HPH double mutant (see Fig. S6 in the supplemental material). In addition, no changes in the glycerol content of the resting and germinated conidia were seen in the parental and mutant strains. In agreement with these data, the expression of genes coding for trehalose and mannitol biosyntheses was similar in resting, swollen, and germinated conidia of the wild type and the AfchoAΔAfbadAΔ::HPH double mutant (see Fig. S7 in the supplemental material, and data not shown). These results showed that there was no interconnection between the metabolism of the osmoprotectant trehalose and polyols and the GB pathway.
The mutants were no more sensitive to other stresses, such as ROS, from which bacteria and plants are known to be protected by GB. For example, no growth differences were seen between the parental strain and the GB mutants of A. fumigatus on solid MM in the presence of reactive oxidants (hydrogen peroxide at 1 to 5 mM and menadione at 5 to 80 μM). Similarly, the growth of the mutant was identical to that of the parental strain on solid MM at pH 2.0 and 9.0 and at constant temperatures between 4 and 50°C. The conidial viability of the mutant was not more altered than that of the parental strain after a freezing shock (24 h at −20°C) or heat shock (5 min at 75°C) or after drying at room temperature for 2 years (data not shown).
Since A. fumigatus CHOA and BADA were expressed in vivo (see Fig. S8A in the supplemental material) and the composition of the lung surfactant is phosphatidylcholine rich, a mutant such as the AfchoAΔ::HPH mutant, which is unable to use choline, could be less fit in a lung environment than its parental strain. In reality, the conidia of the AfchoAΔ::HPH and AfbadAΔ::HPH single mutants and the AfchoAΔAfbadAΔ::HPH double mutant survived in the lungs of immunocompetent mice like the parental strain (see Fig. S8B in the supplemental material). Moreover, the mutants were able to grow on Curosurf, which is a naturally derived surfactant from porcine lung that is rich in phospholipids (mainly phosphatidylcholine) with 1% hydrophobic surfactant proteins B and C (data not shown). These results suggested that the GB pathway is not required for the germination and the establishment of A. fumigatus in the lung. These results also showed that in A. fumigatus, GB was not used as a protectant against various abiotic and biotic stresses.
DISCUSSION
The gene cluster for the synthesis of glycine betaine is unique in A. fumigatus.
In higher plants, the two-enzyme reaction involved in the oxidation of choline to GB is catalyzed by a ferredoxin-dependent choline monooxygenase (EC 1.14.15.7) and an NAD(P)+-dependent betaine aldehyde dehydrogenase (BADH) (EC 1.2.1.8) (29). In mammals and Gram-negative bacteria such as E. coli, P. aeruginosa, or Sinorhizobium meliloti, the two-enzyme biosynthesis of GB is catalyzed by a choline dehydrogenase (CDH) (EC 1.1.99.1) and a betaine aldehyde dehydrogenase (30–32). In Gram-positive bacteria, such as Bacillus subtilis, an alcohol dehydrogenase (EC 1.1.1.1) produces the BA, which is then converted to GB by BADH (33). A choline oxidase activity was previously shown in a few filamentous fungi (34–36), but the enzymatic activity and the encoding genes were not characterized.
AfChoAp is a flavin-dependent choline oxidase, as indicated by the ability of the recombinant enzyme to catalyze the oxidation of choline or BA to GB with molecular oxygen as the oxidizing substrate. The percentage of identity between the choline oxidases of A. fumigatus and Arthrobacter globiformis, which is the only bacterial species that has a unique CHOA gene, was 37% (37). In addition, the amino acids in the catalytic site of the A. globiformis choline oxidase are conserved in A. fumigatus and other fungi, as follows: H99, site of flavin covalent attachment (38); H310, proton relay with H466; E312, substrate binding (39); H351, hydrogen bonding OH of choline (40); V464, substrate positioning and oxygen reactivity (41); H466, positive charge stabilization of the alkoxide intermediate of the reaction (42); S101, facilitating hydride transfer (43); and N510, involved in the reductive and oxidative half reaction (44). Both the fungal and bacterial enzymes display steady-state kinetic mechanisms with formation of a ternary complex with either choline or BA as the substrate, with oxygen reacting with the reduced flavin before the organic product of the reaction is released from the enzyme active site (37).
AfBadAp oxidizes BA to GB with reduction of NAD+ to NADH. The percentage of identity between the AfBadAp and the best-characterized PaBADH bacterial protein from P. aeruginosa was 57%. All relevant amino acids of the active site previously identified from the crystal structure of the PaBADH enzyme were present in BadAp from A. fumigatus and other fungi (45): a catalytic glutamate (E252), a catalytic cysteine (C286), and the NAD+ binding motif (G-x(3)-[ST]-G). The fungal enzyme is also able to utilize NADP+ as the reducing substrate, but NAD+ is clearly preferred as indicated by a (kcat/KNAD+)/(kcat/KNAD(P)+) ratio larger than 300. From a kinetic standpoint, AfBadAp displays a sequential steady-state kinetic mechanism with BA and either NAD+ or NADP+, as previously reported for bacterial betaine aldehyde dehydrogenases (45, 46). AfBadAp is exquisitely efficient in the oxidation reaction, as indicated by the second-order rate constant for substrate capture kcat/Km, with a value of 4 × 105 M−1 s−1. This is essential for the depletion of BA from the intracellular milieu, thereby abating the potential toxic effect of having an aldehyde reacting nonspecifically with a number of intracellular nucleophiles (47).
The GB pathway in A. fumigatus presents several unique features exclusively specific to fungal enzymes: (i) the fungal ChoAp is the only one to use NAD+ as a cofactor, whereas bacterial enzymes preferentially use NADP+; (ii) in contrast to the bacterial enzyme, which retains BA in the active site during catalysis, the fungal enzyme releases it to the solution, where it is further oxidized to GB by AfBadAp; and (iii) AfBadAp is not inhibited by BA, NAD+, or NADP+.
GB is used as a nutrient in fungi.
In plants and bacteria, GB has been shown to enhance tolerance to a variety of stresses, such as high salt concentration, freezing, chilling, drought, or oxidants, because it stabilizes the quaternary structure of enzymes and maintains the highly ordered state of the membranes at nonphysiological temperatures and salt conditions (1, 48–52). All these abiotic stresses also induce an oxidative burst, the damages from which can be reduced by GB (1). In halophilic fungi, such as Penicillium fellutanum, it was suggested that GB was associated with osmotic protection, but there was no biochemical and molecular analysis of the GB pathway in this species (35). In contrast, in A. fumigatus the production of GB does not seem to be associated with a tolerance to stress.
Several reports mention the fact that in bacteria GB has a dual function: it can be used alternatively as an antistress molecule or as an energy source (53–56). Interestingly, A. fumigatus seems to use GB as a sole source of carbon, nitrogen, and energy through successive demethylations to glycine. Choline and GB, with methionine, are the most important carriers of methyl groups in different metabolic pathways. However, methionine is used for protein synthesis, whereas choline serves predominantly for the formation of cell membranes (57–59). Moreover, GB may be directly used as a methyl group donor, whereas choline needs to be converted in a two-step oxidation reaction catalyzed by AfChoAp (57–59). The GB biosynthesis could be a rescue pathway for the use of unusual nitrogen (or carbon) sources under nutrient starvation conditions. Transcriptome data for A. fumigatus during early conidial germination indeed revealed an upregulation of genes involved in lipid catabolism (10). The link between lipid catabolism and GB biosynthesis may be relevant for fungal physiology, since use of GB would be a quick catabolic way to mobilize choline for amino acid synthesis, which was shown to be upregulated during early germination (10). However, despite being highly upregulated during germination, catabolism of choline to produce GB during germination does not seem to be essential to this morphological event. Lipidome analysis of the conidial germination should improve our understanding of lipid metabolism during conidial germination and of the interconnections of GB with the other lipid components of the fungus.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported in part by NSF-CAREER grant MCB-0545712 and NSF grant MCB-1121695 (G.G.) and European grants ALLFUNFP7260338 and ESF Fuminomics RNP 06-132 (J.-P.L.).
Footnotes
Published ahead of print 5 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00348-12.
REFERENCES
- 1. Chen THH, Murata N. 2011. Glycinebetaine protects plants against abiotic stress: mechanisms and biotechnological applications. Plant Cell Environ. 34:1–20 [DOI] [PubMed] [Google Scholar]
- 2. Giri J. 2011. Glycinebetaine and abiotic stress tolerance in plants. Plant Signal Behav. 6:1746–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fitzsimmons LF, Hampel KJ, Wargo MJ. 2012. Cellular choline and glycine betaine pools impact osmoprotection and phospholipase C production in Pseudomonas aeruginosa. J. Bacteriol. 194:4718–4726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peddie BA, Lever M, Randall K, Chambers ST. 1999. Osmoprotective activity, urea protection, and accumulation of hydrophilic betaines in Escherichia coli and Staphylococcus aureus. Antonie Van Leeuwenhoek 75:183–189 [DOI] [PubMed] [Google Scholar]
- 5. Lucht JM, Bremer E. 1994. Adaptation of Escherichia coli to high osmolarity environments: osmoregulation of the high-affinity glycine betaine transport system proU. FEMS Microbiol. Rev. 14:3–20 [DOI] [PubMed] [Google Scholar]
- 6. Wemekamp-Kamphuis HH, Wouters JA, Sleator RD, Gahan CG, Hill C, Abee T. 2002. Multiple deletions of the osmolyte transporters BetL, Gbu, and OpuC of Listeria monocytogenes affect virulence and growth at high osmolarity. Appl. Environ. Microbiol. 68:4710–4716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Price CT, Bukka A, Cynamon M, Graham JE. 2008. Glycine betaine uptake by the ProXVWZ ABC transporter contributes to the ability of Mycobacterium tuberculosis to initiate growth in human macrophages. J. Bacteriol. 190:3955–3961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Al-Bader N, Vanier G, Liu H, Gravelat FN, Urb M, Hoareau CM, Campoli P, Chabot J, Filler SG, Sheppard DC. 2010. Role of trehalose biosynthesis in Aspergillus fumigatus development, stress response, and virulence. Infect. Immun. 78:3007–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Puttikamonkul S, Willger SD, Grahl N, Perfect JR, Movahed N, Bothner B, Park S, Paderu P, Perlin DS, Cramer RA., Jr 2010. Trehalose 6-phosphate phosphatase is required for cell wall integrity and fungal virulence but not trehalose biosynthesis in the human fungal pathogen Aspergillus fumigatus. Mol. Microbiol. 77:891–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lamarre C, Sokol S, Debeaupuis JP, Henry C, Lacroix C, Glaser P, Coppee JY, Francois JM, Latge JP. 2008. Transcriptomic analysis of the exit from dormancy of Aspergillus fumigatus conidia. BMC Genomics 9:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. da Silva Ferreira ME, Kress MR, Savoldi M, Goldman MH, Hartl A, Heinekamp T, Brakhage AA, Goldman GH. 2006. The akuB(KU80) mutant deficient for nonhomologous end joining is a powerful tool for analyzing pathogenicity in Aspergillus fumigatus. Eukaryot. Cell 5:207–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lamarre C, Ibrahim-Granet O, Du C, Calderone R, Latge JP. 2007. Characterization of the SKN7 ortholog of Aspergillus fumigatus. Fungal Genet. Biol. 44:682–690 [DOI] [PubMed] [Google Scholar]
- 13. Lambou K, Lamarre C, Beau R, Dufour N, Latge JP. 2010. Functional analysis of the superoxide dismutase family in Aspergillus fumigatus. Mol. Microbiol. 75:910–923 [DOI] [PubMed] [Google Scholar]
- 14. Whitby LG. 1953. A new method for preparing flavin-adenine dinucleotide. Biochem. J. 54:437–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fan F, Ghanem M, Gadda G. 2004. Cloning, sequence analysis, and purification of choline oxidase from Arthrobacter globiformis: a bacterial enzyme involved in osmotic stress tolerance. Arch. Biochem. Biophys. 421:149–158 [DOI] [PubMed] [Google Scholar]
- 16. Figueroa-Soto CG, Valenzuela-Soto EM. 2000. Kinetic study of porcine kidney betaine aldehyde dehydrogenase. Biochem. Biophys. Res. Commun. 269:596–603 [DOI] [PubMed] [Google Scholar]
- 17. Gadda G. 2003. Kinetic mechanism of choline oxidase from Arthrobacter globiformis. Biochim. Biophys. Acta 1646:112–118 [DOI] [PubMed] [Google Scholar]
- 18. Diaz-Guerra TM, Mellado E, Cuenca-Estrella A, Rodriguez-Tudela JL. 2003. A point mutation in the 14 alpha-sterol demethylase gene cyp51A contributes to itraconazole resistance in Aspergillus fumigatus. Antimicrob. Agents Chemother. 47:1120–1124 (Erratum, 48:1071, 2004.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cove DJ. 1966. The induction and repression of nitrate reductase in the fungus Aspergillus nidulans. Biochim. Biophys. Acta 113:51–56 [DOI] [PubMed] [Google Scholar]
- 20. Punt PJ, Oliver RP, Dingemanse MA, Pouwels PH, van den Hondel CA. 1987. Transformation of Aspergillus based on the hygromycin B resistance marker from Escherichia coli. Gene 56:117–124 [DOI] [PubMed] [Google Scholar]
- 21. Mao J, Jiang L, Jiang B, Liu M, Mao XA. 2010. 1H-14N HSQC detection of choline-containing compounds in solutions. J. Magn. Reson. 206:157–160 [DOI] [PubMed] [Google Scholar]
- 22. Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. 1995. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6:135–140 [DOI] [PubMed] [Google Scholar]
- 23. d'Enfert C, Fontaine T. 1997. Molecular characterization of the Aspergillus nidulans treA gene encoding an acid trehalase required for growth on trehalose. Mol. Microbiol. 24:203–216 [DOI] [PubMed] [Google Scholar]
- 24. Shinohara ML, Correa A, Bell-Pedersen D, Dunlap JC, Loros JJ. 2002. Neurospora clock-controlled gene 9 (ccg-9) encodes trehalose synthase: circadian regulation of stress responses and development. Eukaryot. Cell 1:33–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Avonce N, Mendoza-Vargas A, Morett E, Iturriaga G. 2006. Insights on the evolution of trehalose biosynthesis. BMC Evol. Biol. 6:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krahulec S, Armao GC, Klimacek M, Nidetzky B. 2011. Enzymes of mannitol metabolism in the human pathogenic fungus Aspergillus fumigatus—kinetic properties of mannitol-1-phosphate 5-dehydrogenase and mannitol 2-dehydrogenase, and their physiological implications. FEBS J. 278:1264–1276 [DOI] [PubMed] [Google Scholar]
- 27. Massey V, Hemmerich P. 1977. A photochemical procedure for reduction of oxidation-reduction proteins employing deazariboflavin as catalyst. J. Biol. Chem. 252:5612–5614 [PubMed] [Google Scholar]
- 28. Quaye O, Nguyen T, Gannavaram S, Pennati A, Gadda G. 2010. Rescuing of the hydride transfer reaction in the Glu312Asp variant of choline oxidase by a substrate analogue. Arch. Biochem. Biophys. 499:1–5 [DOI] [PubMed] [Google Scholar]
- 29. Rathinasabapathi B, Burnet M, Russell BL, Gage DA, Liao PC, Nye GJ, Scott P, Golbeck JH, Hanson AD. 1997. Choline monooxygenase, an unusual iron-sulfur enzyme catalyzing the first step of glycine betaine synthesis in plants: prosthetic group characterization and cDNA cloning. Proc. Natl. Acad. Sci. U. S. A. 94:3454–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Velasco-Garcia R, Villalobos MA, Ramirez-Romero MA, Mujica-Jimenez C, Iturriaga G, Munoz-Clares RA. 2006. Betaine aldehyde dehydrogenase from Pseudomonas aeruginosa: cloning, over-expression in Escherichia coli, and regulation by choline and salt. Arch. Microbiol. 185:14–22 [DOI] [PubMed] [Google Scholar]
- 31. Andresen PA, Kaasen I, Styrvold OB, Boulnois G, Strom AR. 1988. Molecular cloning, physical mapping and expression of the bet genes governing the osmoregulatory choline-glycine betaine pathway of Escherichia coli. J. Gen. Microbiol. 134:1737–1746 [DOI] [PubMed] [Google Scholar]
- 32. Mandon K, Osteras M, Boncompagni E, Trinchant JC, Spennato G, Poggi MC, Le Rudulier D. 2003. The Sinorhizobium meliloti glycine betaine biosynthetic genes (betlCBA) are induced by choline and highly expressed in bacteroids. Mol. Plant Microbe Interact. 16:709–719 [DOI] [PubMed] [Google Scholar]
- 33. Boch J, Kempf B, Schmid R, Bremer E. 1996. Synthesis of the osmoprotectant glycine betaine in Bacillus subtilis: characterization of the gbsAB genes. J. Bacteriol. 178:5121–5129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Park YI, Buszko ML, Gander JE. 1999. Glycine betaine: reserve form of choline in Penicillium fellutanum in low-sulfate medium. Appl. Environ. Microbiol. 65:1340–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Park YI, Gander JE. 1998. Choline derivatives involved in osmotolerance of Penicillium fellutanum. Appl. Environ. Microbiol. 64:273–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tani Y, Mori N, Ogata K, Yamada H. 1979. Production and purification of choline oxidase from Cylindrocarpon didymum M-1. Agric. Biol. Chem. 43:815–820 [Google Scholar]
- 37. Gadda G. 2008. Hydride transfer made easy in the reaction of alcohol oxidation catalyzed by flavin-dependent oxidases. Biochemistry 47:13745–13753 [DOI] [PubMed] [Google Scholar]
- 38. Quaye O, Cowins S, Gadda G. 2009. Contribution of flavin covalent linkage with histidine 99 to the reaction catalyzed by choline oxidase. J. Biol. Chem. 284:16990–16997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Quaye O, Lountos GT, Fan F, Orville AM, Gadda G. 2008. Role of Glu312 in binding and positioning of the substrate for the hydride transfer reaction in choline oxidase. Biochemistry 47:243–256 [DOI] [PubMed] [Google Scholar]
- 40. Rungsrisuriyachai K, Gadda G. 2008. On the role of histidine 351 in the reaction of alcohol oxidation catalyzed by choline oxidase. Biochemistry 47:6762–6769 [DOI] [PubMed] [Google Scholar]
- 41. Finnegan S, Agniswamy J, Weber IT, Gadda G. 2010. Role of valine 464 in the flavin oxidation reaction catalyzed by choline oxidase. Biochemistry 49:2952–2961 [DOI] [PubMed] [Google Scholar]
- 42. Ghanem M, Gadda G. 2005. On the catalytic role of the conserved active site residue His466 of choline oxidase. Biochemistry 44:893–904 [DOI] [PubMed] [Google Scholar]
- 43. Yuan H, Gadda G. 2011. Importance of a serine proximal to the c(4a) and n(5) flavin atoms for hydride transfer in choline oxidase. Biochemistry 50:770–779 [DOI] [PubMed] [Google Scholar]
- 44. Rungsrisuriyachai K, Gadda G. 2010. Role of asparagine 510 in the relative timing of substrate bond cleavages in the reaction catalyzed by choline oxidase. Biochemistry 49:2483–2490 [DOI] [PubMed] [Google Scholar]
- 45. Gonzalez-Segura L, Rudino-Pinera E, Munoz-Clares RA, Horjales E. 2009. The crystal structure of a ternary complex of betaine aldehyde dehydrogenase from Pseudomonas aeruginosa provides new insight into the reaction mechanism and shows a novel binding mode of the 2′-phosphate of NADP+ and a novel cation binding site. J. Mol. Biol. 385:542–557 [DOI] [PubMed] [Google Scholar]
- 46. Munoz-Clares RA, Diaz-Sanchez AG, Gonzalez-Segura L, Montiel C. 2010. Kinetic and structural features of betaine aldehyde dehydrogenases: mechanistic and regulatory implications. Arch. Biochem. Biophys. 493:71–81 [DOI] [PubMed] [Google Scholar]
- 47. Maser E. 2007. Enzymology and molecular biology of carbonyl metabolism. Purdue University Press, West Lafayette, IN [Google Scholar]
- 48. Wood JM, Bremer E, Csonka LN, Kraemer R, Poolman B, van der Heide T, Smith LT. 2001. Osmosensing and osmoregulatory compatible solute accumulation by bacteria. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 130:437–460 [DOI] [PubMed] [Google Scholar]
- 49. Caldas T, Demont-Caulet N, Ghazi A, Richarme G. 1999. Thermoprotection by glycine betaine and choline. Microbiology 145:2543–2548 [DOI] [PubMed] [Google Scholar]
- 50. Brigulla M, Hoffmann T, Krisp A, Volker A, Bremer E, Volker U. 2003. Chill induction of the SigB-dependent general stress response in Bacillus subtilis and its contribution to low-temperature adaptation. J. Bacteriol. 185:4305–4314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Diamant S, Rosenthal D, Azem A, Eliahu N, Ben-Zvi AP, Goloubinoff P. 2003. Dicarboxylic amino acids and glycine-betaine regulate chaperone-mediated protein-disaggregation under stress. Mol. Microbiol. 49:401–410 [DOI] [PubMed] [Google Scholar]
- 52. Sheehan VM, Sleator RD, Fitzgerald GF, Hill C. 2006. Heterologous expression of BetL, a betaine uptake system, enhances the stress tolerance of Lactobacillus salivarius UCC118. Appl. Environ. Microbiol. 72:2170–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rozwadowski KL, Khachatourians GG, Selvaraj G. 1991. Choline oxidase, a catabolic enzyme in Arthrobacter pascens, facilitates adaptation to osmotic stress in Escherichia coli. J. Bacteriol. 173:472–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Welsh DT. 2000. Ecological significance of compatible solute accumulation by micro-organisms: from single cells to global climate. FEMS Microbiol. Rev. 24:263–290 [DOI] [PubMed] [Google Scholar]
- 55. Boncompagni E, Osteras M, Poggi MC, le Rudulier D. 1999. Occurrence of choline and glycine betaine uptake and metabolism in the family Rhizobiaceae and their roles in osmoprotection. Appl. Environ. Microbiol. 65:2072–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Serra AL, Mariscotti JF, Barra JL, Lucchesi GI, Domenech CE, Lisa AT. 2002. Glycine betaine transmethylase mutant of Pseudomonas aeruginosa. J. Bacteriol. 184:4301–4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lever M, Slow S. 2010. The clinical significance of betaine, an osmolyte with a key role in methyl group metabolism. Clin. Biochem. 43:732–744 [DOI] [PubMed] [Google Scholar]
- 58. Ueland PM, Holm PI, Hustad S. 2005. Betaine: a key modulator of one-carbon metabolism and homocysteine status. Clin. Chem. Lab. Med. 43:1069–1075 [DOI] [PubMed] [Google Scholar]
- 59. Barak AJ, Tuma DJ. 1983. Betaine, metabolic by-product or vital methylating agent. Life Sci. 32:771–774 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.