

Abstract
Intestinal epithelial activation of nuclear factor kappa B (NF-κB) exerts both detrimental and beneficial functions in response to various luminal insults, including ones associated with mucosa-associated pathogens. Gastrointestinal infection with enteropathogenic Escherichia coli (EPEC) causes severe injuries in epithelial integrity and leads to watery diarrhea. The present study was conducted to investigate the prolonged epithelial responses to persistent EPEC infection via NF-κB activation. EPEC infection led to sustained activation of NF-κB signal in mouse intestinal epithelial cells in vivo and in vitro, which was positively associated with a type III secretion system, whereas early NF-κB is regulated. Moreover, prolonged NF-κB activation was found to be a part of macrophage inhibitory cytokine 1 (MIC-1)-mediated signaling activation, a novel link between NF-κB signaling and infection-associated epithelial stress. EPEC infection induced gene expression of MIC-1, a member of the transforming growth factor β (TGF-β) superfamily, which then activated TGF-β-activated kinase 1 and consequently led to NF-κB activation. Functionally, both EPEC-induced MIC-1 and NF-κB signaling mediated epithelial survival by enhancing the expression of cyclin D1, a target of NF-κB. In summary, the results of the present study suggest that MIC-1 serves as a mediator of prolonged NF-κB activation, which is critical in maintaining gut epithelial integrity in response to infection-induced injuries.
INTRODUCTION
Intestinal epithelial cells (IECs) express a variety of pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), either constitutively or after induction by various stimuli (1). Nuclear factor kappa B (NF-κB) is one of the main downstream effectors of bacterial PRR activation. Increased activation of NF-κB leads to detrimental effects by inducing the expression of proinflammatory mediators with subsequent tissue damage (2–4). However, it is hard to distinguish between molecular patterns of commensal bacteria and those of primary pathogenic bacteria via PRRs. To prevent overstimulation of NF-κB activation by commensal bacteria in the gut intestinal epithelial barrier and maintain homeostasis, IECs have developed various regulatory machineries. Disruption of these regulatory factors can lead to persistently elevated proinflammatory signaling, including that mediated by NF-κB due to enteric bacteria, and subsequent chronic inflammation. This involves sustained production of proinflammatory mediators such as cytokines, chemokines, and oxidative radicals. On the other hand, transient NF-κB activation can be beneficial by triggering antigen sampling and leukocyte recruitment, resulting in bacterial clearance. The beneficial function of NF-κB has been also observed in epithelial cells (2–4). In particular, epithelial NF-κB contributes to epithelial barrier reestablishment and host homeostasis by inducing the transcription of genes involved in epithelial cell survival, wound healing, and tissue remodeling after injury due to pathogens and inflammation.
Infection with enteropathogenic Escherichia coli (EPEC) is common among infants and young children in developing countries (5), yet the pathophysiology of the resulting diarrhea remains unclear. Moreover, mucosa-associated Escherichia coli including EPEC is frequently observed in the intestinal surface of chronically diseased patients such as inflammatory bowel disease (IBD) and colon cancer patients (6–9). Frequently, patients with these chronic diseases can suffer from superinfection with EPEC (6, 9). E. coli strains can be found in the mucosa of some IBD patients that are able to activate NF-κB similar to known pathogenic strains (7). EPEC strains are not invasive microorganisms and do not produce recognized enterotoxins (10, 11). While the precise mechanism by which EPEC causes diarrhea is presently unknown, numerous studies have addressed pathogen-specific effects on host epithelial cells (12–14). It has generally been presumed that diarrhea results from the direct interaction of EPEC with the small intestinal epithelium. EPEC adheres to enterocytes and produces a characteristic “attaching and effacing” (A/E) lesion in the brush border membrane (15, 16). The pathogenic bacteria uses a type III secretion system (TTSS) to deliver the toxic effectors to the host epithelial cells, whose absorptive microvilli are lost (effacement) (17). EPEC produces proinflammatory molecules including bacterial flagellin (18). In contrast, EPEC also injects anti-inflammatory virulence effector molecules into host epithelial cells via a TTSS (19, 20). One central consequence of EPEC infection is NF-κB activation, which in turn promotes the expression of proinflammatory chemokines such as interleukin-8 (IL-8). Along with proinflammatory stimulation, EPEC can downregulate the proinflammatory response to the bacterial products and host-derived cytokines such as tumor necrosis factor alpha (TNF-α) and IL-1β (14). These suppressive effects of EPEC on an overstimulated inflammatory response might be essential for the survival of these noninvasive bacteria.
Macrophage inhibitory cytokine 1 (MIC-1) is also known as growth differentiation factor 15 (GDF15), placental bone morphogenetic protein (PLAB), placental transforming growth factor β (PTGF-β), or nonsteroidal anti-inflammatory drug-activated protein 1 (NAG-1) (21, 22). It is a member of the transforming growth factor β (TGF-β) superfamily and was first isolated from a subtracted cDNA library enriched for genes associated with macrophage inactivation. MIC-1 is synthesized as a 308-amino-acid (62-kDa) propeptide with an RXXR cleavage site. After being cleaved by a furin-like protease, this factor is secreted as a 25-kDa, disulfide-linked dimer. MIC-1 shares relatively low sequence homology with other TGF-β superfamily members and does not cluster within existing TGF-β families (21, 22). Since MIC-1 is a newly identified factor, its functionality has not been extensively characterized, and the nature of its effects depends on the cellular context (23, 24). The placenta is the only tissue with high levels of MIC-1 expression under normal physiological conditions although most epithelial cells constitutively express a low level of MIC-1 (25). However, pathogenic processes, including inflammation and carcinogenesis, elevate the cellular levels of MIC-1 expression, indicating that this protein plays specific roles in cellular behavior under stressful conditions. MIC-1 mediates apoptosis in cancer cells as well as normal epithelial cells. As one of the major secreted proteins induced by p53, MIC-1 is thought to be important for translating p53-mediated activity associated with cell cycle arrest and apoptosis (26). Moreover, MIC-1 has also been linked to modulating the survival of migrating cells in the extracellular matrix and circulation (27, 28). Although the exact intracellular action of MIC-1 has not been well studied, MIC-1-induced growth inhibition requires a functional TGF-β receptor type II-linked signaling pathway (29).
The purpose of this study was to examine the effects of persistent EPEC infection on NF-κB activation in human intestinal epithelial cells. Our results indicated that intestinal cells exposed to EPEC showed sustained activation of NF-κB signaling which was positively associated with a TTSS. Moreover, the mechanism underlying prolonged NF-κB activation was examined in the context of MIC-1-mediated signaling activation by EPEC, thereby suggesting a new link between sustained NF-κB activation and epithelial stress by infection.
MATERIALS AND METHODS
Ethics statement.
This research was conducted in accordance with the Declaration of Helsinki and/or with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health. All animal experiments were also approved by Pusan National University's Institutional Animal Care and Use Committee (approval number PNU-2010-000189).
Cell culture conditions and reagents.
Intestinal epithelial cell lines HCT-8, HCT-116, IEC-18, and CACO-2 were purchased from the American Type Culture Collection (Manassas, VA, USA). HCT-8, CACO-2, and HCT-116 cells were maintained in RPMI medium (Welgene, Daegu, South Korea), and IEC-18 cells were grown in Dulbecco's modified Eagle's medium (DMEM; Welgene). All media were supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich Chemical Co., St. Louis, MO, USA), 50 unit/ml penicillin (Welgene), 1% essential amino acids (Welgene), and 50 μg/ml streptomycin (Welgene) at 37°C in a 5% CO2 humidified incubator. The number of cells and cell viability were assessed by staining with trypan blue (Sigma-Aldrich Chemical Co.) and use of a hemocytometer. (5Z)-7-Oxozeaenol, a TGF-β-activated kinase 1 (TAK1) inhibitor, was purchased from Tocris Biosciences (Bristol, United Kingdom). All other chemicals were purchased from Sigma-Aldrich.
Bacterial strains and infection.
The wild-type (WT) EPEC used for all experiments was the Escherichia coli O127 strain E2348/69; this strain and EPEC ΔescN were kindly provided by Ilan Rosenshine (The Hebrew University of Jerusalem, Jerusalem, Israel). To visualize infection of mucosa by EPEC under fluorescence microscope, EPEC was transformed with a pGFPuv plasmid containing the green fluorescent protein (GFP) gene (Clontech Laboratories, Inc., Mountain View, CA, USA). Briefly, the bacteria were shaken overnight in LB broth (Duchefa Biochemie, Haarlem, The Netherlands) at 37°C, and EPEC ΔescN cells were maintained in 100 μg/ml kanamycin. Bacteria were further subcultured in antibiotic- and serum-free RPMI 1640 medium supplemented with 1% mannose (Sigma-Aldrich Chemical Co.) at 37°C until the absorbance (optical density [OD]) at 600 nm reached 0.5 to 0.6. EPEC was then added to the apical surface of the HCT-116 cell culture at a ratio of 50:1 (bacteria to cells).
For mouse EPEC infection, 6- to 8-week-old C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and allowed to acclimate for 7 days. All mice were individually housed in ventilated cages with free access to food and water. EPEC or EPEC ΔescN was grown to stationary phase in LB broth. Aliquots of the broth culture (1 ml) were centrifuged, and the bacterial pellet was suspended in 1.25 ml of phosphate-buffered saline (PBS). A suspension containing approximately 2 × 108 E2348/69 or EPEC ΔescN cells in 200 μl of PBS was introduced into the animals by gavage with a curved needle 4 cm in length with a steel ball at the tip. Control animals received 200 μl of sterile PBS. Over the course of infection, the mice were observed daily to assess activity levels and water intake, and body weight was measured. At various times following infection (5, 11, 13, and 15 days), the animals were sacrificed after anesthetization by isoflurane inhalation, and intestinal tissues were processed for further analysis.
Immunohistological assessment.
Mice were euthanized by cervical dislocation. The colon and small intestine were then excised and washed gently in PBS to remove fecal debris. The colon and small intestine were then dissected in half, with one half rolled into a “Swiss roll” formation and fixed in a neutral buffered 4% paraformaldehyde solution. Intestine samples were dehydrated, embedded in paraffin, and cut into 5-μm sections for immunohistochemical analysis. Before being subjected to immunostaining, formalin-fixed paraffin-embedded tissues from mouse intestines were cut (5 μm), deparaffinized, and rehydrated. Tissue slides were heated in 10 mM sodium acetate (pH 9.0) for 5 min at 121°C for antigen retrieval and bathed in a 3% H2O2-PBS solution for 15 min at room temperature in the dark to quench endogenous peroxidase. After samples were washed with Tris-HCl–Tween 0.5%, tissue sections (5 μm) were blocked with 3% bovine serum albumin (BSA) in PBS, incubated at a 1:200 dilution of the primary antibodies overnight at 4°C, and repeatedly washed using PBS. Incubation of the horseradish peroxidase-conjugated secondary antibody was done for 2 h at room temperature, followed by repeated washing with PBS. The bound antibodies were identified using freshly prepared substrate buffer (0.05% diaminobenzidine [DAB; Sigma-Aldrich Chemical Co.], 0.015% H2O2 in PBS) for 2 min. After a final wash in PBS and distilled water, the slides were counterstained with a 50% dilution of hematoxylin (Santa Cruz Biotechnology) for 1 min and dehydrated in graded alcohols (50%, 70%, 80%, 90%, 95%, 100%, and 100%). Sections were examined at various magnifications using an Axio Imager (Carl Zeiss MicroImaging, GmbH, Oberkochen, Germany). Images of normal tissue and lesions were captured and processed using Photoshop, version 7.0 (Adobe Systems, Inc., San Jose, CA, USA) following image acquisition.
Western blot analysis.
Segmented intestines were homogenized in the lysis buffer consisting of 1% (wt/vol) sodium dodecyl sulfate (SDS), 1.0 mM sodium orthovanadate, and 10 mM Tris, pH 7.4, and cultured enterocyte cells were washed with ice-cold phosphate buffer, lysed by boiling in lysis buffer, and sonicated for 5 s. Total cellular proteins in the lysates were quantified using a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA). Fifty micrograms of protein was separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) using a mini-gel apparatus (Bio-Rad, Hercules, CA, USA). The proteins were then transferred onto polyvinylidene fluoride membranes (Amersham Pharmacia Biotech, Piscataway, NJ, USA), and each membrane was blocked with 5% skim milk in Tris-buffered saline plus 0.05% Tween (TBST). The blots were probed with primary antibodies, followed by horseradish peroxidase-conjugated anti-mouse, anti-rabbit, or anti-goat secondary antibody. The primary antibodies used were anti-MIC-1, anti-p65, anti-poly(ADP-ribose) polymerase 1/2 (PARP1/2), anti-actin (all from Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti-phospho-p65 and anti-phospho-TAK1 (both from Cell Signaling Technology). Bands were visualized by an enhanced chemiluminescence kit (Elpis Biotech, Taejon, South Korea) according to the manufacturer's instruction.
Confocal microscopy.
After treatment with bacteria, epithelial cells and tissues were fixed with 4% formaldehyde diluted in PBS (USB, Cleveland, OH, USA). The fixed cells were permeabilized with 0.1% NP-40 in PBS for 10 min. After cells were blocked for 1 h with 3% bovine serum albumin (Sigma-Aldrich Chemical Co.) in PBS, they were stained with 100 ng/ml fluorescein isothiocyanate (FITC; Sigma-Aldrich Chemical Co.) in PBS for 30 min. Fluorescence microscopy images were obtained with a Fluoview 1000 confocal laser scanning microscope (Olympus, Tokyo, Japan) using a single line (520 nm). Images were acquired and processed with FV10-ASW software (version 1.7; Olympus).
Conventional and real-time reverse transcription-PCR (RT-PCR).
Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. An aliquot of RNA (100 ng) from each sample was transcribed into cDNA using Prime RT premix (Genetbio, Nonsan, South Korea). The amplification step was performed using TaKaRa HS Ex Taq DNA polymerase (TaKaRa Bio Inc., Shiga, Japan) in a Mycycler Thermal Cycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with the following program: denaturation at 94°C for 2 min followed by 25 cycles of denaturation at 98°C for 10 s, annealing at 59°C for 30 s, and elongation at 72°C for 45 s. An aliquot of each PCR product was subjected to 1.2% (wt/vol) agarose gel electrophoresis and visualized by staining with ethidium bromide. The 5′ forward- and 3′ reverse-complement primers for PCR amplification of each gene were as follows: human IL-8, 5′-ATG ACT TCC AAG CTG GCC GTG GCT-3′ and 5′-TCT CAG CCC TCT TCA AAA ACT TCT-3′; human cyclin D1, 5′-CCC TGG GTG TCC TAC TTC AA-3′ and 5′-TGG CAT TTT GGA GAG GAA GT-3′; human MIC-1, 5′-ACG CTA CGA GGA CCT GCT AA-3′ (forward) and 5′-AGA TTC TGC CAG CAG TTG GT-3′ (reverse); Aequorea victoria (jellyfish) GFP, 5′-GCC CGA AGG TTA TGT ACA GG-3′ and 5′-AAA GGG CAG ATT GTG TGG AC-3′; and human glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-TCA ACG GAT TTG GTC GTA TT-3′ and 5′-CTG TGG TCA TGA GTC CTT CC-3′.
For real-time PCR, 6-carboxyl-fluorescein (FAM) was used as the fluorescent reporter dye and was conjugated to the 5′ ends of the probes to detect cDNA amplified in an iCycler Thermal Cycler (Bio-Rad). The following program was used: denaturation at 94°C for 2 min and 40 cycles of denaturation at 98°C for 10 s, annealing at 59°C for 30 s, and elongation at 72°C for 45 s. Each experiment was tested in triplicate to ensure statistical significance. Relative gene expression was quantified using the comparative threshold cycle (CT) method. CT is defined as the number of PCR cycles at which a statistically significant increase in fluorescence has occurred. The CT required for FAM intensities to exceed a threshold just above background was calculated for the test and reference reactions. GAPDH was used as the endogenous control for all experiments.
IL-8 ELISA.
IL-8 was quantified from each cell supernatant using an enzyme linked immunosorbent assay (ELISA). HCT-8 cells (3 × 104) were dispensed in each well of a 24-well plate. After infection with EPEC or EPEC ΔescN, the medium was collected and centrifuged to remove cell debris. Levels of IL-8 were determined by ELISA using an OptEIA human IL-8 ELISA kit (BD Biosciences, Franklin Lakes, NJ) according to the manufacturer's instructions. Briefly, capture antibody was coated onto ELISA plates overnight at 4°C. After the plate was washed with phosphate-buffered saline (PBS) containing Tween 20 (PBST) and blocked with PBS supplemented with 10% (vol/vol) FBS overnight at 4°C, plates were incubated with serial dilutions of IL-8 and standards. After treatment with detection antibody and tetramethylbenzidine (TMB) substrate, absorbance was measured at 405 nm using an ELISA reader. The assay detection limit was 3.1 pg/ml of IL-8.
Flow cytometry analysis.
Cells (5 × 105) were plated, incubated, and treated with EPEC. After EPEC treatment, the cells were harvested, washed with PBS, suspended in 200 μl of PBS and 200 μl of FBS, fixed by the slow addition of cold 70% ethanol to a total of 1.6 ml, and stored overnight at 4°C. The fixed cells were pelleted, washed once with PBS, and stained with propidium iodide (PI) supplemented with RNase in PBS for 20 min. Cells were examined by flow cytometry using a FACSort apparatus (BD Biosciences, Franklin Lakes, NJ, USA) equipped with CellQuest software (BD Biosciences) and analyzed by gating on an area-versus-width dot plot to exclude cell debris and aggregates. Apoptosis was measured, using CellQuest software, by the level of subdiploid DNA in the cells after treatment with EPEC 2348/69.
Stable transfection.
An MIC-1 short hairpin RNA (shRNA) expression vector was kindly provided by Jong-Sik Kim (Andong National University, South Korea) and Seong-Joon Baek (University of Tennessee, TN, USA). Transfection was performed using Trans-LT1 transfection reagent (Mirus, Madison, WI, USA) according to the manufacturer's protocol. The cells were cotransfected with an MIC-1 shRNA expression vector (pShMIC-1) construct with pcDNA3.1-neo (Invitrogen). Cyclin D1-specific shRNA targeted to the 5′-TGG CAT TTT GGA GAG GAA GT-3′ sequence was cloned into a pSilencer 4.1-CMV-neo vector (where CMV is cytomegalovirus) (Applied Biosystems/Ambion, Austin, TX, USA). Following transfection, cells underwent 2 weeks of selection with 400 μg/ml G418 (Invitrogen). Single resistant colonies were expanded and maintained in medium supplemented with 200 μg/ml G418. The efficiencies of all transfections were around 50 to 60%, as confirmed by expression of a pMX-enhanced GFP vector.
Fecal sample collection and bacterial DNA extraction.
The fecal samples were collected aseptically, transferred to sterile tubes, and transported to the laboratory in ice coolers. The fecal samples were shipped overnight on dry ice and stored at −70°C until further processing. DNA extraction from fecal samples was performed using a FastDNA spin kit for soil (MP Biomedical, Illkirsh, France) according to the supplier's instructions. The DNA concentrations were measured using a UV spectrophotometer. The DNA extracts were stored at −20°C until further processing.
Statistical analyses.
Data were analyzed using SigmaStat for Windows (Jandel Scientific, San Rafael, CA, USA). For comparative analysis of two groups of data, a Student's t test was performed. For comparative analysis of multiple groups, data were subjected to analysis of variance (ANOVA), and pairwise comparisons were made by the Student-Newman-Keuls (SNK) post hoc ANOVA method. Data not meeting the normality assumption were subjected to Kruskal-Wallace ANOVA on ranks, and then pairwise comparisons were made by the SNK method.
RESULTS
Sustained p65 phosphorylation in EPEC-infected intestinal epithelial cells.
NF-κB activation is a central signaling pattern of the epithelial cellular response to early exposure to bacterial infection. Animal models have been used to investigate the epithelial response to EPEC homologues (30, 31); these models include rabbits infected with rabbit-specific EPEC and mice infected with Citrobacter rodentium. Although diseases due to infection with rabbit-specific EPEC or C. rodentium are similar to other EPEC-induced diseases, these homologue models are limited due to differences in many pathophysiological patterns observed in the EPEC-triggered infection. The present study used an established mouse model of EPEC infection (32, 33). C57BL/6J mice are particularly susceptible to infection by EPEC and serve as a suitable in vivo model for studying epithelial responses to EPEC infection. EPEC primarily causes gastrointestinal illness by colonizing the epithelial lining of the small intestine. To visualize the persistent presence of EPEC in mucosa of the experimental animal, GFP expression vector-introduced EPEC was monitored in the feces and intestinal mucosal tissue of both uninfected and infected mice for 15 days (see Fig. S1A and S1B in the supplemental material), which is consistent with a recent report on the persistent colonization at high densities for up to 31 days in the same model (34). During the 15-day period of infection with EPEC, small and large intestinal epithelia were observed for the activation of NF-κB signaling. NF-κB activation was maximally observed in the cells of the epithelium and glands even at 15 days after infection with EPEC (Fig. 1). As a negative control, an EPEC mutant (EPEC ΔescN) did not trigger extended activation of NF-κB in intestinal mucosa. Considering the general pattern of short-term activation of NF-κB signaling by proinflammatory triggers in many inflammatory models, the in vivo pattern of NF-κB activation observed in the present study seemed relatively prolonged. NF-κB activation was also evaluated in HCT-8 epithelial cell cultures treated with EPEC. HCT-8 cells are a frequently used human epithelial cell culture model for microbial infection and inflammatory diseases (35–37). In terms of p65 activation, confocal analysis of p65 was performed in the epithelial cells infected with wild-type and mutant EPEC. Interestingly, wild-type EPEC infection enhanced total levels of p65 (Fig. 2A), subsequently providing more p65 in the nuclear region (Fig. 2B), but the EPEC mutant did not. Since EPEC can regulate NF-κB signals via a TTSS, as reported in many previous studies (14, 38, 39), the earlier (<2 h after infection) pattern of p65 activation was observed in wild-type EPEC- or EPEC ΔescN-infected cells (Fig. 3). Particularly at 30 min after infection, p65 phosphorylation was higher in EPEC ΔescN-infected cells than in the wild-type EPEC-infected cells, as previously reported, although total p65 expression was upregulated only in the wild-type EPEC-infected cells (Fig. 3A and D). Along with the early p65 activation, subsequent IL-8 induction was also higher in the EPEC ΔescN-infected cells than in the wild-type EPEC-infected cells (Fig. 3B and C). However, differently from the early p65 activation, the phosphorylated p65 and total p65 expression were persistent in the wild-type EPEC-infected cells, whereas p65 activation was transient in EPEC ΔescN-infected cells (Fig. 3A), suggesting that early strong p65 activation may be more crucial in IL-8 induction than the late p65 activation. Taken together, our data show that EPEC induced sustained activation of NF-κB in intestinal epithelial cells in vivo and in vitro.
Fig 1.
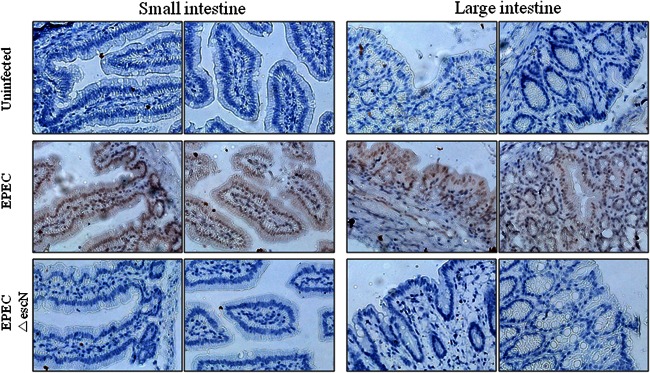
Effects of EPEC on p65 phosphorylation in gut epithelia infected with EPEC. Tissues of ileal and colonic epithelia from C57BL/6J mice infected with wild-type EPEC or EPEC ΔescN were examined by immunohistochemistry. NF-κB activation was visualized with anti-p-p65 antibody (brown) which was counterstained with a 50% dilution of hematoxylin (blue).
Fig 2.

Effects of EPEC on p65 expression and nuclear localization in intestinal epithelial cells infected with EPEC. HCT-8 ileocecal epithelial cells were infected with wild-type EPEC or EPEC ΔescN at a ratio of 50:1 (bacteria to cells) at each time point. EPEC-infected epithelial cells were stained with anti-p65 antibody for fluorescence microscopic observation (A), and the relative density of nuclear p65 was quantified (B). All results are representative of three independent experiments. Values are the means ± standard errors of the means (n = 4 to 6). *, P < 0.05 compared to the wild-type EPEC-infected group at each time point. A Student's t test was used to comparatively analyze the two groups of data. DAPI, 4′,6′-diamidino-2-phenylindole.
Fig 3.

Early effects of EPEC on phosphorylated p65 (p-p65) and IL-8 induction in intestinal epithelial cells infected with EPEC. (A) HCT-8 intestinal epithelial cells were infected with wild-type EPEC or EPEC ΔescN at a ratio of 50:1 (bacteria to cells) at each time point. Total proteins from the epithelial cellular lysate were subjected to Western blot analysis. (B) The levels of IL-8 mRNA were measured by real-time RT-PCR. Values are the means ± standard errors of the means (n = 4 to 6). *, P < 0.05, compared to the wild-type EPEC-infected group at each time point. A Student's t test was used to comparatively analyze the two groups of data. (C) IL-8 secretion in the culture supernatants was measured using an ELISA. Bars with different letters are significantly different (P < 0.05). Pairwise comparisons were performed using SNK post hoc ANOVA. (D) Enterocyte proteins at 0.5 h after infection were compared using Western blot analysis.
Simultaneous induction of MIC-1 expression and NF-κB activation.
As a modulator of sustained NF-κB signaling, macrophage inhibitory cytokine 1 (MIC-1) expression was assessed in the murine infection model. MIC-1 expression was observed together with NF-κB activation in the ileal villus of mice infected with EPEC (Fig. 4A). The coexistence of MIC-1 with activated NF-κB was confirmed in each segmented part of the mouse small intestine (Fig. 4B). Along with findings in the animal experiment, wild-type EPEC infection also enhanced MIC-1 gene expression in cultured epithelial cells, but this was suppressed in EPEC ΔescN-infected cells (Fig. 5A), indicating a positive involvement of the TTSS in MIC-1 induction. The pattern of coexistence of MIC-1 and active p65 in the animal experiment was also observed in EPEC-infected human intestinal epithelial cells including HCT-8, CACO-2, IEC-18, and HCT-116 cells (Fig. 5B). To measure the effect of MIC-1 on NF-κB activation by EPEC infection, p65 phosphorylation was analyzed in cells transfected with an MIC-1-specific shRNA. MIC-1 knockdown attenuated NF-κB activation in EPEC-infected cells, indicating the involvement of MIC-1 in extended NF-κB activation (Fig. 5C). Moreover, the secretion of IL-8, a target of NF-κB, was also partly suppressed by MIC-1 knockdown (Fig. 5D). Taken together, our findings show that NF-κB activation simultaneously occurred with MIC-1 expression in gut epithelia infected with EPEC. In particular, MIC-1 positively affected prolonged NF-κB activation induced by EPEC via the TTSS.
Fig 4.

Effects of EPEC infection on MIC-1 and NF-κB in ileal epithelia in mice. (A) C57BL/6J mice infected with wild-type EPEC or EPEC ΔescN were sacrificed 15 days after infection, and the ileal epithelia were examined by immunofluorescence staining. Proteins were visualized with an anti-MIC-1 antibody (green) and anti-p-p65 (red). (B) Proteins from each small intestine segment of C57BL/6J mice infected with EPEC were analyzed by Western blotting. C, control.
Fig 5.

Roles of MIC-1 in EPEC infection in intestinal epithelial cells. (A) HCT-8 intestinal epithelial cells were infected with wild-type EPEC or EPEC ΔescN at a ratio of 50:1 (bacteria to cells) at each time point. Total proteins from the epithelial cellular lysate were subjected to Western blot analysis. (B) Intestinal epithelial cells (HCT-8, HCT-116, CACO-2, and IEC-18) were infected with EPEC at a ratio of 50:1 (bacteria to cells) for 4 h. Total proteins from epithelial cellular lysates were subjected to Western blot analysis. All results are representative of three independent experiments. (C) Stable cell lines (control and HCT-8 cells stably expressing MIC-1-specific shRNA) were infected with EPEC at a ratio of 50:1 (bacteria to cells) for the indicated times. Total proteins from the epithelial cellular lysate were subjected to Western blot analysis. (D) Stable cell lines (control and HCT-8 cells expressing MIC-1-specific shRNA) were pretreated with vehicle (dimethyl sulfoxide) or 20 μM BAY 11–7082 (an NF-κB inhibitor) and then infected with EPEC for 1 h. IL-8 secreted into the culture supernatant was analyzed using an ELISA. Values are the means ± standard errors of the means (n = 6 to 12). Bars with different letters are significantly different (P < 0.05). Pairwise comparisons were performed using SNK post hoc ANOVA.
MIC-1 induces p65 phosphorylation via TAK1.
The molecular mediator of MIC-1-linked NF-κB activation was addressed in terms of the MIC-1-activated kinase signaling pathway. MIC-1 is a member of the TGF-β superfamily and binds to TGF-β receptor II (29). The TGF-β-activated phosphorylation signaling cascade involves a key step kinase, TGF-β-activated kinase 1 (TAK1), which can lead to NF-κB activation. Therefore, our study was conducted based on the assumption that MIC-1 may activate NF-κB phosphorylation via the TAK1 pathway. First, we observed that EPEC infection triggered TAK1 phosphorylation in human enterocytes. This was decreased by suppressing MIC-1 expression (Fig. 6A), indicating that MIC-1 activates TAK1 signaling in these cells. As direct evidence of the association, administration of recombinant MIC-1 protein was also shown to increase TAK1 phosphorylation (Fig. 6A, right panel). When enzymatic activity of TAK1 was abolished by its specific inhibitor, (5Z)-7-oxozeaenol, EPEC-activated p65 expression and its phosphorylation were suppressed (Fig. 6B), indicating that TAK1 activates NF-κB signaling during EPEC infection. Taken together, our results indicate that MIC-1 activated TAK1 signaling following EPEC infection, which consequently led to NF-κB activation in gut epithelial cells.
Fig 6.
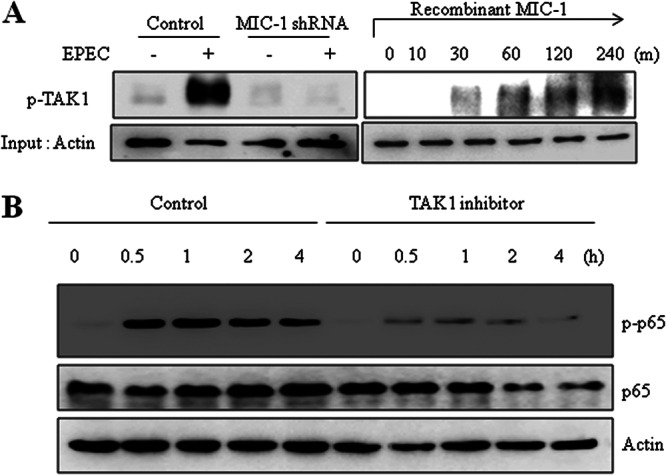
Involvement of TAK1 in MIC-1-linked signaling in EPEC-infected intestinal epithelial cells. (A) Stable cell lines (control and HCT-8 cells expressing MIC-1-specific shRNA) were infected with EPEC at a ratio of 50:1 (bacteria to cells) for 4 h. Phosphorylated TAK1 (p-TAK1) was detected by Western blotting (left). HCT-8 cells were treated with 10 ng/ml recombinant MIC-1 for the indicated times (right). Total proteins from the epithelial cellular lysate were subjected to Western blot analysis. (B) HCT-8 cells were pretreated with vehicle (dimethyl sulfoxide) or 100 nM (5Z)-7-oxozeaenol (5Z-OXO), a TAK1 inhibitor, and then infected with EPEC for the indicated times. Total proteins from the epithelial cellular lysate were subjected to Western blot analysis. All results are representative of three independent experiments.
Cyclin D1, a target of NF-κB, is involved in epithelial survival of infection-induced cytotoxicity.
Functions of extended NF-κB in association with MIC-1 were assessed in terms of epithelial survival of infection-mediated cytotoxicity. EPEC infection triggered epithelial apoptosis, which was measured in terms of PARP1/2 cleavage (Fig. 7A). In contrast, an EPEC mutant (EPEC ΔescN) had fewer proapoptotic effects on the intestinal epithelial cells. Moreover, genetic ablation of MIC-1 using an shRNA increased PARP1/2 cleavage, indicating negative regulation of MIC-1 in EPEC-induced apoptosis. In addition to MIC-1 knockdown, NF-κB inhibition also increased epithelial cell death in response to EPEC infection (Fig. 7B), suggesting the involvement of these two mediators (MIC-1 and NF-κB) in epithelial survival responses. Cell survival was also monitored by measuring cyclin D1, an indicative cell cycle modulator in actively proliferating cells. EPEC infection caused significant induction of cyclin D1 expression as a defense mechanism against cytotoxic infection (Fig. 7C). However, MIC-1 knockdown, NF-κB inhibition, and TAK1 inhibition all attenuated cyclin D1 induction, suggesting positive involvements of MIC-1, NF-κB, and TAK1 in cyclin D1 expression. Cyclin D1 is a critical mediator of epithelial cell survival, and, not surprisingly, suppressing the expression of this factor using shRNA made epithelial cells susceptible to infection-mediated apoptosis (Fig. 7D). This supports our hypothesis that MIC-1 and NF-κB enhance epithelial cell survival through the induction of cyclin D1 expression.
Fig 7.

The role of cyclin D1 in MIC-1-associated survival of EPEC-infected intestinal epithelial cells. (A) HCT-8 intestinal epithelial cells were infected with E. coli K-12, wild-type EPEC, or EPEC ΔescN at a ratio of 50:1 (bacteria to cells) at each time point. Total proteins from the epithelial cellular lysate were subjected to Western blot analysis. (B) Stable cell lines (vector- and shMIC-1-transfected HCT-8 cells) were pretreated with 5 μM BAY 11–7082. Next, the cells were infected with EPEC at a ratio of 50:1 (bacteria to cells) for 12 h. Apoptosis was quantified by staining with propidium iodide (PI) and performing fluorescence-activated cell sorting analysis. All results are representative of three independent experiments. (C) Stable cell lines (vector- and shMIC-1-transfected HCT-8 cells) were pretreated with vehicle (dimethyl sulfoxide), 20 μM BAY11-7082, or 100 nM (5Z)-7-oxozeaenol and then infected with EPEC for 12 h. The total epithelial cellular lysates were subjected to real-time RT-PCR. (D) HCT-8 cells expressing either control or cyclin D1-specific shRNA were infected with EPEC at a ratio of 50:1 (bacteria to cell) for 12 h. Apoptosis was quantified by staining with propidium iodide (PI) and performing fluorescence-activated cell sorting analysis. Values are the means ± standard errors of the means (n = 3 to 6). Bars with different letters are significantly different (P < 0.05). Pairwise comparisons were made using SNK post hoc ANOVA.
DISCUSSION
Prolonged NF-κB activation in epithelial cells was observed in animals infected with EPEC. Similar patterns of prolonged NF-κB signaling were also observed in cultured intestinal epithelial cells exposed to persistent EPEC infection although an EPEC-associated TTSS transmits negative regulators of transient NF-κB activation during the early stage of infection. Moreover, the results in the present study suggest that MIC-1 mediates prolonged activation of the NF-κB signaling pathway, all of which contributed to epithelial survival from pathogen-induced epithelial cell death (Fig. 8).
Fig 8.
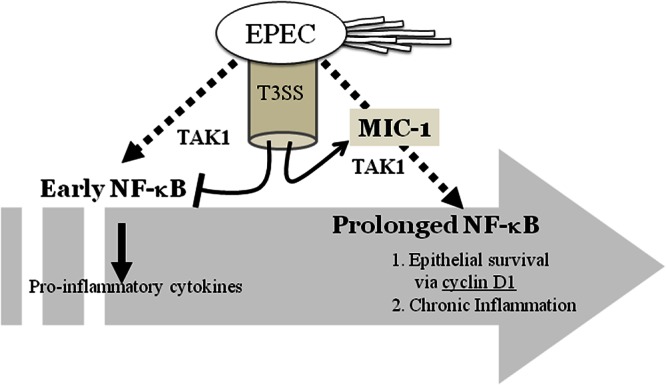
The mechanism underlying sustained activation of NF-κB signaling associated with MIC-1 in intestinal epithelial cells infected with EPEC. EPEC infection can trigger prolonged NF-κB activation. Although the bacteria can regulate transient NF-κB activation and cytokine production via a type III secretion system during the early infection period, it extends p65 activation in a MIC-1-linked signaling pathway, which contributes to epithelial survival from bacteria-induced apoptotic death.
Various pathogen-associated molecular patterns (PAMPs) have been observed during EPEC infection of human epithelia (12, 40, 41). Like other bacteria, EPEC is recognized by innate host receptors such as TLRs and nucleotide-binding oligomerization domain protein (NOD)-like receptors that detect conserved microbial molecules, including flagellin and lipopolysaccharides. Although the EPEC homologue C. rodentium causing attaching and effacing (A/E) lesions induces TLR4-linked colitis and chemokine production via the NF-κB pathway, host defenses are also broken in a TLR4-deficient model. This finding indicates that TLR4-activated NF-κB helps protect against infection. EPEC flagellin is a TLR5 agonist that is also known to trigger the production of proinflammatory mediators, including IL-8, macrophage inflammatory protein 3α, and monocyte chemotactic protein 1 (38, 41, 42). However, at the late phase of infection, flagellin-independent activation of proinflammatory mediators is also observed.
Although EPEC-linked PAMPs, including flagellin, are important in NF-κB activation, various EPEC effector molecules translocated through the TTSS are also known to regulate proinflammatory responses (14, 38, 39). Therefore, an EPEC strain with a mutant TTSS (EPEC ΔescN) produces more proinflammatory stimulation, such as NF-κB activation and subsequent cytokine induction, than the wild-type EPEC. Anti-inflammatory action of EPEC effectors through the TTSS is obvious during the early stage of infection, but EPEC ΔescN had fewer persistent effects on p65 activation than the wild-type EPEC. The prolonged NF-κB activation by the wild-type EPEC was also due to the TTSS. The unidentified bacterial effector molecules contributed to MIC-1 induction and prolonged p65 induction. NF-κB signal can be both detrimental and beneficial to epithelial cells exposed to EPEC. We observed that intestinal NF-κB induced the expression of IL-8, which could trigger the recruitment of inflammatory cells and injure the tissue of the gut. Moreover, sustained NF-κB activation can prolong chemokine production. While early activated NF-κB signals are important for transcriptional activation of proinflammatory cytokine genes, extended NF-κB activation via MIC-1 was shown to be involved in epithelial survival from the cytotoxic EPEC infection. The beneficial effects of NF-κB can be observed in other types of gut epithelial injuries. In particular, epithelial NF-κB contributes to barrier reestablishment by inducing the transcription of genes involved in epithelial cell survival, wound healing, and tissue remodeling after tissue injuries from pathogens and inflammation (43, 44). In the present study, NF-κB protects intestinal epithelial cells against apoptosis induced by toxic insults, which is a defensive function important for maintaining the gut barrier. In addition to microbial stimulation, specific NF-κB activators, such as the bile salt taurodeoxycholate, promote epithelial reconstruction after physical injury (45). A loss-of-function study in an animal model also demonstrated the protective roles of enterocyte-derived NF-κB in the maintenance of intestinal homeostasis after different epithelial insults, including irradiation and ischemia-reperfusion injuries (46, 47). In the present study, NF-κB was found to be potentially crucial for epithelial protection against EPEC-induced barrier disruption and cytotoxicity. Intestinal NF-κB is also important for the spatial organization of the tight junction protein ZO-1, which is vital for maintaining epithelial integrity (48). In response to mucosal microbial infection, prolonged NF-κB activation can be beneficial by promoting epithelial reconstitution and repressing cellular apoptosis, whereas transient NF-κB activation can trigger proinflammatory recruitment.
EPEC infection does not always promote proinflammatory responses. Although mucosal inflammation undoubtedly represents the net effect of EPEC infection, pathogens also develop defensive strategies to evade the host immune response which can recruit immune cells and eliminate bacteria. The suppressive effects of EPEC on overstimulated inflammatory responses are essential for the survival of these noninvasive bacteria. EPEC may exert these effects by injecting into the host cells pathogenic effectors that disrupt the NF-κB pathway, an essential modulator of the early host defense (14, 49). In addition to surface PAMPs, EPEC possesses a TTSS that acts like a molecular syringe which delivers bacterial non-locus of enterocyte effacement (LEE)-encoded effectors (Nle). Recent studies reported that the anti-inflammatory zinc protease NleC disrupts the NF-κB pathway (14, 49) in cooperation with NleE; this accounts for most of the immune suppression caused by EPEC. Originally, MIC-1 was identified in monocytes as a mediator that regulates macrophage activation by limiting proinflammatory cytokine production (22). Although MIC-1 was found in the present study to play critical roles in prolonging of the activation of NF-κB signaling, regulatory functions during the whole process of infection-mediated epithelial inflammation should be examined in other models. MIC-1 in monocyte-derived cells can behave differently from epithelium-produced MIC-1 by inducing anti-inflammatory resolution after infection.
The present study suggested that MIC-1 is a modulator of prolonged NF-κB signaling. Mechanistically, MIC-1-activated TAK1 was found to be a signaling kinase critical for NF-κB activation. This is the first report showing MIC-1-activated proinflammatory signaling via TAK1 phosphorylation. MIC-1 protein binds to the TGF-β receptor II (TGFRII) which can either activate or repress gene expression. In addition to TAK1-associated NF-κB activation, TGFRII-linked Smad proteins can interact with NF-κB proteins, such as p52 (50), leading to modified transcriptional activation of proinflammatory genes. Another EPEC-linked PAMP may stimulate the innate nucleotide oligomerization domain 2 (NOD2) receptors that detect bacterial muramyl dipeptide, a minimal recognition part of bacterial peptidoglycan. Since EPEC contains muramyl dipeptide in the cell wall, these microorganisms can theoretically be sensed by NOD2 although this has not been previously reported. Additional studies should be performed to examine the role of specific EPEC PAMPs in epithelial recognition, particularly in terms of MC-1 gene induction.
Prolonged MIC-1 production and NF-κB signaling in mucosal epithelia do not always positively affect human health. Infection-triggered MIC-1 may be involved in tumor metastasis in cancer patients, and high serum levels of mature-type MIC-1 produce other procarcinogenic effects. In colon cancer, increased MIC-1 expression is associated with the development of colonic adenomas into invasive cancer (27). In terms of mucosal pathogen-induced carcinogenesis, a murine model of EPEC infection using Citrobacter rodentium developed colonic hyperplasia and showed an enhanced response to chemically induced carcinogenesis and genetic susceptibility to epithelial cancer (51, 52). It was recently reported that EPEC suppresses the expression of a DNA repair protein and can therefore play a potential role in human intestinal carcinogenesis (8). Therefore, further investigation of the chronic action of MIC-1 in both normal and transformed gut epithelia is necessary to elucidate the exact effects of mucosal EPEC infection and their potential impact on human health.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a Korea Research Foundation grant funded by the South Korean government (Ministry of Education, Science and Technology) (grant 2012R1A1A2005837).
Footnotes
Published ahead of print 12 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00162-13
REFERENCES
- 1. Imani Fooladi AA, Mousavi SF, Seghatoleslami S, Yazdani S, Nourani MR. 2011. Toll-like receptors: role of inflammation and commensal bacteria. Inflamm. Allergy Drug Targets 10:198–207 [DOI] [PubMed] [Google Scholar]
- 2. Eckmann L, Neish AS. 2011. NF-κB and mucosal homeostasis. Curr. Top. Microbiol. Immunol. 349:145–158 [DOI] [PubMed] [Google Scholar]
- 3. Swamy M, Jamora C, Havran W, Hayday A. 2010. Epithelial decision makers: in search of the “epimmunome.” Nat. Immunol. 11:656–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wullaert A, Bonnet MC, Pasparakis M. 2011. NF-κB in the regulation of epithelial homeostasis and inflammation. Cell Res. 21:146–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Robins-Browne RM. 1987. Traditional enteropathogenic Escherichia coli of infantile diarrhea. Rev. Infect. Dis. 9:28–53 [DOI] [PubMed] [Google Scholar]
- 6. Kallinowski F, Wassmer A, Hofmann MA, Harmsen D, Heesemann J, Karch H, Herfarth C, Buhr HJ. 1998. Prevalence of enteropathogenic bacteria in surgically treated chronic inflammatory bowel disease. Hepatogastroenterology 45:1552–1558 [PubMed] [Google Scholar]
- 7. La Ferla K, Seegert D, Schreiber S. 2004. Activation of NF-κB in intestinal epithelial cells by E. coli strains isolated from the colonic mucosa of IBD patients. Int. J. Colorectal Dis. 19:334–342 [DOI] [PubMed] [Google Scholar]
- 8. Maddocks OD, Short AJ, Donnenberg MS, Bader S, Harrison DJ. 2009. Attaching and effacing Escherichia coli downregulate DNA mismatch repair protein in vitro and are associated with colorectal adenocarcinomas in humans. PLoS One 4:e5517 doi:10.1371/journal.pone.0005517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weber P, Koch M, Heizmann WR, Scheurlen M, Jenss H, Hartmann F. 1992. Microbic superinfection in relapse of inflammatory bowel disease. J. Clin. Gastroenterol. 14:302–308 [DOI] [PubMed] [Google Scholar]
- 10. Long-Krug SA, Weikel CS, Tiemens KT, Hewlett EL, Levine MM, Guerrant RL. 1984. Does enteropathogenic Escherichia coli produce heat-labile enterotoxin, heat-stable enterotoxins a or b, or cholera toxin A subunits? Infect. Immun. 46:612–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Robins-Browne RM, Levine MM, Rowe B, Gabriel EM. 1982. Failure to detect conventional enterotoxins in classical enteropathogenic (serotyped) Escherichia coli strains of proven pathogenicity. Infect. Immun. 38:798–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Edwards LA, Bajaj-Elliott M, Klein NJ, Murch SH, Phillips AD. 2011. Bacterial-epithelial contact is a key determinant of host innate immune responses to enteropathogenic and enteroaggregative Escherichia coli. PLoS One 6:e27030 doi:10.1371/journal.pone.0027030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ruchaud-Sparagano MH, Muhlen S, Dean P, Kenny B. 2011. The Enteropathogenic E. coli (EPEC) Tir effector inhibits NF-κB activity by targeting TNFα receptor-associated factors. PLoS Pathog. 7:e1002414 doi:10.1371/journal.ppat.1002414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sham HP, Shames SR, Croxen MA, Ma C, Chan JM, Khan MA, Wickham ME, Deng W, Finlay BB, Vallance BA. 2011. Attaching and effacing bacterial effector NleC suppresses epithelial inflammatory responses by inhibiting NF-κB and p38 mitogen-activated protein kinase activation. Infect. Immun. 79:3552–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rothbaum R, McAdams AJ, Giannella R, Partin JC. 1982. A clinicopathologic study of enterocyte-adherent Escherichia coli: a cause of protracted diarrhea in infants. Gastroenterology 83:441–454 [PubMed] [Google Scholar]
- 16. Ulshen MH, Rollo JL. 1980. Pathogenesis of escherichia coli gastroenteritis in man—another mechanism. N. Engl. J. Med. 302:99–101 [DOI] [PubMed] [Google Scholar]
- 17. Dean P, Maresca M, Schuller S, Phillips AD, Kenny B. 2006. Potent diarrheagenic mechanism mediated by the cooperative action of three enteropathogenic Escherichia coli-injected effector proteins. Proc. Natl. Acad. Sci. U. S. A. 103:1876–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sampaio SC, Gomes TA, Pichon C, du Merle L, Guadagnini S, Abe CM, Sampaio JL, Le Bouguenec C. 2009. The flagella of an atypical enteropathogenic Escherichia coli strain are required for efficient interaction with and stimulation of interleukin-8 production by enterocytes in vitro. Infect. Immun. 77:4406–4413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao X, Wan F, Mateo K, Callegari E, Wang D, Deng W, Puente J, Li F, Chaussee MS, Finlay BB, Lenardo MJ, Hardwidge PR. 2009. Bacterial effector binding to ribosomal protein s3 subverts NF-κB function. PLoS Pathog. 5:e1000708 doi:10.1371/journal.ppat.1000708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Royan SV, Jones RM, Koutsouris A, Roxas JL, Falzari K, Weflen AW, Kim A, Bellmeyer A, Turner JR, Neish AS, Rhee KJ, Viswanathan VK, Hecht GA. 2010. Enteropathogenic E. coli non-LEE encoded effectors NleH1 and NleH2 attenuate NF-κB activation. Mol. Microbiol. 78:1232–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fairlie WD, Moore AG, Bauskin AR, Russell PK, Zhang HP, Breit SN. 1999. MIC-1 is a novel TGF-β superfamily cytokine associated with macrophage activation. J. Leukoc. Biol. 65:2–5 [DOI] [PubMed] [Google Scholar]
- 22. Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, Zhang HP, Donnellan M, Mahler S, Pryor K, Walsh BJ, Nicholson RC, Fairlie WD, Por SB, Robbins JM, Breit SN. 1997. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-β superfamily. Proc. Natl. Acad. Sci. U. S. A. 94:11514–11519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Whitman M. 1998. Smads and early developmental signaling by the TGFβ superfamily. Genes Dev. 12:2445–2462 [DOI] [PubMed] [Google Scholar]
- 24. Bauskin AR, Brown DA, Kuffner T, Johnen H, Luo XW, Hunter M, Breit SN. 2006. Role of macrophage inhibitory cytokine-1 in tumorigenesis and diagnosis of cancer. Cancer Res. 66:4983–4986 [DOI] [PubMed] [Google Scholar]
- 25. Bottner M, Suter-Crazzolara C, Schober A, Unsicker K. 1999. Expression of a novel member of the TGF-β superfamily, growth/differentiation factor-15/macrophage-inhibiting cytokine-1 (GDF-15/MIC-1) in adult rat tissues. Cell Tissue Res. 297:103–110 [DOI] [PubMed] [Google Scholar]
- 26. Agarwal MK, Hastak K, Jackson MW, Breit SN, Stark GR, Agarwal ML. 2006. Macrophage inhibitory cytokine 1 mediates a p53-dependent protective arrest in S phase in response to starvation for DNA precursors. Proc. Natl. Acad. Sci. U. S. A. 103:16278–16283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brown DA, Ward RL, Buckhaults P, Liu T, Romans KE, Hawkins NJ, Bauskin AR, Kinzler KW, Vogelstein B, Breit SN. 2003. MIC-1 serum level and genotype: associations with progress and prognosis of colorectal carcinoma. Clin. Cancer Res. 9:2642–2650 [PubMed] [Google Scholar]
- 28. Lee DH, Yang Y, Lee SJ, Kim KY, Koo TH, Shin SM, Song KS, Lee YH, Kim YJ, Lee JJ, Choi I, Lee JH. 2003. Macrophage inhibitory cytokine-1 induces the invasiveness of gastric cancer cells by up-regulating the urokinase-type plasminogen activator system. Cancer Res. 63:4648–4655 [PubMed] [Google Scholar]
- 29. Tan M, Wang Y, Guan K, Sun Y. 2000. PTGF-β, a type beta transforming growth factor (TGF-β) superfamily member, is a p53 target gene that inhibits tumor cell growth via TGF-β signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 97:109–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Borenshtein D, McBee ME, Schauer DB. 2008. Utility of the Citrobacter rodentium infection model in laboratory mice. Curr. Opin. Gastroenterol. 24:32–37 [DOI] [PubMed] [Google Scholar]
- 31. Garcia A, Marini RP, Feng Y, Vitsky A, Knox KA, Taylor NS, Schauer DB, Fox JG. 2002. A naturally occurring rabbit model of enterohemorrhagic Escherichia coli-induced disease. J. Infect. Dis. 186:1682–1686 [DOI] [PubMed] [Google Scholar]
- 32. Calderon Toledo C, Arvidsson I, Karpman D. 2011. Cross-reactive protection against enterohemorrhagic Escherichia coli infection by enteropathogenic E. coli in a mouse model. Infect. Immun. 79:2224–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Savkovic SD, Villanueva J, Turner JR, Matkowskyj KA, Hecht G. 2005. Mouse model of enteropathogenic Escherichia coli infection. Infect. Immun. 73:1161–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rhee KJ, Cheng H, Harris A, Morin C, Kaper JB, Hecht G. 2011. Determination of spatial and temporal colonization of enteropathogenic E. coli and enterohemorrhagic E. coli in mice using bioluminescent in vivo imaging. Gut Microbes 2:34–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alcantara Warren C, Destura RV, Sevilleja JE, Barroso LF, Carvalho H, Barrett LJ, O'Brien AD, Guerrant RL. 2008. Detection of epithelial-cell injury, and quantification of infection, in the HCT-8 organoid model of cryptosporidiosis. J. Infect. Dis. 198:143–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sifuentes LY, Di Giovanni GD. 2007. Aged HCT-8 cell monolayers support Cryptosporidium parvum infection. Appl. Environ. Microbiol. 73:7548–7551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thebault S, Deniel N, Marion R, Charlionet R, Tron F, Cosquer D, Leprince J, Vaudry H, Ducrotte P, Dechelotte P. 2006. Proteomic analysis of glutamine-treated human intestinal epithelial HCT-8 cells under basal and inflammatory conditions. Proteomics 6:3926–3937 [DOI] [PubMed] [Google Scholar]
- 38. Khan MA, Bouzari S, Ma C, Rosenberger CM, Bergstrom KS, Gibson DL, Steiner TS, Vallance BA. 2008. Flagellin-dependent and -independent inflammatory responses following infection by enteropathogenic Escherichia coli and Citrobacter rodentium. Infect. Immun. 76:1410–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ruchaud-Sparagano MH, Maresca M, Kenny B. 2007. Enteropathogenic Escherichia coli (EPEC) inactivate innate immune responses prior to compromising epithelial barrier function. Cell Microbiol. 9:1909–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A. 2010. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U. S. A. 107:3076–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Salazar-Gonzalez H, Navarro-Garcia F. 2011. Intimate adherence by enteropathogenic Escherichia coli modulates TLR5 localization and proinflammatory host response in intestinal epithelial cells. Scand. J. Immunol. 73:268–283 [DOI] [PubMed] [Google Scholar]
- 42. Khan MA, Ma C, Knodler LA, Valdez Y, Rosenberger CM, Deng W, Finlay BB, Vallance BA. 2006. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect. Immun. 74:2522–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Karrasch T, Jobin C. 2008. NF-κB and the intestine: friend or foe? Inflamm. Bowel Dis. 14:114–124 [DOI] [PubMed] [Google Scholar]
- 44. Pasparakis M. 2009. Regulation of tissue homeostasis by NF-κB signalling: implications for inflammatory diseases. Nat. Rev. Immunol. 9:778–788 [DOI] [PubMed] [Google Scholar]
- 45. Strauch ED, Yamaguchi J, Bass BL, Wang JY. 2003. Bile salts regulate intestinal epithelial cell migration by nuclear factor-kappa B-induced expression of transforming growth factor-beta. J. Am. Coll. Surg. 197:974–984 [DOI] [PubMed] [Google Scholar]
- 46. Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. 2003. The two faces of IKK and NF-κB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat. Med. 9:575–581 [DOI] [PubMed] [Google Scholar]
- 47. Egan LJ, Eckmann L, Greten FR, Chae S, Li ZW, Myhre GM, Robine S, Karin M, Kagnoff MF. 2004. IκB-kinaseβ-dependent NF-κB activation provides radioprotection to the intestinal epithelium. Proc. Natl. Acad. Sci. U. S. A. 101:2452–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ma TY, Boivin MA, Ye D, Pedram A, Said HM. 2005. Mechanism of TNF-α modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am. J. Physiol. Gastrointest Liver Physiol. 288:G422–430 [DOI] [PubMed] [Google Scholar]
- 49. Yen H, Ooka T, Iguchi A, Hayashi T, Sugimoto N, Tobe T. 2010. NleC, a type III secretion protease, compromises NF-κB activation by targeting p65/RelA. PLoS Pathog. 6:e1001231 doi:10.1371/journal.ppat.1001231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lopez-Rovira T, Chalaux E, Rosa JL, Bartrons R, Ventura F. 2000. Interaction and functional cooperation of NF-κB with Smads. Transcriptional regulation of the junB promoter. J. Biol. Chem. 275:28937–28946 [DOI] [PubMed] [Google Scholar]
- 51. Newman JV, Kosaka T, Sheppard BJ, Fox JG, Schauer DB. 2001. Bacterial infection promotes colon tumorigenesis in ApcMin/+ mice. J. Infect. Dis. 184:227–230 [DOI] [PubMed] [Google Scholar]
- 52. Barthold SW, Jonas AM. 1977. Morphogenesis of early 1, 2-dimethylhydrazine-induced lesions and latent period reduction of colon carcinogenesis in mice by a variant of Citrobacter freundii. Cancer Res. 37:4352–4360 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.