

Abstract
Despite advances in medical device fabrication and antimicrobial treatment therapies, fungal-bacterial polymicrobial peritonitis remains a serious complication for surgery patients, those on peritoneal dialysis, and the critically ill. Using a murine model of peritonitis, we have demonstrated that monomicrobial infection with Candida albicans or Staphylococcus aureus is nonlethal. However, coinfection with these same doses leads to a 40% mortality rate and increased microbial burden in the spleen and kidney by day 1 postinfection. Using a multiplex enzyme-linked immunosorbent assay, we have also identified a unique subset of innate proinflammatory cytokines (interleukin-6, granulocyte colony-stimulating factor, keratinocyte chemoattractant, monocyte chemoattractant protein-1, and macrophage inflammatory protein-1α) that are significantly increased during polymicrobial versus monomicrobial peritonitis, leading to increased inflammatory infiltrate into the peritoneum and target organs. Treatment of coinfected mice with the cyclooxygenase (COX) inhibitor indomethacin reduces the infectious burden, proinflammatory cytokine production, and inflammatory infiltrate while simultaneously preventing any mortality. Further experiments demonstrated that the immunomodulatory eicosanoid prostaglandin E2 (PGE2) is synergistically increased during coinfection compared to monomicrobial infection; indomethacin treatment also decreased elevated PGE2 levels. Furthermore, addition of exogenous PGE2 into the peritoneal cavity during infection overrode the protection provided by indomethacin and restored the increased mortality and microbial burden. Importantly, these studies highlight the ability of fungal-bacterial coinfection to modulate innate inflammatory events with devastating consequences to the host.
INTRODUCTION
In nature, microorganisms rarely exist as single-species communities but instead exist within multispecies consortia, where mutually beneficial, parasitic, and antagonistic interactions may develop (1). Although many recent research efforts have focused on using molecular techniques to survey various species located at biological sites, relatively little is known about the behavior of these communities and, more importantly, how such interactions may impact the human host. Critically, several recent studies have suggested that amplified pathogenic phenotypes may emerge during infection with multiple microbes, leading to infectious synergism, defined as enhanced virulence during polymicrobial versus monomicrobial disease (2–5).
One human infection that is characterized as often being polymicrobial in nature is peritonitis (6, 7). Peritonitis is an inflammatory disease of the lining of the abdominal wall and organs and is most frequently caused by infectious processes resulting from bowel perforation, laparotomy surgery, intestinal hernias, and, most commonly, insertion of medical devices, such as peritoneal dialysis (PD) catheters (8). Crucially, it has been documented that PD-mediated polymicrobial peritonitis results in higher incidences of relapsing infection, catheter loss, a permanent switch to hemodialysis (HD), and mortality than monomicrobial peritonitis, especially peritonitis involving fungi (9–11). Indeed, peritoneal infections involving fungi, namely, the Candida species, are becoming increasingly common in the hospital setting (12). A permanent switch from PD to HD not only negatively impacts patient lifestyle but also results in a significant accumulation of financial burden to the medical community (13). If acute cases of peritonitis are left untreated or misdiagnosed, infecting microorganisms can migrate from local infectious foci into the bloodstream via innate barrier dysfunctions resulting from aggressive host inflammatory responses; hematogenous seeding of microbes often induces full-blown systemic sepsis (14–16). Despite appropriate antimicrobial treatment, sepsis remains a worldwide concern, with mortality rates extending over 60% in severe cases (17). Therefore, a more comprehensive understanding of the etiological agents contributing to polymicrobial peritonitis is warranted in order to develop targeted therapeutic approaches and improve patient quality of life and outcome.
Two of the most commonly isolated organisms from peritonitis episodes are the polymorphic fungus Candida albicans and the ubiquitous bacterial pathogen Staphylococcus aureus (18). Despite representing two distinct phylogenetic domains, S. aureus and Candida spp. share several pathogenic traits, most notably, their ability to cause an array of human diseases, form biofilms on a variety of surfaces, and develop rapid resistance to antimicrobials (19, 20). Importantly, we have previously identified a unique association between these two pathogens, with S. aureus primarily adhering to the hyphal forms of C. albicans during polymicrobial biofilm growth (21, 22). We have also shown that C. albicans and S. aureus can modulate one another's proteomic profile during in vitro biofilm growth, including the expression of several defined and putative virulence factors (22). In addition to peritonitis, C. albicans and S. aureus can be coisolated from a number of infections, including catheter infections, wounds, septicemia, ventilator-associated pneumonia, keratitis, and oral infections, such as denture stomatitis (23).
Although previous studies with these pathogens have demonstrated that fungal-bacterial peritoneal infections result in infectious synergism during coinfection in mice, the precise mechanisms leading to this phenotype and the effects on the host immune response were not defined (24). The “gold standard” model for testing the effects of chronic peritonitis and subsequent sepsis is the cecal-ligation puncture (CLP), in which the cecum is ligated below the ileocecal valve and then pierced with a sterile needle to release the cecal contents into the normally sterile peritoneal cavity (25). This technique has some limitations however, including the inability to distinguish the contribution of individual microbial species to disease, nonstandardized dosing of the infectious inoculum, and incapacity to mimic acute disease. Traditional CLP mimics bowel perforation, overwhelming the host with an infection predominated by Gram-negative bacteria, and may overshadow the important contributions of the Gram-positive bacteria and fungi in polymicrobial peritonitis pathogenesis (26). To that end, we have optimized a murine model of peritonitis to assess the differential pathological effects in the host during polymicrobial versus monomicrobial infection with two commonly isolated peritonitis pathogens, C. albicans and S. aureus. Ultimately, findings from these studies will aid in the development of novel treatment strategies for an increasingly recognized number of polymicrobial diseases and may more broadly apply to the immunopathogenesis of other mixed fungal-bacterial infections.
MATERIALS AND METHODS
Strains and growth conditions.
The methicillin-resistant S. aureus strain NRS383 used in all experiments was obtained from the Network on Antimicrobial Resistance in S. aureus (NARSA) data bank. NRS383 is positive for the toxic shock syndrome toxin (tst) and δ-toxin genes. Frozen stocks were obtained at −80°C and streaked onto Trypticase soy agar (TSA) containing 20 μg/ml nafcillin at 37°C prior to use. A single colony was transferred to 5 ml of Trypticase soy broth (TSB) containing 20 μg/ml nafcillin and shaken at 37°C overnight. On the following day, the overnight culture was diluted 1:100 in fresh TSB containing 20 μg/ml nafcillin and shaken at 37°C for 3 h until the culture reached the log phase of growth. The C. albicans strain used in these experiments was DAY185, a prototrophic control strain that has HIS1, URA3, and ARG4 genes reinserted into strain BWP17, an auxotrophic derivative of strain SC5314 (27). Frozen stocks were obtained at −80°C and streaked onto yeast peptone dextrose (YPD) agar prior to use. A single colony was transferred to 20 ml of YPD broth and shaken at 30°C for 20 h. Prior to infection, both organisms were rinsed 3 times by centrifugation in sterile phosphate-buffered saline (PBS; pH 7.4), counted on a hemocytometer, and diluted in sterile PBS to prepare standardized inocula.
Murine model of peritonitis.
All experiments involving animals were approved by the Louisiana State University Health Sciences Center (LSUHSC) Institutional Animal Care and Use Committee. Mice were given access to food and water ad libitum. In most experiments, groups (n = 5 or 10) of 6-week-old outbred Swiss Webster mice were injected intraperitoneally (i.p.) using a 27-gauge (1/2-in.) needle with either 0.2 ml of saline, 0.2 ml of 3.5 × 107 CFU/ml C. albicans alone (7 × 106 CFU total), 0.2 ml 4 × 108 CFU/ml S. aureus alone (8 × 107 CFU total), or 0.2 ml containing 3.5 × 107 CFU/ml C. albicans and 4 × 108 CFU/ml S. aureus (7 × 106 and 8 × 107 CFU of each microbe, respectively; 8.7 × 107 CFU total). In some experiments, mice were inoculated with 8.7 × 107 CFU of C. albicans or S. aureus alone in 0.2 ml PBS to serve as monomicrobial controls for the increased microbial load encountered during coinfection. After inoculation, mice were initially observed over a 5-day time course for morbidity (hunched posture, inactivity, ruffled fur) and mortality. Because most polymicrobe-induced mortality occurred within 24 h, in subsequent experiments, mice were euthanized at 1 day postinoculation. In some experiments, mice were sacrificed at 8 h postinfection to assess the infection at an early time point. Peritoneal cavities were lavaged by injection of 2 ml of sterile saline containing 0.1% fetal bovine serum (FBS), followed by gentle massaging of the peritoneal cavity. Peritoneal lavage fluid was then removed using a pipette inserted into a small incision in the abdominal cavity. Kidneys and spleens were removed and placed into either 1 ml sterile PBS containing EDTA-free protease inhibitors (Roche, Basel, Switzerland) or 10% phosphate-buffered formalin for histological analysis. Tissues were then weighed and mechanically homogenized (Pro Scientific, Oxford, CT) prior to CFU or cytokine analysis.
Effect of indomethacin on microbial growth in vitro.
C. albicans and S. aureus were grown as described above. On the following day, overnight cultures were diluted 1:100 in fresh medium containing 50 μg/ml indomethacin and 0.28% dimethyl sulfoxide (DMSO) or 0.28% DMSO alone to serve as controls. Because the average peritoneal volume of a Swiss Webster mouse is approximately 2 ml, the concentrations of indomethacin and DMSO were chosen to reflect the physiologically delivered final concentrations. Cells were grown with shaking as described above, and 100-μl aliquots were removed each hour in triplicate and read spectrophotometrically at 600 nm.
Indomethacin treatment.
Indomethacin was prepared freshly as a concentrated stock (35 mg/ml) in DMSO and diluted to a working concentration of 1 mg/ml in sterile PBS (final DMSO concentration, 2.3%). A sham treatment containing only 2.3% DMSO in PBS was also prepared. Groups of mice were intraperitoneally administered 0.1 ml (roughly 5 mg/kg of body weight) of indomethacin or vehicle control 4 h prior to and 8 h after infection. All other procedures were performed as described above.
Exogenous PGE2 administration.
Mice were treated exactly as described above for indomethacin treatment, except that at 1 h prior to and 9 h after infection mice received an i.p. injection of 1 μg 16,16-dimethyl prostaglandin E2 (PGE2; Cayman Chemical, Ann Arbor, MI) in 100 μl PBS. Mice not receiving PGE2 were injected i.p. with 100 μl PBS to serve as the control.
CFU analysis.
Microbial burdens in the kidneys and spleens were enumerated by serial dilution plating of the homogenate onto YPD containing 20 μg/ml nafcillin and 2 μg/ml vancomycin (for C. albicans enumeration) and TSA containing 20 μg/ml nafcillin and 2.5 μg/ml amphotericin B (for S. aureus enumeration) via the drop plate method (28). Plates were incubated overnight at 37°C and left on the benchtop for 5 days to monitor the outgrowth of potentially slow-growing colonies. All CFU counts were then expressed as the number of CFU per gram of homogenized tissue; the limit of detection was approximately 102 CFU per gram tissue.
Cytokine analysis.
After aliquots were removed for CFU analysis, the remaining homogenate was centrifuged at 2,500 rpm for 15 min at 4°C. The supernatant was carefully removed, filter sterilized with a 0.45-μm-pore-size syringe filter to remove tissue debris, and stored at −80°C prior to use. Cytokines were measured using a Bio-Rad Bioplex Pro 23-plex group I mouse cytokine kit and customized 5-plex kits (Bio-Rad, Hercules, CA) according to the manufacturer's protocol. The group I mouse kit measures the following cytokines and chemokines: interleukin-1α (IL-1α), IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12p40, IL-12p70, IL-13, IL-17A, eotaxin, granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), gamma interferon (IFN-γ), keratinocyte chemoattractant (KC), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1α (MIP-1α), MIP-1β, RANTES, and tumor necrosis factor alpha (TNF-α) (assay range, 0.74 pg/ml to 84 ng/ml). The protein concentration of each homogenate supernatant was measured by the bicinchoninic acid assay (Pierce). All homogenates were diluted to 500 μg of total protein in Bioplex sample buffer prior to analysis, and all cytokine concentrations are expressed as pg/ml. Only cytokines showing significant modulation between monomicrobial and polymicrobial infection were reported.
Histological analysis.
Formalin-fixed tissues were sent for histological preparation and were paraffin embedded, sliced, and stained with hematoxylin-eosin (H&E) or periodic acid-Schiff (PAS) (Morphology Imaging Core, LSUHSC). Slides were visualized by standard light microscopy.
Neutrophil analysis.
Neutrophils in both peritoneal lavage fluid and in the spleen and kidneys were identified and quantitated by differential staining and/or immunostaining. Recovered lavage fluid was centrifuged at 500 × g for 5 min at 4°C, and the cell pellet was resuspended in 1 ml PBS. The cells were centrifuged again as described above and finally resuspended in 500 μl PBS containing 5% FBS. Cells were adhered to Vectabond-treated (Vector Laboratories, Burlingame, CA) glass slides by cytospin (2-min spin at 1,000 rpm). Cells were allowed to air dry at room temperature and then immediately fixed in ice-cold acetone for 5 min. Slides were immediately stained or stored at −20°C until needed. Neutrophils were counted on prepared slides by staining with Diff-Quick reagent (IMEB Inc., San Marcos, CA) or immunostained with fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse lymphocyte antigen 6-G (Ly6G) antibody (Biolegend, San Diego, CA) and counterstained with DAPI (4′,6-diamidino-2-phenylindole; Vector Laboratories) for visualization of neutrophils as previously described (64). Similarly, unstained histological slices of kidneys and spleens were deparaffinized, washed, and immunostained with an Ly6G or isotype control antibody and counterstained with DAPI as described above. Slides were visualized by standard light microscopy, or fluorescence was captured on an Olympus Fluoview 1000 confocal microscope using a DAPI/FITC filter set. Ten random fields were examined, and the numbers of neutrophils were counted and averaged per animal.
Prostaglandin quantitation.
Recovered lavage fluid was centrifuged at 500 × g for 5 min, and the supernatant was transferred to clean microcentrifuge tubes. PGE2 is rapidly converted to its 13,14-dihydro-15-keto metabolite in vivo, so a colorimetric prostaglandin E metabolite competitive enzyme immunoassay (EIA) was used to measure PGE2 as a function of its breakdown product, according to the manufacturer's directions (Cayman Chemicals, Ann Arbor, MI). This assay is highly sensitive and detects as little as 2 pg/ml of PGE metabolites.
Statistics.
All experiments used groups of 5 mice and were repeated in duplicate. All assays were repeated in triplicate, and the results were averaged. For cell staining experiments, all slides were prepared from each animal in duplicate, and 10 fields were counted per slide. Overall mortality was determined as significant using Fisher's exact test if P was ≤0.05. Because the data generated from these studies were nonparametric (Shapiro-Wilks normality test, P < 0.05), the Mann-Whitney U test was used to analyze the significance of differences between the monomicrobial and polymicrobial infection groups. Statistical comparisons with a P value of ≤0.05 were considered statistically significant. Values were plotted or denoted as the median, and error bars represent interquartile ranges.
RESULTS
Coinfection enhances mortality and microbial burden.
Coinfection with multiple microbes has been shown to result in enhanced pathogenicity compared to infection with the same microbes individually (2, 4). Therefore, we have optimized a murine model of polymicrobial peritonitis to study the effects of C. albicans-S. aureus coinfection on host survival and the associated immune response. Mice infected with either C. albicans (Fig. 1, light gray) or S. aureus (Fig. 1, black) alone exhibited no mortality (100% survival) at up to 5 days postinoculation and appeared as healthy as the control group receiving only a saline injection (Fig. 1, white). However, coinoculation with the same doses of C. albicans and S. aureus (Fig. 1, striped) resulted in only 40% survival by day 2 postinoculation. Aside from mortality, coinfected mice also appeared moribund (inactivity, hunched posture) and exhibited conjunctivitis-like symptoms. Coinoculated mice received equivalent concentrations of each pathogen used in the monomicrobial inoculations, resulting in an increased total number of microbes compared to the number in those infected with either C. albicans or S. aureus alone. To account for this discrepancy, mice were inoculated with C. albicans (Fig. 1, dark gray) or S. aureus (Fig. 1, hashed) at the same total dose of organisms received during coinfection (8.7 × 107 CFU). These monomicrobial inoculations, using an increased microbial load, also resulted in no mortality at up to 5 days postinfection. Cumulatively, these results demonstrate that enhanced mortality in this model is attributable to inoculation with both pathogens (and not simply the increased microbial burden received during coinoculation) and occurs rapidly, within approximately 24 h of initial infection.
Fig 1.
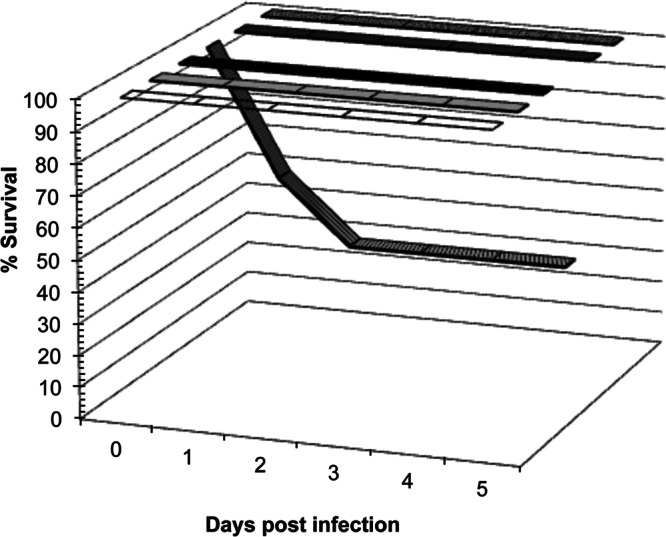
C. albicans-S. aureus coinfection results in mortality by day 1 postinoculation. Mice were injected intraperitoneally with 7 × 106 CFU of C. albicans (light gray bar), 8 × 107 CFU of S. aureus (black bar), or the same doses of each pathogen simultaneously (8.7 × 107 CFU total; striped bar). Mice injected with sterile saline only served as controls (white bar). Mice infected with 8.7 × 107 CFU of C. albicans alone (dark gray bar) or S. aureus alone (hashed bar) served as monomicrobial controls. Overall mortality in the coinfected group was found to be statistically significant using Fisher's exact test (P < 0.05; n = 10).
It has been well documented that both the kidney and spleen support the growth of C. albicans or S. aureus during monomicrobial systemic infection (29, 30). Thus, in order to assess the level of microbial burden associated with monomicrobial versus polymicrobial infection, we enumerated the CFU in the kidney and spleen by standard microbiological culture on differential medium. In mice infected with C. albicans alone, only 1 out of 10 mice had detectable CFU in the kidney (Fig. 2A, closed squares), and no detectable CFU was found in the spleen (Fig. 2B, closed squares). Mice infected with S. aureus alone had a median of 1 × 104 CFU in the kidney (Fig. 2A, closed triangles) and 1 × 104 CFU in the spleen (Fig. 2B, closed triangles). Strikingly, coinoculation with the same doses of C. albicans and S. aureus resulted in a dramatic increase in both C. albicans and S. aureus culture-positive kidney infections compared with monomicrobial inoculation (60% versus 10% and 80% versus 50%, respectively) (Fig. 2A). This corresponds to a median 4-log-unit increase of C. albicans (Fig. 2A, open squares; P < 0.05) and a median 1-log-unit increase of S. aureus (Fig. 2A, open triangles; P < 0.05) in the kidney. Culture-positive spleen infections also increased for both C. albicans and S. aureus in polymicrobial versus monomicrobial inoculations (40% versus 0% and 90% versus 60%, respectively) (Fig. 2B). CFU counts in the spleen were also significantly increased: a 4-log-unit increase for C. albicans (Fig. 2B, open squares; P < 0.05) and a 2-log-unit increase for S. aureus (Fig. 2B, open triangles; P < 0.05) were observed. Therefore, coinfection not only enhances morbidity and mortality but also leads to significant increases in the infection rates and microbial burden found in the kidneys and spleens compared to monomicrobial infection.
Fig 2.

Microbial burden is increased in the target organs during coinfection. Mice were inoculated and sacrificed 1 day later. (A and B) Quantitative counts of both C. albicans (CA) and S. aureus (SA) in kidneys (A) and spleens (B). Mono- and polyinfection groups were compared by the Mann-Whitney U test. *, P < 0.05; horizontal lines, medians. (C) Small staphylococcal abscesses were found growing among a dense network of C. albicans hyphae colonizing the spleen (H&E stain, arrows). Surface colonization of the kidney during polymicrobial infection demonstrates biofilm-like structures with S. aureus attached to a scaffolding of C. albicans hyphae (arrows). The images on the bottom row are magnifications of the boxed areas in the images above.
Histological analysis of these organs confirmed the increased microbial burden in the coinoculated animals, with some bacteria and fungi dispersed throughout the organs. In some cases, severe organ damage was noted; small clusters of staphylococci could be found intermingled among the hyphae of C. albicans (Fig. 2C, spleen). Interestingly, we detected an abundance of polymicrobial structures on the surface of the coinfected organs composed of a scaffolding of C. albicans hyphae with S. aureus (Fig. 2C, kidney) attached. Mice infected with C. albicans or S. aureus alone showed only limited numbers of microbes colonizing the organ surfaces. This indicates that during coinfection, both fungi and bacteria are intimately associated within infected tissues.
Coinfection modulates host innate immunity.
Because C. albicans and S. aureus are known to exhibit distinct immunomodulatory properties in vivo, we hypothesized that coinfection may result in altered or amplified host immune responses compared with monomicrobial infections (31–33). Using a multiplex enzyme-linked immunosorbent assay (ELISA), we simultaneously measured 23 chemokines and cytokines produced in the kidneys and spleens of mice at day 1 postinoculation. Of all 23 cytokines, only 5 showed significant increases in both the kidney (Fig. 3A) and spleen (Fig. 3B) during coinfection compared to monomicrobial infection: IL-6, G-CSF, KC, MCP-1, and MIP-1α. These increases were not incremental: IL-6 levels increased approximately 5-fold in the kidney and 100-fold in the spleen (P < 0.05), G-CSF levels increased ≈100-fold (P < 0.05) in both organs, KC levels increased ≈10-fold in the kidney and spleen (P < 0.05), MCP-1 levels increased ≈5-fold in the kidney and 10-fold in the spleen (P < 0.05), and MIP-1α levels increased ≈10-fold (P < 0.05) in both organs. It is important to note that coinfected mice received only 1.1-fold more organisms than mice infected with S. aureus alone, but the cytokine profiles were upregulated in excess of 5-fold, suggesting a synergistic increase in cytokine production during coinfection. Interestingly, all of these cytokines are involved in the innate proinflammatory response and can be activated within minutes in response to microbe exposure; thus, their induction fits within the relatively short time frame of polymicrobe-enhanced mortality (34). It is not surprising that any of the adaptive cytokines (including IL-4, IL-12, and IFN-γ) were not found to be significantly modulated during coinfection (data not shown), as coinfection-induced death happened before they were likely produced at detectable levels.
Fig 3.
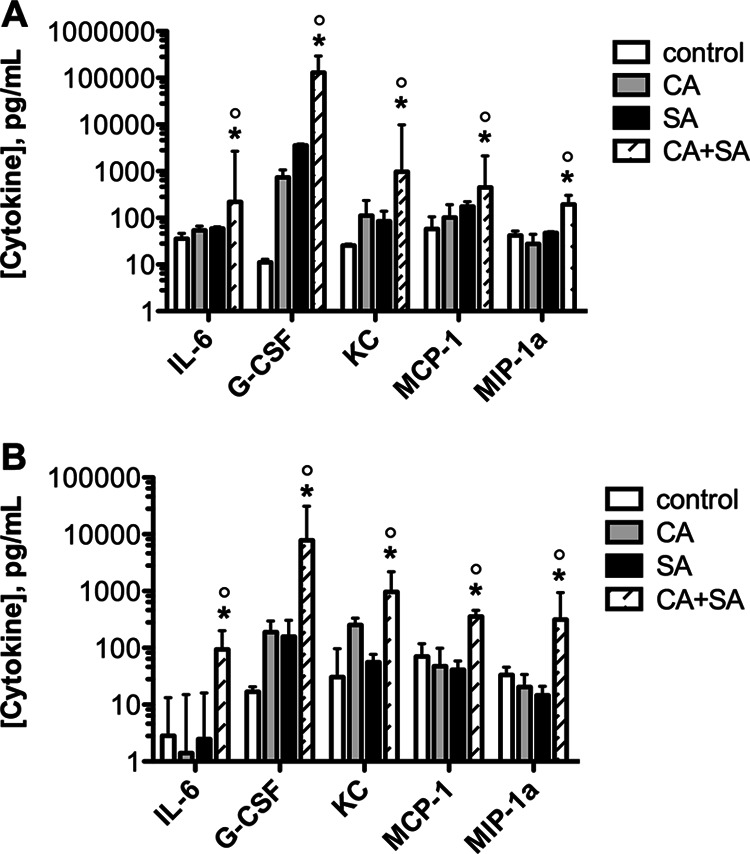
Coinfection with C. albicans and S. aureus modulates host innate immunity. Mice were inoculated and sacrificed 1 day later. The differential expression of 23 cytokines and chemokines in the (A) kidneys and (B) spleens of mice infected with C. albicans alone, S. aureus alone, or both pathogens (striped bar) is shown. Control mice were injected with sterile saline. Significant increases were found for IL-6, G-CSF, KC, MCP-1, and MIP-1α during polymicrobial versus monomicrobial infection. Values were plotted as the median and compared by the Mann-Whitney U test. *, P < 0.05 for C. albicans versus C. albicans plus S. aureus coinfection; °, P < 0.05 for S. aureus versus C. albicans plus S. aureus coinfection. Error bars represent interquartile ranges.
Several of the cytokines found to be upregulated during coinfection, including IL-6, G-CSF, and KC, are known to be involved in activation and recruitment of neutrophils to primary sites of infection (35). Therefore, we monitored neutrophil influx in peritoneal lavage samples at day 1 postinoculation. In uninfected mice, resident monocytes and macrophages predominate in the peritoneal cavity, with few polymorphonuclear leukocytes (PMNs) present, as shown by differential staining (Fig. 4A, control). However, PMNs can rapidly migrate and accumulate in the peritoneum in response to infection and chemical stimuli (36). Indeed, we observed both mature and immature PMNs in mice inoculated with either C. albicans or S. aureus alone (Fig. 4A, C. albicans and S. aureus, white arrows). However, there was a qualitative increase in the presence of PMNs in coinfected mice (Fig. 4A, C. albicans + S. aureus, white arrows). It is important to point out that mature PMNs have multilobed nuclei but developmentally immature PMNs have nuclei that appear bean shaped (metamyelocytes) or band-like (band neutrophils) (37). A significant portion of the peritoneal PMNs from the coinfected mice was of the band variety (Fig. 4A, C. albicans + S. aureus, enlarged panel), suggesting the presence of a potent inflammatory response with possible rapid mobilization of immature neutrophils from the circulation and bone marrow.
Fig 4.
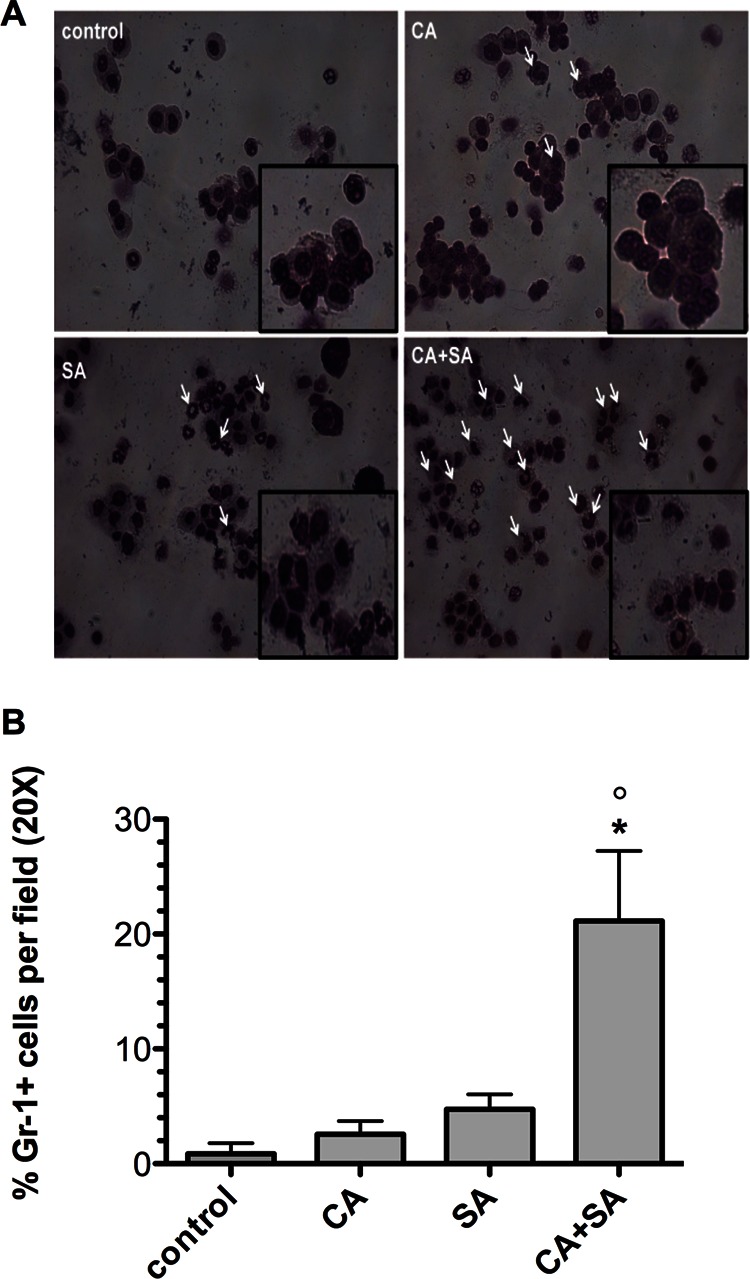
Coinfection results in both mature and immature neutrophil influx into the peritoneal cavity. At 1 day postinfection with C. albicans alone, S. aureus alone, or both pathogens, mice underwent a peritoneal lavage procedure. (A) Cells were stained with Diff-Quick reagent to identify cells containing granules and multilobed nuclei. White arrows, mature and immature (band-like) neutrophils recovered from the peritoneal cavity. (B) Cells were also immunostained with an FITC-conjugated Gr-1/Ly6G primary antibody and counterstained with DAPI to determine polymorphic nuclei. The numbers of Gr-1-positive cells per field were counted and calculated as the total percentage of cells enumerated. Values were plotted as the median. Infection groups were compared by the Mann-Whitney U test. *, P < 0.05 for C. albicans versus C. albicans plus S. aureus coinfection; °, P < 0.05 for S. aureus versus C. albicans plus S. aureus coinfection. Error bars represent interquartile ranges.
In order to more quantitatively assess the number of PMNs in the lavage fluid, we immunostained recovered lavage cells with an FITC-conjugated primary antibody specific for mouse granulocyte differentiation antigen-1/lymphocyte antigen 6-G (Gr-1/Ly6G). Gr-1 is primarily expressed by neutrophils and serves as the hallmark cell surface marker for identifying this cell population in inflammatory infiltrates (38). Gr-1-positive polymorphonuclear cells were expressed as the total number of cells per field. Nearly 20% of the recovered peritoneal cells were polymorphonuclear and Gr-1 positive in coinfected mice, whereas the proportions were 2.5% in mice infected with C. albicans alone and 5% in mice infected with S. aureus alone (Fig. 4B); these results were statistically significant (P < 0.05). Total numbers of cells were similar among infection groups (data not shown). In addition to the peritoneal influx of neutrophils, an increased number of PMNs was detected in the tissues of coinfected mice compared to singly infected mice (data not shown). Although significant numbers of PMNs are recruited to the target organs during coinfection, they are not protective (as demonstrated by increased levels of infection and microbial burden in the kidney and spleen during coinfection; Fig. 2A and B) and may in fact potentially contribute to enhanced morbidity, increased mortality, and polymicrobial pathogenesis.
Inflammatory events during early infection.
The increased inflammatory cytokines observed in the target organs during coinfection may have been due to the increased microbial load at 24 h postinfection. Therefore, we sought to determine whether the same cytokines would be upregulated at an early time point (8 h) in the spleen, when CFU levels may be more equal between the monomicrobial and polymicrobial infection groups. Indeed, CFU levels were not significantly different for C. albicans (Fig. 5A, closed versus open squares) or S. aureus (Fig. 5A, closed versus open triangles) between the monomicrobial or polymicrobial infection groups. On the other hand, while they were not as dramatically increased as they were at the 24-h time point, cytokine levels were still significantly higher (∼4- to 10-fold) in the polymicrobial group than the monomicrobial group at 8 h (Fig. 5B; P < 0.05).
Fig 5.
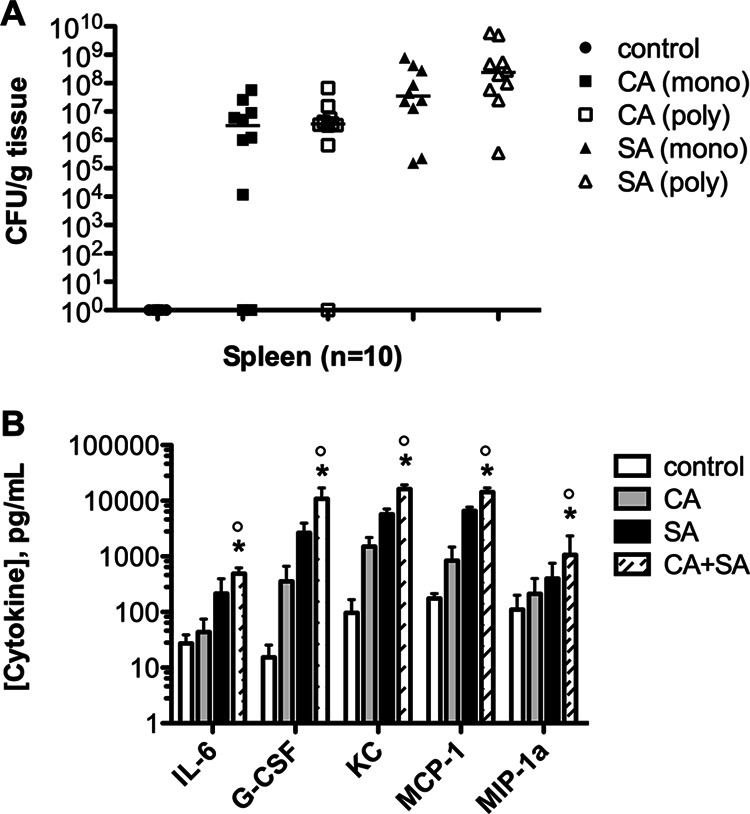
Microbial burden and cytokine production early during infection. (A) Microbial burden between monomicrobial and polymicrobial infections for both organisms in the spleen at 8 h; (B) levels of the 5 cytokines previously identified were assessed in the spleen at 8 h postinoculation. Control mice were injected with sterile saline. Values were plotted as the median and compared by the Mann-Whitney U test. *, P < 0.05 for C. albicans versus C. albicans plus S. aureus coinfection; °, P < 0.05 for S. aureus versus C. albicans plus S. aureus coinfection. Error bars represent interquartile ranges.
Indomethacin protects against the effects of polymicrobial disease.
A previous study demonstrated that indomethacin was partially protective against S. aureus-C. albicans coinfection but failed to identify the protective mechanism (39). Because coinfected mice respond with synergistic increases in innate proinflammatory cytokine production and the influx of nonprotective mature and immature neutrophils, we hypothesized that treatment of these mice with the nonsteroidal anti-inflammatory drug (NSAID) indomethacin may reduce aggressive inflammation and protect against morbidity and mortality. Indeed, administration of 5 mg/kg indomethacin at 4 h preinoculation and 8 h postinoculation protected mice from coinfection-induced mortality, with 100% survival at 2 days postinoculation (Fig. 6A); remarkably, these mice showed no signs of morbidity, appearing healthy throughout infection. In contrast, mice treated with vehicle succumbed to infection rapidly, with 60% mortality at 2 days postinoculation. Importantly, the in vitro growth of both C. albicans and S. aureus in the presence of indomethacin was not inhibited at the physiologically relevant concentrations used in these studies (Fig. 6B). Strikingly, indomethacin had a significant effect on reducing infection rates and the microbial burden in the target organs. Indomethacin treatment significantly reduced C. albicans CFU counts in the kidneys of coinfected mice by nearly 5 log units (Fig. 7A, open inverted triangles; P < 0.05) and nearly 4 log units in the spleen (Fig. 7B, open inverted triangles; P < 0.05). In addition, compared with untreated mice, the percentages of indomethacin-treated mice with detectable C. albicans in kidneys and spleens were dramatically reduced (80% versus 20% and 100% versus 20%, respectively). Similarly, S. aureus CFU counts were also significantly reduced by 3 log units in the kidney (Fig. 7A, open diamonds; P < 0.05) and 6 log units in the spleen (Fig. 7B, open diamonds; P < 0.05).
Fig 6.
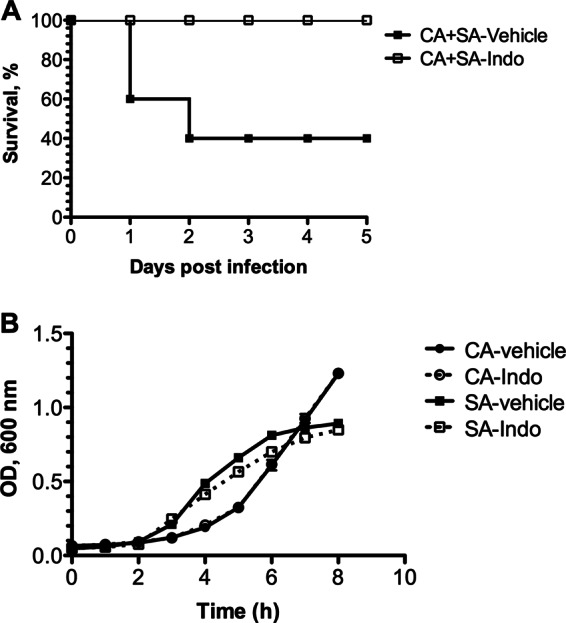
Effect of indomethacin (Indo) on survival and microbial growth. (A) Mice received either vehicle or indomethacin at a dose of 5 mg/kg by i.p. injection 4 h prior to and 8 h after polymicrobial infection. Mortality was assessed for up to 5 days postinfection. Overall mortality was significant using Fisher's exact test (P < 0.05; n = 10). (B) Growth of C. albicans and S. aureus was monitored for up to 8 h in medium supplemented with vehicle alone or indomethacin at physiologically relevant concentrations. OD, optical density.
Fig 7.

Microbial burden and cytokine levels in the target organs during coinfection are decreased by indomethacin administration. Mice were singly or dually infected; control mice were injected with sterile saline only. All mice received indomethacin at a dose of 5 mg/kg or vehicle 4 h prior to and 8 h after infection and sacrificed at 1 day postinoculation. (A and B) Quantitative counts of C. albicans and S. aureus in kidneys (A) and spleens (B) during infection; (C and D) cytokine levels in the kidneys (C) and spleens (D) of coinfected mice treated with indomethacin or vehicle. Infection groups were compared by the Mann-Whitney U test. *, P < 0.05. Horizontal bars and values were plotted as the medians; error bars represent interquartile ranges.
Indomethacin administration also significantly reduced the upregulated proinflammatory cytokine production observed in coinfected mice. A significant reduction of G-CSF and MIP-1α (Fig. 7C, open bars) was achieved in the kidney, while IL-6, G-CSF, and MIP-1α (Fig. 7D, open bars) were significantly reduced in the spleen. The reduction in proinflammatory cytokines was also accompanied by significant decreases in neutrophil infiltrate in the peritoneum and target organs of indomethacin-treated mice. The numbers of Gr-1-positive cells recovered from the peritoneal lavage fluid of indomethacin-treated animals were significantly reduced (P < 0.05) compared to those from animals that received vehicle only (Fig. 8A). The numbers of C. albicans and S. aureus organisms found embedded and on the surface of coinfected organs was greatly reduced compared to those for the organs of mice treated with vehicle alone (Fig. 8B). Interestingly, indomethacin treatment did not have any effect on reducing the microbial burden or infection rates in monomicrobial infections (Fig. 2A and B versus Fig. 7A and B). Together, these results suggest that indomethacin has no direct antimicrobial effect but, rather, indirectly affects the severity of infection by modulating inflammatory responses during polymicrobial infection.
Fig 8.
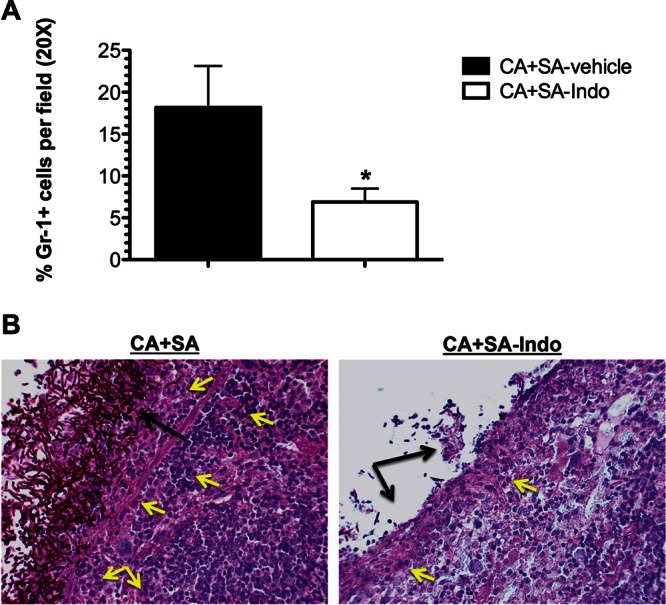
Indomethacin treatment reduces neutrophil influx into the peritoneal cavity and target organs. Mice were dually infected with C. albicans and S. aureus. All mice received either indomethacin at a dose of 5 mg/kg or vehicle 4 h prior to and 8 h after infection and underwent peritoneal lavage immediately following sacrifice at 1 day postinoculation. (A) The numbers of Gr-1-positive polymorphonuclear cells per field were counted and calculated as the total percentage of cells enumerated. Infection groups were compared by the Mann-Whitney U test. *, P < 0.05. Error bars represent interquartile ranges. (B) Periodic acid-Schiff-stained spleen tissue obtained from coinfected mice receiving vehicle (left) or indomethacin (right). Yellow arrows, neutrophils; black arrows, microbial burden.
The NSAID indomethacin is a nonselective cyclooxygenase (COX) inhibitor, inhibiting both constitutive COX-1 and inducible COX-2 enzymatic conversion of free arachidonic acid into prostaglandin H2, a precursor of the type 2 prostaglandins, including the highly immunomodulatory oxylipin PGE2 (40). In particular, PGE2 has been shown to induce inflammatory cytokines, including IL-6, induced by cell wall components of C. albicans or S. aureus (41, 42). Therefore, we hypothesized that polymicrobial infection may induce exacerbated inflammatory responses by induction of PGE2. Therefore, we measured PGE2 production in monomicrobial and polymicrobial infections. Because PGE2 is rapidly degraded in vivo, we utilized a competitive EIA to quantify its stable breakdown product colorimetrically in the peritoneal lavage fluid of infected mice. Coinfected mice demonstrated a >2-fold increase (P < 0.05) in PGE2 production compared to monomicrobially infected mice (Fig. 9). As expected, coinfected mice treated with indomethacin demonstrated a significantly reduced level of PGE2 in the lavage fluid compared to mice receiving vehicle alone (Fig. 9). This indicates that PGE2 may be involved in induction of the nonprotective proinflammatory responses observed during polymicrobial infections.
Fig 9.
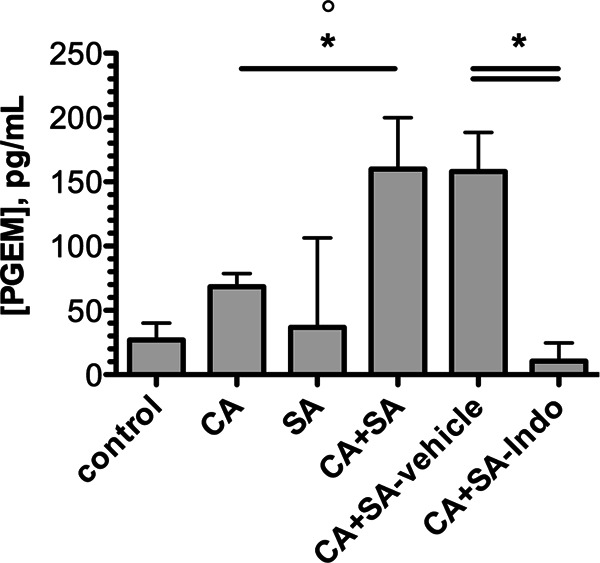
Indomethacin abrogates coinfection-mediated synergistic increases in PGE2 synthesis in the peritoneal cavity. Mice were infected with C. albicans alone, S. aureus alone, or a combination of both pathogens. Control mice were injected with sterile saline only. For experiments testing the efficacy of cyclooxygenase inhibition, coinfected mice received indomethacin at a dose of 5 mg/kg or vehicle alone at 4 h prior to and 8 h after infection. At 1 day after infection, mice were euthanized and immediately underwent peritoneal lavage. Values are plotted as the median. *, P < 0.05 for C. albicans versus C. albicans plus S. aureus coinfection; °, P < 0.05 for S. aureus versus C. albicans plus S. aureus coinfection. Error bars represent interquartile ranges. PGEM, prostaglandin E2 metabolite.
To further test this hypothesis, mice were administered indomethacin during polymicrobial or mock infection as described above. In addition, the same mice were also administered 1 μg exogenous PGE2 or PBS intraperitoneally and followed for 5 days postinfection. Strikingly, 40% of coinfected mice given PGE2 exhibited signs of morbidity and succumbed to infection within 2 days (Fig. 10A, closed circles). Importantly, mice given PGE2 but inoculated only with sterile saline remained healthy (Fig. 10A, closed squares). Mice receiving only indomethacin were completely protected from coinfection-induced death, as shown earlier (Fig. 10A, open circles). PGE2 levels were also monitored in the peritoneal lavage fluid at 1 day postinfection. As expected, PGE2 levels were significantly increased in mice receiving exogenous PGE2 (Fig. 10B, black bar) compared to controls receiving PBS (Fig. 10B, open bar). This was specific to coinfection, as mice receiving PGE2 in the absence of infection exhibited reduced PGE2 levels (Fig. 10B, gray bar). Importantly, PGE2 administration during coinfection also significantly increased both the C. albicans and S. aureus microbial burden in the kidney (Fig. 10C) and spleen (Fig. 10D).
Fig 10.

Addition of PGE2 in the peritoneal cavity increases mortality and microbial burden. All mice were administered indomethacin 4 h before or 8 h after polymicrobial infection. At 1 h prior to and 9 h after infection, mice were administered 1 μg of PGE2 or vehicle by i.p. injection. Uninfected mice receiving both indomethacin and PGE2 served as controls. (A) Survival of mice was significantly decreased in the group receiving PGE2 compared to the group receiving vehicle alone (Fisher's exact test, P < 0.05; n = 10). (B) PGE2 levels were measured by competitive EIA at 1 day postinoculation; groups were compared by the Mann-Whitney U test. *, P < 0.05. Values are plotted as medians. Error bars represent interquartile ranges. (C and D) Microbial burdens of both C. albicans and S. aureus in the kidney (C) and spleen (D) of mice receiving PGE2 (Indo+PGE2) or vehicle (Indo) at 1 day postinoculation. The poly- and monoinfection groups were compared by the Mann-Whitney U test. *, P < 0.05; horizontal lines, medians.
DISCUSSION
The reported incidence of polymicrobial infections likely vastly underrepresents the actual numbers, as routine clinical microbiological analysis does not include screening for hundreds of potential pathogens. As the identification of microbial species through both culture-dependent and -independent techniques becomes more feasible, the number of human diseases labeled polymicrobial in nature will likely increase. Even infections attributed to a single microbial species originate from or are exposed to a complex microbiome (1). Despite this, we still understand relatively little about the dynamics within these microbial communities. Furthermore, even less is known about how these interactions impact human disease in the context of host immunity.
Our studies using a murine model of peritonitis demonstrate that coinfection with two common opportunistic pathogens, C. albicans and S. aureus, results in infectious synergism, as reported previously (24, 43). While no morbidity or mortality was observed with monomicrobial infections, we observed both clinical signs of infection and 60% mortality by day 2 postinoculation. In coinfected mice, morbidity and mortality were associated with both an increased microbial burden and increased rates of infection in the target organs of coinfected mice compared to mice infected with C. albicans or S. aureus alone (Fig. 2A and B). It should be pointed out that while previous studies of Carlson (39) achieved a 100% mortality rate during coinfection, the mortality rate determined in our studies was 60% by day 2 (Fig. 1). There could be several factors explaining this discrepancy, including the strains of mice (CD-1 versus Swiss Webster) or the toxigenic repertoire of the S. aureus isolate used. This is an important point to highlight in our peritonitis model, as not all coinfected mice equally succumbed to infection. While not significantly correlated, there was a trend in which mice demonstrating higher microbial burdens also had higher levels of cytokines. Therefore, the aggressive inflammatory response that we observed in our model may be initiated by an inability of the host to adequately clear the coinfection. Additional experiments are ongoing to further dissect the mechanisms behind polymicrobial synergy.
We also identified structures on the surface of the target organs of coinfected mice (Fig. 2C) that looked very similar to C. albicans-S. aureus in vitro polymicrobial biofilms, with both single staphylococci and staphylococcal microabscesses being preferentially associated with the hyphae of C. albicans (21). We should also mention that organisms were retrieved from infected tissues in a variety of configurations, including infectious foci composed entirely of C. albicans or S. aureus, even during coinfection. However, there was a high propensity for these microbes to associate during polymicrobial peritonitis. Although it is unclear if this association is relevant to an enhanced disease phenotype during polymicrobial peritoneal infection, it is known that several staphylococcal and fungal virulence factors are modulated during coculture growth, including production of the CodY protein, which regulates nutrient acquisition and toxin production (22). Aside from virulence modulation, hypha-associated S. aureus cells may be more difficult for host neutrophils and macrophages to engulf, as Candida hyphae are known to be more resistant to opsonophagocytosis (44). Similarly, staphylococcal production of leukocidins and pore-forming toxins may protect C. albicans from antifungal effector cells (45). Therefore, differentially expressed virulence proteins, as well as the physical association of microbes, may contribute to host immunomodulatory function.
Our studies also demonstrated that coinfection was associated with an acute aggressive inflammatory response, characterized by 5- to 100-fold increases in proinflammatory cytokines and a dramatic influx of neutrophils that were unable to control the infection. Analysis of an early time point during infection demonstrated that the synergistically increased cytokine production during polymicrobial infection compared to the level of production during monomicrobial infection occurred independently of the microbial burden (Fig. 5). Therefore, coinfection-specific host factors or microbe-microbe interactions may potentiate the highly inflammatory phenotype observed. This acute increase in proinflammatory cytokine production, frequently described as a cytokine storm, can result in increased leukopenia, deviated body temperature, increased band neutrophilia, tachycardia, and tachypnea and lead to the catastrophic failure of multiple organ systems, respiratory distress syndrome, central nervous system damage, and gastrointestinal bleeding.
The cytokines found to be most highly upregulated in polymicrobial peritonitis compared to the level of expression during monomicrobial peritonitis were IL-6, G-CSF, KC, MCP-1, and MIP-1α (Fig. 3A and B). IL-6 is an innate proinflammatory cytokine produced by lymphocytes, macrophages, and endothelial cells. High levels of IL-6, also found during human sepsis, strongly correlate with enhanced mortality, while blockade of this cytokine has resulted in protection in murine models of CLP peritonitis (46, 47). Several upregulated cytokines are involved in recruitment and activation of effector cells (KC, MCP-1, MIP-1α, G-CSF) and amplification of inflammatory cytokine production (MIP-1α). KC is a chemokine that plays an important role in the chemotaxis and activation of neutrophils, and its upregulation may partly explain the increased neutrophilia observed in coinfected mice (Fig. 4A and B). The murine KC receptor is homologous to the human IL-8 receptor, and KC, along with MIP-2, is thought to serve as the equivalent of human IL-8; importantly, IL-8 levels are known to be increased during human sepsis (48, 49). Interestingly, polymicrobial infection of nasal epithelial surfaces in vitro with the bacterial pathogens Streptococcus pneumoniae and Haemophilus influenzae results in synergistic production of IL-8 that is dependent on cytolytic toxin activity (50). Because S. aureus possesses a number of toxins, it is possible that this could partially account for the synergistic increases in cytokine production observed in our peritonitis model. The most highly modulated cytokine that was synergistically increased during polymicrobial peritonitis was G-CSF (Fig. 3A and B). Upon pathogen encounter, endothelial cells and macrophages release G-CSF locally, and G-CSF then reaches the bone marrow via systemic circulation. Stimulation of the G-CSF receptor on hematopoietic precursor cells induces proliferation and differentiation of granulocytic progenitors as well as their mobilization to sites of infection (51). Therefore, the increases in both mature and band neutrophils detected during polymicrobial infection could be the result of highly increased production of G-CSF (Fig. 4A).
Bosmann et al., using the same 23-plex ELISA employed in our study, recently found that mice undergoing CLP most highly produced G-CSF, IL-6, KC, MCP-1, and IL-10 compared to the levels produced by controls that underwent sham surgery; these results are strikingly similar to the cytokine results described in this study (52). In addition, human sepsis results in the upregulation of a number of similar proinflammatory cytokines, including IL-6, IL-8, MCP-1, and MIP-1α (53). This begs the question of whether the upregulation of these cytokines is specific to C. albicans-S. aureus infections or whether they are simply activated in response to any polymicrobial peritoneal infection, possibly via simultaneous signaling of innate immunity receptors. Early detection of simultaneous increases in this panel of cytokines may aid in the more rapid diagnosis of polymicrobial infection, guiding appropriate selection of antimicrobial and immunomodulatory therapies, compared to the time to diagnosis by the currently poor means of identification of polymicrobial diseases through conventional culturing techniques (54).
Carlson previously demonstrated that administration of the NSAID indomethacin protected mice from mortality associated with C. albicans-S. aureus polymicrobial peritonitis but never assessed the CFU burden or correlates of protection (39). Indeed, we demonstrated that prophylactic and continued treatment of mice with indomethacin prevents mortality after coinfection without causing any negative consequences for animals infected with either pathogen alone (data not shown). This protection can be negated by delivery of exogenous PGE2 into the peritoneal cavity during coinfection (Fig. 10). Indomethacin, like aspirin and ibuprofen, is a nonselective inhibitor of the COX enzymes; this inhibition impairs production of multiple downstream eicosanoids that are involved in controlling a range of physiologic processes in the host, including pro- and anti-inflammatory responses (55, 56). In our studies, indomethacin inhibited induction of PGE2 during polymicrobial infection, which correlated with significant reductions in several of the proinflammatory cytokines identified to be synergistically increased during polymicrobial infection compared to their level during monomicrobial infection (Fig. 7C and D and 9).
Indomethacin administration also reduced neutrophil infiltration into the peritoneal cavity and target organs (Fig. 8). Previous studies have identified that PGE2 can positively regulate expression of IL-6, KC, G-CSF, and MIP-1α during peritonitis (57–59). Using a casein-induced model of peritonitis, Sugimoto et al. demonstrated that the amounts of PGE2 are significantly increased in the peritoneal cavity (65). This PGE2 increase correlated well with increased G-CSF expression, followed by infiltration of neutrophils into the peritoneal cavity; significantly smaller amounts of PGE2, G-CSF, and neutrophils were found in the peritoneal cavity after administration of indomethacin.
Aside from regulation of pro- and anti-inflammatory cytokine production, PGE2 also has a direct effect on cells of the myeloid lineage. Several studies have shown that PGE2 can decrease the phagocytic capacity of peritoneal macrophages and inhibit the antimicrobial activity (respiratory burst and superoxide generation) of neutrophils (60–62). In some of these cases, normal defense activity could be restored by treatment with indomethacin. Because macrophages are the main cell type resident in the peritoneal cavity, the dramatically increased prostaglandin levels found during coinfection may inhibit this initial phagocytic defense mechanism, allowing colonization and subsequent systemic spread of both C. albicans and S. aureus. Likewise, recruited neutrophils may be rendered ineffective at resolving the infection, further perpetuating polymicrobial pathogenesis.
A highly relevant study recently conducted by von Moltke et al. demonstrated that mice injected intraperitoneally with purified Toll-like receptor agonists or anthrax toxin rapidly (30 min) succumbed to hypovolemic shock as a result of massive prostaglandin and leukotriene release in an inflammasome-dependent manner; the authors dubbed this phenomenon the “eicosanoid storm” (63). Knockout mice lacking functional COX-1 are resistant to these rapid-acting systemic inflammatory stimuli. It was further shown that resident peritoneal macrophages are uniquely primed to precipitously process and release inflammatory eicosanoids. Therefore, targeting PGE2 via inhibition of COX enzymes may be the lynchpin to blocking excessive proinflammatory cytokine production, neutrophil influx, and macrophage dysfunction during acute infectious episodes accompanied by pathological eicosanoid storms.
In summary, these studies have defined the cytokine response to C. albicans-S. aureus coinfection of the murine peritoneal cavity and provide insight into the mechanisms which regulate host innate immunity during simultaneous infection with multiple microbes. While we know that proinflammatory signaling via COX-mediated pathways is important for immunopathogenesis during coinfection, we still do not adequately understand the mechanism of how or why these inflammatory events are aggressively engaged. It is possible that the physical associations or the modulation of virulence proteins during coinfection aberrantly and/or synergistically stimulates nonprotective immune responses; additional studies are warranted and under way in our laboratory. Furthermore, the findings from this investigation will contribute to our understanding of the complex and clinically significant interactions that take place between host and microbe in the context of polymicrobial disease and may aid in the future development of improved diagnostic and therapeutic strategies for clinically relevant coinfections.
ACKNOWLEDGMENTS
We kindly thank Aaron Mitchell (Carnegie Mellon University) for C. albicans strain DAY185 and the NARSA repository for S. aureus strain NRS383 used in this study.
These studies were funded by the National Institutes of Health, National Institute of Allergy and Infectious Diseases, grant R01-A172406.
Footnotes
Published ahead of print 1 April 2013
REFERENCES
- 1. Peters BM, Jabra-Rizk MA, O'May GA, Costerton JW, Shirtliff ME. 2012. Polymicrobial interactions: impact on pathogenesis and human disease. Clin. Microbiol. Rev. 25:193–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Braun LE, Sutter DE, Eichelberger MC, Pletneva L, Kokai-Kun JF, Blanco JC, Prince GA, Ottolini MG. 2007. Co-infection of the cotton rat (Sigmodon hispidus) with Staphylococcus aureus and influenza A virus results in synergistic disease. Microb. Pathog. 43:208–216 [DOI] [PubMed] [Google Scholar]
- 3. Dowd SE, Wolcott RD, Sun Y, McKeehan T, Smith E, Rhoads D. 2008. Polymicrobial nature of chronic diabetic foot ulcer biofilm infections determined using bacterial tag encoded FLX amplicon pyrosequencing (bTEFAP). PLoS One 3:e3326 doi:10.1371/journal.pone.0003326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kuriyama T, Nakagawa K, Kawashiri S, Yamamoto E, Nakamura S, Karasawa T. 2000. The virulence of mixed infection with Streptococcus constellatus and Fusobacterium nucleatum in a murine orofacial infection model. Microbes Infect. 2:1425–1430 [DOI] [PubMed] [Google Scholar]
- 5. Sibley CD, Duan K, Fischer C, Parkins MD, Storey DG, Rabin HR, Surette MG. 2008. Discerning the complexity of community interactions using a Drosophila model of polymicrobial infections. PLoS Pathog. 4:e1000184 doi:10.1371/journal.ppat.1000184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barraclough K, Hawley CM, McDonald SP, Brown FG, Rosman JB, Wiggins KJ, Bannister KM, Johnson DW. 2010. Polymicrobial peritonitis in peritoneal dialysis patients in Australia: predictors, treatment, and outcomes. Am. J. Kidney Dis. 55:121–131 [DOI] [PubMed] [Google Scholar]
- 7. Saini S, Gupta N, Aparna, Lokveer, Griwan MS. 2004. Surgical infections: a microbiological study. Braz. J. Infect. Dis. 8:118–125 [DOI] [PubMed] [Google Scholar]
- 8. Heemken R, Gandawidjaja L, Hau T. 1997. Peritonitis: pathophysiology and local defense mechanisms. Hepatogastroenterology 44:927–936 [PubMed] [Google Scholar]
- 9. Kiernan L, Finkelstein FO, Kliger AS, Gorban-Brennan N, Juergensen P, Mooraki A, Brown E. 1995. Outcome of polymicrobial peritonitis in continuous ambulatory peritoneal dialysis patients. Am. J. Kidney Dis. 25:461–464 [DOI] [PubMed] [Google Scholar]
- 10. Kim GC, Korbet SM. 2000. Polymicrobial peritonitis in continuous ambulatory peritoneal dialysis patients. Am. J. Kidney Dis. 36:1000–1008 [DOI] [PubMed] [Google Scholar]
- 11. Szeto CC, Chow KM, Wong TY, Leung CB, Li PK. 2002. Conservative management of polymicrobial peritonitis complicating peritoneal dialysis—a series of 140 consecutive cases. Am. J. Med. 113:728–733 [DOI] [PubMed] [Google Scholar]
- 12. Carneiro HA, Mavrakis A, Mylonakis E. 2011. Candida peritonitis: an update on the latest research and treatments. World J. Surg. 35:2650–2659 [DOI] [PubMed] [Google Scholar]
- 13. Letsios A. 2011. The effect of the expenditure increase in the morbidity and the mortality of patients with end stage renal disease: the USA case. Hippokratia 15:16–21 [PMC free article] [PubMed] [Google Scholar]
- 14. Cahill RA, Wang JH, Redmond HP. 2007. Enteric bacteria and their antigens may stimulate postoperative peritoneal adhesion formation. Surgery 141:403–410 [DOI] [PubMed] [Google Scholar]
- 15. Karantonis FF, Nikiteas N, Perrea D, Vlachou A, Giamarellos-Bourboulis EJ, Tsigris C, Kostakis A. 2008. Evaluation of the effects of laparotomy and laparoscopy on the immune system in intra-abdominal sepsis—a review. J. Investig. Surg. 21:330–339 [DOI] [PubMed] [Google Scholar]
- 16. Ozmen MM, Col C, Aksoy AM, Tekeli FA, Berberoglu M. 1999. Effect of CO(2) insufflation on bacteremia and bacterial translocation in an animal model of peritonitis. Surg. Endosc. 13:801–803 [DOI] [PubMed] [Google Scholar]
- 17. Levinson AT, Casserly BP, Levy MM. 2011. Reducing mortality in severe sepsis and septic shock. Semin. Respir. Crit. Care Med. 32:195–205 [DOI] [PubMed] [Google Scholar]
- 18. Burke M, Hawley CM, Badve SV, McDonald SP, Brown FG, Boudville N, Wiggins KJ, Bannister KM, Johnson DW. 2011. Relapsing and recurrent peritoneal dialysis-associated peritonitis: a multicenter registry study. Am. J. Kidney Dis. 58:429–436 [DOI] [PubMed] [Google Scholar]
- 19. Al-Fattani MA, Douglas LJ. 2006. Biofilm matrix of Candida albicans and Candida tropicalis: chemical composition and role in drug resistance. J. Med. Microbiol. 55:999–1008 [DOI] [PubMed] [Google Scholar]
- 20. Resch A, Rosenstein R, Nerz C, Gotz F. 2005. Differential gene expression profiling of Staphylococcus aureus cultivated under biofilm and planktonic conditions. Appl. Environ. Microbiol. 71:2663–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harriott MM, Noverr MC. 2009. Candida albicans and Staphylococcus aureus form polymicrobial biofilms: effects on antimicrobial resistance. Antimicrob. Agents Chemother. 53:3914–3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peters BM, Jabra-Rizk MA, Scheper MA, Leid JG, Costerton JW, Shirtliff ME. 2010. Microbial interactions and differential protein expression in Staphylococcus aureus-Candida albicans dual-species biofilms. FEMS Immunol. Med. Microbiol. 59:493–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shirtliff ME, Peters BM, Jabra-Rizk MA. 2009. Cross-kingdom interactions: Candida albicans and bacteria. FEMS Microbiol. Lett. 299:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carlson E. 1982. Synergistic effect of Candida albicans and Staphylococcus aureus on mouse mortality. Infect. Immun. 38:921–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dejager L, Pinheiro I, Dejonckheere E, Libert C. 2011. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. 19:198–208 [DOI] [PubMed] [Google Scholar]
- 26. Rittirsch D, Hoesel LM, Ward PA. 2007. The disconnect between animal models of sepsis and human sepsis. J. Leukoc. Biol. 81:137–143 [DOI] [PubMed] [Google Scholar]
- 27. Davis D, Edwards JE, Jr, Mitchell AP, Ibrahim AS. 2000. Candida albicans RIM101 pH response pathway is required for host-pathogen interactions. Infect. Immun. 68:5953–5959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Donegan K, Matyac C, Seidler R, Porteous A. 1991. Evaluation of methods for sampling, recovery, and enumeration of bacteria applied to the phylloplane. Appl. Environ. Microbiol. 57:51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brieland J, Essig D, Jackson C, Frank D, Loebenberg D, Menzel F, Arnold B, DiDomenico B, Hare R. 2001. Comparison of pathogenesis and host immune responses to Candida glabrata and Candida albicans in systemically infected immunocompetent mice. Infect. Immun. 69:5046–5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kokai-Kun JF, Chanturiya T, Mond JJ. 2007. Lysostaphin as a treatment for systemic Staphylococcus aureus infection in a mouse model. J. Antimicrob. Chemother. 60:1051–1059 [DOI] [PubMed] [Google Scholar]
- 31. Lee LY, Miyamoto YJ, McIntyre BW, Hook M, McCrea KW, McDevitt D, Brown EL. 2002. The Staphylococcus aureus Map protein is an immunomodulator that interferes with T cell-mediated responses. J. Clin. Invest. 110:1461–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin L, Ibrahim AS, Xu X, Farber JM, Avanesian V, Baquir B, Fu Y, French SW, Edwards JE, Jr, Spellberg B. 2009. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog. 5:e1000703 doi:10.1371/journal.ppat.1000703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Noverr MC, Noggle RM, Toews GB, Huffnagle GB. 2004. Role of antibiotics and fungal microbiota in driving pulmonary allergic responses. Infect. Immun. 72:4996–5003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. MacCallum DM, Castillo L, Brown AJ, Gow NA, Odds FC. 2009. Early-expressed chemokines predict kidney immunopathology in experimental disseminated Candida albicans infections. PLoS One 4:e6420 doi:10.1371/journal.pone.0006420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ebong SJ, Call DR, Bolgos G, Newcomb DE, Granger JI, O'Reilly M, Remick DG. 1999. Immunopathologic responses to non-lethal sepsis. Shock 12:118–126 [DOI] [PubMed] [Google Scholar]
- 36. Li FK, Davenport A, Robson RL, Loetscher P, Rothlein R, Williams JD, Topley N. 1998. Leukocyte migration across human peritoneal mesothelial cells is dependent on directed chemokine secretion and ICAM-1 expression. Kidney Int. 54:2170–2183 [DOI] [PubMed] [Google Scholar]
- 37. Berliner N, Hsing A, Graubert T, Sigurdsson F, Zain M, Bruno E, Hoffman R. 1995. Granulocyte colony-stimulating factor induction of normal human bone marrow progenitors results in neutrophil-specific gene expression. Blood 85:799–803 [PubMed] [Google Scholar]
- 38. Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. 2008. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 83:64–70 [DOI] [PubMed] [Google Scholar]
- 39. Carlson EC. 1988. Synergism of Candida albicans and delta toxin producing Staphylococcus aureus on mouse mortality and morbidity: protection by indomethacin. Zentralbl. Bakteriol. Mikrobiol. Hyg. A 269:377–386 [DOI] [PubMed] [Google Scholar]
- 40. Wallace JL. 1999. Distribution and expression of cyclooxygenase (COX) isoenzymes, their physiological roles, and the categorization of nonsteroidal anti-inflammatory drugs (NSAIDs). Am. J. Med. 107:11S–16S [DOI] [PubMed] [Google Scholar]
- 41. Chen BC, Liao CC, Hsu MJ, Liao YT, Lin CC, Sheu JR, Lin CH. 2006. Peptidoglycan-induced IL-6 production in RAW 264.7 macrophages is mediated by cyclooxygenase-2, PGE2/PGE4 receptors, protein kinase A, I kappa B kinase, and NF-kappa B. J. Immunol. 177:681–693 [DOI] [PubMed] [Google Scholar]
- 42. Smeekens SP, van de Veerdonk FL, van der Meer JW, Kullberg BJ, Joosten LA, Netea MG. 2010. The Candida Th17 response is dependent on mannan- and beta-glucan-induced prostaglandin E2. Int. Immunol. 22:889–895 [DOI] [PubMed] [Google Scholar]
- 43. Carlson E. 1983. Effect of strain of Staphylococcus aureus on synergism with Candida albicans resulting in mouse mortality and morbidity. Infect. Immun. 42:285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. d'Ostiani CF, Del Sero G, Bacci A, Montagnoli C, Spreca A, Mencacci A, Ricciardi-Castagnoli P, Romani L. 2000. Dendritic cells discriminate between yeasts and hyphae of the fungus Candida albicans. Implications for initiation of T helper cell immunity in vitro and in vivo. J. Exp. Med. 191:1661–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Diep BA, Otto M. 2008. The role of virulence determinants in community-associated MRSA pathogenesis. Trends Microbiol. 16:361–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Riedemann NC, Neff TA, Guo RF, Bernacki KD, Laudes IJ, Sarma JV, Lambris JD, Ward PA. 2003. Protective effects of IL-6 blockade in sepsis are linked to reduced C5a receptor expression. J. Immunol. 170:503–507 [DOI] [PubMed] [Google Scholar]
- 47. Spittler A, Razenberger M, Kupper H, Kaul M, Hackl W, Boltz-Nitulescu G, Fugger R, Roth E. 2000. Relationship between interleukin-6 plasma concentration in patients with sepsis, monocyte phenotype, monocyte phagocytic properties, and cytokine production. Clin. Infect. Dis. 31:1338–1342 [DOI] [PubMed] [Google Scholar]
- 48. Fujishima S, Sasaki J, Shinozawa Y, Takuma K, Kimura H, Suzuki M, Kanazawa M, Hori S, Aikawa N. 1996. Serum MIP-1 alpha and IL-8 in septic patients. Intensive Care Med. 22:1169–1175 [DOI] [PubMed] [Google Scholar]
- 49. Jerva LF, Sullivan G, Lolis E. 1997. Functional and receptor binding characterization of recombinant murine macrophage inflammatory protein 2: sequence analysis and mutagenesis identify receptor binding epitopes. Protein Sci. 6:1643–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ratner AJ, Lysenko ES, Paul MN, Weiser JN. 2005. Synergistic proinflammatory responses induced by polymicrobial colonization of epithelial surfaces. Proc. Natl. Acad. Sci. U. S. A. 102:3429–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Roberts AW. 2005. G-CSF: a key regulator of neutrophil production, but that's not all! Growth Factors 23:33–41 [DOI] [PubMed] [Google Scholar]
- 52. Bosmann M, Russkamp NF, Patel VR, Zetoune FS, Sarma JV, Ward PA. 2011. The outcome of polymicrobial sepsis is independent of T and B cells. Shock 36:396–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mera S, Tatulescu D, Cismaru C, Bondor C, Slavcovici A, Zanc V, Carstina D, Oltean M. 2011. Multiplex cytokine profiling in patients with sepsis. APMIS 119:155–163 [DOI] [PubMed] [Google Scholar]
- 54. Lin JN, Lai CH, Chen YH, Chang LL, Lu PL, Tsai SS, Lin HL, Lin HH. 2010. Characteristics and outcomes of polymicrobial bloodstream infections in the emergency department: a matched case-control study. Acad. Emerg. Med. 17:1072–1079 [DOI] [PubMed] [Google Scholar]
- 55. Brock TG, McNish RW, Peters-Golden M. 1999. Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J. Biol. Chem. 274:11660–11666 [DOI] [PubMed] [Google Scholar]
- 56. Portanova JP, Zhang Y, Anderson GD, Hauser SD, Masferrer JL, Seibert K, Gregory SA, Isakson PC. 1996. Selective neutralization of prostaglandin E2 blocks inflammation, hyperalgesia, and interleukin 6 production in vivo. J. Exp. Med. 184:883–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hinson RM, Williams JA, Shacter E. 1996. Elevated interleukin 6 is induced by prostaglandin E2 in a murine model of inflammation: possible role of cyclooxygenase-2. Proc. Natl. Acad. Sci. U. S. A. 93:4885–4890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang D, Wang H, Brown J, Daikoku T, Ning W, Shi Q, Richmond A, Strieter R, Dey SK, DuBois RN. 2006. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J. Exp. Med. 203:941–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yu Y, Chadee K. 1998. Prostaglandin E2 stimulates IL-8 gene expression in human colonic epithelial cells by a posttranscriptional mechanism. J. Immunol. 161:3746–3752 [PubMed] [Google Scholar]
- 60. Aronoff DM, Canetti C, Peters-Golden M. 2004. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J. Immunol. 173:559–565 [DOI] [PubMed] [Google Scholar]
- 61. Fernandez-Repollet E, Mittler RS, Tiffany S, Schwartz A. 1982. In vivo effects of prostaglandin E2 and arachidonic acid on phagocytosis of fluorescent methacrylate microbeads by rat peritoneal macrophages. J. Histochem. Cytochem. 30:466–470 [DOI] [PubMed] [Google Scholar]
- 62. Sottile A, Venza M, Venza I, Teti D. 1995. Prostaglandins affect the respiratory burst of human neutrophils. Immunopharmacol. Immunotoxicol. 17:311–321 [DOI] [PubMed] [Google Scholar]
- 63. von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, Brown CR, Krantz BA, Leppla SH, Gronert K, Vance RE. 2012. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 490:107–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu M, Chen K, Yoshimura T, Liu Y, Gong W, Wang A, Gao JL, Murphy PM, Wang JM. 2012. Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci. Rep. 2:786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sugimoto Y, Fukada Y, Mori D, Tanaka S, Yamane H, Okuno Y, Deai K, Tsuchiya S, Tsujimoto G, Ichikawa A. 2005. Prostaglandin E2 stimulates granulocyte colony-stimulating factor production via the prostanoid EP2 receptor in mouse peritoneal neutrophils. J. Immunol. 175:2606–2612 [DOI] [PubMed] [Google Scholar]