

Abstract
Entamoeba histolytica is an intestinal protozoan parasite and is the causative agent of amoebiasis. During invasive infection, highly motile amoebae destroy the colonic epithelium, enter the blood circulation, and disseminate to other organs such as liver, causing liver abscess. Motility is a key factor in E. histolytica pathogenesis, and this process relies on a dynamic actomyosin cytoskeleton. In other systems, phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] is known to regulate a wide variety of cellular functions, including signal transduction, actin remodeling, and cell motility. Little is known about the role of PI(4,5)P2 in E. histolytica pathogenicity. In this study, we demonstrate that PI(4,5)P2 is localized to cholesterol-rich microdomains, lipid rafts, and the actin-rich fractions of the E. histolytica membrane. Microscopy revealed that the trailing edge of polarized trophozoites, uroids, are highly enriched in lipid rafts and their constituent lipid, PI(4,5)P2. Polarization and enrichment of uroids and rafts with PI(4,5)P2 were enhanced upon treatment of E. histolytica cells with cholesterol. Exposure to cholesterol also increased intracellular calcium, which is a downstream effector of PI(4,5)P2, with a concomitant increase in motility. Together, our data suggest that in E. histolytica, PI(4,5)P2 may signal from lipid rafts and cholesterol may play a role in triggering PI(4,5)P2-mediated signaling to enhance the motility of this pathogen.
INTRODUCTION
The intestinal parasite Entamoeba histolytica is known to cause amoebic dysentery and liver abscess. E. histolytica enters the human host via contaminated food or water as an environmentally stable cyst. Excystation leads to the release of trophozoites in the small intestine, which colonize the bowel lumen. From here, the parasite can enter two non-mutually exclusive routes of infection, noninvasive or invasive disease (reviewed in reference 1). In the noninvasive mode, the trophozoite encysts and exits the host, whereas in the invasive mode, the parasite adheres to and destroys the colonic epithelium and enters the circulatory system. This results in extraintestinal infection, of which amoebic liver abscess (ALA) is the most common manifestation. Motility is a key virulence function that enables this parasite to cause extraintestinal infection (2).
Motile amoebae display a polarized morphology with a pseudopod at the leading edge and a uroid (also referred to as a uropod in other eukaryotic cells) at the trailing edge. A key feature of a polarized cell is the differential distribution of proteins and lipids, including cell surface receptors, signaling molecules, and cytoskeletal elements. For example, the pseudopod of E. histolytica is enriched in F actin, myosin IB (3, 4), and signaling molecules like phosphatidylinositol-3,4,5-trisphosphate [PI(3,4,5)P3] (5), while the uroid is enriched in myosin II and signaling molecules, including an important heterotrimeric adhesin, the galactose/N-acetylgalactosamine (Gal/GalNAc) lectin (6), F actin, and various actin-binding proteins (7–9). Such spatial asymmetry helps the cells to generate the necessary forces that are required for migration. For example, polymerization of actin at the leading edge facilitates pseudopod extension. Concurrently, adhesion molecules establish new focal contacts with the substrate, while activation of myosin II motors generates the contractile force required for the detachment of existing focal adhesions at the uroids. Together, these activities result in a net forward movement of the cell body. Consequently, motility in this pathogen has been attributed to its highly dynamic actomyosin cytoskeleton, which promotes the rapid morphological changes required for cell movement (reviewed in reference 10).
Phosphoinositides act as links that connect the actomyosin cytoskeleton to the plasma membrane. In other systems, phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] regulates the strength of the interaction between the cytoskeleton and the plasma membrane, which is required for maintaining cell shape and integrity (11). PI(4,5)P2 belongs to a group of phosphorylated phosphoinositides that act as second messenger molecules by binding to and modulating the activity of various cytoskeleton remodeling proteins. Furthermore, other lipid second messenger molecules can be generated from PI(4,5)P2. For instance, the pleckstrin homology (PH) domain of phospholipase C delta 1 (PLCD1) can bind PI(4,5)P2. PLCD1 hydrolyzes PI(4,5)P2 to yield inositol triphosphate (IP3) and diacylglycerol (DAG) (12, 13). While IP3 stimulates an increase in intracellular calcium (Ca2+) levels (reviewed in references 14 and 15), DAG activates protein kinase C (PKC) (reviewed in reference 16). PI(4,5)P2 can be phosphorylated by phosphoinositide 3-kinases (PI3Ks) to yield yet another signaling molecule, PI(3,4,5)P3 (reviewed in reference 17). All of these signaling molecules, PI(4,5)P2, PI(3,4,5)P3, IP3, DAG, and Ca2+, can interact with other effector proteins to trigger downstream signaling events.
In mammalian cells, PI(4,5)P2 is localized to lipid rafts (18–20). Rafts are specialized membrane microdomains that are cholesterol and sphingolipid rich and are known to organize signaling molecules that participate in a variety of cellular functions, including cell polarization (21, 22), endocytosis (reviewed in reference 23), secretion (reviewed in reference 24), and adhesion (25). Previously, E. histolytica was shown to possess lipid rafts that regulate parasite-host interaction (26, 27). Disruption of rafts with cholesterol-binding agents like methyl-β-cyclodextrin (MβCD) inhibits adhesion of E. histolytica to host cells (26) and host extracellular matrix components (27). On the other hand, cholesterol loading of amoebic membranes results in enhanced adhesion to host cell monolayers, as well as cocompartmentalization of the three subunits of the Gal/GalNAc lectin in lipid rafts (28).
Whether PI(4,5)P2 localizes to rafts in E. histolytica has not been previously studied. Therefore, in the current study, we developed a PI(4,5)P2 biosensor and utilized antibody to PI(4,5)P2 to study its subcellular and submembrane localization, respectively. We demonstrate that PI(4,5)P2 localizes to rafts in E. histolytica and may signal from the raft membrane domain to regulate cell motility.
MATERIALS AND METHODS
Strains and culture conditions.
Entamoeba histolytica trophozoites (strain HM-1:1MSS) were cultured axenically in TYI-S-33 medium (29) in 15-ml glass screw-cap tubes at 37°C.
Exposure to cholesterol or methyl-β-cyclodextrin.
The source of cholesterol used was a bovine cholesterol concentrate (Creative Laboratory Products, Inc., Indianapolis, IN). In all cases, to control for extracellular sources of cholesterol, trophozoites were incubated in serum-free medium (TYI-S-33) for 30 min at 37°C prior to experimentation. After serum starvation, trophozoites were incubated in serum-free medium supplemented with 0 to 3 mg/ml cholesterol or 50 mM methyl-β-cyclodextrin (MβCD; Sigma-Aldrich, St. Louis, MO) for 15 or 60 min at 37°C. Cells treated with 0 mg/ml cholesterol represented the untreated controls for all experiments.
Lipid raft isolation, lipid extraction, and lipid dot blots.
Isolation of a Triton X-100-resistant membrane and resolution of a detergent-resistant membrane (DRM) by sucrose gradient density centrifugation were carried out as previously described (26). Extraction of lipids from the sucrose gradient membrane fractions was carried out as described previously (30). Briefly, methanol–12.1 N HCl (10:1) was added to each fraction at a volumetric ratio of 1:1, and chloroform was added at a volumetric ratio of 2:1 (solvent-membrane fraction) to facilitate phase separation. The organic phase was subsequently extracted with the addition of methanol–1 N HCl (1:1) at a volumetric ratio of 1:1. The organic phase was vacuum dried, and the pellets were used for lipid dot blot analysis. Specifically, lipids were spotted onto a Hybond-C nitrocellulose membrane. The membrane was blocked in 1.5% (wt/vol) fatty acid-free bovine serum albumin (BSA) at room temperature for 1 h and probed with mouse anti-PI(4,5)P2 antibody (1:1,000 dilution; Abcam, Cambridge, MA), followed by incubation with secondary antibody, peroxidase-conjugated goat anti-mouse IgG (1:2,000 dilution; Cappel, ICN Pharmaceuticals, Costa Mesa, CA). Immunoblots were visualized using an enhanced chemiluminescence Western blotting detection system (Pierce Biotechnology, Rockford, IL) according to the manufacturer's instructions. Semiquantitative densitometric analyses of immunoblots were performed using ImageJ software (version 1.42q; National Institutes of Health, Bethesda, MD). The net densitometry values were corrected to reflect the differences in membrane fraction volume. Whole-cell lipid extraction and lipid dot blot analyses were performed as previously described (31).
Generation of GST-tagged PHPLCD1 recombinant protein.
The cDNA encoding the PH domain from PLCD1 (PHPLCD1) was amplified from pEGFP-N1 (SignaGen Laboratories, Gaithersburg, MD) by PCR using the following primer pair: 5′-GGAATTCATGGACTCGGGCCGGGAC-3′ and 5′-GGTCGACTCCTTCAGGAAGTTCTGCAG-3′. The resulting PCR product was cloned into the pCR2.1-TOPO plasmid (Invitrogen, Carlsbad, CA). The DNA sequence encoding PHPLCD1 was excised from pCR2.1-TOPO using EcoRI and SalI endonucleases and was ligated into the polylinker region downstream of and in frame with the sequence encoding glutathione S-transferase (GST) in the pGEX-5X-1 expression vector (Amersham Biosciences, Piscataway, NJ). The authenticity of the DNA construct was confirmed using restriction enzyme analysis and sequencing. Recombinant GST and GST-PHPLCD1 proteins were obtained by expression in Escherichia coli BL21 (Amersham, Piscataway, NJ) according to the manufacturer's protocol. To assess the purity of the eluted proteins, SDS-PAGE and silver staining were carried out as previously described (32). To validate the GST tag, Western blotting was performed as described previously (33) using an anti-GST antibody (Chemicon, Temecula, CA) at a dilution of 1:10,000.
Protein-lipid overlay assay.
A protein-lipid overlay assay was carried out to assess the PI(4,5)P2 binding capability of the GST-PHPLCD1 fusion protein as described previously (34). Briefly, 1.6 to 100 pmol of phosphoinositides (Echelon Biosciences Inc., Salt Lake City, UT), including phosphatidylinositol 3-phosphate (PI3P), phosphatidylinositol 4-phosphate (PI4P), phosphatidylinositol 5-phosphate (PI5P), phosphatidylinositol 3,4-bisphosphate [PI(3,4)P2], phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2], phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2], and phosphatidylinositol 3,4,5-triphosphate [PI(3,4,5)P3], was spotted on a Hybond-C nitrocellulose membrane. The membrane was dried for 1 h at room temperature and incubated in blocking buffer containing 2% (wt/vol) fatty acid-free BSA in Tris-buffered saline (TBS)–Tween (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% [vol/vol] Tween 20) at room temperature for 1 h. The membrane was incubated overnight at 4°C in blocking buffer containing 0.2 μg/ml of either GST or GST-PHPLCD1, washed in TBS-Tween, and incubated in polyclonal anti-GST antibody (1:10,000 dilution) in blocking buffer for 1 h. The membrane was washed, incubated in horseradish peroxidase (HRP)–anti-rabbit IgG (1:5,000 dilution) in TBS-Tween for 1 h, and then washed and developed as described previously (33).
We also tested the ability of GST or GST-PHPLCD1 to bind E. histolytica cell extracts. Whole-cell protein extracts of trophozoites (5 × 105 and 1 × 105 cells) were resolved by SDS-PAGE and electroblotted onto a nitrocellulose membrane. The membrane was washed twice with distilled water and dried for 1 h at room temperature. Serial dilutions of PI(4,5)P2 were spotted on one edge of the same membrane, which served as a positive control for binding of the probe. The membrane was dried and incubated with blocking buffer containing 2% (wt/vol) fatty acid-free BSA in TBS-Tween (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% [vol/vol] Tween 20), followed by incubation with 0.2 μg/ml of either GST or GST-PHPLCD1 probes in the blocking buffer, as described above. The membrane was washed in TBS-Tween and incubated in polyclonal anti-GST antibody (1:10,000 dilution) in blocking buffer, followed by incubation with HRP–anti-rabbit IgG (1:5,000 dilution) in TBS-Tween, and then washed and developed as described previously (33). As a positive control for protein transfer, the membrane was incubated in anti-actin monoclonal antibody (1:2,500 dilution; Abcam) for 1 h. The membrane was washed, incubated in HRP–anti-mouse IgG (1:2,000 dilution) in TBS-Tween for 1 h, and then washed and developed as described previously (33).
Biosensor staining and fluorescence microscopy.
For fluorescence microscopy, E. histolytica trophozoites were plated on a chambered cover glass slide system (Lab-Tek, Christchurch, New Zealand) and fixed with 4% (vol/vol) paraformaldehyde overnight at 37°C. Cells were washed in phosphate-buffered saline (PBS), incubated in 50 mM NH4Cl in PBS for 15 min, and then permeabilized with 0.05% (wt/vol in PBS) saponin for 10 min as previously described (35). All subsequent buffers contained 0.05% (wt/vol in PBS) saponin. This regimen preserves the cellular morphology and permeabilizes cells without removing phosphoinositides from the plasma membrane (5, 35). Cells were blocked with 10% (vol/vol) fetal bovine serum (FBS)–PBS and stained with approximately 10 μg/ml of either affinity-purified GST or GST-PHPLCD1, followed by staining with Alexa Fluor 488-conjugated rabbit anti-GST antibody (1:2,000; Invitrogen) or Texas Red-conjugated goat anti-GST antibody (1:1,000; Rockland Immunochemicals, Gilbertsville, PA), as described previously (35). Stained cells were viewed using a Nikon Eclipse TI-E spectral confocal microscope (Nikon Instruments Inc., Lewisville, TX) or an LSM510 confocal microscope (Carl Zeiss MicroImaging, Thornwood, NY). Images were analyzed using Nikon NIS Elements or LSM Image Browser software, respectively. To assess the effect of cholesterol on localization of PI(4,5)P2, cells were exposed to either 0 or 3 mg/ml cholesterol for 1 h and then fixed and stained as described above.
For lipid raft staining, E. histolytica trophozoites were fixed with 4% (vol/vol) paraformaldehyde overnight at 37°C. Dialkyindocarbocyanine (DiIC16; Invitrogen) staining of lipid rafts was carried out as described previously (27).
To determine if PI(4,5)P2 and lipid rafts localized to uroids, capping was induced by incubation of amoebae with Alexa Fluor 488-conjugated concanavalin A (ConA; Invitrogen) for 1 h on ice, followed by incubation on cover glass chambered slides for 15 min at 37°C as described previously (6). PI(4,5)P2 or lipid raft staining was carried out as described above.
Measurement of motility.
To test the effect of cholesterol on parasite motility, a motility assay was employed according to the methods of Zaki et al. (36). Briefly, cells were serum starved for 30 min at 37°C and exposed to a range of cholesterol concentrations (0 to 3 mg/ml) in serum-free medium for 30 min at 37°C. Eight milliliters of complete TYI-S-33 medium supplemented with 0.75% (wt/vol) agarose was poured into 60-mm petri dishes and allowed to solidify at room temperature. A trough (2 by 30 mm) was cut into the solidified medium to serve as the motility chamber. Following exposure to cholesterol, trophozoites (5 × 105) were placed in the trough of the motility chamber. A coverslip (22 by 40 mm) was placed over the trough and the plate was incubated at 37°C for 3 h in 5% CO2. Images were captured using confocal microscopy. The migration distance was measured using Zeiss LSM510 image analysis software.
Measurement of intracellular Ca2+ levels.
To measure the effect of cholesterol treatment on intracellular Ca2+ levels, a Ca2+ assay was employed as described previously (31). Briefly, cells were incubated in serum-free medium supplemented with the fluorescent Ca2+ indicator Fluo-4/AM (Invitrogen) or dimethyl sulfoxide (DMSO; diluent control) for 30 min at 37°C. Then, cells were incubated with a range of concentrations of cholesterol (0 to 3 mg/ml) for either 15 min or 1 h at 37°C. Subsequently, 1 × 105 trophozoites were pipetted into the wells of an uncoated 96-well plate, and after 5 min the plates were transferred to a fluorescence microplate reader for 30 min. To account for the background fluorescence, the fluorescence values for cells incubated in DMSO were subtracted from the fluorescence values for cells incubated in Fluo-4/AM. The fluorescence value of the untreated control cells at the 35-min time point was arbitrarily set equal to 100%, and the values at other time points were reported as a percentage of this value.
Statistical analyses.
All values are given as means ± standard deviations (SDs) of at least 3 trials. To compare means, statistical analyses were performed using GraphPad Instat v.3 software with an unpaired t test, Welch corrected (two-tail P value), or with one-way analysis of variance (ANOVA) and a Tukey-Kramer multiple-comparison test. In all cases, P values of less than 0.001 were considered highly statistically significant, while P values of less than 0.01 or 0.05 were considered statistically significant.
RESULTS
PI(4,5)P2 compartmentalizes in lipid rafts and actin-rich pellet fractions of the DRM in E. histolytica.
In mammalian cells, PI(4,5)P2 has been shown to localize to lipid rafts (18, 19). Therefore, we determined if E. histolytica exhibits similar compartmentalization of PI(4,5)P2. The detergent-resistant membrane (DRM), which consists of lipid rafts and the actin-rich membrane, was isolated as described previously (26). Sucrose gradient density centrifugation was used to resolve these two membrane types. Total phosphoinositides were extracted from each of the gradient fractions and subjected to a lipid dot blot analysis using antibodies to PI(4,5)P2 (Fig. 1A). We previously demonstrated that membrane fractions 9 to 14 possess high levels of cholesterol and contain lipid rafts (31). Less buoyant fractions (fractions 17 to 19) and the pellet fraction (fraction 20) are actin rich (26, 28, 31). Similar to the enrichment seen in mammalian cells, we observed an enrichment of PI(4,5)P2 in lipid rafts (fractions 9 to 14) (Fig. 1A and B). However, the actin-rich pellet fraction also possessed high levels of this phospholipid (Fig. 1A and B). Thus, there may be two pools of PI(4,5)P2 in E. histolytica, one pool in lipid rafts and another pool interacting directly with the actin cytoskeleton. However, we cannot rule out the possibility that PI(4,5)P2 in rafts also interacts with the actin cytoskeleton in live cells.
Fig 1.
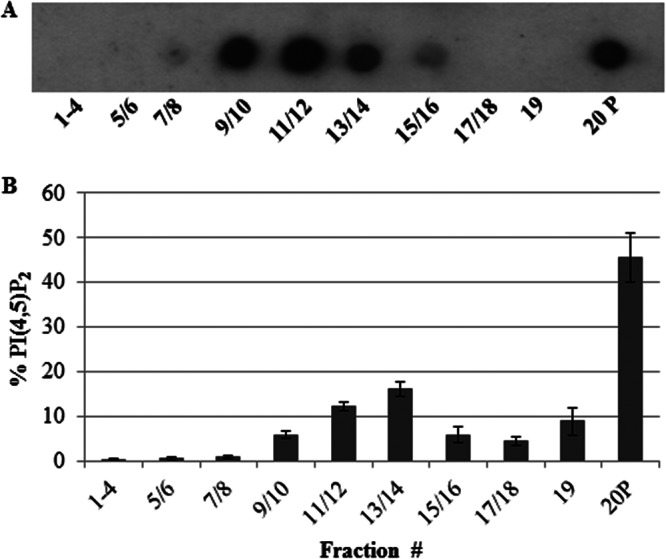
PI(4,5)P2 compartmentalizes in lipid rafts and actin-rich pellet fractions. The DRM was isolated from Entamoeba histolytica trophozoites and subjected to sucrose gradient density fractionation. PI(4,5)P2 was extracted from these fractions, and lipid dot blots were performed using antibodies specific to PI(4,5)P2. (A) A representative blot is shown. Cells displayed PI(4,5)P2 enrichment in lipid raft fractions (fractions 9 to 14) and in an actin-rich pellet fraction (fraction 20 P). (B) Semiquantitative scanning densitometry was used to analyze the distribution of PI(4,5)P2 in membrane fractions, correcting for the difference in the starting volume of each membrane fraction. The total level of PI(4,5)P2 in the DRM was calculated, and the percentage of PI(4,5)P2 in each fraction (± SD; n = 3) is presented.
Exposure to cholesterol increases levels of PI(4,5)P2 in the DRM.
Previous studies in mammalian cells have revealed that the level of membrane cholesterol influences the localization of PI(4,5)P2 in the DRM (18). Therefore, we tested the effect of exposure to cholesterol or to the cholesterol-sequestering agent MβCD on the localization and overall levels of PI(4,5)P2 in E. histolytica trophozoites. On average, cells treated with 0.05 mg/ml cholesterol displayed a slight increase in PI(4,5)P2 levels in the lipid raft fractions compared to control untreated cells; however, there was no difference in the level of PI(4,5)P2 in the total DRM (Fig. 2A to C). The most dramatic changes in the level of PI(4,5)P2 in the total DRM were observed when the cells were exposed to 0.75 and 3 mg/ml cholesterol, with an approximately 60% increase (Fig. 2A). In contrast, MβCD caused a 56% decrease in the level of PI(4,5)P2 from that in the total DRM (Fig. 2A). Furthermore, treatment with 0.75 and 3 mg/ml cholesterol increased the level of PI(4,5)P2 specifically in lipid rafts (Fig. 2B and C). To determine if these changes were due to overall increases or decreases in PI(4,5)P2, we also quantified total PI(4,5)P2 in whole cells. We did not find a significant difference in the total cellular level of PI(4,5)P2 after cholesterol or MβCD exposure (Fig. 2D). Thus, PI(4,5)P2 may be moving between membrane compartments in a cholesterol-dependent manner. However, we cannot rule out the possibility that there is a rapid turnover of PI(4,5)P2, where continuous synthesis and hydrolysis maintain a constant total level.
Fig 2.

Exposure to cholesterol increases the levels of PI(4,5)P2 in the DRM and specifically in lipid raft fractions. Trophozoites were serum starved and exposed to cholesterol (0 to 3 mg/ml) or the cholesterol-depleting agent MβCD. Cells treated with no cholesterol (0 mg/ml) served as the untreated control. (A to C) The DRM was isolated and subjected to sucrose gradient density fractionation. PI(4,5)P2 was extracted from these fractions, and lipid dot blots were performed using antibodies specific to PI(4,5)P2. Levels of PI(4,5)P2 were analyzed by semiquantitative scanning densitometry. In all cases, values were corrected for differences in starting volumes. (A) The total level of PI(4,5)P2 in the DRM was calculated and reported as a percentage of that for the untreated control. Cells treated with cholesterol (0.75 or 3 mg/ml) and MβCD displayed an approximately 60% increase and 56% decrease in PI(4,5)P2 in the total DRM, respectively, compared to the levels for the control (n ≥ 3) (*, P < 0.5; **, P < 0.01). (B) A representative lipid dot blot of the sucrose gradient density fraction is shown. Cells treated with cholesterol (0.75 and 3 mg/ml) displayed enhanced enrichment of PI(4,5)P2 in lipid rafts (fractions 9 to 14) compared to the control, while MβCD-treated cells showed a decrease in PI(4,5)P2 from all fractions of the DRM. (C) The data represent the percentage of PI(4,5)P2 in each fraction compared to the level of PI(4,5)P2 in the total DRM of the untreated control cells (± SD; n ≥ 3). (D) Phosphoinositides were extracted from whole-cell lysates, and PI(4,5)P2 levels were measured using dot blots with antibodies specific to PI(4,5)P2. The level of PI(4,5)P2 for each treatment was quantified by scanning densitometry and reported as a percentage of the level for the untreated control. The total cellular PI(4,5)P2 level remained unchanged upon treatment with cholesterol or MβCD. Data are the means ± SDs for ≥3 independent experiments.
A GST-tagged PH probe reveals localization of PI(4,5)P2 to plasma membrane and uroids.
We have previously determined the subcellular localization of PI3P (35) and PI(3,4,5)P3 (5) using a GST-tagged FYVE finger domain and a GST-tagged pleckstrin homology (PH) domain of Bruton's tyrosine kinase (PHBTK), respectively. We used a similar approach to determine the subcellular localization of PI(4,5)P2 using a recombinant GST-tagged version of the PH domain from PLCD1 (PHPLCD1). This protein domain has been shown to specifically bind PI(4,5)P2, and the GST-tagged chimera has been used to study the localization of PI(4,5)P2 in mammalian cells (37).
Recombinant GST (control) and GST-PHPLCD1 were expressed in bacteria and affinity purified. SDS-PAGE demonstrated that the proteins were 28 kDa and 48 kDa in size, which are consistent with the predicted sizes of GST and GST-PHPLCD1, respectively (Fig. 3A). The authenticity of the GST tag was confirmed by Western blotting with antibodies specific for GST (Fig. 3B). To test the binding specificity of GST-PHPLCD1, we employed a protein-lipid overlay assay as described previously (34, 37). The probe bound to PI(4,5)P2 in a dose-dependent manner and did not display any significant capacity for binding to other related phosphoinositides (Fig. 3C). GST alone (control) did not bind any of the phosphoinositides (data not shown). We also determined if the probes bound nonspecifically to E. histolytica proteins. Whole-cell protein extract was resolved by SDS-PAGE, electroblotted onto a nitrocellulose membrane, and decorated with GST or GST-PHPLCD1. Neither GST nor GST-PHPLCD1 displayed binding to E. histolytica proteins (Fig. 3D). Together, these data suggest that the GST-PHPLCD1 probe is highly specific for PI(4,5)P2 and does not cross-react with other cellular lipids or proteins. Therefore, it could be utilized for subcellular localization of PI(4,5)P2.
Fig 3.
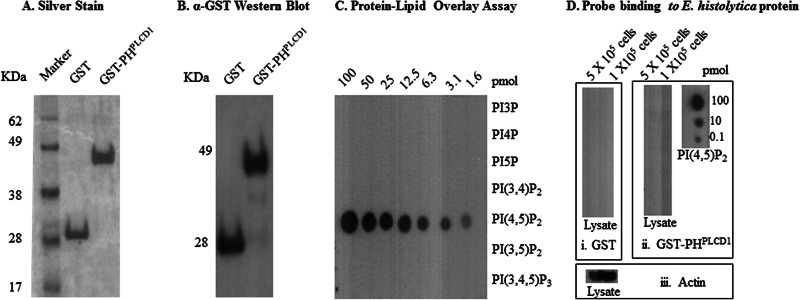
The purified GST-PHPLCD1 biosensor binds with high affinity to PI(4,5)P2. (A) SDS-PAGE and silver staining of GST and GST-PHPLCD1 fusion proteins after affinity purification. The molecular mass markers are shown. (B) Western blot analysis of purified GST and GST-PHPLCD1 using anti-GST antibody. (C) Serial dilutions (100 to 1.6 pmol) of the indicated phosphoinositides were spotted onto a nitrocellulose membrane and incubated with 0.2 μg/ml of GST-PHPLCD1. Binding of the protein was detected with anti-GST antibodies. The probe exhibited binding specificity to PI(4,5)P2 alone. (D) Whole-trophozoite extracts (5 × 105 and 1 × 105 cells) were resolved by SDS-PAGE and electroblotted onto a Hybond-C nitrocellulose membrane. Individual lanes of lysate were decorated with GST (i), GST-PHPLCD1 (ii), or anti-actin antibody (positive control for protein transfer) (iii). (ii) As an additional control for binding of the GST-PHPLCD1 probe, the membrane was washed after the protein transfer and serial dilutions of PI(4,5)P2 were spotted on the same membrane and incubated with GST-PHPLCD1. Binding of the probes was detected with anti-GST antibodies. Neither GST nor GST-PHPLCD1 probe bound to the whole-cell protein extract, further supporting the specificity of the probe.
E. histolytica trophozoites were fixed, permeabilized, and stained with GST or GST-PHPLCD1, followed by staining with a fluorescence-labeled anti-GST antibody. Confocal microscopy revealed that cells stained with control GST exhibited minimal fluorescence (Fig. 4A and D). In contrast, the GST-PHPLCD1 probe uniformly decorated the plasma membrane, giving most cells a ring-like appearance (Fig. 4B and E). This was not surprising, as PI(4,5)P2 is known to account for 1% of plasma membrane phospholipids and >99% of all doubly phosphorylated phospholipids and is maintained at a high steady-state level in mammalian cells (reviewed in references 17, 20, and 38). Interestingly, in some cells, PI(4,5)P2 was also enriched on one side of the cell, opposite apparent forming pseudopods (Fig. 4C and F).
Fig 4.

The PI(4,5)P2 biosensor GST-PHPLCD1 decorates the plasma membrane of E. histolytica. Trophozoites were stained with GST or GST-PHPLCD1 and fluorescent anti-GST antibody, and immunofluorescence confocal microscopy was performed. (A) Cells stained with the control, GST, exhibited minimal staining. (B) GST-PHPLCD1 uniformly decorated the plasma membrane of the trophozoites, giving the cells a ring-like appearance. (C) In some instances, there was an enrichment of GST-PHPLCD1staining on one edge of the cell (arrow), opposite an apparent pseudopod (arrowhead). (D to F) The differential interference contrast (DIC) images corresponding to the images in panels A to C, respectively, are shown.
Studies in other systems (18) and our biochemical analysis (Fig. 2A to C) indicated that cholesterol levels impact the localization of PI(4,5)P2 in the DRM. Therefore, we wanted to determine the subcellular localization of PI(4,5)P2 in whole cells after exposure to cholesterol or after cholesterol depletion. Interestingly, there was an increase in the number of cells displaying PI(4,5)P2 localization at one edge upon cholesterol treatment (Fig. 5B and E) compared to the number for the untreated control (Fig. 5A and D). Image analysis revealed that there was a 3.4-fold increase in the number of cells displaying this morphology after cholesterol treatment compared to the number of control cells (Fig. 5G). In contrast, cholesterol depletion with MβCD disrupted the localization of PI(4,5)P2 at the plasma membrane, in that it was observed in the interior of the cell (Fig. 5C and F). This suggested a requirement of cholesterol richness for proper localization of PI(4,5)P2 to the plasma membrane.
Fig 5.

Treatment with cholesterol enhances the number of cells displaying PI(4,5)P2 enrichment at the edge opposite the forming pseudopod. Trophozoites were serum starved and exposed to either 0 mg/ml cholesterol (control), 3 mg/ml cholesterol, or the lipid raft-disrupting agent MβCD. Cells were stained with GST-PHPLCD1 and fluorescent anti-GST antibody, and immunofluorescence confocal microscopy was performed. (A and B) Cholesterol-treated cells displayed an increased PI(4,5)P2 localization on one edge of the cell compared to untreated control cells. (C) In MβCD-treated cells, PI(4,5)P2 staining was no longer confined to the plasma membrane. (D to F) The DIC images corresponding to the images in panels A to C, respectively, are shown. (G) The percentage of apparently polarized cells in control cells or after cholesterol treatment was quantified by visually scoring cells at ×20 magnification. There was a 3.4-fold increase in the number of cells displaying PI(4,5)P2 localization on one edge after cholesterol treatment compared to the number for untreated control cells. Data are the means ± SDs of 3 independent experiments (***, P < 0.001).
Cholesterol treatment enhances motility.
In other systems, PI(4,5)P2 plays an essential role in cell motility. Since PI(4,5)P2 became asymmetrically distributed after exposure to cholesterol (Fig. 5B and E) and since asymmetry is a hallmark of motile cells, we hypothesized that exposure to cholesterol was enhancing motility. To test this, we utilized a previously published under-agar motility assay (36) to determine the effect of cholesterol exposure on cell movement. Upon exposure to cholesterol, the ability of the trophozoites to move out of the wells increased significantly in a dose-dependent fashion (Fig. 6A). After exposure to cholesterol, not only did cells traverse longer distances but also there was an apparent increase in the number of motile cells (Fig. 6B and C). This suggests a possible role for cholesterol in mediating motility in E. histolytica.
Fig 6.
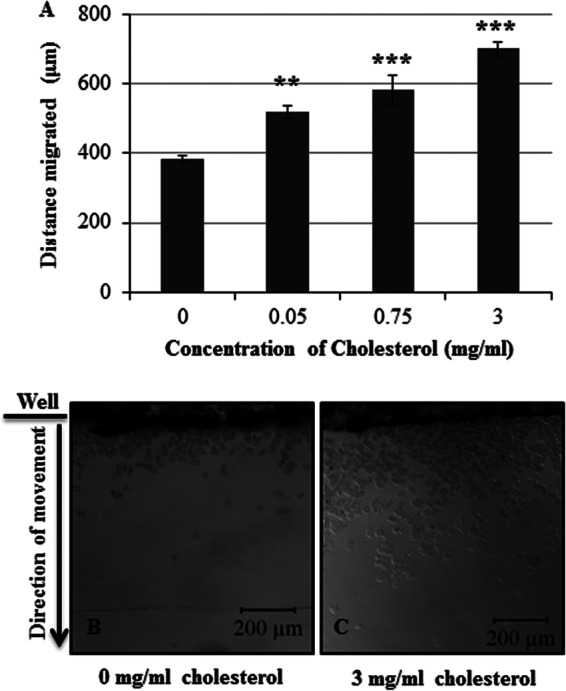
Exposure to cholesterol enhances motility in E. histolytica cells. E. histolytica trophozoites were serum starved and exposed to 0 to 3 mg/ml cholesterol. A motility assay was performed, and images were captured using confocal microscopy. The distance migrated by trophozoites was measured. (A) Trophozoites exhibited a dose-dependent and statistically significant increase in motility after exposure to cholesterol (**, P < 0.01; ***, P < 0.001). The data represent the means ± SDs from ≥3 independent experiments. (B and C) Cholesterol treatment also increased the number of motile cells compared to the number of untreated control cells entering the agar. Representative ×10 magnification fields of control cells and cells treated with 3 mg/ml cholesterol are shown.
PI(4,5)P2 localizes to uroids in ConA-capped E. histolytica cells.
In apparently polarized cells, PI(4,5)P2 appeared to be enriched on one edge opposite that of the forming pseudopods. This could represent localization of PI(4,5)P2 to the trailing edge (uroid). Therefore, we employed a polyvalent ligand, ConA, which is known to interact with and cap cell surface receptors at the trailing edge of the trophozoites (39). Thus, labeled ConA serves as a uroid marker. PI(4,5)P2 colocalized with fluorescent ConA in uroids (Fig. 7A to D) and with patches of ConA in trophozoites in which capping was initiated but was not complete (data not shown).
Fig 7.
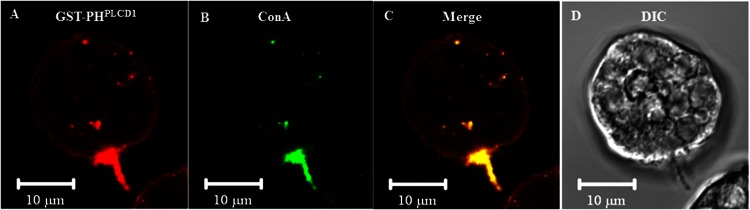
Fluorescence confocal microscopy of ConA-capped E. histolytica reveals localization of PI(4,5)P2 to uroids. (A to D) Trophozoites were incubated with fluorescein isothiocyanate-ConA (green) for 1 h on ice, followed by incubation at 37°C for 15 min to initiate uroid formation. Cells were fixed and stained with GST-PHPLCD1 and anti-GST antibody (red).
Lipid rafts localize to uroids.
Since our biochemical analysis demonstrated that lipid rafts were enriched in PI(4,5)P2 and since microscopy revealed that PI(4,5)P2 localized to uroids, we wanted to determine if, like in mammalian cells (40), the uroids of E. histolytica were enriched in lipid rafts. Therefore, we employed a lipid raft stain, DiIC16, on fixed E. histolytica trophozoites as previously described (27). Cells with no apparent pseudopod formation (nonpolarized) displayed a uniform plasma membrane raft staining (Fig. 8A and B), whereas apparently polarized cells also showed an enrichment of raft staining at one edge opposite the forming pseudopod (Fig. 8C and D). Colocalization of DiIC16 with ConA revealed that the uroids were indeed enriched in lipid rafts (Fig. 8E to H). Similar to the results seen for PI(4,5)P2 staining, trophozoites with incomplete uroid formation exhibited DiIC16 staining that colocalized with lateral patches of ConA (data not shown).
Fig 8.

Fluorescence confocal microscopy of ConA-capped E. histolytica reveals localization of lipid rafts to uroids. The lipid raft localizes to uroids in E. histolytica. (A to D) Trophozoites were fixed and stained with the lipid raft marker DiIC16. Nonpolarized cells displayed uniform plasma membrane raft staining, while apparent polarized cells exhibited an enriched raft staining on one edge (arrowheads) opposite the pseudopod (arrows). (E to H) E. histolytica trophozoites were incubated with fluorescein isothiocyanate-ConA (green) for 1 h on ice, followed by incubation at 37°C for 15 min to initiate uroid formation. Cells were fixed and stained with the raft marker DiIC16 (red). There was a colocalization of ConA and DiIC16 staining at the uroid of E. histolytica, further supporting the finding that lipid rafts are enriched at uroids in polarized cells.
Cholesterol treatment enhances intracellular Ca2+ levels in E. histolytica.
In other systems, PLCD1 hydrolyzes PI(4,5)P2 into IP3 and DAG, which facilitates Ca2+ signaling (13, 41). Since PLCD1 also localizes to rafts (42), it is reasonable to hypothesize that the accumulation of PI(4,5)P2 in rafts after cholesterol treatment might lead to an increase in intracellular Ca2+. Therefore, we measured intracellular Ca2+ levels after cholesterol treatment using a previously published protocol (31). Several studies have demonstrated that bona fide ligands of E. histolytica, such as collagen and fibronectin themselves, induce signaling leading to increased intracellular Ca2+ (31, 43). Therefore, to test the effect of cholesterol alone on intracellular Ca2+ levels, we utilized uncoated 96-well plates for our studies. After 1 h of pretreatment with cholesterol, we observed a significant dose-dependent increase in Ca2+ (Fig. 9A). Since signaling events take place immediately following stimulation, we also tested a shorter time of exposure to cholesterol. After 15 min of pretreatment with 0.75 and 3 mg/ml cholesterol, we observed similar increases in Ca2+, of which some were statistically significant (Fig. 9B). Together, these data indicate the possibility of a cholesterol-dependent, PI(4,5)P2-mediated rise in intracellular Ca2+ levels.
Fig 9.
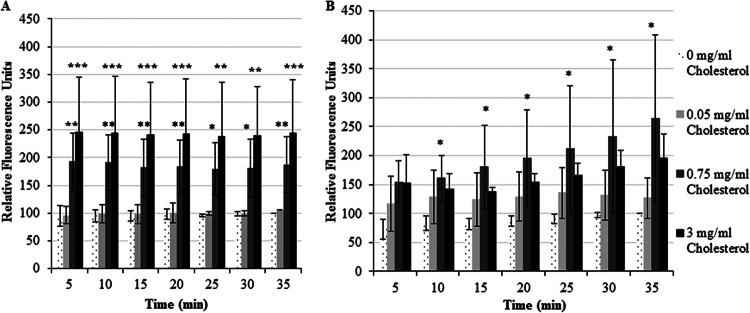
Exposure to cholesterol increases intracellular Ca2+ levels in E. histolytica. Trophozoites were serum starved and exposed to cholesterol at concentrations of 0 (control) to 3 mg/ml for either 60 or 15 min. Intracellular Ca2+ levels were measured after treatment over a 35-min time interval. Intracellular Ca2+ levels at the 35-min time point for the untreated control cells were arbitrarily set equal to 100%, and the values at the other time points were reported as a percentage of this final value. The data are the means ± SDs for ≥3 independent experiments. (A) Sixty minutes of pretreatment with 0.75 or 3 mg/ml cholesterol resulted in a significant increase in the intracellular Ca2+ level compared to that for untreated controls (*, P < 0.5; **, P < 0.01; ***, P < 0.001). (B) Similarly, an increase in intracellular Ca2+ levels was observed with a 15-min cholesterol pretreatment (*, P < 0.5).
DISCUSSION
This study represents the first analysis of the submembrane localization of PI(4,5)P2 in E. histolytica. Biochemical analyses showed that PI(4,5)P2 is compartmentalized in lipid raft and actin-rich pellet fractions of the DRM. Microscopy demonstrated that PI(4,5)P2 uniformly decorated the plasma membrane in a majority of the cells but also colocalized with lipid rafts at the uroids in polarized cells. The connection between PI(4,5)P2 and rafts in E. histolytica was supported by our observations that cholesterol treatment increased the level of PI(4,5)P2 in the DRM, lipid rafts, and raft-rich uroids and that MβCD depleted the DRM and plasma membrane of PI(4,5)P2. Finally, treatment with cholesterol resulted in an increase in intracellular Ca2+ [a downstream effector of PI(4,5)P2] and a concomitant increase in motility. Therefore, in E. histolytica, PI(4,5)P2 may signal from rafts and cholesterol may play a role in triggering PI(4,5)P2-dependent signaling.
Various studies have supported the existence of different cellular pools of PI(4,5)P2 to explain the complex roles of PI(4,5)P2 in cells (reviewed in reference 38). In E. histolytica, we observed compartmentalization of PI(4,5)P2 in lipid rafts as well as in the actin-rich pellet fraction at steady state. We also showed that exposure to cholesterol resulted in enrichment of PI(4,5)P2 in the DRM and, specifically, in lipid rafts without an increase in the total cellular level of PI(4,5)P2. One explanation for this is that PI(4,5)P2 translocates between membrane subcompartments in a cholesterol-dependent fashion. The parasite likely encounters different extracellular cholesterol levels depending on the location in the human host (e.g., liver versus intestine). Therefore, simple translocation of PI(4,5)P2 between membrane domains would represent a rapid way to regulate lipid signaling throughout infection. Ca2+ is a downstream effector of PI(4,5)P2 and accumulates upon hydrolysis of PI(4,5)P2 to IP3 and DAG. Since we also observed an increase in the intracellular Ca2+ level upon cholesterol treatment, it is also possible that there is rapid hydrolysis and synthesis of PI(4,5)P2 in rafts, which would maintain the apparent constant total cellular level of this lipid. In other systems, in addition to PLCD1 (42), enzymes that synthesize PI(4,5)P2, such as phosphatidylinositol-4-phosphate-5-kinase (PIP5K), are also recruited to rafts upon stimulation (44, 45). Thus, it is conceivable that rapid turnover of PI(4,5)P2 could occur in rafts.
We cannot rule out the possibility that it is actually the rise in intracellular Ca2+ that induces segregation of PI(4,5)P2 into rafts. Using an in vitro lipid monolayer system, Levental et al. (46) showed that the lateral organization of PI(4,5)P2 is sensitive to small changes in Ca2+ concentration. However, it is not known if Ca2+ can influence the submembrane distribution of PI(4,5)P2 in vivo. Regardless of whether there is a Ca2+-dependent step, the outcome of exposure of trophozoites to cholesterol is enrichment of PI(4,5)P2 in lipid rafts. This provides significant insight into signaling in this parasite.
In the present study, we demonstrated that PI(4,5)P2 uniformly decorates the plasma membrane of this parasite. We also showed that in polarized cells, both lipid rafts and the constituent signaling lipid, PI(4,5)P2, localize to the uroids. In mammalian cells, there is increasing evidence that lipid rafts play a vital role in delivering signaling molecules to uroids during cell migration. For example, Millán et al. (40) showed that the uroids of polarized T lymphocytes are enriched in cholesterol and disruption of rafts leads to loss of uroids and subsequent inhibition of cell migration. Furthermore, segregation of membrane proteins to uroids and pseudopods is inhibited by disruption of lipid rafts in T lymphocytes (40, 47) and neutrophils (21). Another study of polarized neutrophils revealed that DiIC16-stained raft fractions progress from a uniform distribution to discrete patches that coalesce at uroids to form caps (48). Furthermore, cap formation in these cells relies on the interaction of transmembrane proteins of the DRM with both the lipids of the DRM and the proteins of the actomyosin cytoskeleton (48). Since we observed the localization of lipid rafts and PI(4,5)P2 in uroids and in ConA-positive, laterally localized membrane patches, it is possible that a similar lipid raft-mediated delivery of PI(4,5)P2 to uroids occurs in E. histolytica. Another mechanism by which PI(4,5)P2 could become enriched in uroids is by local synthesis. A number of reports in other systems have shown that key PI(4,5)P2-synthesizing enzymes also localize to uroids (49–52). However, whether these enzymes localize to uroids of E. histolytica remains to be determined.
PI(4,5)P2 can function as a second messenger and bind various actin-binding proteins. A proteomic analysis of E. histolytica uroids identified the presence of actin-binding proteins that have been reported to interact with PI(4,5)P2 in other eukaryotic cells, including the spectrin-family proteins (e.g., filamin 2, an actinin-like protein) and a novel protein, filopodin (53). In other systems, PI(4,5)P2 has been shown to regulate actin cytoskeleton by binding and modulating filamin (54) and α-actinin (55). Filopodin is an ezrin-radixin-moesin (ERM) domain-containing protein. Members of the ERM family bind PI(4,5)P2 and are known to regulate uroid formation in T cells (56). Thus, PI(4,5)P2 may serve as a bridge at the uroid, connecting the cytoskeleton to the plasma membrane through various actin-binding proteins.
PI(4,5)P2 can also generate a variety of secondary messenger molecules. For instance, at the leading edge of Dictyostelium discoideum cells, PI(4,5)P2 is converted to PI(3,4,5)P3 by PI3K; PI(3,4,5)P3 binds to and recruits several proteins to promote pseudopod formation (reviewed in reference 52). Likewise, PI(3,4,5)P3 localizes to pseudopods of motile E. histolytica (5). At the trailing edge of mammalian cells, hydrolysis of PI(4,5)P2 to DAG, which activates PKC, is essential for T cell chemotaxis (57). IP3, another product of PI(4,5)P2 hydrolysis, stimulates an increase in intracellular Ca2+ levels, and increases in Ca2+ correlate with enhanced motility in neutrophils (58, 59). Ca2+ is a potent stimulator of motility through activation of myosin light-chain kinase (MLCK), leading to an increase in myosin II-based contractility at the uroid (60). In the current study, an increase in intracellular Ca2+ was accompanied by a robust augmentation of motility after exposure to cholesterol. Thus, a similarly potent PI(4,5)P2 to Ca2+ to MLCK system may exist in this pathogen. In support of this, myosin II is found to localize to the uroids of E. histolytica (2).
On average, we observed a modest increase in PI(4,5)P2 levels in rafts after treatment with 0.05 mg/ml cholesterol, but there was no difference in the total PI(4,5)P2 levels in the DRM. One explanation for observing a statistically significant increase in motility after treatment with the same concentration of cholesterol could be that motility is very sensitive to the submembrane position of PI(4,5)P2 and even small increases in PI(4,5)P2 in rafts can cause enhanced cell movement. Our observation that an increase in PI(4,5)P2 in rafts and uroids is accompanied by increases in intracellular Ca2+ and motility is suggestive of raft-mediated signaling from PI(4,5)P2. However, it remains to be seen if the effect of PI(4,5)P2 on motility after exposure to cholesterol is direct or indirect. One way to test this would be to generate E. histolytica cells with reduced levels of enzymes that are responsible for PI(4,5)P2 synthesis (e.g., by knockdown of PIP5K). In E. histolytica, these enzymes have not yet been definitively identified. Genome data suggest the existence of a putative E. histolytica PIP5K gene (GI:67481998), and a BLASTP analysis of the translated protein sequence revealed a 41% identity to human phosphatidylinositol-4-phosphate-5-kinase type 1β. However, it remains to be seen if this an authentic PI(4,5)P2-synthesizing enzyme.
Cholesterol is a major constituent of the eukaryotic plasma membrane (61). Aley et al. (62) demonstrated that E. histolytica possesses a high plasma membrane content of cholesterol, with a cholesterol/phospholipid molar ratio of 0.87. There are intriguing correlations between E. histolytica virulence and cholesterol. For example, cholesterol has been shown to enhance virulence functions in E. histolytica, including adhesion (27, 28), expression of ConA binding sites on the cell surface (63), and hemolytic and erythrophagocytic activity (64). Several studies have demonstrated that it is possible to revive the virulence of avirulent E. histolytica strains by supplying medium with cholesterol (65, 66) or by repeated passage through liver, which is a major site of cholesterol synthesis (67). Finally, long-term culture of E. histolytica in medium supplemented with liposomes carrying phosphatidylcholine and cholesterol can preserve virulence (63). Our study is the first to show that cholesterol impacts the submembrane and subcellular localization of PI(4,5)P2 in E. histolytica and could be key to unraveling the molecular mechanism by which cholesterol regulates virulence.
ACKNOWLEDGMENTS
We thank Brenda H. Welter and Amanda M. Goldston for critical reading of the manuscript and helpful discussions. We thank Terri F. Bruce (Clemson University, Clemson, SC) and the Jordan Hall Imaging Facility (JHIF) for microscopy support. We also thank Kimberly Paul for advice regarding lipid extraction.
The project described was supported by grant no. R01AI046414 from the National Institute of Allergy and Infectious Diseases to L.A.T. and a JHIF Mini Grant to A.B.K. Research reported in this publication was also supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant no. P20 GM103444. This material is based upon work supported by CSREES/USDA under project number SC-1700312 (technical contribution no. 6087 of the Clemson University Experiment Station).
The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the views of the National Institute of Allergy and Infectious Diseases, the National Institutes of Health, or the USDA.
Footnotes
Published ahead of print 1 April 2013
REFERENCES
- 1. Laughlin RC, Temesvari LA. 2005. Cellular and molecular mechanisms that underlie Entamoeba histolytica pathogenesis: prospects for intervention. Expert Rev. Mol. Med. 7:1–19 [DOI] [PubMed] [Google Scholar]
- 2. Tavares P, Rigothier MC, Khun H, Roux P, Huerre M, Guillén N. 2005. Roles of cell adhesion and cytoskeleton activity in Entamoeba histolytica pathogenesis: a delicate balance. Infect. Immun. 73:1771–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tavares P, Sansonetti P, Guillén N. 2000. Cell polarization and adhesion in a motile pathogenic protozoan: role and fate of the Entamoeba histolytica Gal/GalNAc lectin. Microb. Infect. 2:643–649 [DOI] [PubMed] [Google Scholar]
- 4. Voigt H, Olivo JC, Sansonetti P, Guillén N. 1999. Myosin IB from Entamoeba histolytica is involved in phagocytosis of human erythrocytes. J. Cell Sci. 112:1191–1201 [DOI] [PubMed] [Google Scholar]
- 5. Byekova YA, Powell RR, Welter BH, Temesvari LA. 2010. Localization of phosphatidylinositol (3,4,5)-trisphosphate to phagosomes in Entamoeba histolytica achieved using glutathione S-transferase- and green fluorescent protein-tagged lipid biosensors. Infect. Immun. 78:125–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arhets P, Gounon P, Sansonetti P, Guillén N. 1995. Myosin II is involved in capping and uroid formation in the human pathogen Entamoeba histolytica. Infect. Immun. 63:4358–4367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tavares P, Sansonetti P, Guillén N. 2000. The interplay between receptor capping and cytoskeleton remodeling in Entamoeba histolytica. Arch. Med. Res. 31:S140–S142 [DOI] [PubMed] [Google Scholar]
- 8. Vargas M, Sansonetti P, Guillén N. 1996. Identification and cellular localization of the actin-binding protein ABP-120 from Entamoeba histolytica. Mol. Microbiol. 22:849–857 [DOI] [PubMed] [Google Scholar]
- 9. Marion S, Tavares P, Arhets P, Guillén N. 2004. Signal transduction through the Gal-GalNAc lectin of Entamoeba histolytica involves a spectrin-like protein. Mol. Biochem. Parasitol. 135:31–38 [DOI] [PubMed] [Google Scholar]
- 10. Guillén N. 1996. Role of signalling and cytoskeletal rearrangements in the pathogenesis of Entamoeba histolytica. Trends Microbiol. 4:191–197 [DOI] [PubMed] [Google Scholar]
- 11. Raucher D, Stauffer T, Chen W, Shen K, Guo S, York JD, Sheetz MP, Meyer T. 2000. Phosphatidylinositol 4,5-bisphosphate functions as a second messenger that regulates cytoskeleton-plasma membrane adhesion. Cell 100:221–228 [DOI] [PubMed] [Google Scholar]
- 12. Berridge MJ. 1984. Inositol trisphosphate and diacylglycerol as second messengers. Biochem. J. 220:345–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB. 1995. Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell 83:1037–1046 [DOI] [PubMed] [Google Scholar]
- 14. Hilgemann DW, Feng S, Nasuhoglu C. 2001. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE 2001(111):re19 doi:10.1126/stke.2001.111.re19 [DOI] [PubMed] [Google Scholar]
- 15. Rohacs T. 2009. Phosphoinositide regulation of non-canonical transient receptor potential channels. Cell Calcium 45:554–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hurley JH, Misra S. 2000. Signaling and subcellular targeting by membrane-binding domains. Sci. STKE 29:49–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Payrastre B, Missy K, Giuriato S, Bodin S, Plantavid M, Gratacap MP. 2001. Phosphoinositides: key players in cell signalling, in time and space. Cell. Signal. 13:377–387 [DOI] [PubMed] [Google Scholar]
- 18. Liu Y, Casey L, Pike LJ. 1998. Compartmentalization of phosphatidylinositol 4,5-bisphosphate in low-density membrane domains in the absence of caveolin. Biochem. Biophys. Res. Commun. 245:684–690 [DOI] [PubMed] [Google Scholar]
- 19. Johnson CM, Chichili GR, Rodgers W. 2008. Compartmentalization of phosphatidylinositol 4,5-bisphosphate signaling evidenced using targeted phosphatases. J. Biol. Chem. 283:29920–29928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson CM, Rodgers W. 2008. Spatial segregation of phosphatidylinositol 4,5-bisphosphate (PIP2) signaling in immune cell functions. Immunol. Endocr. Metab. Agents Med. Chem. 8:349–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bodin S, Welch MD. 2005. Plasma membrane organization is essential for balancing competing pseudopod- and uropod-promoting signals during neutrophil polarization and migration. Mol. Biol. Cell 16:5773–5783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sánchez-Madrid F, Serrador JM. 2009. Bringing up the rear: defining the roles of the uropod. Nat. Rev. Mol. Cell Biol. 10:353–359 [DOI] [PubMed] [Google Scholar]
- 23. Parton RG, Richards AA. 2003. Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic 4:724–738 [DOI] [PubMed] [Google Scholar]
- 24. Goldston AM, Powell RR, Temesvari LA. 2012. Sink or swim: lipid rafts in parasite pathogenesis. Trends Parasitol. 28:417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harris TJC, Awrey DE, Cox BJ, Ravandi A, Tsang A, Siu CH. 2001. Involvement of a Triton-insoluble floating fraction in Dictyostelium cell-cell adhesion. J. Biol. Chem. 276:18640–18648 [DOI] [PubMed] [Google Scholar]
- 26. Laughlin RC, McGugan GC, Powell RR, Welter BH, Temesvari LA. 2004. Involvement of raft-like plasma membrane domains of Entamoeba histolytica in pinocytosis and adhesion. Infect. Immun. 72:5349–5357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mittal K, Welter BH, Temesvari LA. 2008. Entamoeba histolytica: lipid rafts are involved in adhesion of trophozoites to host extracellular matrix components. Exp. Parasitol. 120:127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Welter BH, Goldston AM, Temesvari LA. 2011. Localization to lipid rafts correlates with increased function of the Gal/GalNAc lectin in the human protozoan parasite, Entamoeba histolytica. Int. J. Parasitol. 41:1409–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Diamond LS, Harlow DR, Cunnick CC. 1978. A new medium for the axenic cultivation of Entamoeba histolytica and other Entamoeba. Trans. R. Soc. Trop. Med. Hyg. 72:431–432 [DOI] [PubMed] [Google Scholar]
- 30. Hope H, Pike L. 1996. Phosphoinositides and phosphoinositide-utilizing enzymes in detergent-insoluble lipid domains. Mol. Biol. Cell 7:843–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goldston AM, Powell RR, Koushik AB, Temesvari LA. 2012. Exposure to host ligands correlates with colocalization of Gal/GalNAc lectin subunits in lipid rafts and PIP2 signaling in Entamoeba histolytica. Eukaryot. Cell 11:743–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Powell RR, Temesvari LA. 2004. Involvement of a Rab8-like protein of Dictyostelium discoideum, Sas1, in the formation of membrane extensions, secretion and adhesion during development. Microbiology 150:2513–2525 [DOI] [PubMed] [Google Scholar]
- 33. Welter BH, Laughlin RC, Temesvari LA. 2002. Characterization of a Rab7-like GTPase, EhRab7: a marker for the early stages of endocytosis in Entamoeba histolytica. Mol. Biochem. Parasitol. 121:254–264 [DOI] [PubMed] [Google Scholar]
- 34. Dowler S, Kular G, Alessi DR. 2002. Protein lipid overlay assay. Sci. STKE 2002(129):pl6 doi:10.1126/stke.2002.129.pl6 [DOI] [PubMed] [Google Scholar]
- 35. Powell RR, Welter BH, Hwu R, Bowersox B, Attaway C, Temesvari LA. 2006. Entamoeba histolytica: FYVE-finger domains, phosphatidylinositol 3-phosphate biosensors, associate with phagosomes but not fluid filled endosomes. Exp. Parasitol. 112:221–231 [DOI] [PubMed] [Google Scholar]
- 36. Zaki M, Andrew N, Insall RH. 2006. Entamoeba histolytica cell movement: a central role for self-generated chemokines and chemorepellents. Proc. Natl. Acad. Sci. U. S. A. 103:18751–18756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Watt SA, Kular G, Fleming IN, Downes CP, Lucocq JM. 2002. Subcellular localization of phosphatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of phospholipase C delta1. Biochem. J. 363:657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McLaughlin S, Wang J, Gambhir A, Murray D. 2002. PIP2 and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31:151–175 [DOI] [PubMed] [Google Scholar]
- 39. Espinosa-Cantellano M, Martinez-Palomo A. 1994. Entamoeba histolytica: mechanism of surface receptor capping. Exp. Parasitol. 79:424–435 [DOI] [PubMed] [Google Scholar]
- 40. Millán J, Montoya MC, Sancho D, Sánchez-Madrid F, Alonso MA. 2002. Lipid rafts mediate biosynthetic transport to the T lymphocyte uropod subdomain and are necessary for uropod integrity and function. Blood 99:978–984 [DOI] [PubMed] [Google Scholar]
- 41. Berridge MJ, Irvine RF. 1984. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature 312:315–321 [DOI] [PubMed] [Google Scholar]
- 42. Yamaga M, Kawai K, Kiyota M, Homma Y, Yagisawa H. 2008. Recruitment and activation of phospholipase C (PLC)-delta1 in lipid rafts by muscarinic stimulation of PC12 cells: contribution of p122RhoGAP/DLC1, a tumor-suppressing PLCdelta1 binding protein. Adv. Enzyme Regul. 48:41–54 [DOI] [PubMed] [Google Scholar]
- 43. Carbajal ME, Manning-Cela R, Pina A, Franco E, Meza I. 1996. Fibronectin-induced intracellular calcium rise in Entamoeba histolytica trophozoites: effect on adhesion and the actin cytoskeleton. Exp. Parasitol. 82:11–20 [DOI] [PubMed] [Google Scholar]
- 44. Saito K, Tolias KF, Saci A, Koon HB, Humphries LA, Scharenberg A, Rawlings DJ, Kinet JP, Carpenter CL. 2003. BTK regulates PtdIns-4,5-P2 synthesis: importance for calcium signaling and PI3K activity. Immunity 19:669–677 [DOI] [PubMed] [Google Scholar]
- 45. Szymańska E, Korzeniowski M, Raynal P, Sobota A, Kwiatkowska K. 2009. Contribution of PIP-5 kinase Iα to raft-based FcγRIIA signaling. Exp. Cell Res. 315:981–995 [DOI] [PubMed] [Google Scholar]
- 46. Levental I, Christian DA, Wang Y, Madara JJ, Discher DE, Janmey PA. 2009. Calcium-dependent lateral organization in phosphatidylinositol (4,5) bisphosphate (PIP2)-and cholesterol-containing monolayers. Biochemistry 48:8241–8248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gómez-Móuton C, Abad JL, Mira E, Lacalle RA, Gallardo E, Jiménez-Baranda S, Illa I, Bernad A, Mañes S, Martínez-A C. 2001. Segregation of leading-edge and uropod components into specific lipid rafts during T cell polarization. Proc. Natl. Acad. Sci. U. S. A. 98:9642–9647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Seveau S, Eddy RJ, Maxfield FR, Pierini LM. 2001. Cytoskeleton-dependent membrane domain segregation during neutrophil polarization. Mol. Biol. Cell 12:3550–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lacalle RA, Peregil RM, Albar JP, Merino E, Martínez-A C, Mérida I, Mañes S. 2007. Type I phosphatidylinositol 4-phosphate 5-kinase controls neutrophil polarity and directional movement. J. Cell Biol. 179:1539–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lokuta MA, Senetar MA, Bennin DA, Nuzzi PA, Chan KT, Ott VL, Huttenlocher A. 2007. Type Iγ PIP kinase is a novel uropod component that regulates rear retraction during neutrophil chemotaxis. Mol. Biol. Cell 18:5069–5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heit B, Robbins SM, Downey CM, Guan Z, Colarusso P, Miller BJ, Jirik FR, Kubes P. 2008. PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat. Immunol. 9:743–752 [DOI] [PubMed] [Google Scholar]
- 52. Iijima M, Huang YE, Devreotes P. 2002. Temporal and spatial regulation of chemotaxis. Dev. Cell 3:469–478 [DOI] [PubMed] [Google Scholar]
- 53. Marquay Markiewicz J, Syan S, Hon CC, Weber C, Faust D, Guillén N. 2011. A proteomic and cellular analysis of uropods in the pathogen Entamoeba histolytica. PLoS Negl. Trop. Dis. 5:e1002 doi:10.1371/journal.pntd.0001002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Furuhashi K, Inagaki M, Hatano S, Fukami K, Takenawa T. 1992. Inositol phospholipid-induced suppression of F-actin-gelating activity of smooth muscle filamin. Biochem. Biophys. Res. Commun. 184:1261–1265 [DOI] [PubMed] [Google Scholar]
- 55. Fukami K, Endo T, Imamura M, Takenawa T. 1994. Alpha-actinin and vinculin are PIP2-binding proteins involved in signaling by tyrosine kinase. J. Biol. Chem. 269:1518–1522 [PubMed] [Google Scholar]
- 56. Lee JH, Katakai T, Hara T, Gonda H, Sugai M, Shimizu A. 2004. Roles of p-ERM and Rho-ROCK signaling in lymphocyte polarity and uropod formation. J. Cell Biol. 167:327–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cronshaw DG, Kouroumalis A, Parry R, Webb A, Brown Z, Ward SG. 2006. Evidence that phospholipase C-dependent, calcium-independent mechanisms are required for directional migration of T lymphocytes in response to the CCR4 ligands CCL17 and CCL22. J. Leukoc. Biol. 79:1369–1380 [DOI] [PubMed] [Google Scholar]
- 58. Lawson MA, Maxfield FR. 1995. Ca2+- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature 377:75–79 [DOI] [PubMed] [Google Scholar]
- 59. Mandeville JTH, Maxfield FR. 1996. Calcium and signal transduction in granulocytes. Curr. Opin. Hematol. 3:63–70 [DOI] [PubMed] [Google Scholar]
- 60. Gao Y, Ye LH, Kishi H, Okagaki T, Samizo K, Nakamura A, Kohama K. 2001. Myosin light chain kinase as a multifunctional regulatory protein of smooth muscle contraction. IUBMB Life 51:337–344 [DOI] [PubMed] [Google Scholar]
- 61. Bansal D, Bhatti HS, Sehgal R. 2005. Role of cholesterol in parasitic infections. Lipids Health Dis. 4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Aley SB, Scott WA, Cohn Z. 1980. Plasma membrane of Entamoeba histolytica. J. Exp. Med. 152:391–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Serrano-Luna J, Gutiérrez-Meza M, Mejía-Zepeda R, Galindo-Gómez S, Tsutsumi V, Shibayama M. 2010. Effect of phosphatidylcholine-cholesterol liposomes on Entamoeba histolytica virulence. Can. J. Microbiol. 56:987–995 [DOI] [PubMed] [Google Scholar]
- 64. Saxena A, Kaul D, Vinayak V. 1986. Amoebic erythrophagocytosis: significance of membrane cholesterol to phospholipid ratio. IRCS Med. Sci. 14:330–331 [Google Scholar]
- 65. Sharma R. 1959. Effect of cholesterol on the growth and virulence of Entamoeba histolytica. Trans. R. Soc. Trop. Med. Hyg. 53:278–281 [DOI] [PubMed] [Google Scholar]
- 66. Singh B, Srivastava R, Dutta G. 1971. Virulence of strains of Entamoeba histolytica to rats and the effect of cholesterol, rat caecal and hamster liver passage on the virulence of non-invasive strains. Indian J. Exp. Biol. 9:21–27 [PubMed] [Google Scholar]
- 67. Diamond LS, Phillips BP, Bartgis IL. 1974. A comparison of the virulence of nine strains of axenically cultivated E. histolytica in hamster liver. Arch. Invest. Med. (Mex.) 5(Suppl 2):423–426 [PubMed] [Google Scholar]