

Abstract
The study of human intestinal pathogens has been limited by the lack of methods for the long-term culture of primary human intestinal epithelial cells (PECs). The development of infection models with PECs would allow a better understanding of host-parasite interactions. The objective of this study was to develop a novel method for prolonged in vitro cultivation of PECs that can be used to study Cryptosporidium infection. We isolated intact crypts from human intestines removed during weight loss surgery. The fragments of intestinal layers were cultivated with culture medium supplemented with growth factors and antiapoptotic molecules. After 7 days, the PECs formed self-regenerating cell clusters, forming villi that resemble intestinal epithelium. The PECs proliferated and remained viable for at least 60 days. The cells expressed markers for intestinal stem cells, epithelial cells, and mature enterocytes. The PECs were infected with Cryptosporidium. In contrast to older models in which parasite numbers decay, the burden of parasites increased for >120 h. In summary, we describe here a novel method for the cultivation of self-regenerating human epithelial cells from small intestinal crypts, which contain both intestinal stem cells and mature villus cells. We present data that suggest these cells support Cryptosporidium better than existing cell lines. PECs should provide an improved tool for studying host-parasite interactions involving Cryptosporidium and other intestinal pathogens.
INTRODUCTION
The intestinal epithelium provides a barrier to invasive pathogens but is also the major target for a number of intestinal pathogens, including rotavirus, norovirus, Cryptosporidium, Giardia, Salmonella, and some strains of Escherichia coli (1–4). Together, these pathogens are major causes of acute and persistent watery diarrhea, which remains a major cause of morbidity and mortality worldwide (5). Understanding the interaction between epithelial cells and parasites would facilitate the identification of targets or drugs needed to treat them. However, progress in research on these organisms has been hampered by the limitations of current experimental models (6). Because animal models are suboptimal for some human infections, human colonic cell lines have been used as an alternative to study intestinal pathogens (7, 8). The utility of cell lines is limited by the fact they are not derived from the small intestine and are transformed. In addition, they do not readily support some of the major pathogens, e.g., Cryptosporidium spp., which undergoes incomplete replication, and norovirus, which has not been propagated in vitro (9–11). The culture of primary human intestinal epithelial cells (PECs) that retain human characteristics would be an alternative for studying human intestinal infections. Short-term-cultured PECs have been used to study cryptosporidiosis (12). However, cultured PECs are short-lived cells and undergo apoptosis when cultured in vitro (13). Recent studies have demonstrated the feasibility for the long-term culture of stem cells obtained from adult human intestines; these stem cells can be expanded in a basement membrane matrix such as Matrigel and cultured for long-term periods (14, 15). However, although these models form villi, Matrigel is not optimal for Cryptosporidium infection since the pore size of this matrix limits the access of the sporozoites to target cells. We hypothesized that using intact crypts cultured in medium but supplemented with growth and differentiation factors with antiapoptotic molecules would allow the establishment of a system for long-term cultures of PECs that support the infection of intestinal pathogens. In the present study, we show the feasibility of long-term cultured PECs that can be infected with Cryptosporidium, providing an improved method for in vitro cultivation of the organism.
MATERIALS AND METHODS
PEC culture media.
Wash medium consisted of DMEM/F-12 (Dulbecco modified Eagle medium plus nutrient mixture F-12) supplemented with 5% fetal bovine serum (FBS), and antibiotic-antimycotic (1×) solution (Life Technologies, Carlsbad, CA). Transport medium consisted of wash medium supplemented with recombinant osteoprotegerin (OPG; 100 ng/ml; R&D Systems, Minneapolis, MN). Maintenance medium was transport medium supplemented with epithelial growth factor (50 ng/ml; Life Technologies), noggin (100 ng/ml; R&D Systems), R-spondin (1 μg/ml; R&D Systems), and OPG (100 ng/ml; R&D Systems). Differentiation medium consisted of enterocyte differentiation medium containing DMEM plus butyrate (Becton Dickinson, Franklin Lakes, NJ) supplemented with epithelial growth factor, noggin, and R-spondin.
Isolation and culture of PECs.
Jejunal tissues were removed during gastric bypass surgery performed for weight reduction. Jejunal tissues that would otherwise have been discarded were obtained with IRB approval. In the operating suite tissue was placed in a 50-ml conical tube containing 25 ml of transport medium and then transported immediately to the laboratory on ice. Within 3 h, the tissue was washed, the tube was inverted gently to decant the supernatant, and washing medium was added (three times). The tissue was dissected with a scalpel, the mucosal surface was exposed, and the superficial mucus was removed mechanically with a cell scraper (Corning Life Sciences, Manassas, VA). The mucosal surface was covered with 5 mM EDTA–1× phosphate-buffered saline for 3 min at room temperature; sheets of epithelial cells, including intact crypts, were removed mechanically with a scalpel; and 10 ml of washing medium was added to obtain the layers of cells. The medium and cells were aspirated and transferred to conical tubes. After centrifugation (40 × g, 10 min), the supernatant containing isolated cells was discarded. The pellet was resuspended in 5 ml of red blood cell lysis solution (8.3 g of ammonium chloride/liter in 0.01 M Tris-HCl for 10 min at room temperature). After incubation, 20 ml of washing medium was added to dilute the lysis buffer. The sample was centrifuged (40 × g, 10 min), and then the supernatant was discarded. The pellet was resuspended in 20 ml of washing medium and centrifuged (40 × g, 10 min) three times, and the sheets containing epithelial cells and crypts were gently scraped from the bottom and aspirated with a micropipette tip (size, 1 ml) with the end of the tip previously cut with a scalpel. The cells were gently resuspended in 5 ml of maintenance medium and plated in 12-well culture plates in 250 μl/well (37°C, 5% CO2, 7 days). The maintenance medium was carefully replaced daily. After 7 days, the cells were harvested by aspiration and centrifugation (40 × g, 10 min). The supernatant containing dead cells was discarded, and the epithelial cell layers were concentrated and plated in 96-well round-bottom plates (∼100 fragments in 50 μl of maintenance medium per well; 37°C, 5% CO2. Additional maintenance medium (25 μl) was added every other day for up to 60 days. For the characterization experiments, maintenance medium was replaced with 25 μl of differentiation medium 48 h before the cells were harvested for characterization.
Viability assays, histology, and electron microscopy.
The viability of the PECs was assessed by a two-color fluorescence cell viability assay using a Live/Dead viability kit (Life Technologies) to detect calcein (live cells) and ethidium homodimer (dead cells) by fluorescence microscopy at 495 nm. For histological analysis, the PECs were fixed in 10% formalin and embedded in paraffin wax; for infection experiments, the PECs were challenged C. parvum sporozoites, and then the infected PECs were fixed as described above. Sections were stained with methylene blue or hematoxylin-eosin and examined by fluorescence. For transmission electron microscopy, the PECs were fixed (2.5% paraformaldehyde and 0.1% glutaraldehyde in 0.05 M cacodylate buffer [pH 7.3] and with 0.03% trinitrophenol and 0.03% CaCl2), washed (0.1 M cacodylate buffer), scraped off the flasks, and pelleted. The pellets were postfixed (1% OsO4 in 0.1 M cacodylate buffer), stained (1% uranyl acetate in 0.1 M maleate buffer), dehydrated in ethanol, and embedded (Poly/Bed 812; Polysciences, Warrington, PA). Ultrathin sections were cut using a Reichert-Leica Ultracut S ultramicrotome, stained with lead citrate, and examined by transmission electron microscopy using a Philips 201 or CM-100 electron microscope at 60 kV.
Immunofluorescence and ELISA.
The PECs were characterized by immunofluorescence using fluorescein isothiocyanate (FITC) anti-pan-cytokeratin as a marker of epithelial cells (Sigma-Aldrich, St. Louis, MO) and FITC anti-vimentin as a marker for fibroblasts (Sigma-Aldrich). For parasite detection, we used the C. parvum gp900 and gp40 antibody (GenWay Biotech, San Diego, CA) at a working dilution of 1:50 and phalloidin-Alexa Fluor 568 (Life Technologies) at a concentration of 1:1,000. Cell proliferation was evaluated using Alexa Fluor 647-mouse anti-human Ki67 (BD Biosciences). The production of alkaline phosphatase was measured in the supernatants of cells from days 30 to 35 in culture using the SensoLyte pNPP alkaline phosphatase enzyme-linked immunosorbent assay (ELISA) kit according to the instructions of the manufacturer (AnaSpec, Fremont, CA).
RNA extraction and PCR assays.
PECs were harvested by centrifugation (40 × g, 10 min), the supernatant was discarded, and the pellets were stored frozen (−20 C°). The frozen cells were suspended in guanidine buffer and disrupted with QIAshredder columns (Qiagen, Valencia, CA). RNA was isolated using the RNeasy Kit Plus (Qiagen). The purity and concentration were analyzed by spectrophotometry (NanoDrop 1000; Thermo Scientific, Pittsburgh, PA). The presence of RNA of epithelial cell markers was analyzed by using a one-step real-time reverse transcription-PCR (RT-PCR) Sybr green kit (Life Technologies). Epithelial cell RNA (25 ng) extracted from 30-day-old PECs, freshly isolated intestinal cells, HCT-8 cells, and fibroblasts was analyzed using previously described primers for cytokeratin 8, vimentin, aldolase, mucin, lysozyme, LgR5, Ki67, and human 18S rRNA (16–18). After reverse transcription (50°C, 15 min), denaturing (95°C, 5 min), and 40 cycles of melting (95°C, 30 s) and annealing-extension (60°C, 1 min), the amplicons were detected by electrophoresis in agarose gels at 1% (Fig. 2). For infection experiments, the parasite burden was quantified by RT-PCR from infected PECs obtained at different time points (see below). For RT-PCR amplification, we used 100 ng of RNA as a template, and we used the method and the specific primers previously reported to detect 18S rRNA (19). To analyze the number of parasites, we used the ABI Prism software using as a reference the threshold cycle (CT) values obtained from a standard curve prepared with known numbers of C. parvum oocysts ranging from 102 to 106 organisms. To compare infection efficiencies, HCT-8 cells were infected as described above, and the numbers of parasites were determined as detailed above. Heat-killed (95°C) Cryptosporidium sporozoites were used as negative controls, and human 18S rRNA was used as a reference gene to normalize the samples.
Fig 2.
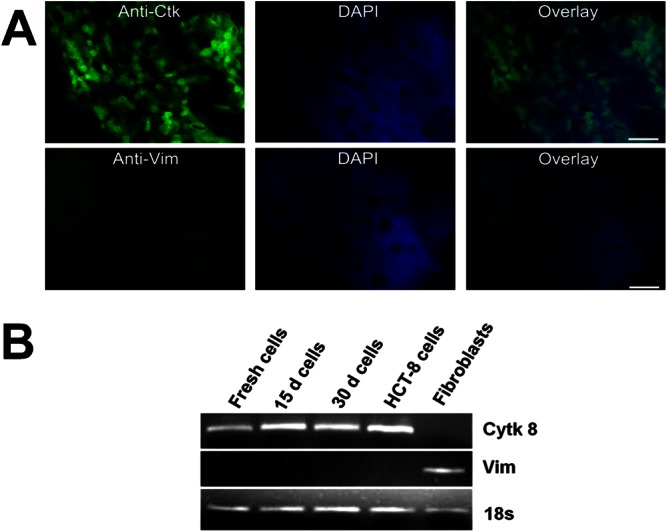
Cytokeratin expression in cultured PECs as determined by immunofluorescence and RT-PCR. (A) The expression of cytokeratin (Ctk) and vimentin (Vim) was analyzed by using immunofluorescence. Cultured PECs stained with anti-cytokeratin-FITC (top row, left) or anti-vimentin FITC (bottom row, left) were counterstained with DAPI (4′,6′-diamidino-2-phenylindole; top and bottom center); overlaid images are shown at the top and bottom right. Scale bar, ∼40 μm. (B) RT-PCR analysis in an 1% agarose gel shows the expression of cytokeratin 8 (Ctk 8), vimentin (Vim), and 18S rRNA (18s) over time.
C. parvum infection.
C. parvum oocysts, Iowa strain (Sterling Parasitology Laboratory, University of Arizona, Tucson, AZ), were stained with CFSE (carboxyfluorescein diacetate succinimidyl ester; CellTrace CFSE cell proliferation kit; Life Technologies) and excysted by using acidified water followed by bile salts (7). After intact oocysts by were removed centrifugation (500 × g, 3 min), sporozoites were suspended in culture media, and filtered through a 3-μm filter (Millipore, Billerica, MA). No oocysts were seen in the filtered medium by microscopy. Sporozoites (2 × 104) were added to each well containing PECs and incubated (5% CO2, 37°C); the supernatant was removed after 3 h of infection, and then fresh infection medium (maintaining medium supplemented with 0.8% taurocholate) was added daily up to harvesting. For fluorescence microscopy, unfixed cells from the supernatant were analyzed 120 h after the infection. The parasite burden was assessed by RT-PCR, as we described previously, using total RNA extracted from infected PECs (including supernatant) at 0, 24, 48, 72, 96, and 120 h after infection.
RESULTS
PECs remain viable for up to 60 days and display an epithelial structure.
We were able to detect viable PECs from surgical specimens in the vast majority of cases (replicated >15 times). During the beginning of cell isolation, a large number of dead cells and small PEC fragments were found in the supernatant, but these were discarded during subsequent washing steps. Then, after 1 week of culture, the layers containing PECs were concentrated in 96-well plates, and the viability was monitored. After 2 months of culture, the viability assay carried out with attached, unfixed PECs showed that the cells all stained with the vital dye (Fig. 1A), which indicates that the cells remained viable for at least 60 days. In contrast, heat-killed cells did not stain with the vital dye but stained with the dye for dead cells (Fig. 1B). Histological sections of the cultured PECs confirmed the presence of crypts and villi (Fig. 1C) with typical enterocytes and goblet cells (Fig. 1D), which suggested that PECs contained a mixed population of epithelial cells.
Fig 1.
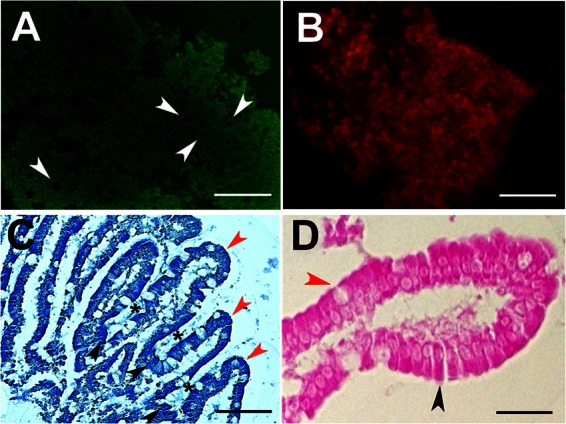
Viability and morphology of cultured PECs. (A) The viability of cultured PECs was determined by a Live/Dead fluorescence assay and by examining unfixed cells costained with the vital dye calcein (green) and the marker of dead cells ethidium homodimer (red) at 488 nm (no dead cells are observed). (B) As a control, heat-killed cells are also shown. Scale bars, ∼100 μm. (C) Histological analysis of cultured cells. Paraffin wax sections of the PECs stained in methylene blue, villus (red arrowheads), and crypts (black arrowheads) are shown. The space corresponding to the mouth of the crypts is indicated (black asterisks). Scale bar, ∼50 μm. (D) Morphologies of PECs, enterocytes with a typical cylinder shape (black), and goblet cells with the typical rounded shape (red) are indicated by arrowheads. Scale bar, ∼30 μm.
Cultured PECs show epithelial cell characteristics.
To confirm that the PECs were in fact epithelial cells, we stained the cultured PECs with fluorescent antibodies to cytokeratin and vimentin. Nearly all of the cells stained for cytokeratin, a marker for epithelial cells (Fig. 2A, top) but not for vimentin, a marker for fibroblasts (Fig. 2A, bottom). As shown in Fig. 2B, cytokeratin expression persisted at similar levels throughout the time in culture, which indicates that the daughter cells produced during cultivation maintain an epithelial phenotype (Fig. 2B). The levels of cytokeratin were similar to those observed in fresh PECs and HCT-8 cells.
Cultured PECs shows proliferation and differentiation markers.
By electron microscopy, we showed that the cultured PECs expressed both tight junctions and microvilli (Fig. 3A). Within the cultured PECs, some cells express the proliferation marker Ki67 (Fig. 3B). We detected markers of stem cells (LGR5) and proliferating cells (Ki67) by PCR (Fig. 3C). We also detected markers for differentiated villus cells, including aldolase, mucin, and lysozyme (Fig. 3C). The supernatants of 30-day-old PECs contained alkaline phosphatase, as is expected of mature epithelial cells (Fig. 3D). Thus, the cultured cells express markers for both long-lived stem cells and differentiated cells.
Fig 3.
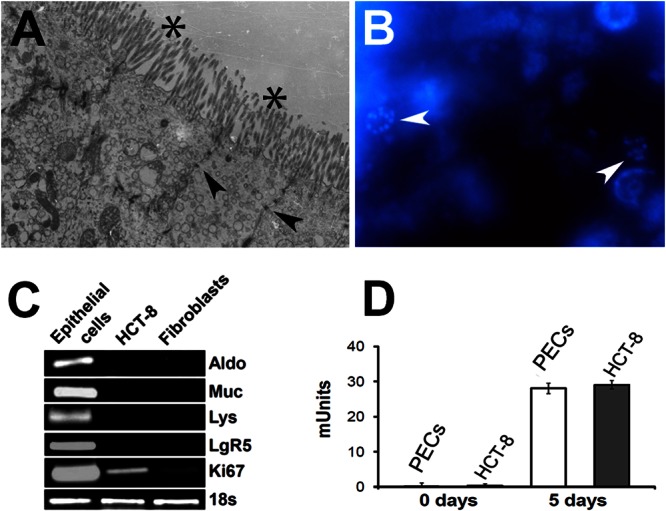
Markers of proliferation and differentiation in cultured PECs. (A) An electron micrograph of cultured cells shows the presence of tight junctions (arrowheads) and microvilli (asterisks). (B) The nuclear proliferation marker Ki67 was detected by immunofluorescence using the monoclonal antibody anti-Ki67-PE (arrowheads). Cells were counterstained with DAPI. Scale bar, ∼10 μm. (C) Differentiation and proliferation markers detected by PCR using aldolase (Aldo), mucin (Muc), lysozyme (Lys), leucine-rich receptor (LgR5), Ki67, and 18S rRNA (18s). (D) Alkaline phosphatase production by cultured PECs. The alkaline phosphatase activity expressed in milliunits from supernatants collected at day 0 (baseline) or day 5 from 30-day-old cultured PECs was determined.
C. parvum infection.
We challenged the PECs with Cryptosporidium sporozoites previously stained with CFSE in order to track the formation of later stages and, as shown in Fig. 4A, no oocyst-like structures were observed after filtration and before the inoculation. As expected because of the low numbers in the initial inoculum, we observed very few intracellular forms during the first few days of infection (Fig. 4H); however, as evident in Fig. 4B, we show that the supernatant obtained after 5 days of infection contained a large number of parasites positively stained for CFSE (Fig. 4B). Since sporozoites die few hours after the excystation, we concluded that the detection of stained intracellular forms and stained parasites in the supernatant must be the result of parasite proliferation. Microscopic analysis of the forms found in the supernatant revealed the different morphologies among stained forms; we detected parasites with shapes and sizes similar to those found in sporozoites and merozoites (Fig. 4C), and we detected non-nucleated parasites with characteristics similar to those of trophozoites (Fig. 4D). We observed parasites multinucleated with a crescent shape, which could correspond to a meronte-like structure (Fig. 4D). Oocysts, the infective product of sexual reproductive, were also noted in the supernatants (Fig. 4F). Since Cryptosporidium RNA is stable for just a few hours after the parasites die, this finding has been used to determine parasite viability (20); increases in the RNA level over the time should thus be the result of parasite proliferation. Therefore, to determine the growth characteristics of the parasites, we compared the parasite burdens by quantitative reverse transcriptase real-time PCR for parasite RNA. Infection of HCT-8 cells (the cell line said to best support Cryptosporidium) resulted in a gradual decay of the amount of parasite RNA. In contrast, parasite RNA increased after the infection of primary epithelial cells for periods of at least 120 h (Fig. 4G). Immunofluorescence (IF) staining with anti-cryptosporidium antibody showed the presence of intracellular forms in sections of PECs cultured for 120 h (Fig. 4H) and confirmed the presence of Cryptosporidium in cultured PECs. The parasite staining was similar to that observed in previous reports of HTC-8 cells infected with Cryptosporidium (21).
Fig 4.

PECs infected with C. parvum. (A) Sporozoites were filtered and stained with CFSE before infection. (B) After 5 days of infection, the supernatant showed (objective; ×40 magnification) abundant stained parasites containing oocyst-like structures. (C to F) The supernatant was analyzed in detail (objective; ×100 magnification) showing the presence of merozoites (C), trophozoites (D), meronts (E), and mature oocysts (F). Scale bars: 10 μm, A and B; 1 μm, C to F. (G) Quantification of the number of parasites in infected PECs by real-time RT-PCR. Low inocula of filtered sporozoites (102) were added to 2-week-old cultured PECs (red and blue) and HCT-8 cells (black). The numbers of parasites were evaluated for up to 5 days. Red and blue lines indicate the means of two independent experiments (standard deviations of PCR triplicates are not observed in the figure). The total numbers of parasites in the sample were determined using a standard curve with a known number of parasites. (H) Infection of PECs was confirmed by immunofluorescence by detecting anti-Cryptosporidium monoclonal antibody, indicated in green.
DISCUSSION
The first objective in this study was to develop a system to establish a long-term culture of PECs that retains the phenotypic properties of an epithelial layer, including progenitor cells, proliferative cells, differentiated cells, and senescent cells. A substantial obstacle in the cultivation of intestinal epithelial cells is the onset of apoptosis (anoikis) when epithelial cells are detached from the basement membrane (22). Previous studies have shown the utility of antiapoptotic molecules to extend the life of intestinal primary cells after detaching them from the intestinal tissue (14, 23). Thus, here we added recombinant osteoprotegerin (OPG) during the transport of the tissue and immediately after PEC isolation to prevent anoikis, OPG is a tumor necrosis factor alpha receptor family member, which acts as a decoy by binding TRAIL and TRANCE, blunting the apoptosis pathway (24–26). We previously demonstrated that intestinal epithelial cells produce OPG in response to Cryptosporidium infection (25). We reasoned that OPG might improve the survival of cells, especially during the first 48 h of culture. Although we observed a high rate of death among cells on days 1 to 3, we observed over time that after OPG addition, a large number of PECs survived beyond several days of culture. Thus, the dying cells observed in the first several days are likely terminal differentiated enterocytes. On the other hand, undifferentiated cells or cells in the early process of differentiation survived. Thus, OPG might be a critical molecule to consider for preventing apoptosis in the beginning of long-term PEC cultures. Despite the addition of antiapoptotic factors, normal epithelial cells turn over every 7 days. Thus, the presence of viable cells observed in prolonged culture (after 1 week) is likely to be related to proliferation rather than to extension of the life span of partially differentiated cells due to OPG. During the isolation of PECs, we isolated intact crypts. We hypothesized that the cultured clusters of PECs should contain mixed populations of epithelial cells, including stem cells. Intestinal stem cells can be expanded in vitro when cultured in the presence of Wnt pathway activators such as noggin and R-spondin (14, 23, 27, 28). Therefore, we added here recombinant noggin and R-spondin proteins to induce proliferation in the cultured PECs. We confirmed the presence of stem cells in long-term-cultured PECs by staining and PCR for LGR5 and Ki67. Intestinal stem cells have the capacity to produce all four types of intestinal cells (29). We hypothesized that differentiation factors could be used to direct and control the differentiation of expanded populations of stem cells present in the culture. We used medium containing butyrate to induce differentiation. Butyrate is a short-chain fatty acid normally produced by commensal bacteria that enhances the functional differentiation of enterocytes (30) via the PTEN/phosphoinositide 3-kinase pathway (31). In cells cultured with butyrate, we noted the presence of live cells with crypt and villus structures, which expressed markers of differentiated cells, including microvilli, tight junctions, and alkaline phosphatase production. The histology showed the presence of well-differentiated enterocytes, as well as goblet cells. This contrasts with studies of epithelial cells differentiated from stem cells. In the latter case, the cells grow in organoids (cyst-like structures). The latter have some markers of mature cells but do not form complete villus structures. Thus, we demonstrated here that PECs can be successfully propagated in vitro for a period of at least 60 days and also produce markers of mature villi.
The second objective in the present study was to demonstrate the feasibility of infecting long-term-cultured PECs with the intestinal parasite Cryptosporidium. PECs have a number of advantages over cancer cell lines since they may more accurately reflect in vivo conditions than do immortalized cells. In particular, primary cells allow direct and meaningful examination ex vivo of species tropism and the importance of specific-receptor ligand interaction. In previous studies we used human intestinal tissues as explants as an alternative for studying Cryptosporidium infection (25, 32). However, we have observed a low rate of infection even when large numbers of parasites are used, and we have also observed a high rate of apoptosis even in uninfected cells after 48 h of cultivation. We hypothesized that improvement in PEC culture could lead to an improved in vitro model for cryptosporidiosis. To demonstrate that PECs support Cryptosporidium infection, we challenged the PECs with low numbers of filtered C. parvum sporozoites in order to detect larger amounts of parasites after culture. Since it is difficult to characterize the infection by electron microscopy because of the low numbers of infected cells (due to the inocula) especially during the first few days of infection, we used more sensitive methods such as RT-PCR to track the infection over the time (Fig. 4G). In contrast to cell lines, we noted proliferation, as demonstrated by drastically increasing parasite RNA levels for >120 h. The quantity of parasite mRNA was twice as high as in HCT-8 cells, the cell line that best supports Cryptosporidium in vitro (33). In addition, in contrast to previous models, we present evidence here that Cryptosporidium is completing its life cycle; this evidence is based on the fact that the infection is initiated with just sporozoites (prestained with a tracking dye), and then we observed stained asexual forms, such as merozoites and trophozoites (probably detached from death cells), and sexual forms, including thin wall oocysts in the supernatant after 120 h of infection (Fig. 4C to F). In conclusion, we were able to produce a stable system to culture PECs that are able to persist in culture for at least 60 days. Our model supports Cryptosporidium better than previous in vitro models. Since the cultured PECs retained intestinal characteristics, we anticipate that the method described here should provide an improved tool for studying host-parasite interactions involving Cryptosporidium and other intestinal pathogens.
ACKNOWLEDGMENTS
We thank Mary K. Estes and Don Powell for their valuable suggestions and comments regarding the manuscript.
This study was conducted with the support of the Institute for Translational Sciences (ITS) at the University of Texas Medical Branch at Galveston, supported in part by a Clinical and Translational Science Award (UL1TR000071) from the National Center for Advancing Translational Sciences, National Institutes of Health.
Footnotes
Published ahead of print 18 March 2013
REFERENCES
- 1. Sartor RB. 2008. Microbial influences in inflammatory bowel diseases. Gastroenterology 134:577–594 [DOI] [PubMed] [Google Scholar]
- 2. Deng M, Rutherford MS, Abrahamsen MS. 2004. Host intestinal epithelial response to Cryptosporidium parvum. Adv. Drug Deliv. Rev. 56:869–884 [DOI] [PubMed] [Google Scholar]
- 3. Ciarlet M, Estes MK. 2001. Interactions between rotavirus and gastrointestinal cells. Curr. Opin. Microbiol. 4:435–441 [DOI] [PubMed] [Google Scholar]
- 4. Eckmann L. 2003. Mucosal defences against Giardia. Parasite Immunol. 25:259–270 [DOI] [PubMed] [Google Scholar]
- 5. Hodges K, Gill R. 2010. Infectious diarrhea: cellular and molecular mechanisms. Gut Microbes 1:4–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kothavade RJ. 2011. Challenges in understanding the immunopathogenesis of Cryptosporidium infections in humans. Eur. J. Clin. Microbiol. Infect. Dis. 30:1461–1472 [DOI] [PubMed] [Google Scholar]
- 7. Feng H, Nie W, Sheoran A, Zhang Q, Tzipori S. 2006. Bile acids enhance invasiveness of Cryptosporidium spp. into cultured cells. Infect. Immun. 74:3342–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang YL, Buck GA, Widmer G. 2010. Cell sorting-assisted microarray profiling of host cell response to Cryptosporidium parvum infection. Infect. Immun. 78:1040–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Karanis P, Aldeyarbi HM. 2011. Evolution of Cryptosporidium in vitro culture. Int. J. Parasitol. 41:1231–1242 [DOI] [PubMed] [Google Scholar]
- 10. Duizer E, Schwab KJ, Neill FH, Atmar RL, Koopmans MP, Estes MK. 2004. Laboratory efforts to cultivate noroviruses. J. Gen. Virol. 85:79–87 [DOI] [PubMed] [Google Scholar]
- 11. King BJ, Keegan AR, Robinson BS, Monis PT. 2011. Cryptosporidium cell culture infectivity assay design. Parasitology 138:671–681 [DOI] [PubMed] [Google Scholar]
- 12. Hashim A, Mulcahy G, Bourke B, Clyne M. 2006. Interaction of Cryptosporidium hominis and Cryptosporidium parvum with primary human and bovine intestinal cells. Infect. Immun. 74:99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kaeffer B. 2002. Mammalian intestinal epithelial cells in primary culture: a mini-review. In Vitro Cell Dev. Biol. Anim. 38:123–134 [DOI] [PubMed] [Google Scholar]
- 14. Jung P, Sato T, Merlos-Suarez A, Barriga FM, Iglesias M, Rossell D, Auer H, Gallardo M, Blasco MA, Sancho Clevers EH, Batlle E. 2011. Isolation and in vitro expansion of human colonic stem cells. Nat. Med. 17:1225–1227 [DOI] [PubMed] [Google Scholar]
- 15. Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, Hoskins EE, Kalinichenko VV, Wells SI, Zorn AM, Shroyer NF, Wells JM. 2011. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470:105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. George MD, Wehkamp J, Kays RJ, Leutenegger CM, Sabir S, Grishina I, Dandekar S, Bevins CL. 2008. In vivo gene expression profiling of human intestinal epithelial cells: analysis by laser microdissection of formalin fixed tissues. BMC Genomics 9:209 doi:10.1186/1471-2164-9-209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Murayama M, Okamoto R, Tsuchiya K, Akiyama J, Nakamura T, Sakamoto N, Kanai T, Watanabe M. 2009. Musashi-1 suppresses expression of Paneth cell-specific genes in human intestinal epithelial cells. J. Gastroenterol. 44:173–182 [DOI] [PubMed] [Google Scholar]
- 18. Rochelle PA, De Leon R, Stewart MH, Wolfe RL. 1997. Comparison of primers and optimization of PCR conditions for detection of Cryptosporidium parvum and Giardia lamblia in water. Appl. Environ. Microbiol. 63:106–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cai X, Woods KM, Upton SJ, Zhu G. 2005. Application of quantitative real-time reverse transcription-PCR in assessing drug efficacy against the intracellular pathogen Cryptosporidium parvum in vitro. Antimicrob. Agents Chemother. 49:4437–4442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fontaine M, Guillot E. 2003. Study of 18S rRNA and rDNA stability by real-time RT-PCR in heat-inactivated Cryptosporidium parvum oocysts. FEMS Microbiol. Lett. 226:237–243 [DOI] [PubMed] [Google Scholar]
- 21. Tosini F, Agnoli A, Mele R, Gomez Morales MA, Pozio E. 2004. A new modular protein of Cryptosporidium parvum, with ricin B and LCCL domains, expressed in the sporozoite invasive stage. Mol. Biochem. Parasitol. 134:137–147 [DOI] [PubMed] [Google Scholar]
- 22. Hofmann C, Obermeier F, Artinger M, Hausmann M, Falk W, Schoelmerich J, Rogler G, Grossmann J. 2007. Cell-cell contacts prevent anoikis in primary human colonic epithelial cells. Gastroenterology 132:587–600 [DOI] [PubMed] [Google Scholar]
- 23. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. 2009. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459:262–265 [DOI] [PubMed] [Google Scholar]
- 24. Vidal K, Serrant P, Schlosser B, van den Broek P, Lorget F, Donnet-Hughes A. 2004. Osteoprotegerin production by human intestinal epithelial cells: a potential regulator of mucosal immune responses. Am. J. Physiol. Gastrointest Liver Physiol. 287:G836–G844 [DOI] [PubMed] [Google Scholar]
- 25. Castellanos-Gonzalez A, Yancey LS, Wang HC, Pantenburg B, Liscum KR, Lewis DE, White AC., Jr 2008. Cryptosporidium infection of human intestinal epithelial cells increases expression of osteoprotegerin: a novel mechanism for evasion of host defenses. J. Infect. Dis. 197:916–923 [DOI] [PubMed] [Google Scholar]
- 26. Toruner M, Fernandez-Zapico M, Sha JJ, Pham L, Urrutia R, Egan LJ. 2006. Antianoikis effect of nuclear factor-κB through upregulated expression of osteoprotegerin, BCL-2, and IAP-1. J. Biol. Chem. 281:8686–8696 [DOI] [PubMed] [Google Scholar]
- 27. Ootani A, Li X, Sangiorgi E, Ho QT, Ueno H, Toda S, Sugihara H, Fujimoto K, Weissman IL, Capecchi MR, Kuo CJ. 2009. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med. 15:701–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Haramis AP, Begthel H, van den Born M, van Es J, Jonkheer S, Offerhaus GJ, Clevers H. 2004. De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science 303:1684–1686 [DOI] [PubMed] [Google Scholar]
- 29. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. 2007. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449:1003–1007 [DOI] [PubMed] [Google Scholar]
- 30. Peng L, Li ZR, Green RS, Holzman IR, Lin J. 2009. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 139:1619–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bai Z, Zhang Z, Ye Y, Wang S. 2010. Sodium butyrate induces differentiation of gastric cancer cells to intestinal cells via the PTEN/phosphoinositide 3-kinase pathway. Cell Biol. Int. 34:1141–1145 [DOI] [PubMed] [Google Scholar]
- 32. Dann SM, Wang HC, Gambarin KJ, Actor JK, Robinson P, Lewis DE, Caillat-Zucman S, White AC., Jr 2005. Interleukin-15 activates human natural killer cells to clear the intestinal protozoan cryptosporidium. J. Infect. Dis. 192:1294–1302 [DOI] [PubMed] [Google Scholar]
- 33. Upton SJ, Tilley M, Brillhart DB. 1994. Comparative development of Cryptosporidium parvum (Apicomplexa) in 11 continuous host cell lines. FEMS Microbiol. Lett. 118:233–236 [DOI] [PubMed] [Google Scholar]