

Abstract
We previously demonstrated that bacterial flagellar motility is a fundamental mechanism by which host phagocytes bind and ingest bacteria. Correspondingly, loss of bacterial motility, consistently observed in clinical isolates from chronic Pseudomonas aeruginosa infections, enables bacteria to evade association and ingestion of P. aeruginosa by phagocytes both in vitro and in vivo. Since bacterial interactions with the phagocyte cell surface are required for type three secretion system-dependent NLRC4 inflammasome activation by P. aeruginosa, we hypothesized that reduced bacterial association with phagocytes due to loss of bacterial motility, independent of flagellar expression, will lead to reduced inflammasome activation. Here we report that inflammasome activation is reduced in response to nonmotile P. aeruginosa. Nonmotile P. aeruginosa elicits reduced IL-1β production as well as caspase-1 activation by peritoneal macrophages and bone marrow-derived dendritic cells in vitro. Importantly, nonmotile P. aeruginosa also elicits reduced IL-1β levels in vivo in comparison to those elicited by wild-type P. aeruginosa. This is the first demonstration that loss of bacterial motility results in reduced inflammasome activation and antibacterial IL-1β host response. These results provide a critical insight into how the innate immune system responds to bacterial motility and, correspondingly, how pathogens have evolved mechanisms to evade the innate immune system.
INTRODUCTION
Pseudomonas aeruginosa is a Gram-negative bacterium that causes acute and chronic infections in immunocompromised humans. P. aeruginosa is well characterized to cause chronic pulmonary infections in cystic fibrosis (CF) patients which lead to inflammation and progressive pulmonary damage, with proinflammatory cytokines, including interleukin1β (IL-1β), elevated in the bronchoalveolar lavage fluid and the sputum (1). IL-1β leads to inflammatory cell recruitment and amplification of proinflammatory cytokines that result in bactericidal responses to P. aeruginosa. Correspondingly, eradication of P. aeruginosa leads to a decrease in the levels of IL-1β (1).
The potent IL-1β response to P. aeruginosa is triggered by the NLRC4 (nucleotide-binding domain, leucine-rich repeat-containing family, caspase-associated recruitment domain 4) inflammasome (2–4). The NLRC4 inflammasome is a multiprotein complex comprised of the proteins NLRC4, ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain), and procaspase-1 (5, 6). Inflammasome activation leads to the cleavage of procaspase-1 to activated caspase-1 that induces the processing of pro-IL-1β and pro-IL-18, which are released as mature IL-1β and IL-18 (6). NLRC4 inflammasome activation also leads to an inflammatory cell death known as pyroptosis. Although NLRC4-dependent cytokine processing in response to P. aeruginosa is dependent on ASC, pyroptosis is independent of ASC (2, 3).
Many Gram-negative pathogens encode a molecular syringe-like complex called the type three secretion system (T3SS), used for injecting effector proteins into the host cell cytosol to promote virulence (7, 8). The T3SS genes of P. aeruginosa are induced upon contact with host cells (9, 10), and the NLRC4 inflammasome subsequently responds to T3SS-dependent injection of FliC, the monomeric subunit of the flagellum, and PscI, the T3SS basal body rod component, into the host cell (2–4, 11). Pilin, a major component of the type IV bacterial pilus, has also been proposed to lead to inflammasome activation via the T3SS (12). Since T3SS-dependent activation of the NLRC4 inflammasome relies upon bacterial apposition with the macrophage cell surface, we hypothesized that mechanisms which would reduce bacterial association with macrophages would lead to reduced activation of the inflammasome.
Loss of flagellar motility by either downregulation or loss of genes required for flagellar motility leads to persistence of P. aeruginosa in chronically infected patients (13, 14). We recently demonstrated that loss of flagellar motility, independent of loss of flagellar structure, enables bacteria to evade cell surface interactions and subsequent phagocytosis by macrophages (15). In this work, we report that loss of flagellar motility by P. aeruginosa leads to reduced IL-1β production, caspase-1 activation, and cell death. We show that inflammasome activation is dependent on bacterial contact with host cells, and cytokine processing is independent of bacterial phagocytosis. We further show that loss of flagellar motility leads to reduced caspase-1 activation in peritoneal macrophages. Using a peritonitis model, we demonstrate that nonmotile P. aeruginosa elicits a reduced IL-1β response in vivo in comparison to that induced by wild-type (WT) motile P. aeruginosa. Our data provide new insights into the role of flagellar motility in the modulation of inflammatory responses and, specifically, in induction of inflammasome activation.
MATERIALS AND METHODS
Mice.
Six- to 8-week-old mice on the C57BL/6 background were used for all experiments. C57BL/6 WT mice were obtained from the National Cancer Institute (Bethesda, MD). ASC−/− mice have been described previously (16) and were obtained from V. Dixit (Genentech, CA). Caspase-1−/− mice have been described previously (17). This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Research Council (18). The protocol was approved by the Dartmouth IACUC.
Materials.
The DuoSet enzyme-linked immunosorbent assay (ELISA) kit for mouse IL-1β, which contains antibodies raised against recombinant IL-1β, was purchased from R&D Systems (Minneapolis, MN). Lipopolysaccharide (LPS), ATP disodium salt hydrate (ATP), cytochalasin D, and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) were purchased from Sigma-Aldrich (St. Louis, MO). FAM-YVAD-FMK FLICA caspase-1 stain was purchased from ImmunoChemistry Technologies (Bloomington, MN). Propidium iodide was purchased from MP Biomedicals (Solon, OH). Allophycocyanin (APC)-conjugated anti-mouse CD45 antibody (clone 30-F11) was purchased from eBioscience (San Diego, CA). Phycoerythrin (PE)-conjugated anti-mouse Ly6G antibody (clone 1A8), APC-conjugated anti-mouse F4/80 antibody (clone BM8), and anti-β-actin antibody (clone 2F1-1) were purchased from BioLegend (San Diego, CA). Polyclonal anti-mouse IL-1β (AF-401-NA) was purchased from R&D Systems, and polyclonal anti-mouse caspase-1 (SC-514) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Fetal bovine serum was purchased from Atlanta Biologicals (Lawrenceville, GA).
Cells.
Bone marrow-derived dendritic cells (BMDC) were cultured using a modification of the protocol of Inaba et al. (19) as previously described (20). For isolation of murine macrophages, naive C57BL/6 mice were intraperitoneally injected with 1 ml of 4% thioglycolate solution. Mice were sacrificed 3 to 4 days later, and macrophages were harvested by peritoneal lavage. The lavage fluid was centrifuged, and cells were resuspended in ammonium chloride and potassium lysis buffer to lyse red blood cells. Cells were washed twice using phosphate-buffered saline (PBS), and pelleted cells were resuspended in antibiotic-free RPMI 1640 supplemented with 10% fetal bovine serum.
Bacteria.
P. aeruginosa strains on the PA14 background were provided by G. O'Toole and D. Hogan (Dartmouth Medical School, Hanover, NH). PA14 motAB motCD and fliC mutants have been described previously (21, 22). The construct used for the generation of the P. aeruginosa PAO1 popB mutant has been described previously (23) and was cloned into P. aeruginosa strain PA14. P. aeruginosa strains on the PAK background were provided by B. Kazmierczak (Yale University, New Haven, CT) and R. Ramphal and J. Jyot (University of Florida, Gainesville, FL). The PAK motAB motCD mutant has previously been described (24). Bacteria were cultured overnight and subsequently subcultured for 3 h for in vitro bacterial infection. For in vivo infection, bacteria either were grown for 12 to 14 h, washed twice, and resuspended in PBS for infection or were subcultured for 3 h after overnight culture and subsequently washed and used for infection.
In vitro bacterial infection.
Six- or 7-day-old BMDC were harvested and washed twice. A total of 2.5 × 105 cells were seeded per well in a 24-well plate for 4 h at 37°C. Subcultured and washed bacteria were added at the multiplicity of infection (MOI) as indicated. Bacteria were coincubated for 3 h at 37°C. For experiments in which macrophages were used for cytokine expression analysis, cells were harvested as described above and, where indicated, ATP was added at a final concentration of 5 mM at the time of bacterial coincubation. Cell-free supernatants were collected and analyzed by ELISA. For phagocytosis experiments, BMDC were treated with either 10 μM cytochalasin D suspended in dimethyl sulfoxide (DMSO) or DMSO control for 10 min prior to infection with bacteria. For inducing forced contact, after the addition of bacteria to BMDC, plates were centrifuged at 500 × g for 5 min and coincubated for 3 h at 37°C. Cell-free supernatants were collected as described above and analyzed by ELISA.
Active caspase-1 assay and immunofluorescence microscopy.
A total of 2.5 × 105 BMDC were seeded per well in a 24-well plate for 4 h at 37°C. Subcultured and washed bacteria were added at an MOI of 50. Bacteria were coincubated for 3 h, and FAM-YVAD-FMK FLICA caspase-1 stain was added 1 h before collection. Cells were washed and treated as per the manufacturer's instructions. In some experiments, BMDC were further stained with APC-conjugated anti-mouse CD45 antibody after Fc receptor block with monoclonal antibody 2.4G2.
For microscopy, thioglycolate-stimulated peritoneal macrophages were washed twice after harvesting using serum free Hanks balanced salt solution (HBSS) without ACK lysis and seeded at 2 × 105 cells/coverslip for 1 h at 37°C on 18-mm2 coverslips in a 12-well plate. Nonadherent cells were aspirated and adherent macrophages were infected with the indicated genotype of live subcultured PA14 at an MOI of 50. At 1 h postinfection, the culture medium and extracellular bacteria were aspirated and cells were stained with YVAD-FMK FLICA caspase-1 stain for 1 h at 37°C. Cells were washed twice using PBS and fixed using 4% paraformaldehyde in PBS for 15 min at 37°C. Cells were washed thrice using PBS and treated with 10 mM Tris at neutral pH in PBS for 5 min at room temperature. Cells were again washed with PBS, stained with DAPI for 5 min at room temperature, washed with PBS, and mounted for microscopy. Images were acquired at magnifications of ×20 and ×63 using Axio Observer.Z1 (Carl Zeiss, Inc.). Immunofluorescence data are representative of at least two independent experiments.
PI staining assay.
A total of 2.5 × 105 BMDC were seeded per well in a 24-well plate or 1.5-ml microcentrifuge tubes for 4 h at 37°C. Subcultured and washed bacteria were added at the MOI indicated below. At 3 h postinfection, cells were harvested and washed twice with PBS. Cells were stained with 1:200 propidium iodide (PI) diluted in PBS for 5 min on ice and analyzed by flow cytometry.
Immunoblotting.
A total of 1 × 106 BMDC (for IL-1β) or 5 × 105 BMDC (for caspase-1) were seeded per well in a 12-well plate for 4 h at 37°C. Subcultured and washed bacteria were added at an MOI of 50. As a positive control, BMDC were prestimulated for 4 h with LPS at 50 ng/ml, followed by the addition of ATP at a final concentration of 5 mM at the time of bacterial coincubation. At 3 h postinfection, cell-free supernatants were collected and each sample was precipitated with methanol. Cell lysates were collected using Laemmli buffer for β-actin analysis. Proteins were separated by electrophoresis on a 15% polyacrylamide gel. Data using C57BL/6 BMDC are representative of at least two (caspase-1) or three (IL-1β) independent experiments.
In vivo bacterial infection.
WT mice were injected intraperitoneally (i.p.) with 3 × 106 CFU of the indicated genotype of PA14. Mice were sacrificed 4 h postinfection, and peritoneal lavage fluid was collected. Blood samples were collected by cardiac puncture or from the inferior vena cava. Both peritoneal lavage and blood samples were centrifuged for 10 min at 14,000 rpm, and supernatants were collected. Serum and peritoneal lavage supernatant samples were analyzed by ELISA. For analysis of infiltrating cells, peritoneal lavage fluid was collected and cells were counted by hemocytometry. The cells were further stained with PE-conjugated anti-mouse Ly6G antibody and APC-conjugated anti-mouse F4/80 antibody after Fc receptor block with monoclonal antibody 2.4G2. Recovered CFU from the peritoneal lavage fluid were determined by plating bacteria and normalizing the recovered CFU to the input CFU.
Statistical analyses.
Means and standard deviations are shown for each graph. These are derived from multiple independent experiments performed with technical duplicates or triplicates per condition. Unpaired Student's t test with Welch's correction was performed to analyze statistical significance within the different groups using Prism 4.0a (GraphPad Software, Inc.). Statistical significance is represented in figures by asterisks.
RESULTS
Loss of P. aeruginosa flagellar motility leads to reduced IL-1β production.
We previously reported that bacterial flagellar motility is a fundamental mechanism by which host phagocytes bind and ingest bacteria (15). Since loss of bacterial motility leads to reduced association with phagocytes, we tested the hypothesis that loss of bacterial motility leads to reduced inflammasome activation due to the reduced association with host cells. P. aeruginosa strains PA14 and PAK were used for testing this hypothesis. Genetic deletion of the MotAB and MotCD flagellar stator complexes in P. aeruginosa leads to the assembly of an intact flagellum but loss of flagellar rotation and swimming motility (21). When the motAB motCD mutant was compared to the swimming-competent wild-type PA14, we found that loss of flagellar motility leads to reduced IL-1β production in response to P. aeruginosa in both peritoneal macrophages and BMDC (Fig. 1A and B). The PA14 motAB motCD mutant elicits IL-1β production comparable to that elicited by the fliC mutant, which is deficient for flagellin synthesis and flagellar assembly. Therefore, loss of bacterial motility, regardless of flagellar assembly, leads to reduced activation of the inflammasome and IL-1β production (Fig. 1A and B). The popB mutant, which fails to form the bacterial T3SS translocon pore complex, was used as a negative control. Consistent with previous reports, the popB mutant elicited reduced inflammasome activation (Fig. 1A and B). These results were independently confirmed with the P. aeruginosa PAK strain, which also leads to reduced IL-1β production elicited by the respective motAB motCD mutant in comparison to its WT motile counterpart (Fig. 1A and B). As a control for specificity, we utilized ASC knockout (ASC−/−) cells, since loss of ASC abrogates the inflammasome-dependent IL-1β response to P. aeruginosa (2, 3). As expected, ASC−/− BMDC displayed a marked reduction in IL-1β production in response to P. aeruginosa (Fig. 1C). Thus, we show that loss of motility in P. aeruginosa specifically leads to reduced IL-1β production. Interestingly, we observed a dose-dependent increase in secreted IL-1β in response to P. aeruginosa that was maximal at an MOI of 20 to 50 and subsequently decreased at an MOI of 100 (Fig. 1D). Since P. aeruginosa is known to induce both pyroptotic and nonpyroptotic cell death (10), we suspected that at high MOIs the cells were dying prior to the IL-1β response. To test this, we infected BMDC at titrated MOIs from 1 to 100 (results for MOIs of 5 and 100 are shown) and analyzed cell death. We found that an MOI of 100 leads to increased cell death (Fig. 1D), and this corresponds with reduced IL-1β production, likely due to cell death preceding cytokine processing. Based on these analyses, an MOI of 5 to 50 was used for our experiments.
Fig 1.

Motile P. aeruginosa leads to more robust inflammasome activation. Peritoneal macrophages (A) and BMDC (B) from C57BL/6 mice and BMDC from ASC−/− mice (C) were infected with P. aeruginosa strains PA14 and PAK, as indicated, at an MOI of 5. Macrophages were infected in the presence of 5 mM ATP (A), whereas BMDC were not (B and C). Culture supernatants were collected 3 h postinfection and analyzed by ELISA for IL-1β production. (D) BMDC from C57BL/6 mice were infected with PA14 at the indicated MOI, and cells and culture supernatants were collected 3 h postinfection. IL-1β was analyzed by ELISA, and cellular cytotoxicity was analyzed by propidium iodide staining followed by flow cytometric analyses. Bacteria were gated out based on scatter controls. Results are representative of at least three (B, n ≥ 9) and two (A and C, n ≥ 6; D, n ≥ 5) independent experiments and expressed as means ± standard deviations. ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05; ns, not significant.
Enhanced cell surface contact, but not phagocytosis, increases inflammasome activation elicited by P. aeruginosa.
Phagocytosis has been reported to play a role in the activation of alternative inflammasomes, including NLRP3 (25). We have previously shown that flagellar motility by P. aeruginosa facilitates both cell surface interactions with macrophages and subsequent phagocytosis (15). Therefore, to test whether phagocytosis plays a role in inflammasome activation, BMDC were treated with cytochalasin D, an inhibitor of actin polymerization and phagocytosis, and subsequently infected with wild-type PA14. We found that cytochalasin D treatment of the cells did not alter the IL-1β response to P. aeruginosa (Fig. 2A), from which we infer that phagocytic uptake of P. aeruginosa was not necessary and that activation of the inflammasome was sufficiently triggered by translocation of effectors during cell surface contact between the bacteria and the host cells.
Fig 2.
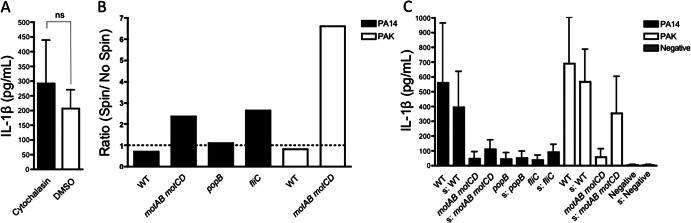
IL-1β production in response to P. aeruginosa is independent of phagocytosis and increases with induction of bacterium-BMDC contact. (A) BMDC from C57BL/6 mice were infected with PA14 (WT) at an MOI of 50 in the presence of either 10 μM cytochalasin D or a vehicle control. Culture supernatants were collected 3 h postinfection and analyzed by ELISA for IL-1β production. (B and C) BMDC from C57BL/6 mice were infected with the indicated WT or isogenic mutant strain of P. aeruginosa at an MOI of 5. Bacteria were either centrifuged at 500 × g for 5 min to force contact with BMDC (denoted by “s”) or not centrifuged as controls (unmarked). Culture supernatants were collected 3 h postinfection and analyzed by ELISA for IL-1β production. The ratio of IL-1β production after centrifugation to IL-1β production without centrifugation (B) and the raw values of elicited IL-1β (C) are shown. Data are representative of two independent experiments (A, n = 5; C, n = 6) and expressed as means ± standard deviations.
Since cell surface contact sufficed to elicit an IL-1β response, we hypothesized that promotion of association between host cells and bacteria would lead to increased inflammasome activation regardless of motility. To test this, we forced contact between the BMDC and bacteria by centrifugation of the bacteria onto the cells. The nonmotile motAB motCD and fliC mutants induced substantially higher IL-1β responses upon forced contact (Fig. 2B and C), in support of our hypothesis that association with host cells is required for inducing IL-1β release in response to P. aeruginosa. Moreover, this shows that the nonmotile bacteria are capable of eliciting an IL-1β response if association with host cells is artificially enabled. Thus, phagocytosis is not essential for activation of the inflammasome in response to P. aeruginosa, whereas association of bacteria on the host cell surface leads to increased inflammasome activation.
In vitro caspase-1 activation is reduced in response to nonmotile P. aeruginosa.
Since IL-1β responses to nonmotile P. aeruginosa were reduced, we hypothesized that inflammasome-dependent responses would be reflective of, and could be validated by, correspondingly reduced caspase-1 activity. To test if nonmotile P. aeruginosa leads to reduced caspase-1 activation in comparison to the motile wild-type strain, we employed a fluorescent caspase-1-specific probe, FAM-YVAD-FMK, which binds to the active site of caspase-1. For flow cytometric analyses, bacteria were gated out and only the BMDC populations were used for further analyses. Caspase-1 was activated at higher levels in response to motile P. aeruginosa strain PA14, whereas it was activated at reduced levels in response to the motAB motCD and popB mutants (Fig. 3A and B). Caspase-1 activation levels were also significantly reduced in response to the PAK motAB motCD mutant in comparison to its wild-type counterpart. In parallel, we added ATP to these incubations since ATP is commonly used to potentiate the inflammasome response (26). Addition of ATP elicited greater overall inflammasome activation; however, significantly higher caspase-1 activation was still observed in response to motile P. aeruginosa compared to the motAB motCD and popB mutants (Fig. 3C). Thus, caspase-1 activation is reduced in response to nonmotile P. aeruginosa in BMDC.
Fig 3.

Loss of flagellar motility causes reduced caspase-1 activation. BMDC from C57BL/6 mice were infected with the indicated genotype of P. aeruginosa at an MOI of 50. Bacteria were gated out based on scatter controls, and the percentage of active caspase-1+ cells was determined. (A) Representative flow cytometry dot plot for the BMDC gating scheme (i) and gated populations analyzed for caspase-1 and CD45 expression (ii, iii, and iv). The dot plots are representative of BMDC infected with wild-type PA14 (ii) or the motAB motCD (iii) or popB (iv) mutant. (B and C) Quantitative analysis of the percentage of caspase-1+ cells following infection of BMDC with the indicated wild-type or isogenic mutant strains of P. aeruginosa in the absence (B) or presence (C) of ATP. Data are representative of at least two independent experiments (B and C, n ≥ 5) and expressed as means ± standard deviations. ***, P ≤ 0.001. (D) Macrophages were uninfected (i) or infected with wild-type PA14 (ii and v) or the motABmotCD (iii and vi) or popB (iv) mutant at an MOI of 50 for 2 h. Cells were stained for active caspase-1 (with FAM-YVAD-FLICA) and DNA (with DAPI). Images were acquired at magnifications of ×20 (i to iv) and ×63 (v and vi). Images are representative of at least two independent experiments.
To confirm the flow cytometric analyses, we performed immunofluorescence microscopy with the use of primary murine macrophages infected with wild-type PA14 or the motAB motCD and popB mutants and stained for caspase-1 activation. As observed with the BMDC, macrophages infected with wild-type PA14 led to higher caspase-1 activation than that obtained with the motAB motCD and popB mutants (Fig. 3D). Active caspase-1 staining was observed around nuclei and toward the periphery of the cytoplasm in activated macrophages infected with WT PA14, whereas this staining was diminished in the motAB motCD and popB mutant-infected macrophages. These data confirm that loss of motility leads to reduced caspase-1 activation levels in BMDC and macrophages in vitro.
Nonmotile P. aeruginosa elicits reduced cell death, and cleavage of caspase-1 and IL-1β, in comparison to motile P. aeruginosa.
Loss of flagellar motility decreases both cell surface association with and retention of P. aeruginosa to phagocytes (15), and nonmotile P. aeruginosa elicits lesser T3SS-dependent inflammasome responses (Fig. 1 and 3). Therefore, we hypothesized that nonmotile bacteria would be less efficient at induction of host cell death through reduced induction of the T3SS (9, 10), engagement with the phagocyte, and delivery of cytotoxic molecules. Cytotoxicity was measured using propidium iodide staining (27), bacteria were gated out, and the percentage of PI+ cells was assessed. At an MOI of 5, there was no significant difference between cytotoxicity induced by wild-type PA14 and the motAB motCD mutant (Fig. 4A). This indicated that the differential IL-1β response elicited by BMDC at this MOI was not due to differential cytotoxicity by the WT and the motAB motCD mutant. However, at an MOI of 50, wild-type PA14 led to >2-fold-larger amounts of cell death in comparison to the motAB motCD mutant, and wild-type PAK led to 2-fold-larger amounts of cell death in comparison to its respective motAB motCD mutant (Fig. 4B and C). PA14 motAB motCD led to cell death at levels comparable to the PA14 fliC mutant. Cytotoxicity, however, was triggered at higher levels in response to PA14 fliC and PA14 motAB motCD in comparison to the PA14 popB mutant, which is known to activate the inflammasome at much reduced levels (2). These observations support our guiding hypothesis that P. aeruginosa motility enables a cell surface engagement that leads to inflammation and, in this case, cytotoxicity.
Fig 4.

Motility-deficient P. aeruginosa is less efficient at induction of cell death. (A) BMDC from C57BL/6 mice were infected with wild-type PA14 or the motAB motCD mutant at an MOI of 5. Bacteria were gated out, and the percentage of PI+ cells was determined. (B and C) Gated PI+ cells upon infection with WT or the indicated deletion mutant of P. aeruginosa at an MOI of 50. (B) Representative histograms of data represented in panel C showing gated PI+ cells when BMDC were not infected (filled gray) or were infected with wild-type PA14 (solid black line) or motAB motCD bacteria (dashed line) at an MOI of 50. Data are representative of at least two independent experiments (n ≥ 4) and expressed as means ± standard deviations. ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05.
Since the wild-type and the motAB motCD mutant of P. aeruginosa induced differential cytotoxicity at the higher MOI (Fig. 4B and C), we wanted to verify that the IL-1β released was the cleaved, active form. To do so, we performed Western analyses for active IL-1β and caspase-1 released in the supernatant following infection. In confirmation of our observations depicted in Fig. 1 to 3, nonmotile P. aeruginosa led to both reduced IL-1β and caspase-1 maturation and release into the supernatant at an MOI of 50 (Fig. 5). This demonstrates that following infection with equal numbers of motile and nonmotile P. aeruginosa organisms, and with equal numbers of phagocytes, the nonmotile P. aeruginosa elicits a reduced caspase-1 response (consistent with Fig. 3) and, accordingly, a reduced release of mature, active IL-1β.
Fig 5.
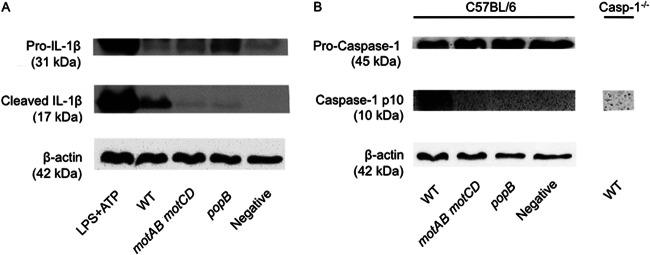
Loss of flagellar motility leads to reduced release of active IL-1β and caspase-1. BMDC from C57BL/6 mice and, where indicated, caspase-1−/− BMDC were infected with wild-type or the indicated mutant of PA14 at an MOI of 50. The uncleaved pro- and biologically active cleaved forms of IL-1β (A) and caspase-1 (B) were analyzed in the supernatants by Western analyses. Cell lysates for the corresponding samples were analyzed for β-actin. BMDC treated with LPS and ATP were included as a positive control.
Loss of P. aeruginosa motility leads to reduced IL-1β responses in vivo.
In order to test the relevance of our findings in vivo and to analyze whether loss of bacterial motility leads to reduced IL-1β production in vivo, we employed an established P. aeruginosa peritonitis model (2). C57BL/6 mice were injected intraperitoneally with the indicated genotype of live PA14, and peritoneal lavage and blood serum samples were collected 4 h postinfection and analyzed for IL-1β release (Fig. 6A and B). Wild-type PA14 elicited significantly greater IL-1β in the peritoneal exudates than elicited by the motAB motCD and popB mutants (Fig. 6A). The amount of IL-1β produced in the peritoneum in response to the motAB motCD strain was comparable to that produced in response to the popB strain. This trend for IL-1β production was observed distally in the blood sera as well, although statistical significance was lost. However, it should be noted that the trend of higher IL-1β levels released in response to WT PA14 and progressively lesser IL-1β released in response to the motAB motCD and popB strains, respectively, was preserved (Fig. 6B). There was no significant difference between recovered CFU from the peritoneal lavage fluid of mice infected with wild-type PA14 or the motAB motCD mutant (Fig. 6C). Fluorescence-activated cell sorter (FACS) analyses of the number of total leukocytes in the peritoneum upon infection with wild-type PA14 or the motAB motCD mutant were also found to be similar (Fig. 6D). FACS analyses of the cellular populations by the neutrophil and macrophage markers Ly6-G and F4/80, respectively, indicated that the increase in total leukocytes could be due to neutrophil influx (Fig. 6E and F). Although not statistically significant, the total percentages of F4/80+ peritoneal macrophages were similar in mice infected with WT and nonmotile Pseudomonas (Fig. 6F). Thus, loss of bacterial swimming motility leads to a reduced IL-1β response to an in vivo P. aeruginosa infection. These data support a model in which loss of flagellar motility leads to reduced activation of the inflammasome in response to P. aeruginosa and whereby the bacteria could exploit host-selective pressure to evade the potent IL-1β inflammatory response.
Fig 6.

Nonmotile P. aeruginosa elicits reduced IL-1β responses in vivo. (A and B) C57BL/6 mice were injected intraperitoneally with 3 × 106 CFU of the indicated isogenic strain of PA14 (WT, n = 22; motAB motCD mutant, n = 10; popB mutant, n = 8; PBS control, n = 6). Mice were sacrificed 4 h postinfection, and peritoneal lavage and blood samples were collected. Peritoneal lavage (A) and blood serum (B) samples were analyzed by ELISA for IL-1β production. (C) The peritoneal lavage fluid at this time point was analyzed for the total number of recovered CFU, normalized to CFU of input bacteria (WT, n = 9; motAB motCD mutant, n = 9; PBS control, n = 3). (D) Total number of leukocytes in the peritoneal lavage fluid 4 h postinfection (WT, n = 4; motAB motCD mutant, n = 4; PBS control, n = 3). (E and F) Percentages of Ly6G+ (E) and F4/80+ (F) cells out of total leukocytes shown in panel D are shown. Data are expressed as means ± standard deviations. ***, P ≤ 0.001; **, P ≤ 0.01; *, P < 0.05.
DISCUSSION
Loss of bacterial flagellar motility has been consistently described for a variety of systems and diseases to be required for bacterial persistence within the host. One pertinent example of this is the temporal loss of P. aeruginosa flagellar motility observed during chronic infections of patients with CF (13, 14). It is unclear whether the observed progressive loss of motility is due to selective pressure or to bacterial responses to the host environment, such as the bacterial flagellar hook protein being cleaved by neutrophil elastase (28), though these are not mutually exclusive. However, it is evident that decreases in motility contribute to the bacterial evasion of the host innate immune response, and we have previously demonstrated that even minor decreases in flagellar motility result in measurable phagocytic resistance. Loss of flagellar motility has previously been demonstrated to alter the host inflammatory response: in the burned-mouse model, an established acute infection model for systemic sepsis, and in a heterologous zebrafish system, flagellar motility was shown to be required for full virulence of P. aeruginosa (24, 29).
We have previously shown that flagellar motility, independent of the presence of the flagella, is required for binding and phagocytosis of P. aeruginosa by innate immune cells of the host. Based on this observation and the previously reported differential inflammation in response to nonmotile bacteria, we hypothesized that inflammatory responses that were dependent upon bacterial interactions with the cell surfaces of macrophages and other host cells may be modulated by bacterial motility. Here we provide the first report of the role bacterial motility plays in subsequent activation of the inflammasome.
There has been considerable interest in the activation of the inflammasome by P. aeruginosa and its role in pathogenesis (30). Although flagellin itself leads to the activation of the inflammasome, flagellin-independent inflammasome activation has been reported (2, 31). We extended the inquiry of the role of the flagellum with the use of motility mutants that lack the two stator complexes required to generate flagellar torque. With the use of this bacterial mutant deficient in flagellar motility, we have shown that loss of flagellar motility leads to reduced activation of the inflammasome in response to P. aeruginosa. This is supported by reduced caspase-1 activation and IL-1β production in vitro, as well as reduced IL-1β production in vivo. The simplest mechanistic explanation for this outcome is that loss of motility diminishes cell surface interactions between the bacteria and the host cells, as demonstrated by induction of IL-1β upon forced contact (Fig. 2). Microarray analysis has shown that the lack of these stator proteins does not lead to significantly altered expression of known immunogenic effectors of P. aeruginosa (15), and the motAB motCD mutant encodes inflammasome-activating ligands as well as the T3SS.
Intriguingly, previous reports have not identified bacterial motility as a key factor for inflammasome activation (2–4, 31). A key aspect that could lead to the difference between these studies and ours is the experimental methodology as well as the bacteria used. Miao and colleagues used Salmonella enterica serovar Typhimurium, an intracellular pathogen, for their studies (31). In addition, for some of their experiments, bone marrow-derived macrophages (BMM) were prestimulated with LPS or bacteria were centrifuged onto the BMM to promote contact between phagocytes and bacteria. S. Typhimurium is an intracellular pathogen, and the mechanism for inflammasome activation has been shown to be different for intracellular and extracellular bacteria depending on the requirement of the P2X7 receptor (26). Similarly, studies by the Núñez and Flavell labs often used stimulation of macrophages with LPS prior to infection and centrifugation of P. aeruginosa onto the macrophages to promote infection (2, 3). Since we did not employ LPS prestimulation to induce the production of pro-IL-1β for our experiments, this could be the reason for the discrepancy observed between our studies. Since centrifugation promotes contact between phagocytes and bacteria, prolonged contact between them can lead to the induction of the bacterial T3SS (9) and the injection of effector proteins. In another study of IL-1β responses to P. aeruginosa, macrophages were stimulated with LPS to induce pro-IL-1β (4). Although cellular activation in response to the flgK mutant, which lacks both a full structural flagellum and flagellar motility due to the loss of the flagellar hook-associated protein FlgK, was observed, it should be borne in mind that the flgK mutant can secrete monomeric flagellin and could lead to caspase-1 activation. In this respect, we have observed that prestimulation with purified LPS leads to lesser difference in IL-1β processing between the nonmotile strains and motile strains than does stimulation by bacteria alone (Y. R. Patankar and B. Berwin, unpublished data). Regardless of the bacterial ligand leading to inflammasome activation in these studies (2–4), it was shown that the T3SS was crucial for the activation of the inflammasome in P. aeruginosa via secreted effector proteins (2–4). Likewise, we observed higher IL-1β production by BMDC upon forced contact of nonmotile bacteria and cells (Fig. 2B and C), which is consistent with the aforementioned reports. These observations reconcile the discrepancies in the field on the role of FliC, motility, and the T3SS in inflammasome activation. In accordance with the reduced activation of caspase-1 and IL-1β production in response to nonmotile P. aeruginosa, cell death was reduced in comparison to that elicited by the wild-type motile P. aeruginosa (Fig. 4A and B). Caspase-1 and IL-1β maturation and host cell death are distinctive features of inflammasome activation. Additionally, IL-1β production was significantly reduced in the peritoneum of mice infected with flagellar motility mutants as opposed to those infected with wild-type bacteria, which was independent of differential bacterial clearance from the peritoneum (Fig. 6A and C). Although the difference was more pronounced in the peritoneum, we observed that serum IL-1β levels showed a similar trend, with lower IL-1β production in mice infected with swimming-deficient P. aeruginosa (Fig. 6B). Although there was no difference in the number of total leukocytes in the peritoneum 4 h postinfection (Fig. 6D), there was a trend, albeit not statistically significant, that fewer Ly6G+ neutrophils were recruited in response to nonmotile PA14 at this time point (Fig. 6E; P = 0.0766). We speculate that over longer periods of infection, this may emerge as a significant difference and, if so, that this may reflect the differential IL-1β production which is subsequently integral to neutrophil recruitment.
Since loss of motility leads to reduced activation of the inflammasome, we propose that the loss-of-motility phenotype could be used by P. aeruginosa for selective resistance and persistence in the host by evasion of proinflammatory, bactericidal cytokine responses. CF patients with chronic lung infection by P. aeruginosa have increased levels of proinflammatory cytokines in the lung, including IL-1β (1). However, these levels likely reflect sufficient inflammation to chronically damage the lung while, in concert with other reported innate immunodeficiencies observed during CF disease (1, 32–35), being insufficient to clear the bacterial infection and leaving the host prone to temporary exacerbation of IL-1β levels as a result of endotoxin release from bacteria or by coinfection with other microbes.
A recent concept that is an extension of the pathogen-associated molecular pattern (PAMP) theory is termed patterns of pathogenesis, and it postulates that the immune system responds to conserved molecular features of pathogens in the context of their presentation to host cells (36). Bacterial flagellar motility is shared among a variety of Gram-negative bacteria, is a trait unlike any present in healthy host cells and is therefore a candidate for pattern-like recognition, and is lost during effective chronic infections, which suggests a benefit to the residual bacteria. However, precisely how motility confers association with phagocytes is unclear. Our previously published data, supported by the data presented here, indicate that bacterial motility confers greater association and retention on the surfaces of phagocytes (15). Another possibility, that is not mutually exclusive, is that flagellar motility could trigger an unidentified mechanosensory receptor, leading to host-promoted association of bacteria on the cell surface and eventual phagocytosis (15). Alternatively, motility may enable receptor clustering on the host cell surface, which could amplify the downstream signals and eventually lead to a phagocytic event and subsequent inflammatory cytokine production. Regardless of the signaling mechanisms, motility could be considered a pattern of pathogenesis, since it is an essential virulence factor for many pathogens (37–40) and serves as a trigger for the immune system to recognize the presence of bacteria and lead to activation of the appropriate innate immune defenses.
In conclusion, we demonstrate that loss of flagellar motility in two strains of P. aeruginosa leads to reduced IL-1β production as well as caspase-1 activation by dendritic cells and macrophages. Inflammasome activation is independent of phagocytosis of bacteria and is increased upon promotion of contact with host cells. Consistent with the differential bacterial and T3SS engagement of motile and nonmotile bacteria with the phagocytes, loss of flagellar motility leads to reduced host cell death. Loss of bacterial motility further leads to reduced IL-1β production in mice. These data provide novel insights into the contribution of P. aeruginosa motility to innate immune responses, and why loss of motility confers a benefit to the bacteria during chronic infection.
ACKNOWLEDGMENTS
We thank the Leib Lab (Dartmouth Medical School, NH) for technical assistance with microscopy and George O'Toole and Deborah Hogan (Dartmouth Medical School, NH), Vishva Dixit (Genentech, CA), Reuben Ramphal (University of Florida, FL), and Denise Monack and Petr Broz (Stanford University, CA) for reagents and advice.
This research was supported by National Institutes of Health (NIH) grants COBRE P20RR016437 and RO1 AI067405, NIH training grants GM008704 and AI0073634, and training funds from the Cystic Fibrosis Foundation Research Development Program (STANTO011RO).
Footnotes
Published ahead of print 25 March 2013
REFERENCES
- 1. Hartl D, Gaggar A, Bruscia E, Hector A, Marcos V, Jung A, Greene C, McElvaney G, Mall M, Doring G. 2012. Innate immunity in cystic fibrosis lung disease. J. Cyst. Fibros. 11:363–382 [DOI] [PubMed] [Google Scholar]
- 2. Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. 2007. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204:3235–3245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Franchi L, Stoolman J, Kanneganti TD, Verma A, Ramphal R, Núñez G. 2007. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur. J. Immunol. 37:3030–3039 [DOI] [PubMed] [Google Scholar]
- 4. Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. 2008. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc. Natl. Acad. Sci. U. S. A. 105:2562–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Medzhitov R. 2007. Recognition of microorganisms and activation of the immune response. Nature 449:819–826 [DOI] [PubMed] [Google Scholar]
- 6. Davis BK, Wen H, Ting JP. 2011. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29:707–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Diaz MR, King JM, Yahr TL. 2011. Intrinsic and extrinsic regulation of type III secretion gene expression in Pseudomonas aeruginosa. Front. Microbiol. 2:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. 2010. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11:1136–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dasgupta N, Ashare A, Hunninghake GW, Yahr TL. 2006. Transcriptional induction of the Pseudomonas aeruginosa type III secretion system by low Ca2+ and host cell contact proceeds through two distinct signaling pathways. Infect. Immun. 74:3334–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hauser AR. 2009. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 7:654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A. 2010. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U. S. A. 107:3076–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arlehamn CS, Evans TJ. 2011. Pseudomonas aeruginosa pilin activates the inflammasome. Cell. Microbiol. 13:388–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luzar MA, Thomassen MJ, Montie TC. 1985. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: relationship to patient clinical condition. Infect. Immun. 50:577–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mahenthiralingam E, Campbell ME, Speert DP. 1994. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect. Immun. 62:596–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lovewell RR, Collins RM, Acker JL, O'Toole GA, Wargo MJ, Berwin B. 2011. Step-wise loss of bacterial flagellar torsion confers progressive phagocytic evasion. PLoS Pathog. 7:e1002253 doi:10.1371/journal.ppat.1002253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. 2004. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430:213–218 [DOI] [PubMed] [Google Scholar]
- 17. Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. 1995. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 267:2000–2003 [DOI] [PubMed] [Google Scholar]
- 18. National Research Council 1996. Guide for the care and use of laboratory animals. National Academies Press, Washington, DC [Google Scholar]
- 19. Inaba K, Inaba M, Deguchi M, Hagi K, Yasumizu R, Ikehara S, Muramatsu S, Steinman RM. 1993. Granulocytes, macrophages, and dendritic cells arise from a common major histocompatibility complex class II-negative progenitor in mouse bone marrow. Proc. Natl. Acad. Sci. U. S. A. 90:3038–3042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amiel E, Alonso A, Uematsu S, Akira S, Poynter ME, Berwin B. 2009. Pivotal advance: Toll-like receptor regulation of scavenger receptor-A-mediated phagocytosis. J. Leukoc. Biol. 85:595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Toutain CM, Zegans ME, O'Toole GA. 2005. Evidence for two flagellar stators and their role in the motility of Pseudomonas aeruginosa. J. Bacteriol. 187:771–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang S, McCormack FX, Levesque RC, O'Toole GA, Lau GW. 2007. The flagellum of Pseudomonas aeruginosa is required for resistance to clearance by surfactant protein A. PLoS One 2:e564 doi:10.1371/journal.pone.0000564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cisz M, Lee PC, Rietsch A. 2008. ExoS controls the cell contact-mediated switch to effector secretion in Pseudomonas aeruginosa. J. Bacteriol. 190:2726–2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arora SK, Neely AN, Blair B, Lory S, Ramphal R. 2005. Role of motility and flagellin glycosylation in the pathogenesis of Pseudomonas aeruginosa burn wound infections. Infect. Immun. 73:4395–4398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shimada T, Park BG, Wolf AJ, Brikos C, Goodridge HS, Becker CA, Reyes CN, Miao EA, Aderem A, Gotz F, Liu GY, Underhill DM. 2010. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe 7:38–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Franchi L, Kanneganti TD, Dubyak GR, Nunez G. 2007. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J. Biol. Chem. 282:18810–18818 [DOI] [PubMed] [Google Scholar]
- 27. Miao EA, Rajan JV, Aderem A. 2011. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 243:206–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sonawane A, Jyot J, During R, Ramphal R. 2006. Neutrophil elastase, an innate immunity effector molecule, represses flagellin transcription in Pseudomonas aeruginosa. Infect. Immun. 74:6682–6689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rawls JF, Mahowald MA, Goodman AL, Trent CM, Gordon JI. 2007. In vivo imaging and genetic analysis link bacterial motility and symbiosis in the zebrafish gut. Proc. Natl. Acad. Sci. U. S. A. 104:7622–7627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lavoie EG, Wangdi T, Kazmierczak BI. 2011. Innate immune responses to Pseudomonas aeruginosa infection. Microbes Infect. 13:1133–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. 2006. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 7:569–575 [DOI] [PubMed] [Google Scholar]
- 32. Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. 2004. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 10:487–493 [DOI] [PubMed] [Google Scholar]
- 33. Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. 1998. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 95:1005–1015 [DOI] [PubMed] [Google Scholar]
- 34. Zhou Z, Duerr J, Johannesson B, Schubert SC, Treis D, Harm M, Graeber SY, Dalpke A, Schultz C, Mall MA. 2011. The ENaC-overexpressing mouse as a model of cystic fibrosis lung disease. J. Cyst. Fibros. 10(Suppl 2):S172–S182 [DOI] [PubMed] [Google Scholar]
- 35. Regnis JA, Robinson M, Bailey DL, Cook P, Hooper P, Chan HK, Gonda I, Bautovich G, Bye PT. 1994. Mucociliary clearance in patients with cystic fibrosis and in normal subjects. Am. J. Respir. Crit. Care Med. 150:66–71 [DOI] [PubMed] [Google Scholar]
- 36. Vance RE, Isberg RR, Portnoy DA. 2009. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6:10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Drake D, Montie TC. 1988. Flagella, motility and invasive virulence of Pseudomonas aeruginosa. J. Gen. Microbiol. 134:43–52 [DOI] [PubMed] [Google Scholar]
- 38. Freter R, Jones GW. 1976. Adhesive properties of Vibrio cholerae: nature of the interaction with intact mucosal surfaces. Infect. Immun. 14:246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pier GB, Meluleni G, Goldberg JB. 1995. Clearance of Pseudomonas aeruginosa from the murine gastrointestinal tract is effectively mediated by O-antigen-specific circulating antibodies. Infect. Immun. 63:2818–2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Siitonen A, Nurminen M. 1992. Bacterial motility is a colonization factor in experimental urinary tract infection. Infect. Immun. 60:3918–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]