

Abstract
Immunomodulatory agents potentially represent a new class of broad-spectrum antimicrobials. Here, we demonstrate that prophylaxis with immunomodulatory cytosine-phosphate-guanidine (CpG) oligodeoxynucleotide (ODN), a toll-like receptor 9 (TLR9) agonist, confers protection against Yersinia pestis, the etiologic agent of plague. The data establish that intranasal administration of CpG ODN 1 day prior to lethal pulmonary exposure to Y. pestis strain KIM D27 significantly improves survival of C57BL/6 mice and reduces bacterial growth in hepatic tissue, despite paradoxically increasing bacterial growth in the lung. All of these CpG ODN-mediated impacts, including the increased pulmonary burden, are TLR9 dependent, as they are not observed in TLR9-deficient mice. The capacity of prophylactic intranasal CpG ODN to enhance survival does not require adaptive immunity, as it is evident in mice lacking B and/or T cells; however, the presence of T cells improves long-term survival. The prophylactic regimen also improves survival and reduces hepatic bacterial burden in mice challenged intraperitoneally with KIM D27, indicating that intranasal delivery of CpG ODN has systemic impacts. Indeed, intranasal prophylaxis with CpG ODN provides significant protection against subcutaneous challenge with Y. pestis strain CO92 even though it fails to protect mice from intranasal challenge with that fully virulent strain.
INTRODUCTION
Yersinia pestis, the etiologic agent of plague, is a Gram-negative facultative intracellular bacterium that is endemic in rodent populations and can be spread to humans by the bite of an infected flea. Transmission by fleabite commonly results in bacterial growth in the draining lymph node, which manifests as bubonic plague (1–3). In some instances, fleabite infections progress directly to septicemic plague, in which case the bacteria disseminate systemically (4, 5). Septicemic plague can also result from bubonic or pneumonic infections. Pneumonic plague, which may occur either as a primary infection through inhalation of Y. pestis bacteria or as a secondary consequence of septicemic plague, can be transmitted from person to person via infectious droplets produced by coughing, perhaps making this form of plague the greatest threat to public health (4, 6, 7).
Y. pestis has caused three major pandemics throughout recorded history and has been responsible for hundreds of millions of human deaths (1, 2). Although cases of plague are less frequent today due to advances in treatments and improvements in public health surveillance and sanitation, isolated Y. pestis infections and small outbreaks still occur globally, especially in developing countries (3, 8–10). Antibiotics can be used to treat plague but fail to prevent lethality unless treatment begins within a short time after symptoms appear (4, 6). In addition to naturally acquired infections, a major current concern is the potential use of Y. pestis as a bioweapon (6, 8, 9).
Despite substantial efforts over the past century, there is no safe and effective vaccine for plague (reviewed in references 8 and 11). Subunit vaccines based upon the F1 and LcrV antigens of Y. pestis show promise (reviewed in references 12 and 13) but fail to induce adequate protection in every nonhuman primate model of plague (14). The cynomolgus macaque and African green monkey, for example, both produce antibodies in response to vaccination; however, the African green monkey is less effectively protected from pneumonic plague challenge. The reason(s) for the discrepant results in these two animals models is currently unclear (8, 14). Due to the lack of an approved vaccine and the small window of opportunity afforded to infected individuals for treatment with antibiotics, researchers continue to search for new plague countermeasures.
Y. pestis is known to suppress and evade innate immunity using a variety of mechanisms, including secretion of immunomodulatory proteins that reduce cytokine production, prevent phagocytosis, and deplete cell populations that are key to mounting an effective innate immune response (reviewed in references 9 and 15–17). Y. pestis also produces a tetra-acylated form of lipopolysaccharide (LPS) that poorly stimulates Toll-like receptor 4 (TLR4) (18–20). Y. pestis strains engineered to constitutively express a more inflammatory, TLR4-activating, hexa-acylated form of LPS display greatly reduced virulence, and their inoculation induces protective immunity against challenge with the virulent parental strains (18, 21).
The fact that engineering Y. pestis to activate TLR4 results in attenuation (18, 21) suggests that stimulation of the innate immune system via an exogenous source prior to or during the early stages of infection may promote survival of Y. pestis-infected individuals. Indeed, treating mice with mimetics of the inflammatory forms of LPS lipid A protect against pulmonary Y. pestis challenge in a TLR4-dependent manner (22). Bacterial DNA can also be immunostimulatory due to the presence of unmethylated cytosine-phosphate-guanidine (CpG) repeats, which stimulate TLR9-dependent cytokine production and lymphocyte activation (23, 24). Synthetic oligodeoxynucleotides (ODN) containing these CpG motifs are immunomodulatory as well and have been used both as adjuvants and as stimulators of innate immunity in animal infection/vaccination models (25–33). Treatment with CpG ODN can protect mice against lethal infections caused by Gram-negative Francisella tularensis, Klebsiella pneumoniae, and Burkholderia pseudomallei (25, 28, 30–33) but also can exacerbate infections with Salmonella enterica (34).
In this report, we investigate the capacity of CpG ODN to protect mice against Y. pestis infection. Pigmentation locus (pgm)-negative Y. pestis strains, such as KIM D27, retain substantial virulence when mice are inoculated intranasally or intraperitoneally, but they are highly attenuated when inoculated subcutaneously. In contrast, pgm-positive strains, such as CO92, display high virulence when inoculated intranasally, intraperitoneally, or subcutaneously. The cause of mortality in mice infected intranasally with pgm-negative and -positive strains also differs, with pgm-negative strains causing bacteremia and sepsis while pgm-positive strains cause fulminant pneumonia, in addition to bacteremia and sepsis (35, 36). We demonstrate that intranasal prophylaxis with CpG ODN confers protection against intranasal or intraperitoneal infection with KIM D27. We also demonstrate that CpG ODN prophylaxis fails to protect against intranasal infection with fully virulent Y. pestis strain CO92, even though it does protect against subcutaneous infection with that fully virulent strain.
MATERIALS AND METHODS
Mice.
Wild-type C57BL/6 mice, B-cell-deficient μMT mice (B6.129S2-Igh-6tm1Cgn), T-cell-deficient TCRbd-deficient mice (B6.129P2-Tcrbtm1Mom Tcrdtm1Mom), RAG2-deficient mice, and TLR9-deficient mice on the C57BL/6 background were purchased from The Jackson Laboratory (Bar Harbor, ME) and then bred in the specific-pathogen-free Trudeau Institute Animal Breeding Facility. All mice used in these experiments were between 6 and 9 weeks old and were age and sex matched within each individual experiment. Unless otherwise indicated, experiments were conducted with male mice. Mice were cared for according to the Trudeau Institute Animal Care and Use Committee guidelines.
Bacteria.
For studies using conditionally attenuated pigmentation-negative Y. pestis strain KIM D27 (37), bacteria from frozen glycerol stocks were grown overnight at 26°C with continuous shaking in Bacto heart infusion broth (Becton, Dickinson and Company) supplemented with 2.5 mM CaCl2. After dilution to an optical density at 620 nm (OD620) of 0.1 to 0.2, they were regrown for 3 h at 26°C and then washed with saline before inoculating mice. The median lethal dose (MLD) of strain KIM D27 for intranasal and intraperitoneal infection is approximately 1 × 104 and 1 × 103 CFU, respectively (38). For intranasal infections, mice lightly anesthetized with isoflurane received approximately 2 × 105 CFU (20 MLD) applied to the nares in a volume of 30 μl saline. For intraperitoneal infections, mice were inoculated with approximately 2 × 104 CFU (20 MLD) in a volume of 100 or 200 μl saline.
The fully virulent pigmentation-positive Y. pestis strain CO92 NR-641 was obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, NIAID. Bacteria from frozen glycerol stocks were grown overnight at 26°C with continuous shaking in Bacto heart infusion broth, diluted to an OD620 of approximately 0.1, and regrown for 2.5 to 3 h at 26°C. Preliminary studies established that the MLD of strain CO92 for subcutaneous and intranasal infection is approximately 3 CFU and 1 × 103 CFU, respectively, when grown as described above. For subcutaneous infections, isoflurane-anesthetized mice received approximately 10 CFU in a volume of 100 μl saline, which was injected into the scruff of the neck. For intranasal infections, mice lightly anesthetized with isoflurane received approximately 1 × 104 CFU (10 MLD) applied to the nares in a volume of 25 μl saline.
CpG ODN.
Unless indicated otherwise in the figure legend, mice were treated intranasally with 50 μg CpG ODN 1826 (39). The ODN were administered in a volume of 25 μl to the nares of mice lightly anesthetized with isoflurane. Control mice were left untreated or received sham treatments of phosphate-buffered saline (PBS), as indicated in the figure legends. The ODN were supplied by IDT (Coralville, IA).
Survival endpoints and bacterial burden.
In all survival studies, recumbent animals were considered moribund and were euthanized. For measurement of bacterial burden, mice were euthanized by carbon dioxide narcosis at the indicated day after initiating infection. CFU were determined by homogenizing liver or lung tissues in saline and plating serial dilutions on blood agar plates, which were incubated at 26°C for 48 h (38, 40). Bronchial alveolar lavage (BAL) fluid was collected from euthanized mice in 1 ml of PBS, and bacterial burden was determined by plating serial dilutions on blood agar plates.
Flow cytometry.
Lungs were perfused with saline containing heparin, minced, and then digested with collagenase and DNase (38). Red blood cells were lysed with red blood cell lysing buffer (Sigma), and viable cells were counted using trypan blue exclusion. Cells were treated with 2.4G2 monoclonal antibody (Bio X cell) to block Fc receptors and then stained with anti-CD11b-fluorescein isothiocyanate (FITC) (clone M1/70), anti-F4/80-phycoerythrin (PE) (clone BM8), anti-CD11c-allophycocyanin (APC) (clone HL3), and anti-Ly6G-Pacific Blue (clone 1A8). Data were collected on a Becton Dickinson FACSCanto II and analyzed using FlowJo software (version 8.8; Tree Star). In all experiments, forward- and side-scatter gating was used to exclude dead cells and debris, and forward-scatter area of the peak versus forward-scatter height of the peak were used to exclude doublets. Cell subsets were classified using modified versions of gating strategies described by Hall et al. and Judy et al. (30, 41). Anti-F4/80-PE was purchased from eBioscience (San Diego, CA), anti-Ly6G-Pacific Blue was purchased from Biolegend (San Diego, CA), and anti-CD11b-FITC and CD11c-APC were purchased from BD Pharmingen (San Diego, CA).
Quantification of intracellular bacteria in lungs.
Gentamicin protection assays were based on published procedures (42), with modification for use with nonadherent cells. Briefly, whole lung cell suspensions were prepared from collagenase/DNase-digested lungs of infected mice as described above (excluding red blood cell lysis) and treated with 40 μg/ml gentamicin in Dulbecco's modified Eagle medium (DMEM) for 2 h at 37°C. Following the incubation, cells were pelleted using gentle centrifugation (550 × g), rinsed twice with PBS, and lysed with 200 μl of 0.1% Triton X-100 in PBS. Bacterial CFU were enumerated by plating the lysates onto blood agar.
Cytospins and immunofluorescence.
Cytospin smears of 100-μl aliquots of BAL fluid were fixed in 75% acetone–25% ethanol for 15 min, air dried, and then rehydrated in 0.1% Triton X-100 in PBS. After blocking for 30 min in 5% normal mouse serum in PBS, the slides were stained with anti-F1-488 (Y. pestis), anti-F4/80-Alexa Fluor647 (AbD Serotech), and Hoechst. The anti-F1 antibody was conjugated to Dylight 488 using the Dylight 488 microscale antibody labeling kit from Thermo Fisher and used at a 1:200 dilution. Anti-F4/80-Alexa Fluor 647 was used at a 1:75 dilution. Cytospin smears were imaged using a Leica TCS SP5 confocal microscope and analyzed using LAS AF software, version 2.2.1.
Statistics.
Survival data were analyzed by log-rank tests, CFU data were analyzed by Mann-Whitney tests or by analysis of variance (ANOVA) when comparing more than two groups, and flow cytometry data were analyzed by Student's t tests (Prism 4.0; GraphPad Software). For graphical presentation and for assessments of statistical significance, CFU measurements that fell below the limit of our assays were assigned a value lower than the detection limit.
RESULTS
Intranasal prophylaxis with CpG ODN protects against lethal pulmonary challenge with Y. pestis strain KIM D27.
To determine whether immunomodulatory ODN could protect C57BL/6 mice from Y. pestis challenge, 50 μg of CpG ODN was administered intranasally to mice at different time points relative to intranasal infection with 20 MLD Y. pestis strain KIM D27. Markedly improved survival was observed when the ODN were administered 24 h prior to infection, in which case 80% of the infected mice survived (Fig. 1A). The degree of protection was more modest when CpG ODN were administered 4 days prior to infection and absent when administered 7 days prior to infection or 24 h postinfection. These data indicate that the protective effects of CpG ODN treatment are prophylactic and not therapeutic.
Fig 1.

Intranasal treatment of mice with CpG ODN can protect against Y. pestis challenge. (A) Survival of wild-type C57BL/6 mice treated intranasally with 50 μg CpG ODN at day −7, −4, −1, or +1 relative to the time of intranasal infection with 20 MLD Y. pestis KIM D27 (day 0; n = 4 to 5 mice per group). (B) Survival of wild-type C57BL/6 mice treated intranasally with the indicated doses of CpG ODN 1 day prior to infection with 20 MLD Y. pestis KIM D27 (n = 5 mice per group). (C) Intranasal administration of 50 μg CpG ODN 1 day prior to infection protects mice from intranasal Y. pestis challenge. Survival data pooled from 4 independent experiments (n = 20 mice per group).
Experiments were next conducted to determine whether protection from Y. pestis challenge could be induced using decreased amounts of CpG ODN. While some degree of protection was observed with either 10 or 2 μg of CpG ODN, the greatest improvement in survival was observed with 50 μg (Fig. 1B). The compiled data from four independent studies confirm that treatment with 50 μg CpG ODN 1 day prior to infection confers highly significant protection (P < 0.0001) (Fig. 1C). This regimen was used for the remainder of the experiments described in this report.
Intranasal prophylaxis with CpG ODN decreases levels of Y. pestis KIM D27 bacteria in the liver but transiently increases levels of bacteria in the lung.
To measure the effect of CpG ODN prophylaxis on bacterial burden, mice were treated intranasally with CpG ODN, infected intranasally with 20 MLD Y. pestis KIM D27, and euthanized 3 days later. Untreated control mice and control mice treated with PBS displayed similar bacterial burdens (Fig. 2). Treatment with CpG ODN reduced the number of bacterial CFU in the liver by more than 1,000-fold but increased the number of CFU in the lung by nearly 10-fold compared to levels in control mice (Fig. 2).
Fig 2.

Intranasal prophylaxis with CpG ODN reduces the hepatic bacterial burden despite increasing the pulmonary burden. Wild-type C57BL/6 mice were infected with 20 MLD Y. pestis KIM D27. CpG indicates mice treated intranasally with 50 μg CpG ODN 1 day prior to infection (n = 35). None indicates mice that received no treatment prior to infection (n = 20). PBS indicates mice that were treated with PBS diluent prior to infection (n = 15). The data were pooled from 7 independent experiments and expressed as box-and-whisker plots depicting the maximum, minimum, and median as well as 25th and 75th percentiles. ***, P value below 0.001; ns, the difference was not significant. The dashed line indicates the limit of detection.
In light of the surprising observation that treatment with CpG ODN increased the pulmonary burden at day 3 after challenge, kinetic studies were undertaken to determine the impact of CpG ODN treatment on bacterial burden at earlier and later time points. The levels of bacterial CFU detected in the lungs of CpG ODN-treated and control animals were indistinguishable at 6 h after challenge and at day 1 after challenge (Fig. 3A). A difference in burden became apparent on day 2, when the number of CFU in the lungs of CpG ODN-treated mice reached significantly higher levels than that of control animals. The pulmonary burden in mice treated with CpG ODN remained significantly higher than that of control mice through day 3 but began to decrease by day 4, at which time it no longer differed significantly from control mice.
Fig 3.
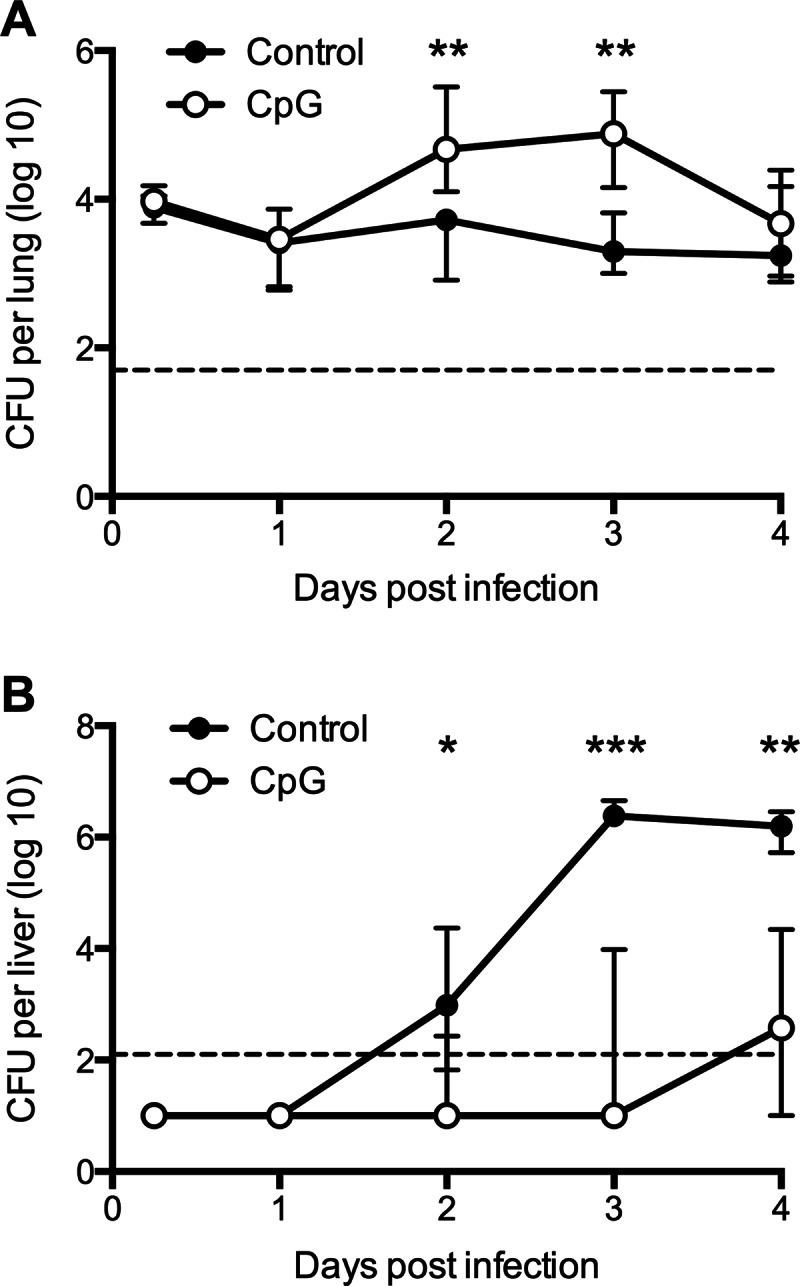
Kinetic analysis of Y. pestis bacterial burden in mice treated prophylactically with CpG ODN. CFU data for lung (A) and liver (B) tissue of wild-type C57BL/6 mice treated with 50 μg CpG ODN 1 day prior to intranasal infection with 20 MLD Y. pestis KIM D27. Control mice were left untreated or were treated with PBS. The data were pooled from 3 independent experiments and expressed as the medians and interquartile ranges (n = 9 to 15 mice per group per time point). ***, P value below 0.001; **, P value between 0.01 and 0.001; *, P value between 0.05 and 0.01. The dashed line indicates the limit of detection.
A different trend was observed in the liver (Fig. 3B). At 6 h and at day 1 after challenge, the bacterial burden was near or below the limit of detection in both CpG ODN-treated and control mice. The median burden of the control mice increased above the detection limit on day 2, peaked at day 3, and remained high at day 4. In striking contrast to the pulmonary burden, the hepatic burden in animals treated with CpG ODN was significantly lower than that of control mice at all times.
By day 6 after challenge, many of the control mice had succumbed to plague. While some of the mice treated with CpG ODN also succumbed, the majority of these mice lived until day 14. When examined at this time point, no bacteria were detected in the livers of any of the mice. Although 30% of the day 14 mice still had detectable CFU in their lungs, most had cleared the bacteria by this time (data not shown).
Intranasal prophylaxis with CpG ODN protects against Y. pestis KIM D27 challenge in the absence of adaptive immunity.
CpG ODN can stimulate both B-cell and T-cell proliferation (24, 43). B cells are indispensable for CpG ODN-induced protection against F. tularensis (28), and B cells and antibodies can play important roles in the clearance of Y. pestis (44–49). To determine whether the CpG ODN-induced protection during Y. pestis infection is B cell dependent, μMT mice, which lack functional B cells, were treated with CpG ODN and then infected intranasally with 20 MLD Y. pestis KIM D27. Pretreatment with ODN significantly increased the survival of μMT mice (P = 0.01) (Fig. 4A), indicating that the protection induced by CpG ODN does not require B cells.
Fig 4.

CpG ODN-induced protection against Y. pestis infection is not dependent on adaptive immunity. Mice were treated intranasally with 50 μg CpG ODN 1 day prior to infection with 20 MLD Y. pestis KIM D27. Control mice were left untreated or were treated with PBS diluent. (A) Survival of B-cell-deficient μMT mice. The data were pooled from 2 independent experiments (n = 9 to 10 mice per group). (B) Survival of TCRbd-deficient mice. The data were pooled from 2 independent experiments (n = 10 mice per group). (C) Survival of RAG2-deficient mice. The data were pooled from 2 independent experiments (n = 11 to 12 mice per group).
To determine whether the observed protection was dependent on T cells, TCRbd-deficient mice were treated with CpG ODN 1 day prior to infection with KIM D27. Pretreatment with ODN extended the median survival time of infected TCRbd-deficient mice from 6.5 to 16 days (P = 0.0001) (Fig. 4B), indicating that T cells were not required for CpG ODN to exert a protective effect.
The findings described above indicate that prophylaxis with CpG ODN confers significant protection in the absence of either B or T cells. To determine whether this regimen confers protection when both arms of the adaptive immune system are absent, RAG-deficient mice, which lack both B and T cells, were treated with CpG ODN and then infected intranasally with 20 MLD Y. pestis KIM D27. Similar to the results obtained with TCRbd-deficient mice, pretreatment with CpG ODN significantly delayed the time to death of infected RAG-deficient mice, extending their median survival time from 7 to 16.5 days (P = 0.0009) (Fig. 4C). Together, these results indicate that CpG ODN can exert a protective effect against Y. pestis KIM D27 infection in the absence of adaptive immunity.
TLR9 is required for CpG ODN-induced protection against challenge with Y. pestis KIM D27.
CpG ODN can stimulate the immune system through interactions with TLR9 (23, 50, 51). To determine whether CpG ODN confers protection against plague through a TLR9-dependent pathway, TLR9-deficient mice were treated with CpG ODN and then infected intranasally with KIM D27. Consistent with a critical role for TLR9, treatment with CpG ODN was unable to improve the survival of TLR9-deficient mice (Fig. 5A), and the numbers of bacteria detected in the livers of CpG ODN-treated TLR9-deficient mice were not statistically different from those detected in control wild-type mice or control TLR9-deficient mice (Fig. 5B). Notably, CpG ODN treatment failed to increase the number of CFU in the lungs of the TLR9-deficient mice, indicating that the CpG ODN-induced increase in lung bacterial burden that is observed in wild-type animals is also TLR9 dependent.
Fig 5.
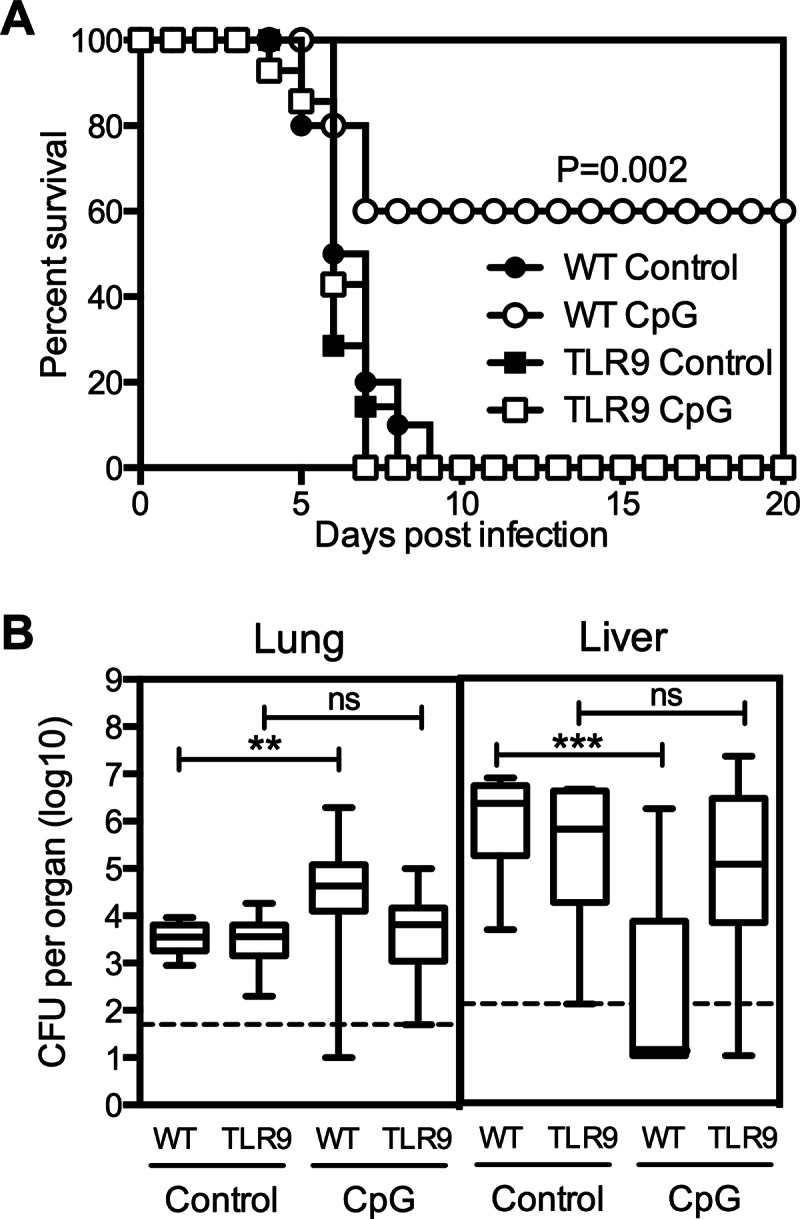
TLR9 is required for CpG ODN-induced protection against Y. pestis. Wild-type and TLR9-deficient mice were treated intranasally with 50 μg CpG ODN 1 day prior to infection with 20 MLD Y. pestis KIM D27. (A) Survival (n = 10 to 14 male and female mice per group). (B) Bacterial burden data pooled from 3 independent experiments and expressed as box-and-whisker plots depicting the maximum, minimum, and median as well as 25th and 75th percentiles (n = 13 to 16 mice per group). All mice used in these studies were male. Samples were collected at day 3 after challenge. ***, P value below 0.001; **, P value between 0.01 and 0.001; ns, the difference is not significant. The dashed line indicates the limit of detection.
Intranasal prophylaxis with CpG ODN protects mice against intraperitoneal challenge with Y. pestis KIM D27.
To assess whether the protective effects of intranasal prophylaxis with CpG ODN extend to systemic challenge, mice were treated intranasally with CpG ODN and then challenged intraperitoneally with KIM D27. The CpG ODN treatment significantly increased the survival of infected mice (P = 0.003) (Fig. 6A) and reduced the number of bacterial CFU in liver tissue (P = 0.003) (Fig. 6B). No significant difference in pulmonary burden was observed between CpG ODN-treated and control mice after intraperitoneal infection with Y. pestis (Fig. 6B).
Fig 6.
Intranasal prophylaxis with CpG ODN protects mice against intraperitoneal challenge with Y. pestis. Mice were treated intranasally with 50 μg CpG ODN 1 day prior to intraperitoneal infection with approximately 2 × 104 CFU Y. pestis KIM D27. Control mice were treated with PBS diluent. (A) Survival data pooled from 4 independent experiments (n = 25 mice per group). (B) Bacterial burden data pooled from 3 independent experiments and expressed as box-and-whisker plots depicting the maximum, minimum, and median as well as 25th and 75th percentiles (n = 15 mice per group). Samples were collected at day 3 after challenge. **, P value between 0.01 and 0.001. The dashed line indicates the limit of detection.
Intranasal treatment with CpG ODN alters the lung leukocyte population and increases the number of cell-associated Y. pestis KIM D27 bacteria in the lung.
Treatment with CpG ODN can induce the recruitment of leukocytes to lung tissue (30, 52, 53). To investigate whether CpG ODN treatment alters pulmonary leukocyte populations in the model described here, lung cells were prepared and analyzed by flow cytometry. Treatment with CpG ODN did not impact the number of Ly6G+ neutrophils in lung tissue but significantly increased the number of F4/80−/CD11b+/CD11c+ dendritic cells and F4/80+/CD11bhi macrophages (Fig. 7A to E). The number of F4/80+/CD11c+ alveolar macrophages decreased significantly in CpG ODN-treated animals (Fig. 7F). These changes occurred both in the presence and the absence of infection with KIM D27, indicating that the effects of CpG ODN on the lung leukocyte populations are not dependent on the presence of Y. pestis.
Fig 7.

Intranasal administration of CpG ODN alters lung leukocyte populations. Mice were treated intranasally with 50 μg CpG ODN 1 day prior to intranasal infection with 20 MLD Y. pestis KIM D27. Control mice were treated with PBS diluent. Lung leukocytes were analyzed at day 3 after challenge. Leukocytes in uninfected mice were analyzed at day 4 after treatment with CpG or PBS. (A and B) Gating strategy used to identify lung cell populations of PBS-treated (A) or CpG ODN-treated (B) mice infected with Y. pestis. F4/80+ cells that were CD11c+ but CD11b− were classified as alveolar macrophages, F4/80+ cells that had high expression of CD11b were classified as CD11bhi macrophages, F4/80− cells that were CD11c− but CD11b+ and Ly6G+ were classified as neutrophils, and F4/80− cells that were both CD11b+ and CD11c+ were classified as CD11b+/CD11c+ dendritic cells. (C to F) Numbers of F4/80−/CD11c−/CD11b+/Ly6G+ neutrophils (C), F4/80+/CD11bhi macrophages (D), F4/80−/CD11c+/CD11b+ dendritic cells (E), and F4/80+/CD11c+/CD11b− alveolar macrophages (F). Data were pooled from 2 independent experiments and expressed as box-and-whisker plots depicting the maximum, minimum, and median as well as 25th and 75th percentiles (n = 8 to 10 mice per group). ***, P value below 0.001; **, P value between 0.01 and 0.001; *, P value between 0.05 and 0.01.
Experiments were also conducted to investigate the impact of CpG ODN treatment on the location of bacteria within the lungs of infected mice. Three days after intranasal infection with KIM D27, bacterial CFU were enumerated in BAL fluid and lung tissue. The number of CFU in the BAL fluid from the majority of the PBS-treated mice was near or below the limit of detection, despite there being detectable levels of bacteria in the lung tissue of these same animals (Fig. 8A). Pretreatment with CpG ODN increased the bacterial burden in the lung tissue and also strikingly increased the number of CFU in BAL fluid (Fig. 8A). Consistent with results shown in Fig. 2, CpG ODN treatment dramatically reduced the hepatic bacterial burden despite increasing the bacterial burden in the lung (Fig. 8A).
Fig 8.

Intranasal prophylaxis with CpG ODN increases the bacterial burden of BAL fluid as well as the incidence of cell-associated bacteria. Mice were treated intranasally with 50 μg CpG ODN 1 day prior to intranasal infection with 20 MLD Y. pestis KIM D27. Control mice were treated with PBS diluent. (A) Bacterial burden data of lung tissue, BAL fluid, and liver pooled from 2 independent experiments and expressed as box-and-whisker plots depicting the maximum, minimum, and median as well as 25th and 75th percentiles (n = 19 to 20 mice per group). Samples were collected at day 3 after challenge. ***, P value below 0.001. The dashed line indicates the limit of detection. (B) Numbers of viable bacteria recovered from whole lung cell suspensions of either CpG-ODN- or PBS-treated mice after treatment with gentamicin for 2 h. The data are pooled from 3 independent experiments and are expressed as box-and-whisker plots depicting the maximum, minimum, and median as well as 25th and 75th percentiles (n = 15 mice per group). Samples were collected at day 3 after challenge. ***, P value below 0.001. The dashed line indicates the limit of detection. (C) Micrographs of immunostained cytospin smears prepared from BAL fluid samples collected from CpG ODN- or PBS-treated mice on day 3 after KIM D27 challenge. Samples were stained with anti-F4/80 (blue), anti-F1 antigen (green), and Hoechst (white). Arrows indicate leukocytes associated with bacteria. Scale bars indicate a distance of 50 μm.
To determine whether intranasal CpG ODN treatment increases the number of intracellular bacteria, gentamicin protection assays were performed using cells that were isolated from the lungs of both CpG ODN- and PBS-treated mice 3 days after challenge with KIM D27 (42). Cell suspensions were treated with 40 μg/ml gentamicin for 2 h to kill extracellular bacteria, after which the cells were lysed and plated to enumerate surviving bacteria. Significantly more bacterial CFU were recovered from cell suspensions prepared from CpG ODN-treated mice than from those of the PBS-treated counterparts, indicating that the lungs of CpG ODN-treated animals contained more intracellular bacteria. Consistent with these data is the observation that cell-associated bacteria were more readily detected by immunofluorescence in cytospin smears of BAL fluid isolated from CpG-treated mice than they were in BAL fluid from PBS-treated mice (Fig. 8B). Bacteria were found to associate with both macrophages and polymorphonuclear neutrophils, which were identified by F4/80 staining and nuclear morphology, respectively (Fig. 8B and data not shown). Despite their increase in CpG ODN-treated lungs, intracellular bacteria accounted for <0.3%, on average, of the total bacteria present in the lungs of CpG-treated mice (data not shown). This finding, together with the observation that Triton X-100-mediated lysis of whole BAL fluid samples did not significantly increase the overall number of CFU detected by plating (data not shown), demonstrates that most of the bacteria in the lungs of CpG-treated animals are extracellular.
Intranasal prophylaxis with CpG ODN protects mice against subcutaneous, but not pulmonary, challenge with Y. pestis strain CO92.
The increased bacterial load in the lungs of CpG ODN-treated mice infected with KIM D27 suggested that CpG ODN prophylaxis might fail to protect against infections that cause fulminant pneumonia. To investigate the impact of CpG ODN prophylaxis during fulminant plague pneumonia, CpG ODN-treated mice were inoculated intranasally with 10 MLD Y. pestis strain CO92, a fully virulent pgm-positive strain. In contrast to the findings with KIM D27 (Fig. 1), pretreatment with CpG ODN failed to significantly protect against intranasal challenge with CO92 (Fig. 9A). Nevertheless, intranasal prophylaxis with CpG ODN enhanced peripheral host defense against fully virulent Y. pestis, as this treatment significantly protected mice against a lethal subcutaneous challenge with CO92 (P = 0.002) (Fig. 9B).
Fig 9.

Intranasal prophylaxis with CpG ODN can protect against subcutaneous but not intranasal challenge with Y. pestis strain CO92. (A) Survival of mice treated intranasally with 50 μg CpG ODN 1 day prior to intranasal infection with approximately 1 × 104 CFU Y. pestis CO92. Control mice were treated with PBS diluent. The data were pooled from 2 independent experiments (n = 15 mice per group). (B) Survival of mice treated intranasally with 50 μg CpG ODN 1 day prior to subcutaneous infection with 10 CFU Y. pestis CO92. Control mice were treated with PBS diluent. The data were pooled from 2 independent experiments (n = 20 mice per group). (C) Survival of mice treated with 50 μg CpG ODN at the indicated time points relative to subcutaneous infection with 10 CFU Y. pestis CO92. Control mice were treated with PBS diluent at both time points, and PBS was administered as an additional sham treatment to mice receiving only one CpG ODN treatment. The data were pooled from 2 independent experiments (n = 19 to 20 mice per group).
To investigate whether protection against subcutaneous CO92 challenge could be augmented and/or extended by repeated administration of CpG ODN, studies were conducted in which mice received a second intranasal treatment with CpG ODN 2 days after infection. These mice were not significantly more protected than mice receiving the pretreatment alone (Fig. 9C). Furthermore, no significant protection against subcutaneous CO92 challenge was observed in mice receiving only the postchallenge treatment, although it did extend the median survival time from 6.5 to 8 days. This finding is consistent with the observation that CpG ODN administration is unable to exert a significant protective effect against intranasal Y. pestis D27 challenge when administered therapeutically (Fig. 1A).
DISCUSSION
This study demonstrates that prophylactic intranasal administration of immunomodulatory CpG ODN confers mice with significant protection against plague. The protective response appears to be mediated primarily by impacts of CpG ODN on innate immunity, independent of adaptive immunity, since it is measurable in RAG-deficient mice, which lack lymphocytes. While prophylaxis with CpG ODN significantly extends the life span of TCRbd- and RAG-deficient mice infected with the KIM D27 strain of Y. pestis, it only modestly increases their overall survival (Fig. 4). A more dramatic improvement in overall survival is observed in CpG ODN-treated μMT mice, which lack functional B cells but can mount a Y. pestis-specific T-cell response. Thus, T cells appear to enhance the protective impact of prophylactic CpG ODN treatment. Prior studies have shown that T cells can contribute to protection against Y. pestis infection (9, 38, 54–58).
Our findings expand upon prior work demonstrating that stimulation of innate immunity can enhance protection against pulmonary Y. pestis challenge. Previous studies established that modifying the LPS structure of Y. pestis or treating mice with lipid A mimetics can stimulate TLR4-dependent innate defense against plague (18, 21, 22). Our studies extend these observations to treatment with CpG ODN, a mimetic of bacterial DNA that stimulates immunity via TLR9. Paradoxically, treatment with CpG ODN increases the numbers of bacteria in the lungs of KIM D27-infected mice, despite decreasing the bacterial burden in the liver and spleen and increasing overall survival (Fig. 1C and 2 and data not shown). All of the impacts of CpG ODN in this model are dependent on TLR9, including the increased lung bacterial burden (Fig. 5). This is not the first instance in which CpG ODN treatment has been shown to increase bacterial burden. Wong et al. reported that treating mice with CpG ODN at the time of infection with Salmonella enterica increases the number of splenic bacterial CFU on day 5 of infection (34). Unlike the Y. pestis KIM D27 data reported here, however, CpG ODN treatment exacerbated disease in the Salmonella-infected mice, resulting in greater mortality (34). Conversely, treatment of mice with CpG ODN reduces lung bacterial burden and improves survival during lethal respiratory infections with K. pneumoniae and B. pseudomallei (25, 31, 32, 59). Therefore, our KIM D27 plague model appears to be unique in that CpG ODN treatment increases the pulmonary bacterial burden while simultaneously promoting the survival of the host.
Further studies are required to define the TLR9-dependent mechanisms by which intranasal prophylaxis with CpG ODN transiently increases the pulmonary burden of KIM D27. The increase is not evident until 2 days after initiating the infection, suggesting that treatment with CpG ODN does not influence the ability of KIM D27 to colonize the lung. The paucity of detectable CFU in BAL fluid collected from PBS-treated mice at day 3 indicates that most KIM D27 CFU normally reside in the lung tissue and not in the alveolar space (Fig. 8). Treatment with CpG ODN significantly increases the number of CFU in BAL fluid, but presently it is unclear whether this increase results from bacterial replication in the alveolar space or from the egress of bacteria growing within the lung tissue.
We also found increased numbers of pulmonary F4/80−/CD11b+/CD11c+ dendritic cells and F4/80+/CD11bhi macrophages in mice treated with CpG ODN (Fig. 7D and E). These leukocytes may provide a protective niche for intracellular replication of KIM D27, since Y. pestis can survive and replicate within phagocytes (60–63), and CpG ODN pretreatment increases the capacity of S. enterica to grow within macrophages (34). The lungs of CpG ODN-treated mice were found to contain more intracellular bacteria (Fig. 8); however, the majority of bacteria appear to reside extracellularly in both the CpG-treated mice and controls. Alternatively, the transient increase in lung burden may result from the CpG ODN-mediated decrease in the number of pulmonary F4/80+/CD11c+/CD11b+ alveolar macrophages (Fig. 7F), particularly if these cells normally contribute to the clearance of Y. pestis.
Despite its ability to infect and colonize lung tissue, KIM D27 does not cause fulminant pneumonia in mice; rather, the infected animals appear to succumb to sepsis resulting from the systemic spread of bacteria (35, 36). This raises the possibility that the benefit of CpG ODN treatment primarily derives from its ability to control or prevent infection at extrapulmonary sites. For example, CpG ODN treatment may inhibit the egress of bacteria from the lung, thereby reducing the dissemination of KIM D27. Alternatively, intranasal treatment with CpG ODN may reduce the susceptibility of visceral organs to infection by KIM D27, a possibility supported by the observations that intranasal prophylaxis with CpG ODN reduces the hepatic burden in mice challenged intraperitoneally with KIM D27 (Fig. 6) and protects against subcutaneous challenge with the fully virulent strain CO92 (Fig. 9B).
Our data establish that intranasal prophylaxis with CpG ODN confers mice with significant protection against Y. pestis infection but also highlight the limits of this protection. The CpG ODN treatment failed to protect against low-dose pulmonary challenge with Y. pestis strain CO92, even though it did protect against subcutaneous challenge with the fully virulent strain. We anticipate that the efficacy of CpG ODN prophylaxis will be improved by changing the vehicle used for its delivery; for example, the window of efficacy for B. pseudomallei infection was extended by incorporating CpG ODN into cationic liposomes (64).
There is a significant need for countermeasures that protect humans from acutely lethal diseases caused by highly virulent bacterial pathogens. Y. pestis is one of the world's most virulent bacterial pathogens, and it has the potential to be weaponized (4, 6). Given that prophylaxis with CpG ODN confers mice with significant protection against Y. pestis infection, and given the relative ease with which intranasal prophylaxis could be delivered to humans, further studies aimed at improving the protection against plague afforded by CpG ODN prophylaxis are merited.
ACKNOWLEDGMENTS
We thank Robert Brubaker for the KIM D27 strain of Y. pestis, Ron LaCourse for technical assistance with flow cytometry, and the dedicated staff of the Trudeau Institute animal facilities for the expert care of mice. We are grateful to the Trudeau Institute Select Agent Program for enabling studies with fully virulent Y. pestis, and we thank Lawrence Johnson for general advice and review of the manuscript.
This work was supported by the Trudeau Institute and Public Health Service grants R01-AI061577 (S. T. Smiley), R01-AI071295 (S. T. Smiley), and U54-AI057158 (W. I. Lipkin) from the National Institute of Allergy and Infectious Disease.
Footnotes
Published ahead of print 1 April 2013
REFERENCES
- 1. Perry RD, Fetherston JD. 1997. Yersinia pestis–etiologic agent of plague. Clin. Microbiol. Rev. 10:35–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pollitzer R. 1954. Plague. Monogr. Ser. World Health Organ. 22:1–698 [PubMed] [Google Scholar]
- 3. Prentice MB, Rahalison L. 2007. Plague. Lancet 369:1196–1207 [DOI] [PubMed] [Google Scholar]
- 4. Butler T. 1983. Plague and other Yersinia infections. Plenum Press, New York, NY [Google Scholar]
- 5. Sebbane F, Jarrett CO, Gardner D, Long D, Hinnebusch BJ. 2006. Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc. Natl. Acad. Sci. U. S. A. 103:5526–5530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Inglesby TV, Dennis DT, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Koerner JF, Layton M, McDade J, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Schoch-Spana M, Tonat K. 2000. Plague as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA 283:2281–2290 [DOI] [PubMed] [Google Scholar]
- 7. Kool JL. 2005. Risk of person-to-person transmission of pneumonic plague. Clin. Infect. Dis. 40:1166–1172 [DOI] [PubMed] [Google Scholar]
- 8. Smiley ST. 2008. Current challenges in the development of vaccines for pneumonic plague. Expert Rev. Vaccines 7:209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smiley ST. 2008. Immune defense against pneumonic plague. Immunol. Rev. 225:256–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ratsitorahina M, Chanteau S, Rahalison L, Ratsifasoamanana L, Boisier P. 2000. Epidemiological and diagnostic aspects of the outbreak of pneumonic plague in Madagascar. Lancet 355:111–113 [DOI] [PubMed] [Google Scholar]
- 11. Titball RW, Williamson ED. 2004. Yersinia pestis (plague) vaccines. Expert Opin. Biol. Ther. 4:965–973 [DOI] [PubMed] [Google Scholar]
- 12. Williamson ED, Packer PJ, Waters EL, Simpson AJ, Dyer D, Hartings J, Twenhafel N, Pitt ML. 2011. Recombinant (F1+V) vaccine protects cynomolgus macaques against pneumonic plague. Vaccine 29:4771–4777 [DOI] [PubMed] [Google Scholar]
- 13. Hart MK, Saviolakis GA, Welkos SL, House RV. 2012. Advanced development of the rF1V and rBV A/B vaccines: progress and challenges. Adv. Prev. Med. 2012:731604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pitt M. 2004. Non-human primates as a model for pneumonic plague. Animals Models and Correlates of Protection for Plague Vaccines Workshop, Gaithersburg, MD, 13 to 14 October 2004 [Google Scholar]
- 15. Cornelis GR. 2002. Yersinia type III secretion: send in the effectors. J. Cell Biol. 158:401–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Viboud GI, Bliska JB. 2005. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol. 59:69–89 [DOI] [PubMed] [Google Scholar]
- 17. Heesemann J, Sing A, Trulzsch K. 2006. Yersinia's stratagem: targeting innate and adaptive immune defense. Curr. Opin. Microbiol. 9:55–61 [DOI] [PubMed] [Google Scholar]
- 18. Montminy SW, Khan N, McGrath S, Walkowicz MJ, Sharp F, Conlon JE, Fukase K, Kusumoto S, Sweet C, Miyake K, Akira S, Cotter RJ, Goguen JD, Lien E. 2006. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat. Immunol. 7:1066–1073 [DOI] [PubMed] [Google Scholar]
- 19. Kawahara K, Tsukano H, Watanabe H, Lindner B, Matsuura M. 2002. Modification of the structure and activity of lipid A in Yersinia pestis lipopolysaccharide by growth temperature. Infect. Immun. 70:4092–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rebeil R, Ernst RK, Gowen BB, Miller SI, Hinnebusch BJ. 2004. Variation in lipid A structure in the pathogenic yersiniae. Mol. Microbiol. 52:1363–1373 [DOI] [PubMed] [Google Scholar]
- 21. Szaba FM, Kummer LW, Wilhelm LB, Lin JS, Parent MA, Montminy-Paquette SW, Lien E, Johnson LL, Smiley ST. 2009. D27-pLpxL, an avirulent strain of Yersinia pestis, primes T cells that protect against pneumonic plague. Infect. Immun. 77:4295–4304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Airhart CL, Rohde HN, Bohach GA, Hovde CJ, Deobald CF, Lee SS, Minnich SA. 2008. Induction of innate immunity by lipid A mimetics increases survival from pneumonic plague. Microbiology 154:2131–2138 [DOI] [PubMed] [Google Scholar]
- 23. Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, Wagner H, Lipford GB. 2001. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. U. S. A. 98:9237–9242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. 1995. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 374:546–549 [DOI] [PubMed] [Google Scholar]
- 25. Deng JC, Moore TA, Newstead MW, Zeng X, Krieg AM, Standiford TJ. 2004. CpG oligodeoxynucleotides stimulate protective innate immunity against pulmonary Klebsiella infection. J. Immunol. 173:5148–5155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krug A, Rothenfusser S, Hornung V, Jahrsdorfer B, Blackwell S, Ballas ZK, Endres S, Krieg AM, Hartmann G. 2001. Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur. J. Immunol. 31:2154–2163 [DOI] [PubMed] [Google Scholar]
- 27. Ballas ZK, Krieg AM, Warren T, Rasmussen W, Davis HL, Waldschmidt M, Weiner GJ. 2001. Divergent therapeutic and immunologic effects of oligodeoxynucleotides with distinct CpG motifs. J. Immunol. 167:4878–4886 [DOI] [PubMed] [Google Scholar]
- 28. Elkins KL, Rhinehart-Jones TR, Stibitz S, Conover JS, Klinman DM. 1999. Bacterial DNA containing CpG motifs stimulates lymphocyte-dependent protection of mice against lethal infection with intracellular bacteria. J. Immunol. 162:2291–2298 [PubMed] [Google Scholar]
- 29. Klinman DM. 2004. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev. Immunol. 4:249–258 [DOI] [PubMed] [Google Scholar]
- 30. Judy BM, Taylor K, Deeraksa A, Johnston RK, Endsley JJ, Vijayakumar S, Aronson JF, Estes DM, Torres AG. 2012. Prophylactic application of CpG oligonucleotides augments the early host response and confers protection in acute melioidosis. PLoS One 7:e34176 doi:10.1371/journal.pone.0034176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Easton A, Haque A, Chu K, Patel N, Lukaszewski RA, Krieg AM, Titball RW, Bancroft GJ. 2011. Combining vaccination and postexposure CpG therapy provides optimal protection against lethal sepsis in a biodefense model of human melioidosis. J. Infect. Dis. 204:636–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Waag DM, McCluskie MJ, Zhang N, Krieg AM. 2006. A CpG oligonucleotide can protect mice from a low aerosol challenge dose of Burkholderia mallei. Infect. Immun. 74:1944–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wongratanacheewin S, Kespichayawattana W, Intachote P, Pichyangkul S, Sermswan RW, Krieg AM, Sirisinha S. 2004. Immunostimulatory CpG oligodeoxynucleotide confers protection in a murine model of infection with Burkholderia pseudomallei. Infect. Immun. 72:4494–4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wong CE, Sad S, Coombes BK. 2009. Salmonella enterica serovar Typhimurium exploits Toll-like receptor signaling during the host-pathogen interaction. Infect. Immun. 77:4750–4760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lathem WW, Crosby SD, Miller VL, Goldman WE. 2005. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc. Natl. Acad. Sci. U. S. A. 102:17786–17791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee-Lewis H, Anderson DM. 2010. Absence of inflammation and pneumonia during infection with nonpigmented Yersinia pestis reveals a new role for the pgm locus in pathogenesis. Infect. Immun. 78:220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lahteenmaki K, Virkola R, Saren A, Emody L, Korhonen TK. 1998. Expression of plasminogen activator pla of Yersinia pestis enhances bacterial attachment to the mammalian extracellular matrix. Infect. Immun. 66:5755–5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Parent MA, Berggren KN, Kummer LW, Wilhelm LB, Szaba FM, Mullarky IK, Smiley ST. 2005. Cell-mediated protection against pulmonary Yersinia pestis infection. Infect. Immun. 73:7304–7310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jakob T, Walker PS, Krieg AM, von Stebut E, Udey MC, Vogel JC. 1999. Bacterial DNA and CpG-containing oligodeoxynucleotides activate cutaneous dendritic cells and induce IL-12 production: implications for the augmentation of Th1 responses. Int. Arch. Allergy Immunol. 118:457–461 [DOI] [PubMed] [Google Scholar]
- 40. Parent MA, Wilhelm LB, Kummer LW, Szaba FM, Mullarky IK, Smiley ST. 2006. Gamma interferon, tumor necrosis factor alpha, and nitric oxide synthase 2, key elements of cellular immunity, perform critical protective functions during humoral defense against lethal pulmonary Yersinia pestis infection. Infect. Immun. 74:3381–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hall JD, Woolard MD, Gunn BM, Craven RR, Taft-Benz S, Frelinger JA, Kawula TH. 2008. Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect. Immun. 76:5843–5852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Patel AA, Lee-Lewis H, Hughes-Hanks J, Lewis CA, Anderson DM. 2012. Opposing roles for interferon regulatory factor-3 (IRF-3) and type I interferon signaling during plague. PLoS Pathog. 8:e1002817 doi:10.1371/journal.ppat.1002817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bendigs S, Salzer U, Lipford GB, Wagner H, Heeg K. 1999. CpG-oligodeoxynucleotides co-stimulate primary T cells in the absence of antigen-presenting cells. Eur. J. Immunol. 29:1209–1218 [DOI] [PubMed] [Google Scholar]
- 44. Anderson GW, Jr, Worsham PL, Bolt CR, Andrews GP, Welkos SL, Friedlander AM, Burans JP. 1997. Protection of mice from fatal bubonic and pneumonic plague by passive immunization with monoclonal antibodies against the F1 protein of Yersinia pestis. Am. J. Trop. Med. Hyg. 56:471–473 [DOI] [PubMed] [Google Scholar]
- 45. Eisele NA, Anderson DM. 2009. Dual-function antibodies to Yersinia pestis LcrV required for pulmonary clearance of plague. Clin. Vaccine Immunol. 16:1720–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Green M, Rogers D, Russell P, Stagg AJ, Bell DL, Eley SM, Titball RW, Williamson ED. 1999. The SCID/Beige mouse as a model to investigate protection against Yersinia pestis. FEMS Immunol. Med. Microbiol. 23:107–113 [DOI] [PubMed] [Google Scholar]
- 47. Hill J, Copse C, Leary S, Stagg AJ, Williamson ED, Titball RW. 2003. Synergistic protection of mice against plague with monoclonal antibodies specific for the F1 and V antigens of Yersinia pestis. Infect. Immun. 71:2234–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hill J, Eyles JE, Elvin SJ, Healey GD, Lukaszewski RA, Titball RW. 2006. Administration of antibody to the lung protects mice against pneumonic plague. Infect. Immun. 74:3068–3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Motin VL, Nakajima R, Smirnov GB, Brubaker RR. 1994. Passive immunity to yersiniae mediated by anti-recombinant V antigen and protein A-V antigen fusion peptide. Infect. Immun. 62:4192–4201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408:740–745 [DOI] [PubMed] [Google Scholar]
- 51. Takeshita F, Leifer CA, Gursel I, Ishii KJ, Takeshita S, Gursel M, Klinman DM. 2001. Cutting edge: role of Toll-like receptor 9 in CpG DNA-induced activation of human cells. J. Immunol. 167:3555–3558 [DOI] [PubMed] [Google Scholar]
- 52. Tasaka S, Kamata H, Miyamoto K, Nakano Y, Shinoda H, Kimizuka Y, Fujiwara H, Hasegawa N, Fujishima S, Miyasho T, Ishizaka A. 2009. Intratracheal synthetic CpG oligodeoxynucleotide causes acute lung injury with systemic inflammatory response. Respir. Res. 10:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pesce I, Monaci E, Muzzi A, Tritto E, Tavarini S, Nuti S, De Gregorio E, Wack A. 2010. Intranasal administration of CpG induces a rapid and transient cytokine response followed by dendritic and natural killer cell activation and recruitment in the mouse lung. J. Innate Immun. 2:144–159 [DOI] [PubMed] [Google Scholar]
- 54. Philipovskiy AV, Smiley ST. 2007. Vaccination with live Yersinia pestis primes CD4 and CD8 T cells that synergistically protect against lethal pulmonary Y. pestis infection. Infect. Immun. 75:878–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lin JS, Kummer LW, Szaba FM, Smiley ST. 2011. IL-17 contributes to cell-mediated defense against pulmonary Yersinia pestis infection. J. Immunol. 186:1675–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chattopadhyay A, Park S, Delmas G, Suresh R, Senina S, Perlin DS, Rose JK. 2008. Single-dose, virus-vectored vaccine protection against Yersinia pestis challenge: CD4+ cells are required at the time of challenge for optimal protection. Vaccine 26:6329–6337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Levy Y, Flashner Y, Tidhar A, Zauberman A, Aftalion M, Lazar S, Gur D, Shafferman A, Mamroud E. 2011. T cells play an essential role in anti-F1 mediated rapid protection against bubonic plague. Vaccine 29:6866–6873 [DOI] [PubMed] [Google Scholar]
- 58. Wang S, Goguen JD, Li F, Lu S. 2011. Involvement of CD8+ T cell-mediated immune responses in LcrV DNA vaccine induced protection against lethal Yersinia pestis challenge. Vaccine 29:6802–6809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rozak DA, Gelhaus HC, Smith M, Zadeh M, Huzella L, Waag D, Adamovicz JJ. 2010. CpG oligodeoxyribonucleotides protect mice from Burkholderia pseudomallei but not Francisella tularensis Schu S4 aerosols. J. Immune Based Ther. Vaccines 8:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pujol C, Bliska JB. 2003. The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect. Immun. 71:5892–5899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. O'Loughlin JL, Spinner JL, Minnich SA, Kobayashi SD. 2010. Yersinia pestis two-component gene regulatory systems promote survival in human neutrophils. Infect. Immun. 78:773–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Noel BL, Lilo S, Capurso D, Hill J, Bliska JB. 2009. Yersinia pestis can bypass protective antibodies to LcrV and activation with gamma interferon to survive and induce apoptosis in murine macrophages. Clin. Vaccine Immunol. 16:1457–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Straley SC, Harmon PA. 1984. Yersinia pestis grows within phagolysosomes in mouse peritoneal macrophages. Infect. Immun. 45:655–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Puangpetch A, Anderson R, Huang YY, Sermswan RW, Chaicumpa W, Sirisinha S, Wongratanacheewin S. 2012. Cationic liposomes extend the immunostimulatory effect of CpG oligodeoxynucleotide against Burkholderia pseudomallei infection in BALB/c mice. Clin. Vaccine Immunol. 19:675–683 [DOI] [PMC free article] [PubMed] [Google Scholar]