

Abstract
Shiga toxin (Stx) is a major virulence factor of enterohemorrhagic Escherichia coli that occasionally causes fatal systemic complications. We recently developed a tetravalent peptide (PPP-tet) that neutralizes the cytotoxicity of Stx2 using a multivalent peptide library approach. In this study, we used this technique to identify a series of tetravalent peptides that bound to Stx1, another major Stx family member, with high affinity by targeting one receptor-binding site of the B subunit. One peptide, MMA-tet, markedly inhibited Stx1 and Stx2 cytotoxicity with greater potency than PPP-tet. After forming a complex with Stx1 through its specific receptor-binding region, MMA-tet did not affect vesicular transport of the toxin to the endoplasmic reticulum but substantially rescued inhibition of the protein synthesis induced by Stx1. Oral application of MMA-tet protected mice from a fatal dose of an E. coli O157:H7 strain producing both toxins. MMA-tet may be a promising therapeutic agent against the infection.
INTRODUCTION
Infection by enterohemorrhagic Escherichia coli (EHEC) causes bloody diarrhea and hemorrhagic colitis in humans that is occasionally followed by fatal systemic complications, such as acute encephalopathy and hemolytic-uremic syndrome (1–4). Because Shiga toxin (Stx) is a major virulence factor of EHEC, Stx neutralizers can be practical therapeutic agents against EHEC infections.
Stx can be classified into two subgroups, Stx1 and Stx2 (5). Stx consists of a catalytic A subunit and a B-subunit pentamer. The former has 28S rRNA N-glycosidase activity and inhibits eukaryotic protein synthesis, whereas the latter is responsible for the high-affinity binding of the toxin to the functional cell surface receptor Galα(1-4)-Galβ(1-4)-Glcβ-ceramide (Gb3) (4, 6, 7). The crystal structure of Stx reveals the presence of three distinctive binding sites for the trisaccharide moiety of Gb3 (i.e., sites 1, 2, and 3 on each B-subunit monomer) (8, 9). Highly selective and potent binding of Stx to Gb3 is mainly attributed to the multivalent interaction of the B-subunit pentamer with the trisaccharide moiety of Gb3. This is occasionally referred to as the “clustering effect.” On the basis of this, several synthetic Stx neutralizers with clustered trisaccharides that can bind to Stx with high affinity and inhibit its cytotoxicity have been developed (10–15). However, the clinical application of these neutralizers has been substantially hampered by the synthetic complexity of the trisaccharide moiety.
Recently, we developed a multivalent peptide library technique and identified a novel peptide-based neutralizer against Stx2 (16, 17), which is more closely related to the severity of EHEC infections (18, 19). In this approach, a library of novel tetravalent peptides designed to exert the clustering effect was screened for high-affinity binding to one of the trisaccharide binding sites, site 3, of the Stx2 B subunit. We identified four tetravalent peptides that bind to Stx2 with high affinity and effectively inhibit its cytotoxicity. One of the tetravalent peptides, named PPP-tet, protected mice from a fatal dose of E. coli O157:H7 (16) and, furthermore, inhibited the lethal effect of intravenously administered Stx2 in a nonhuman primate model (20). In stark contrast to the Stx neutralizers with assembled trisaccharides, which competitively inhibit the binding of Stx to target cells, PPP-tet did not inhibit Stx binding, but instead, it induced the aberrant intracellular transport of the toxin (16). After binding to Gb3, Stx is first transported in a retrograde manner to the Golgi complex and then to the endoplasmic reticulum (ER), where the catalytic A subunit is released into the cytosol to inhibit protein synthesis (21). After forming a complex with Stx2, PPP-tet specifically inhibits the process of vesicular transport of Stx2 from the Golgi complex to the ER, followed by the effective degradation of Stx2 in an acidic compartment (16). Recently, other synthetic compounds or chemicals that similarly affect the intracellular transport of Stx have been reported (22–24), further confirming the usefulness of these transport modulators as Stx neutralizers.
PPP-tet, however, did not efficiently inhibit the cytotoxicity of Stx1, another major Stx family member, indicating an urgent need to identify a peptide-based neutralizer against Stx1. In this study, using the multivalent peptide library approach, we identified a tetravalent peptide that markedly inhibited the cytotoxicity of both Stx1 and Stx2. This tetravalent peptide, named MMA-tet, rescued mice from the lethality of E. coli O157:H7 infection with marked efficacy. We also elucidated the unique mechanism by which MMA-tet exerts its inhibitory effect on the toxins in target cells.
MATERIALS AND METHODS
Materials.
Recombinant Stx1 and Stx2, histidine-tagged Stx1 B subunit (1BH), histidine-tagged Stx2 B subunit (2BH), 1BH with single amino acid substitutions (1BH-D17E, 1BH-D18E, 1BH-F30A, 1BH-G62A, and 1BH-W34A), 1BH with double amino acid substitutions (1BH-D17E/W34A, 1BH-D18E/G62A, and 1BH-G62A/W34A), and 1BH with a triple amino acid substitution (1BH-D17E/G62A/W34A) were prepared as described previously (14, 16). The Gb3 polymer and rabbit anti-Stx antiserum were obtained as described previously (15, 16). AlphaScreen reagent and l-[4,5-3H(N)]Leu were purchased from PerkinElmer (Tokyo, Japan).
Peptides and peptide library screening.
Tetravalent peptides and tetravalent peptide libraries were synthesized as described previously (16). Recombinant 1BH or 1BH-F30A (0.5 mg of protein) bound to Ni2+ beads was incubated with 300 μg of a given library peptide in phosphate-buffered saline (PBS) overnight at 4°C. After extensive washing, bound peptides were sequenced on an Applied Biosystems model 477A protein sequencer. To calculate the relative amino acid preference at each degenerate position, the corrected quantities of amino acids in the peptides recovered from the 1BH beads were compared with those of amino acids in the peptides recovered from the 1BH-F30A beads to calculate the abundance ratios of amino acids (16).
Kinetic analysis of binding between inhibitory peptides and immobilized Stx B subunit.
The binding of tetravalent peptides to immobilized 1BH or 2BH was quantified using a Biacore T100 system instrument (GE Healthcare Sciences) as described previously (16). The resonance unit is an arbitrary unit used by the Biacore system.
Cytotoxicity assay.
Subconfluent Vero cells cultured in a 96-well plate in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum were treated with Stx1 or Stx2 (1 pg/ml) in the absence or presence of a given tetravalent peptide for 72 h at 37°C. Each peptide was added simultaneously with Stx1 or Stx2. The relative number of living cells was determined using a Cell Counting Kit-8 (Dojindo, Japan), which allows the sensitive determination of cell viability based on the production of an orange formazan dye from 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-8), by the intracellular dehydrogenases.
Analysis of binding between 1BH or a 1BH mutant and inhibitory peptides by AlphaScreen assay.
The indicated amounts of the biotinylated tetravalent peptide and 1BH or its mutant (10 μg/ml) were incubated in each well of an OptiPlate-384 microplate (PerkinElmer) for 1 h at room temperature. The plate was incubated with nickel chelate acceptor donor beads (20 μg/ml) for 30 min, followed by incubation with streptavidin donor beads (20 μg/ml) for 1 h at room temperature under protection from light. After excitation at 680 nm, the emission at 615 nm was measured by an EnVision system (PerkinElmer). The signal intensity (cps) is an arbitrary unit used by the system.
ELISA of the binding between 1BH and inhibitory peptides.
The indicated tetravalent peptides (1.7 μM dissolved in PBS) were coated onto each well of a 96-well enzyme-linked immunosorbent assay (ELISA) plate and incubated for 24 h at 4°C. After blocking, the plate was incubated with 1BH (1 μg/ml) for 1 h at room temperature. Bound 1BH was detected using rabbit anti-Stx1 antiserum as described previously (16).
125I-Stx binding assay.
Vero cells cultured in a 24-well plate were treated with 125I-Stx1 or 125I-Stx2 (1 × 106 to 2 × 106 cpm/μg protein, 1 μg/ml) in the absence or presence of a given compound for 30 min at 4°C. Each compound was added simultaneously with 125I-Stx1 or 125I-Stx2. After extensive washing, the recovered radioactivity was measured by a gamma counter.
Intracellular localization of Stx1.
Subconfluent Vero cells in a glass base dish (35 mm) were treated with Stx1 (1 μg/ml) in the absence or presence of MMA-tet (52 μM) for 1 h at 37°C. MMA-tet was added simultaneously with Stx1. After incubation, the cells were fixed with 3% paraformaldehyde. Immunostaining of Stx1, GM130, Vti1a, GS28, HSP47, calnexin, and ribophorin was performed using rabbit anti-Stx1 polyclonal antibody, mouse anti-GM130 IgG monoclonal antibody (BD Biosciences, NJ), mouse anti-Vti1a IgG monoclonal antibody (BD Biosciences), mouse anti-GS28 monoclonal antibody (BD Biosciences), mouse anti-HSP47 IgG monoclonal antibody (Enzo Life Sciences, Inc., NY), rabbit anticalnexin polyclonal antibody (Santa Cruz Biotechnology, Inc., CA), and goat antiribophorin IgG polyclonal antibody (Santa Cruz Biotechnology), respectively, followed by detection using Alexa Fluor-labeled secondary antibodies. The cells were analyzed by confocal laser scanning microscopy (Olympus, Tokyo, Japan).
Assay for protein synthesis inhibition.
Vero cells cultured in a 96-well plate in DMEM supplemented with 10% fetal calf serum were pretreated with the indicated amount of MMA-tet for 1 h and then treated with Stx1 (100 pg/ml) for 1.5 h. After extensive washing, the cells were incubated with 2 μCi/well [3H]Leu in Leu-free minimal essential medium Eagle (MEME; Sigma, Tokyo, Japan) at 37°C for 30 min, and the radioactivity incorporated into cellular proteins was counted as described previously (25).
Mouse infection protocol.
Specific-pathogen-free 3-week-old female C57BL/6 mice (Charles River Breeding Laboratories, Wilmington, MA) were maintained on a low-protein diet to induce calorie malnutrition (26). At 5 weeks of age, mice were infected intragastrically with 2 × 106 CFU of E. coli O157:H7 strain N-9 as described elsewhere (26). The indicated amount of the acetylated form of MMA-tet (Ac-MMA-tet) or saline was administered intragastrically to the mice twice a day from day 2 to day 5. Data were analyzed by Kaplan-Meier survival analysis or by Fisher's exact test when no mice had died by the end of the observation period. All animal experiments were approved by the animal ethics committee of Nara Medical University prior to their commencement.
RESULTS
Tetravalent peptide library screening identified peptide motifs with high affinity for the Stx1 B subunit.
In this study, we used a tetravalent peptide library composed of tetravalent peptides containing a polylysine core bifurcating at both ends with four randomized peptides (16). The tetravalent peptide library was screened for the capability to bind to wild-type 1BH but not to 1BH-F30A, which has a mutation in receptor-binding site 1, because this site has been demonstrated to play an essential role in the receptor binding of Stx1 (27). A tetravalent peptide library with Arg fixed at position 4 (XRX library) was used for the first round of selection on the basis of the previous observation that the Stx2 neutralizer PPP-tet, which has clustered Args in its motif, can also bind to the Stx1 B subunit (data not shown). As shown in Fig. 1A, Arg was strongly selected at positions 5 to 7 and hydrophobic amino acids were preferred at positions 1 to 3. On the basis of this result, second sets of tetravalent peptide libraries with clustered Args (RXR and XRR libraries) were screened to further refine peptide selection. Preferred selection of Arg at positions 5 and 7 was confirmed with the RXR library. Furthermore, Met was strongly selected at positions 1 and 2 with both libraries, and Ala was relatively preferred at position 3 with the XRR library. Based on these results, we identified four candidate motifs: MMARRRR, MAARRRR, AMARRRR, and AAARRRR. Tetravalent forms of these peptides with the same core structure, which were referred to as MMA-tet, MAA-tet, AMA-tet, and AAA-tet, respectively, were synthesized and examined for their capability to bind to 1BH and 2BH. As shown in Fig. 1B, all of these tetravalent peptides, especially AAA-tet and MMA-tet, bound to both B subunits with high affinity, whereas MA-tet, which has the same core structure but lacks any Stx binding motifs, did not bind to either B subunit.
Fig 1.
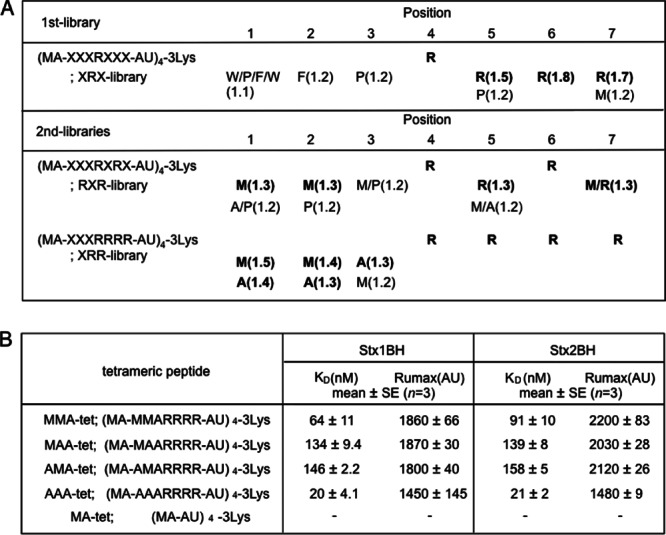
Identification of peptide motifs with high affinity for the Stx B subunit using tetravalent peptide library screening. (A) The tetravalent peptide library was composed of tetravalent peptides with a polylysine core bifurcating at both ends with four randomized peptides. The peptide library for the first screening has a sequence of Met-Ala-X-X-X-R-X-X-X-Ala-U, in which U indicates aminohexanoic acid and X indicates any amino acid except Cys. Screening of the library was performed to identify tetravalent peptides that bound to 1BH but not to 1BH-F30A. For the second screening, a peptide library with fixed Args at positions 4 and 6 (RXR library) or fixed Args at positions 4 to 7 (XRR library) was used. Values in parentheses indicate the relative selectivities for the amino acids. Boldface letters indicate amino acids that were strongly selected. Each screening was performed twice; representative values are shown. (B) The kinetics of the binding of each tetravalent peptide with each identified binding motif to immobilized 1BH or 2BH was analyzed using the Biacore system. KD, dissociation constant; Rumax, maximum resonance unit. −, binding was not detected.
MMA-tet efficiently inhibits the cytotoxicity of both Stx1 and Stx2.
The ability of the four tetravalent peptides to inhibit the cytotoxicity of Stx1 and Stx2 in Vero cells was examined (Fig. 2). Among the tetravalent peptides, MMA-tet inhibited the cytotoxicity of both toxins with the highest potency, followed by AMA-tet. MAA-tet and AAA-tet displayed less potency. Interestingly, MMA-tet inhibited Stx2 more efficiently than did PPP-tet, indicating that this tetravalent peptide is promising as a universal neutralizer against both toxins.
Fig 2.
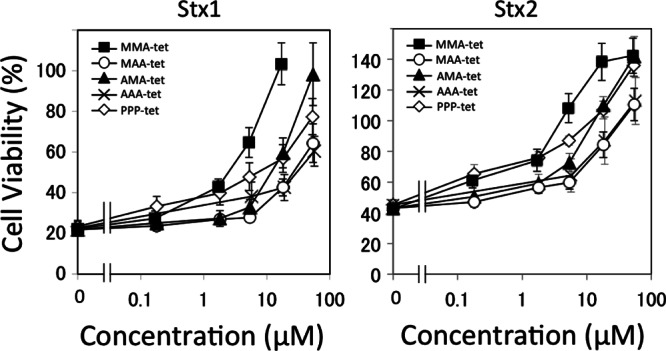
Inhibitory effects of the tetravalent peptides on the cytotoxicity of Stx1 or Stx2 in Vero cells. The effects of the tetravalent peptides on the cytotoxicity of Stx1 (1 pg/ml) or Stx2 (1 pg/ml) in Vero cells were examined by the cytotoxicity assay. Data are presented as a percentage of the control value (mean ± standard error, n = 3).
MMA-tet binds to 1BH through specific receptor-binding regions.
To elucidate the mechanism by which MMA-tet binds to 1BH, the binding between MMA-tet and a series of 1BH mutants with mutations in the trisaccharide binding sites was examined using the AlphaScreen assay. Using this system, we detected high-affinity binding between MMA-tet and 1BH (Fig. 3A). Under the same conditions, the maximum binding of MMA-tet to 1BH-G62A, a site 2 mutant, was 65% of that of 1BH, whereas the levels of binding of MMA-tet to 1BH-F30A and 1BH-D17E, both of which are site 1 mutants, were markedly reduced to 11 and 10%, respectively, consistent with the fact that MMA-tet was identified by targeting Phe30 in site 1. Interestingly, the maximum binding of MMA-tet to 1BH-D18E and 1BH-W34A, both of which are site 3 mutants, was also markedly reduced to the same level (Fig. 3A, left). Furthermore, the maximum binding of MMA-tet to 1BH-D18E/G62A, a site 2 and 3 double mutant, was reduced to 18%, and the binding of MMA-tet to 1BH-D17E/W34A (a site 1 and 3 double mutant), 1BH-G62A/W34A (a site 2 and 3 double mutant), and 1BH-D17E/G62A/W34A (a site 1, 2, and 3 triple mutant) was completely diminished (Fig. 3A, right). These results indicate the substantial contribution of both sites 1 and 3, but not site 2, to the binding of MMA-tet to the Stx1 B subunit.
Fig 3.
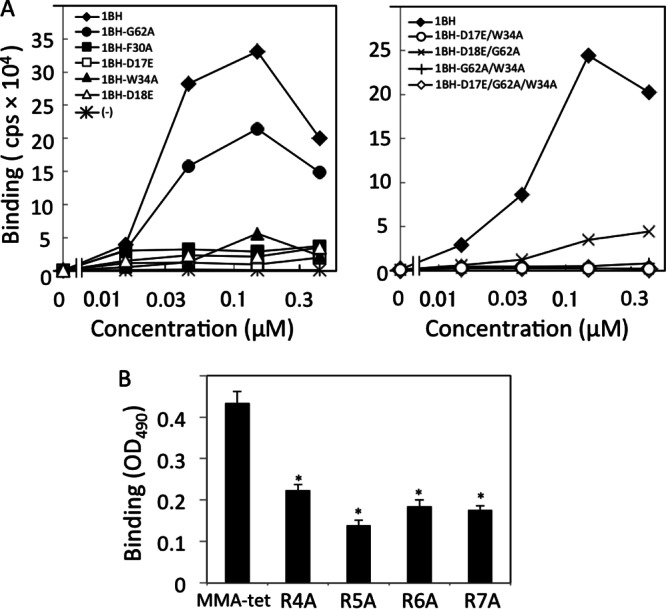
Analysis of the binding of MMA-tet with the Stx1 B subunit. (A) The binding between MMA-tet and a series of 1BH mutants (10 μg/ml) was examined using the AlphaScreen assay. Data are presented as signal intensity (cps). Each experiment was performed three times, and representative data are shown. (B) The binding of 1BH (1 μg/ml) with MMA-tet or Ala-substituted MMA-tet was examined using ELISA (mean ± standard error, n = 3; *, P < 0.001, Tukey's test). R4A, R5A, R6A, and R7A, MMA-tet with a substitution of Arg to Ala at positions 4, 5, 6, and 7, respectively. OD490, optical density at 490 nm.
To evaluate the importance of the Arg cluster present in MMA-tet, the effect of mutating each Arg to Ala on the binding of MMA-tet to 1BH was examined. As shown in Fig. 3B, all of these substitutions, especially substitution of the second Arg, significantly reduced the binding of MMA-tet to 1BH, suggesting the possible electrostatic interaction of the Arg cluster with the acidic amino acid cluster present on the receptor-binding surface of the B subunit, including Asp16, Asp17, and Asp18. In fact, among them, Asp17 and Asp18, which constitute sites 1 and 3, respectively, have been demonstrated to have essential roles in the binding of MMA-tet to the B subunit.
MMA-tet did not inhibit the uptake of Stx and its subsequent vesicular transport to the ER.
MMA-tet and the other identified tetravalent peptides did not inhibit the cell surface binding of 125I-Stx1 or 125I-Stx2, although the Gb3 polymer (15), an Stx neutralizer with clustered trisaccharides, efficiently inhibited this binding (Fig. 4A). We previously demonstrated that PPP-tet does not inhibit the binding of Stx2 to the cell surface receptor, but it induces the aberrant cellular transport of Stx2, allowing it to exert its inhibitory effect (16). Thus, the effect of MMA-tet on the intracellular transport of Stx1 was examined. The retrograde transport of Stx1 to the Golgi complex and then to the ER was confirmed by the colocalization of Stx1 with GM130 (a cis-Golgi complex marker), Vti1a (a trans-Golgi complex marker), GS28 (a trans-Golgi complex marker), and ER markers, such as HSP47, calnexin, and ribophorin. As shown in Fig. 4B, MMA-tet did not affect the colocalization of Stx1 with any of these markers, suggesting that even in a complex with MMA-tet, Stx1 can be transported to the ER through the Golgi complex.
Fig 4.
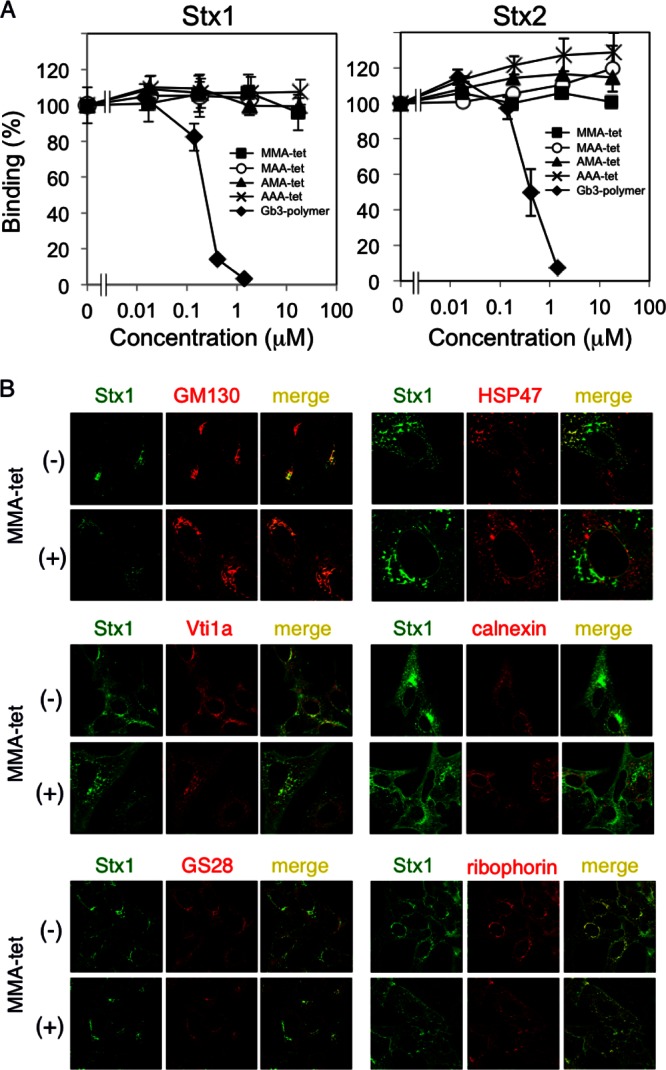
MMA-tet did not affect the uptake and subsequent intracellular transport of Stx1 to the ER. (A) Binding of 125I-Stx1 or 125I-Stx2 (1 μg/ml) to Vero cells. Data are presented as a percentage of the control value (mean ± standard error, n = 3). (B) Colocalization of Stx1 (1 μg/ml) with GM130, Vti1a, GS28, HSP47, calnexin, and ribophorin in the absence or presence of MMA-tet (52 μM) was examined by immunocytochemical staining.
MMA-tet rescued the inhibition of protein synthesis caused by Stx1 following its ER localization.
The Stx1 A subunit has been demonstrated to translocate from the ER to the cytosol to exert its 28S rRNA N-glycosidase activity, resulting in the inhibition of protein synthesis. After 1.5 h of incubation of Vero cells with Stx1, i.e., during the early stage of this translocation, [3H]Leu uptake into newly synthesized proteins was inhibited by 30% (Fig. 5). The inhibition of [3H]Leu uptake was substantially recovered by the presence of MMA-tet in a dose-dependent manner, suggesting that MMA-tet negatively affects the translocation of the A subunit from the ER to the cytosol.
Fig 5.
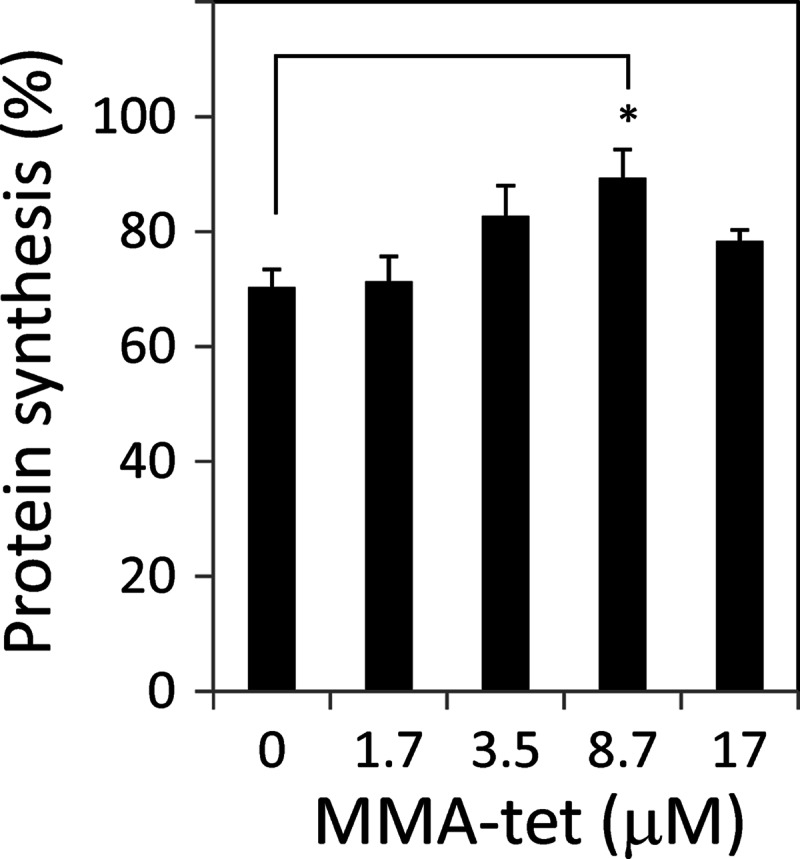
Effect of MMA-tet on Stx1-induced protein synthesis inhibition. Vero cells cultured in DMEM supplemented with 10% fetal calf serum were pretreated with each amount of MMA-tet for 1 h and then treated with Stx1 (100 pg/ml) for 1.5 h. After extensive washing, the cells were incubated with 2 μCi/well [3H]Leu in Leu-free MEME at 37°C for 30 min. The relative amounts of radioactivity incorporated into cellular proteins are presented as percentages of the control value without Stx1 (mean ± standard error, n = 3; *, P < 0.05, Tukey's test).
Acetylated MMA-tet protected mice from the lethality caused by E. coli O157:H7 infections.
The inhibitory effects of MMA-tet on the lethality of E. coli O157:H7 infections in mice with protein-calorie malnutrition (26), which are very susceptible to infection, were examined. To prevent proteolytic degradation in the gastrointestinal tract, an acetylated form of MMA-tet (Ac-MMA-tet) was synthesized. Intragastrically administered Ac-MMA-tet completely inhibited the lethality of E. coli O157:H7 infections at an amount of 4.2 nmol/g (P < 0.0001) and rescued mice with high potency at an amount of 1.25 nmol/g (P < 0.005) (Fig. 6).
Fig 6.
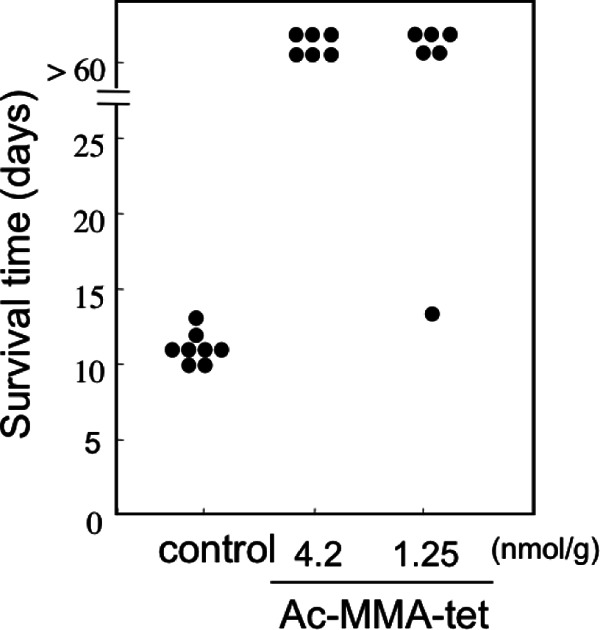
Ac-MMA-tet protects mice from the lethality of E. coli O157:H7 infection. Mice with protein-calorie malnutrition were infected intragastrically with a fatal dose of E. coli O157:H7 strain N-9 on day 0. The indicated amount of Ac-MMA-tet was administered intragastrically twice a day from day 2 to day 5. The survival time of each animal after Ac-MMA-tet (1.25 nmol/g of body weight, n = 6; 4.2 nmol/g of body weight, n = 6) or saline (n = 8) administration is shown.
DISCUSSION
Using a tetravalent peptide library approach, we identified four tetravalent peptides that exhibit high affinities for the Stx1 B subunit and inhibit Stx1 cytotoxicity by targeting one of the receptor-binding sites, namely, site 1. Interestingly, one of these tetravalent peptides, MMA-tet, markedly inhibited both Stx1 and Stx2 with even more potency than PPP-tet, a previously identified Stx2 neutralizer (16). In the previous study, it was revealed that both sites 1 and 2 of Stx1 play essential roles in receptor binding, whereas site 3 has a lower contribution to the binding but cooperatively enhances effective binding through site 1 and/or site 2 (27). In this study, MMA-tet was found to bind to the Stx1 B subunit through both sites 1 and 3, possibly contributing to the potent inhibitory effect of MMA-tet against cytotoxicity. Conversely, AAA-tet, which has the lowest KD (dissociation constant) value for 1BH binding among the identified tetravalent peptides, did not efficiently inhibit cytotoxicity. Binding analysis between AAA-tet and 1BH by ELISA indicated that the contribution of site 3, including Asp18 and Trp34, is less than that of site 1 (K. Tsutsuki unpublished data), further supporting the importance of occupation of both sites 1 and 3 to function as an effective Stx1 neutralizer.
All four Args present in MMA-tet were revealed to be involved in its effective binding to the Stx1 B subunit, suggesting an electrostatic interaction of these Args with the acidic amino acid cluster present on the receptor-binding surface of the Stx1 B subunit (8). The superior inhibitory effect of MMA-tet on Stx1 compared to the effects of the other tetravalent peptides with the same clustered Args may be explained by the presence of the consecutive Mets at positions 1 and 2, both of which could be involved in forming hydrophobic interactions with the B subunit through Phe30 (site 1) and Trp34 (site 3), as mentioned previously. AMA-tet and MAA-tet, each of which lacks one of these Mets, inhibited the cytotoxicity of Stx1 with less potency than MMA-tet, confirming the importance of the hydrophobic interaction for the inhibitory efficacy, in addition to the electrostatic interaction through clustered Args.
Although MMA-tet did not inhibit the uptake of Stx1 or its subsequent retrograde transport to the ER through the Golgi complex, the inhibition of protein synthesis caused by Stx1 was substantially recovered in the presence of MMA-tet. Because the translocation of the A subunit from the ER to the cytosol is considered crucial for the inhibition of protein synthesis (28, 29), MMA-tet may prevent or retard the translocation of the A subunit, which is mediated by the retrotranslocation system (21, 30, 31). As described previously, MMA-tet binds to the Stx1 B subunit through both sites 1 and 3, possibly allowing site 2 to facilitate retrograde transport of the Stx1/MMA-tet complex to the ER. In addition, as previously demonstrated with PPP-tet (16), the Arg cluster present in MMA-tet may also contribute to the effective binding of the complex to target cells due to its cell-permeant nature (32, 33); conversely, in the absence of the peptides, any dysfunction of each receptor-binding site results in a marked reduction of the cell association of Stx1 (14, 27). This type of interaction of the complex with target cells, however, may impair signal transduction, affecting the subsequent retrotranslocation of the A subunit. Although the precise mechanism remains to be elucidated, some of the signal transduction pathways induced by the association of the Stx1 B subunit (34–38), such as the mitogen-activated protein kinase pathway, were found to be impaired in the presence of MMA-tet (Y. Takenaka unpublished data), further supporting this contention.
Finally, Ac-MMA-tet, a stable form of MMA-tet, was found to completely protect mice from a fatal dose of E. coli O157:H7 strain N-9, which produces both Stx1 and Stx2, even when administered after an established infection. This marked protective effect of Ac-MMA-tet may be attributed to its unique property to function in its target cells through its cell-permeant nature, broadening the window for active treatment. Thus, the approach of screening a multivalent peptide library for activity against the specific receptor-binding region of Stx1 successfully identified a peptide-based Stx neutralizer functioning against both Stx1 and Stx2 with remarkable therapeutic potency exceeding that of the previously identified Stx2 neutralizer PPP-tet.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Center for Global Health and Medicine, Japan (21S106); a grant from the MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2012-2016; grants from the Ministry of Education, Culture, Sports, Science and Technology, Japan (scientific research grants 10017944 and 24390035); and a grant from the Ministry of Health, Labor and Welfare, Japan (scientific research grant 10103178).
Footnotes
Published ahead of print 1 April 2013
REFERENCES
- 1. Karmali MA, Steele BT, Petric M, Lim C. 1983. Sporadic cases of haemolytic-uraemic syndrome associated with fecal cytotoxin and cytotoxin-producing Escherichia coli in stools. Lancet i:619–620 [DOI] [PubMed] [Google Scholar]
- 2. Riley LW, Remis RS, Helgerson SD, McGee HB, Wells JG, Davis BR, Hebert RJ, Olcott ES, Johnson LM, Hargrett NT, Blake PA, Cohen ML. 1983. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 308:681–685 [DOI] [PubMed] [Google Scholar]
- 3. O'Brien AD, Holmes RK. 1987. Shiga and Shiga-like toxins. Microbiol. Rev. 51:206–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paton JC, Paton AW. 1998. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin. Microbiol. Rev. 11:450–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scheutz F, Teel LD, Beutin L, Piérard D, Buvens G, Karch H, Mellmann A, Cprioli A, Tozzoli R, Morabito S, Strockbine NA, Melton-Celsa AR, Sanchez M, Persson S, O'Brien AD. 2012. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 50:2951–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karmali MA, Petric M, Lim C, Fleming PC, Arbus GS, Lior H. 1985. The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J. Infect. Dis. 151:775–782 [DOI] [PubMed] [Google Scholar]
- 7. Melton-Celsa AR, O'Brien AD. 1998. Structure, biology, and relative toxicity of Shiga toxin family members for cells and animals, p 121–128 In Kaper JB, O'Brien AD. (ed.), Escherichia coli O157:H7 and other Shiga toxin-producing E. coli strains. ASM Press, Washington, DC [Google Scholar]
- 8. Ling H, Boodhoo A, Hazes B, Cummings MD, Armstrong GD, Brunton JL, Read RJ. 1998. Structure of the Shiga-like toxin I B-pentamer complexed with an analogue of its receptor Gb3. Biochemistry 37:1777–1788 [DOI] [PubMed] [Google Scholar]
- 9. Fraser ME, Fujinaga M, Cherney MM, Melton-Celsa AR, Twiddy EM, O'Brien AD, James MNG. 2004. Structure of Shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J. Biol. Chem. 279:27511–27517 [DOI] [PubMed] [Google Scholar]
- 10. Kitov PI, Sadowska JM, Mulvey G, Armstrong GD, Ling H, Pannu NS, Read RJ, Bundle DR. 2000. Shiga-like toxins are neutralized by tailored multivalent carbohydrate ligands. Nature 403:669–672 [DOI] [PubMed] [Google Scholar]
- 11. Paton AW, Morona R, Paton JC. 2000. A new biological agent for treatment of Shiga toxigenic Escherichia coli infections and dysentery in humans. Nat. Med. 6:265–270 [DOI] [PubMed] [Google Scholar]
- 12. Nishikawa K, Matsuoka K, Kita E, Okabe N, Mizuguchi M, Hino K, Miyazawa S, Yamasaki C, Aoki J, Takashima S, Yamakawa Y, Nishijima M, Terunuma D, Kuzuhara H, Natori Y. 2002. A therapeutic agent with oriented carbohydrates for treatment of infections by Shiga toxin-producing Escherichia coli O157:H7. Proc. Natl. Acad. Sci. U. S. A. 99:7669–7674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mulvey GL, Marcato P, Kitov PI, Sadowska J, Bundle DR, Armstrong GD. 2003. Assessment in mice of the therapeutic potential of tailored, multivalent Shiga toxin carbohydrate ligands. J. Infect. Dis. 187:640–649 [DOI] [PubMed] [Google Scholar]
- 14. Nishikawa K, Matsuoka K, Watanabe M, Igai K, Hino K, Hatano K, Yamada A, Abe N, Terunuma D, Kuzuhara H, Natori Y. 2005. Identification of the optimal structure required for a Shiga toxin neutralizer with oriented carbohydrates to function in the circulation. J. Infect. Dis. 191:2097–2105 [DOI] [PubMed] [Google Scholar]
- 15. Watanabe M, Matsuoka K, Kita E, Igai K, Higashi N, Miyagawa A, Watanabe T, Yanoshita R, Samejima Y, Terunuma D, Natori Y, Nishikawa K. 2004. Oral therapeutic agents with highly clustered globotriose for treatment of Shiga toxigenic Escherichia coli infections. J. Infect. Dis. 189:360–368 [DOI] [PubMed] [Google Scholar]
- 16. Nishikawa K, Watanabe M, Kita E, Igai K, Omata K, Yaffe MB, Natori Y. 2006. A multivalent peptide library approach identifies a novel Shiga toxin inhibitor that induces aberrant cellular transport of the toxin. FASEB J. 20:2597–2599 [DOI] [PubMed] [Google Scholar]
- 17. Watanabe-Takahashi M, Sato T, Dohi T, Noguchi N, Kano F, Murata M, Hamabata T, Natori Y, Nishikawa K. 2010. An orally applicable Shiga toxin neutralizer functions in the intestine to inhibit the intracellular transport of the toxin. Infect. Immun. 78:177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ostroff SM, Tarr PI, Neill MA, Lewis JH, Hargrett-Bean N, Kobayashi JM. 1989. Toxin genotypes and plasmid profiles as determinants of systemic sequelae in Escherichia coli O157:H7 infections. J. Infect. Dis. 160:994–998 [DOI] [PubMed] [Google Scholar]
- 19. Tesh VL, Burris JA, Owens JW, Gordon VM, Wadolkowski EA, O'Brien AD, Samuel JE. 1993. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 61:3392–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stearns-Kurosawa DJ, Collins V, Freeman S, Debord D, Nishikawa K, Oh SY, Leibowitz CS, Kurosawa S. 2011. Rescue from lethal Shiga toxin 2-induced renal failure with a cell-permeable peptide. Pediatr. Nephrol. 26:2031–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johannes L, Römer W. 2010. Shiga toxins—from cell biology to biomedical applications. Nat. Rev. Microbiol. 8:105–116 [DOI] [PubMed] [Google Scholar]
- 22. Spooner RA, Watson P, Smith DC, Boal F, Amessou M, Johannes L, Clarkson GJ, Lord JM, Stephens DJ, Roberts LM. 2008. The secretion inhibitor Exo2 perturbs trafficking of Shiga toxin between endosomes and the trans-Golgi network. Biochem. J. 414:471–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stechmann B, Bai SK, Gobbo E, Lopez R, Merer G, Pinchard S, Panigai L, Tenza D, Raposo G, Beaumelle B, Sauvaire D, Gillet D, Johannes L, Barbier J. 2010. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 141:231–242 [DOI] [PubMed] [Google Scholar]
- 24. Mukhopadhyay S, Linstedt AD. 2012. Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science 335:332–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamasaki C, Nishikawa K, Zeng XT, Katayama Y, Natori Y, Komatsu N, Oda T, Natori Y. 2004. Induction of cytokines by toxins that have an identical RNA N-glycosidase activity: Shiga toxin, ricin, and modeccin. Biochim. Biophys. Acta 1671:44–50 [DOI] [PubMed] [Google Scholar]
- 26. Kurioka T, Yunou Y, Kita E. 1998. Enhancement of susceptibility to Shiga toxin-producing Escherichia coli O157:H7 by protein calorie malnutrition in mice. Infect. Immun. 66:1726–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Soltyk AM, MacKenzie CR, Wolski VM, Hirama T, Kitov PI, Bundle DR, Brunton JL. 2002. A mutational analysis of the globotriaosylceramide-binding sites of verotoxin VT1. J. Biol. Chem. 277:5351–5359 [DOI] [PubMed] [Google Scholar]
- 28. LaPointe P, Wei X, Gariépy J. 2005. A role for the protease-sensitive loop region of Shiga-like toxin 1 in the retrotranslocation of its A1 domain from the endoplasmic reticulum lumen. J. Biol. Chem. 280:23310–23318 [DOI] [PubMed] [Google Scholar]
- 29. Tam PJ, Lingwood CA. 2007. Membrane cytosolic translocation of verotoxin A1 subunit in target cells. Microbiology 153:2700–2710 [DOI] [PubMed] [Google Scholar]
- 30. Tsai B, Ye Y, Rapoport TA. 2002. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 3:246–255 [DOI] [PubMed] [Google Scholar]
- 31. Li S, Spooner RA, Hampton RY, Lord JM, Roberts LM. 2012. Cytosolic entry of Shiga-like toxin A chain from the yeast endoplasmic reticulum requires catalytically active Hrd1p. PLoS One 7:e41119 doi:10.1371/journal.pone.0041119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Derossi D, Chassaing G, Prochiantz A. 1998. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 8:84–87 [PubMed] [Google Scholar]
- 33. Wadia JS, Dowdy SF. 2002. Protein transduction technology. Curr. Opin. Biotechnol. 13:52–56 [DOI] [PubMed] [Google Scholar]
- 34. Katagiri YU, Mori T, Nakajima H, Katagiri C, Taguchi T, Takeda T, Kiyokawa N, Fujimoto J. 1999. Activation of Src family kinase Yes induced by Shiga toxin binding to globotriaosyl ceramide (Gb3/CD77) in low density, detergent-insoluble microdomains. J. Biol. Chem. 274:35278–35282 [DOI] [PubMed] [Google Scholar]
- 35. Mori T, Kiyokawa N, Katagiri YU, Taguchi T, Suzuki T, Sekino T, Sato N, Ohmi K, Nakajima H, Takeda T, Fujimoto J. 2000. Globotriaosyl ceramide (CD77/Gb3) in the glycolipid-enriched membrane domain participates in B-cell receptor-mediated apoptosis by regulating Lyn kinase activity in human B cells. Exp. Hematol. 28:1260–1268 [DOI] [PubMed] [Google Scholar]
- 36. Lauvrak SU, Wälchli S, Iversen TG, Slagsvold HH, Torgersen ML, Spilsberg B, Sandvig K. 2006. Shiga toxin regulates its entry in a Syk-dependent manner. Mol. Biol. Cell 17:1096–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Torgersen ML, Wälchli S, Grimmer S, Skånland SS, Sandvig K. 2007. Protein kinase Cδ is activated by Shiga toxin and regulates its transport. J. Biol. Chem. 282:16317–16328 [DOI] [PubMed] [Google Scholar]
- 38. Tesh VL. 2012. Activation of cell stress response pathways by Shiga toxins. Cell. Microbiol. 14:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]